
Abstract
Cell competition describes the process in which cells of greater fitness are capable of sensing and instructing elimination of lesser fit mutant cells. Since its discovery in Drosophila, cell competition has been established as a critical regulator of organismal development, homeostasis, and disease progression. It is therefore unsurprising that stem cells (SCs), which are central to these processes, harness cell competition to remove aberrant cells and preserve tissue integrity. Here, we describe pioneering studies of cell competition across a variety of cellular contexts and organisms, with the ultimate goal of better understanding competition in mammalian SCs. Furthermore, we explore the modes through which SC competition takes place and how this facilitates normal cellular function or contributes to pathological states. Finally, we discuss how understanding of this critical phenomenon will enable targeting of SC-driven processes, including regeneration and tumor progression.

Similar content being viewed by others
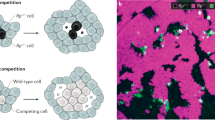

Introduction
Cell death is one of the most critical processes regulating tissue physiology [1,2,3,4,5,6,7]. Through unveiling the modes in which cells undergo death within their environments, it has become evident that much of this is dependent upon relative fitness— what deems a cell fit in one environment may instigate its elimination in another [4, 8, 9]. Pioneering work investigating how cells of varying fitness differentially contribute to the adult organism resulted in the discovery of cell competition— the sensing and active elimination of relatively unfit cells by superior neighbors within a population [8] (Fig. 1). This phenomenon, which is distinct from passive clonal fitness selection, was first described in the Drosophila imaginal wing disc, a larval epithelial structure that segregates during embryogenesis and undergoes proliferation and differentiation to give rise to the adult wing [10]. Evaluation of genetic mosaics revealed that cells bearing mutations in Minute (M) genes, which encode ribosomal proteins (Rp) [11], exhibit decreased proliferation, developmental delays [11], and do not persist into the adult, instead being selectively eliminated by wild type (WT) counterparts [8]. Interestingly, while M homozygosity results in lethality, heterozygous animals (M/+) remain viable [12], highlighting the role of cell competition in preserving tissue integrity by selecting against aberrant cells that would otherwise contribute to the adult organism [12].

a Depiction of classical cell competition; a wild type (WT) cell senses relatively impaired fitness in a neighboring mutant cell and behaves as a “winner” by inducing apoptotic elimination of the mutant cell. On a population scale (bottom panel), this promotes competitive removal of aberrant cells followed by WT population expansion to compensate for the eliminated cells and maintain tissue integrity. Example depicts Minute (M) cells actively eliminating M+/− cells. b Depiction of super competition; one or more mutations confer enhanced fitness to a cell and enable it to behave as a “winner” by inducing apoptotic elimination of an otherwise viable WT cell. On a population scale (bottom panel), this promotes clonal expansion of aberrant “winner” cells, characteristic of tumorigenesis. Example depicts mutant cells with elevated levels of Myc actively eliminating WT cells with lower Myc expression.
Competitive elimination of M mutants was later revealed to be mediated by Brk elevation and subsequent c-Jun amino-terminal kinase (JNK) pathway activation [13]. As Brk inhibits the pro-survival and proliferative Dpp signal, M mutants were proposed to exhibit slower proliferation and Brk upregulation due to diminished responsiveness to Dpp [13]. This inspired examination as to the effects of proliferation-enhancing Myc mutations [14, 15] and exposed another side of cell competition, or “super competition [14],” in which mutant cells bestowed with enhanced fitness eliminate otherwise healthy WT neighbors [14, 15]. Interestingly, although high-expressing Myc cells over-proliferated, competitive elimination of WT cells prevented tissue abnormalities [14, 15], suggesting a role for competition in regulating organ size [15].
Together, these seminal works in Drosophila established the foundation for elucidating the physiological roles of cell competition and uncovering molecular mechanisms governing this phenomenon [8, 9, 13, 14]. A large body of research has since implicated cell competition in processes including development, homeostasis, and tumorigenesis [4, 5], with much of it similarly conducted in Drosophila due its amenability to physical and genetic manipulation as well as regenerative capacity [16]. For a comprehensive overview of these studies, we refer the reader to the following reviews [4, 5, 17]. Here, we evaluate the ways in which cell competition shapes cellular cooperation across various organisms and physiological contexts, placing greater emphasis on mammalian SC-dependent processes (Table 1 and Fig. 2).

a Elimination through apoptotic induction of loser cell by winner cell. b Elimination through induced necroptosis of loser cell by winner cell. c Elimination through induced senescence of loser cell by winner cell. d Elimination through induced differentiation of loser cell by winner cell. e Elimination through mechanical extrusion of loser cell apically by winner cells (extruded cells may subsequently undergo apoptosis, necroptosis, or differentiation). Curved arrows represent mechanical tension. f Elimination through entosis and subsequent autophagy of loser cell by engulfing entotic winner cell. g Elimination through phagocytosis of loser cell by engulfing phagocytic winner cell.
Stem cell competition in development
Stem cells (SCs) possess the unique capacity for self-renewal and differentiation [18]. The degree to which a SC can give rise to various cell types within an organism, or potency, is a primary feature distinguishing different SCs [18]. While resident adult SCs exhibit uni- or multipotency, contributing to their respective tissues in a lineage-restricted manner, embryonic SCs are pluripotent, differentiating into all three germ layers comprising an embryo, as suggestive of their name [19]. Collectively, SCs bear the responsibility of ensuring normal development and tissue establishment, upon which organismal function relies. Protection of SC integrity is therefore critical for these processes and regulated by various intrinsic and extrinsic factors, which converge to promote cellular coordination.
Unsurprisingly, cell competition has been established as an important facilitator in maintaining SC integrity during embryogenesis and tissue development, eliminating cells rendered unfit for further contribution [4]. As such, changes in factors regulating proliferation, SC potency, metabolism, and cellular stress can provoke competition [4]. One such factor is the tumor suppressor p53, which functions to trigger intrinsic cell death under stress-inducing conditions such as DNA damage and cell cycle regulation [20, 21]. This was evidenced in tetraploid (4n) cells within mouse embryo chimeras, which were removed by apoptosis following gastrulation [22, 23]. Using co-cultures, this competitive removal was later found to be p53-dependent—as 4n cells exhibiting increased p53 levels were eliminated by 2n cells with relatively lower levels, while knockdown of p53 reversed this phenotype [24]. Corroborating this, Bmpr1a-mutant cells underwent apoptotic elimination by WT cells in mouse embryos and co-cultures as a result of elevated p53 and subsequent inhibition of mTOR signaling [25], with mutations in the negative regulators of p53 also disadvantageous to cells [26]. These findings suggest that in addition to engaging intrinsic cellular checks, changes in p53 expression are harnessed for population fitness sensing and cell competition to help ensure normal embryogenesis [26]. Furthermore, a genetic knockdown screen striving to identify genes advantageous during embryonic development revealed that cells with p53 downregulation can displace WT cells when co-injected into blastocysts and under differentiation conditions in vitro [27]. Intriguingly, this did not result in any apparent negative consequences to the organism, indicating that cellular cooperation can be achieved through competition to facilitate successful organismal development [27].
Consistent with the role of differential Myc in driving competition in Drosophila [14, 15], decreased Myc levels resulting from impaired BMP-signaling in mouse embryonic SCs (ESCs) prompted elimination through factors secreted by WT cells [28]. Given the importance of Myc for maintaining pluripotency and driving proliferation in early developmental stages, such competitive elimination may also serve as a safeguard by removing cells with lower Myc and defective proliferation arising from mutations acquired during cell division [28]. In agreement, examination of mouse epiblasts found that mosaicism arising from differential Myc expression results in apoptotic elimination of cells with lower levels of Myc when in the vicinity of higher-expressing cells [29]. As Myc was found to function downstream of TEAD1 and YAP to maintain pluripotency in the epiblast [30], this enabled removal of unspecified cells of lesser potency to guard against premature differentiation [30, 31]. Similar findings were made in the developing mouse epidermis, in which slow-dividing progenitors exhibiting lower Myc expression were eliminated via apoptosis and engulfed by faster proliferating neighbors, which was proposed to play a critical morphogenetic role in normal skin development and function [32]. As phagocytic engulfment is also employed by basal epithelial cells to clear dying cells during hair follicle regression [33], it would be interesting to investigate whether this process is mediated through Myc-dependent cell competition or another means, ultimately enabling the retention of a select pool of SCs with optimal fitness.
Reflecting overall cellular status and fitness, mitochondrial function has also been associated with competition during development [34]. This can be observed in the elimination of 35% of epiblast cells prior to gastrulation [34]. Using single-cell transcriptional profiling, cells eliminated across this period in embryogenesis exhibited molecular changes reflecting defects in mitochondrial function [34]. Furthermore, introduction of non-pathological changes to mitochondrial function were sufficient to trigger competition, indicating that cell competition is critical in ensuring optimal metabolic fitness early in development [34]. A role for oncogenic RasV12, which has been demonstrated to increase mitochondrial metabolism, has also been unveiled while investigating whether cell competition contributes to the developing mammalian nervous system [35]. In neuroepithelial co-cultures of WT and RasV12 neural progenitors, mutant cells were eliminated by WT neighbors through apoptotic induction followed by phagocytosis [36]. Notably, the cell-competition conditions suppressed juvenescence markers in RasV12 cells, which displayed reduced proliferative potential and increased senescence. Investigation of whether this similarly promotes competition between neuronal SCs in vivo can therefore be valuable for understanding neurodevelopmental abnormalities.
Cellular changes resulting in proteotoxic, endoplasmic reticulum (ER) and oxidative stress have also been identified as underlying causes of cell elimination across distinct loser genotypes in Drosophila development [37,38,39,40,41,42,43,44,45,46]. This is evident in transcriptional profiling, which revealed activation of oxidative stress across various loser genotypes, with Nrf2-dependent activation of oxidative stress pathways sufficient for eliciting loser status [37]. Defective protein translation in cells mutant for Hel25a, an mRNA splicing and nuclear export regulator, was also reported to underlay competition-induced autophagy and subsequent apoptosis bordering WT cells via NFκB and JNK signaling [38]. However, other studies instead reported proteotoxic stress as the driver for loser status, with autophagy conferring cytoprotective effects [39, 40]. Additional work reported that proteotoxic stress converges through expression of Xrp1 [41], a transcription factor also necessary for Minute-induced competition [42, 43], causing eIF2α phosphorylation and decreased cellular fitness [41, 44, 45]. Xrp1 activation itself can thereby induce proteotoxic stress, partaking in a feed-forward loop that triggers the oxidative stress response and confers loser status [41]. Furthermore, ER stress and mutations in Hel25a and Rp were found to drive competition and alter protein synthesis through this manner [44], although downstream translational changes alone were insufficient for inducing competition in the absence of Xrp1 and subsequent eIF2α phosphorylation [44, 45]. The importance of eIF2 in competition is also evidenced in cell elimination triggered by abnormal eIF2γ or Rp gene dosage as a result of aneuploidy, thereby illustrating a critical role in preventing developmental abnormalities [46].
Describing another consequential role for cell competition, work in Drosophila testes revealed that spermatogonial SCs with a fitness-enhancing mutation in chinmo can actively evict WT SCs from the niche and thereby cause gene drives [47]. Through ectopic secretion of Pcan, mutant SCs actively remodeled their surrounding extracellular matrix (ECM) and upregulated ECM-binding proteins, selectively removing WT SCs [47]. Therefore, despite parental heterogeneity (chinmo+/-), a majority of Drosophila progeny inherited a chinmo−/− genotype [47]. As declining chinmo levels promoted aging, this provides an interesting molecular insight as to how aberrant SC competition benefiting individual cellular fitness may potentially be disadvantageous to long-term organismal fitness. Expanding upon these critical insights, contribution of similar physiological and environmental factors to cell competition in mammalian development can be investigated.
Collectively, these studies exemplify the versatile functions of cell competition during development—from enabling resolution of cellular disturbances in proliferation, stress, and metabolism and maintaining SC potency to remodeling of the cellular environment and driving organismal inheritance.
Stem cell competition in cellular and tissue maintenance
To fulfill their role in tissue maintenance and disease prevention, adult SCs employ cell competition in response to various environmental cues [3,4,5, 17]. As such, various factors have been identified in enabling SCs to facilitate competition within these contexts.
Similarly to development, differential p53 expression can serve as an environmental stressor sensor to maintain homeostasis [48,49,50,51,52]. This role was observed in mouse bone marrow chimeras, in which transplantation of p53-deficient hematopoietic cells conferred selective advantage to these cells only when subjected to post-irradiation stress [48]. In contrast, non-irradiated p53-deficient cells were outcompeted by WT cells during co-transplantation, thereby preventing the clonal expansion of mutants under normal conditions [48]. Complementing these findings, cells expressing elevated p53 in response to irradiation-induced stress underwent competition that resulted in a loser phenotype of cellular senescence [49]. These findings illustrate the contextual manner in which p53 promotes cell competition to benefit organismal fitness. p53 has further been implicated in promoting modes of cell competition independent of apoptotic induction [50,51,52]. This is evident in epithelial cells with p53 mutation, which underwent necroptosis followed by mechanical extrusion [50]. However, as subsequent mutation in Ras instead resulted in accumulation of epithelial mutants, this explains the occurrence of a mutational order during disease progression and suggests that this may be mediated by cell competition [50]. Similarly, in epithelial cells silenced for Scribble (Scr), a tumor suppressor regulating cellular polarity and adhesion, elevation of p38-MAPK signaling resulted in apoptosis induced by surrounding WT cells [51]. Later investigations correlated these findings with elevated p53 levels in mutant cells, which caused hypersensitivity to compaction through cellular crowding [52]. Sensing mechanical stress, mutant cells activated Rho-associated kinase (ROCK) and p38 signaling, further increasing p53 levels and ultimately triggering mechanical extrusion by surrounding WT cells [52].
To preserve tissue integrity within the mammalian epidermis, SCs additionally rely on cell competition. Proliferating SCs of the basal epidermis undergo either symmetrical division, yielding two SCs that are retained basally, or asymmetrical division, yielding one SC and one suprabasal cell that differentiates as it gradually migrates upwards through the epidermis [53, 54]. Elimination of damaged SCs and retention of SCs with greater fitness within the basal layer therefore helps preserve skin integrity [53]. Studies of epidermal SC fate after induction of double-stranded DNA breaks uncovered yet another alternative role for p53 in cell competition [55]. DNA damage and concomitant activation of p53 resulted in downstream p21 and Notch signaling, which together regulate cellular differentiation and adhesion to neighboring cells [55]. While damaged SCs underwent asymmetric division, thereby differentiating out of the niche via relative loss of integrin−β1, undamaged SCs continued contributing to the niche and underwent clonal expansion to protect tissue integrity from aberrant mutations [55]. Similarly, higher expression of the hemidesmosome component collagen 17 (Col17a1) enabled epidermal SCs to remain attached and continue dividing symmetrically in the basal epidermis. However, stressed or damaged SCs gradually lost Col17a1 expression and detached, or were induced to differentiate through asymmetric division by outcompeting clones with higher Col17a1 levels [56]. Showing promising translational application, interference through forced maintenance of Col17a1 prevented competition and reversed aging, which otherwise occurred due to Col17a1 loss and SC exhaustion over time [56].
Regulatory changes altering the cellular state have additionally been correlated with competition during cellular homeostasis in vitro [57]. Investigation of immortalized mammalian cell lines has shown that unique clones stochastically arising in cell cultures can actively eliminate each other under co-culture conditions due to factors such as oxygen availability and metabolism, proliferation, and protein but not RNA synthesis [57]. Highlighting the contextual dependency of cell competition, clones behaving as “winners” in one co-culture combination could become “losers” in another. To investigate whether relative levels of cellular fitness can therefore be a reflection of a gain or loss of information within cells, cell fusion experiments generating heterokaryons composed of “winner” and “loser” cell combinations were performed [57]. Interestingly, the outcomes in heterokaryon behavior varied from winner to loser or non-competitor status, suggesting that cellular fitness depends on the integration of various factors within the cell [57]. Continuous sensing of cellular changes with respect to the environment can thereby guide competition to promote a fitness standard throughout the local cellular community.
Stem cell competition in disease prevention
In conjunction with its homeostatic role, cell competition has unsurprisingly been implicated in prevention of tumorigenesis [58,59,60,61,62,63,64,65]. In a diethylnitrosamine (DEN)-induced tumor model of the esophageal epithelium, encircling of early tumors by Notch1-mutant cells encouraged tumor elimination [58]. As most nascent tumors regressed, deep sequencing attributed this to the distinct mutational landscape of early tumors, amongst which Notch1 mutation was prevalent [58]. Thus, early fitness pressure resulting in positive competitive selection of these mutants ultimately enabled tumor cell elimination [58]. This underlines a cooperative nature of cell competition, enabling alteration of the genetic landscape for ultimate preservation of tissue integrity.
Such cellular coordination during competition has also been observed in other tissues. In mouse hair follicles and skin, hypertrophy caused by activated β-catenin or HrasV12 was found to regress in chimeras, in which cells expressing mutant genes competed with WT counterparts [59]. Remarkably, although the skin expectedly exhibited hyperproliferation and developed abnormal growths, mutant cells at the core of the aberrant growths became fully surrounded and expelled from the tissue by surrounding WT cells, and disruption of Wnt ligand production by mutant cells prevented WT cells from encapsulating mutants [59]. Comparably, a tumor-suppressive role of cell competition was found when investigating WT hepatocytes surrounding Notch1-Akt activation driven tumors in the mouse liver [60]. While endogenous or hyperactivation of Hippo signaling in surrounding WT hepatocytes drove regression of tumors, inhibition of the Hippo pathway effectors YAP/TAZ in WT or activation in accelerated tumor growth [60]. Interestingly, tumor cells required YAP/TAZ activation to survive when surrounded by WT but not YAP/TAZ deficient hepatocytes [60]. Furthermore, although endogenous signaling was not observed around liver tumors generated through grafting in a metastatic liver tumor model, induction of YAP/TAZ signaling in peripheral hepatocytes remarkably attenuated tumor load [60]. These results emphasize the dependency of tumor prevention on the plasticity of cells in sensing and responding to relative differences within the environment.
Stressing this notion, cell competitiveness in the thymus was shown to facilitate the substitution of old resident progenitors with young bone marrow derived progenitors, with disruption of this process resulting in a phenotype resembling T-lineage acute lymphoblastic leukemias (T-ALL) [61]. Despite being genetically identical, cells underwent competition as a result of distinct gene expression patterns in old and young progenitors, with old progenitors exhibiting reduced Bcl2 expression correlated with impaired IL-7r signaling [61]. The occurrence of cell competition has also indirectly been revealed in the form of genetic mosaicism arising in somatic cells, as well as the prevalence of mutant clones within tissues over time [62,63,64,65]. This has been observed in single clones bearing specific advantageous mutations that still retain normal function have been found in the aging hematopoietic system [62], skin and esophagus [63, 64], as well as various other tissues [65].
Representing another specific form of tumor prevention, the process of epithelial defense against cancer (EDAC) is highly reliant upon cell competition [66]. EDAC describes the sensing of fitness differences amongst epithelial cells, resulting in cytoskeletal changes and mechanical extrusion of mutant cells by WT neighbors [66, 67]. This has been extensively modeled using the Madin–Darby canine kidney (MDCK) epithelial cell line, which can form an epithelial layer [68]. Employing this model, various gene mutations were identified in provoking EDAC-associated mechanical extrusion, including RasV12 [69], v-SRC [70], ERBB2 [71], constitutively active YAP [72], as well as the binding partner of the tumor suppressor gene Lgl, MAHJ [73]. While the underlying molecular changes facilitating EDAC for each of these cases remain unclear, apoptosis of Mahj silenced cells was found to be induced as a result of c-Jun N-terminal kinase (JNK) activation, implicating loser JNK signaling in mechanical cell competition [73]. Moreover, investigation of EDAC arising from RasV12 revealed that WT cells surrounding mutants accrue filamin, thereby inducing transformed cells to accumulate the actin-binding protein Eplin [74]. These changes in turn stimulate protein kinase A (PKA) and myosin II activity, which together enable competitive elimination of mutants via mechanical extrusion [67, 74, 75]. Examining the underlying basis for the cytoskeletal and cellular state alterations in RasV12-EDAC, surrounded mutant cells were found to exhibit metabolic changes including increased glucose uptake, Eplin accumulation, and secretion of lactate, ultimately provoking their elimination via apical extrusion [76]. Apical extrusion of RasV12 mutants by WT cells was similarly observed in intestinal organoids [76] and mouse pancreas [77]. Moreover, RasV12 mutants were found to exhibit aberrant lysosomal processing and defective autophagic flux, although complete abolishment of autophagic activity prevented apical extrusion of mutants by WT cells [77]. Further studies should determine whether these molecular features pertain specifically to RasV12-driven EDAC, or whether other genetic alterations converge to facilitate mechanical competition during EDAC in a similar manner.
Collectively, these examples underscore the critical regulatory role of SC competition in homeostasis and tumor prevention, whether it is mediated through apoptosis, induced differentiation, or mechanical extrusion.
Cell competition and tumorigenesis
Given the role of cell competition in ensuring normal development and homeostasis, it is critical to consider how dysregulation of this tumor-suppressive mechanism contributes to disease progression. As cell competition is utilized to maintain tissue architecture and integrity, this implies that aberrant cells must overcome competitive elimination to facilitate disease progression.
Such behavior is exemplified during super-competitive removal of viable WT cells by superfit mutant neighbors [14, 15]. In addition to Myc, many super-competitive genotypes have since been observed in Drosophila, including mutations in tumor suppressor genes of the Hippo pathway, or cells overexpressing the YAP/TAZ homolog Yki [78]. Validating super competition in a mammalian system, co-culture of WT or TEAD-overexpressing mouse embryonic fibroblasts resulted in WT cell elimination, as TEAD activity directly resulted in elevated Myc expression [79]. This Myc-driven super competition is also consistent with findings in which cells with attenuated Myc expression were eliminated by WT neighbors [28, 29, 32]. Inspecting human tumor contexts, Myc-upregulated cells were continuously found adjacent to apoptotic cells within the tumor parenchyma and at the tumor-stroma interface [80]. Strikingly, co-cultures pairing various cancer cell lines of distinct genetic backgrounds and differential Myc expression exhibited super competition, which was abrogated upon Myc inhibition [80]. This suggests that Myc-driven super competition does not necessarily result from Myc mutations, but rather, Myc expression serves as a cellular fitness state readout of upstream molecular changes.
Competition arising from abnormal proliferation has also been observed in human pluripotent SC (hPSC) cultures, where faster growing cells that have acquired genetic abnormalities could outcompete WT cells [81]. In this case, elimination occurred as a result of enclosure and mechanical compression of loser cells by WT cells [81]. Investigating the molecular mechanism, this compression was found to be facilitated by a redistribution of F-actin, causing WT cells to sequester yes-associated protein (YAP) in the cytoplasm and undergo apoptosis, while neighboring mutant cells are able to retain nuclear YAP and remaining proliferative [81]. As tumor cells typically exhibit greater proliferative capacity, it is relevant to evaluate whether these changes promote competition during tumorigenesis in vivo.
Additional molecular factors underlying aberrant cell competition, have been elucidated using tumor samples and 3D models of tumorigenesis. To investigate whether active cell competition can help account for the prevalence of APC mutants in human colorectal cancers, WT and APC−/− intestinal organoid co-cultures were established [82]. As APC associates with other proteins to form the “destruction complex” that binds and targets β-catenin for destruction, absence of APC promotes nuclear translocation of β-catenin and expression of Wnt [83]. Remarkably, APC mutants not only exhibited elevated Wnt levels but actively outcompeted WT cells by secreting Wnt antagonists that selectively induced differentiation of WT cells [82]. This was corroborated in vivo as Apc−/− mice treated with lithium chloride, which desensitizes WT cells to the Wnt antagonists, prevented mutant cell expansion and adenoma formation [82]. Alteration of signals in the environment therefore exposes an alternative way in which tumor cells drive super competition. Furthermore, in intestinal organoids comprised of WT and cancer cells of various genetic backgrounds, tumorigenic cells eliminated WT neighbors by inducing apoptosis via JNK signaling, resulting in the loss of stemness [84]. However, treatment with stemness-promoting factors increased fitness and prevented elimination of WT cells [84]. Additionally, physiological changes causing dysregulation of lipid metabolism and chronic inflammation have been implicated in competition-dependent tumorigenesis [85]. In mice with low-induction of RasV12 mutations, administration of a high fat diet (HFD) enabled mutants to evade competitive apical extrusion by WT neighbors, thereby resulting in small intestinal and pancreatic epithelial lesions [85].
On a tissue scale, changes promoting competitive clonal expansion can result in “field cancerization,” in which early mutational changes lacking immediate morphological alterations can prime the tissue landscape towards future tumor initiation [86, 87]. Previously, we mentioned differentiation-induced elimination of stressed epithelial cells through Notch [55], whose tumor suppressor function is also frequently disrupted in many squamous tumors [88]. Investigating the esophageal epithelium, depletion of Notch in a subset of cells similarly promoted differentiation of adjacent WT cells after division [89]. Although these mutant cells were able to reestablish homeostasis after eventually replacing WT cells throughout the epithelium, this laid the foundation for future transformation, as exposure to additional chemical mutagens or oncogenic mutations significantly accelerated tumor development [89]. This additionally illustrates how through cell competition, mutational order may culminate in field changes of diverse consequences.
The role of clonal expansion in tumor initiation was further investigated by observing in vivo dynamics between WT and mutant intestinal SCs [90], which undergo neutral competition during homeostasis [91]. Interestingly, although Kras- and Apc-mutant SCs displayed a distinct clonal advantage, a majority of these cells were replaced by WT SCs over time and prevented from clonally overtaking or “fixing” their respective crypt [90]. However, aberrant clonal fixation occurred at greater frequency with an increase in the number of SCs bearing a mutational hit in Apc [90]. Furthermore, Tp53 mutants that failed to clonally expand under normal conditions exhibited a striking competitive advantage under conditions recapitulating chronic colitis [90]. This illuminates a critical role for tissue architecture and physiological context during cell competition, rather than individual cell status, in shaping tissue fate. Additional work has also demonstrated the potential of Kras-mutant SCs in small intestine tumor initiation, further revealing that clonally-fixed mutant crypts exhibit enhanced crypt fission [92]. As fission results in the establishment of two crypts from one, this work helps elucidate how clonal expansion facilitated by SC competition can promote field change within the tissue and in turn, tumorigenesis [92].
Molecular fitness fingerprints of super competition have also been detected in various isoforms of the calcium channel gene Flower (Fwe), and found to play a critical role in preventing developmental abnormalities, delaying aging, and promoting regeneration of tissues through cell competition in Drosophila [93,94,95,96]. Cells expressing two of the Fwe isoforms (2 and 4) convey a fitness advantage when in the presence of cells expressing the other two isoforms (1 and 3) [93]. Examining whether human Fwe (hFwe) isoform expression can be detected in mammalian cancers, benign and malignant tumors from breast and colon cancer were found to exhibit winning isoform combinations, whereas the surrounding stromal cells expressed the losing isoform combinations [97]. Recapitulation of loser-associated isoform expression in breast tumor cells resulted in increased tumor volume upon grafting into mice, whereas in colon and prostate tumor xenografts, silencing all Fwe isoforms reduced tumor growth and metastasis [97]. Further research can therefore elucidate how these tumor-promoting expression patterns arise as well as their association with super competition.
Alongside the elimination of healthy target cells through induction of apoptosis, differentiation, or mechanical extrusion, another fascinating mode of cell competition has been reported [4, 5, 17, 98] (Figs. 2 and 3). Through entosis, live epithelial cells are first internalized and subsequently degraded by neighboring cells [98]. Prior to this, cell competition arising from Minute mutations in Drosophila imaginal discs was reported to be dependent on the ability of WT cells to engulf loser cells upon their death [99]. This was further suggested to be reliant on the activity of the engulfment genes draper and wasp, rac1, mbc, and the phosphatidylserine receptor [99]. However, later work in Drosophila reported these genes to be dispensable to winner cell status, with a majority of engulfment being performed by hemocytes following loser cell extrusion, rather than WT winner cells [100]. Despite conflicting evidence, these data were ultimately followed by investigation of whether engulfment-dependent cell competition can account for the “cell-in-cell” structures detected in some human tumors [98, 101]. Using mammary epithelial cells known to possess phagocytic properties, the engulfment of viable but matrix-detached cells followed by lysosomal digestion, termed entosis, was unveiled [98]. This process is distinct from phagocytosis, as cells are cleared independently of apoptotic activation and exposure of phosphatidylserine [102]. Furthermore, this cellular invasion was found to be driven by Rho and ROCK activity, with myosin II-associated differences in contractile force at cellular adherens junctions causing compaction of one cell into its neighbor [98]. Validating these findings, cell cannibalism in human tumors was later found to result from actomyosin and RhoA-dependent differences in mechanical deformability, as tumor cells preferentially engulfed neighbors with relatively low deformability [103]. Furthermore, in these winner tumor cells, activation of Kras and Rac signaling resulted in downregulation of myosin, thereby allowing internalization of less fit neighboring cells. Intervening with this process, exogenous expression of epithelial cadherins in human breast tumor cells induced entosis, thereby preventing tumor growth [103]. This corresponded to variability in the distribution of Rho activity within entotic cells, with inhibition of this leading to a reduction in entosis and an increase in growth of tumor cells that were provided with exogenous cadherins [103]. Interestingly, the occurrence of entosis itself has been found to have distinct consequences for tumorigenesis in a p53-dependent manner [104, 105]. Following engulfment, host cells undergoing mitosis often exhibited aberrant division resulting in aneuploidy or other forms of DNA damage [104, 105]. However, while p53 null cells underwent cell death as a result of this damage, cells bearing mutant p53 endured and exhibited genomic instability that promoted tumorigenesis upon xenografic transplantation in vivo [104, 105]. This indicates that entosis can serve as a tumor preventive mechanism in normal cells, yet catalyze tumor progression when occurring in cells that have undergone transformation [104].

a Mechanical forces (gray) from winner (purple) cells can result in loser (blue) cell elimination through apical extrusion followed by apoptosis, necroptosis, or differentiation. b Membrane bound ligand (red) and receptor interactions between winner-loser cells can result in loser cell elimination through either apoptosis, necroptosis, differentiation, or senescence. c Secreted signals from winner cell bind receptors on loser cell, which can result in loser cell elimination through apoptosis, necroptosis, differentiation, or senescence. d Mechanical forces (gray) from winner cell and diminished adhesion (maroon) in loser cell can result in asymmetric division and loser cell elimination through differentiation. e Mechanical forces (gray) from winner cell can result in loser cell compaction and elimination through entosis.
Further unraveling of the molecular, cellular, and cooperative nature through which cells exploit competition will illuminate novel perspectives into tumorigenesis. Coupled with this, manipulation of the molecular players and factors in the surrounding environment that render tumor cells as super competitors can in turn enable development of approaches for preventing and intervening with tumorigenesis.
Integrating signals
As cells rely on intricate communication within their networks to respond to extrinsic signals, it is important to consider the non-autonomous ramifications of cell elimination. The biological response of a cell in consequence to events occurring within adjacent cells is referred to as the bystander effect [106, 107]. This was used to describe the ability of irradiated cells to induce apoptosis of adjacent unirradiated cells, thereby reflecting an amplification of signals within the environment (Fig. 4). Since a majority of loser cells are eliminated through apoptotic activation during cell competition, it is expected that this bears repercussions within the surrounding environment. Dying cells have previously been shown to secrete factors that can instruct processes such as proliferation and apoptosis in the neighboring environment [1, 108]. Understanding how cellular environments integrate processes such as apoptosis-induced compensatory proliferation and apoptosis-induced apoptosis with cell competition can therefore shed additional light onto mechanisms of tissue integrity maintenance and disease progression.

a Apoptosis-induced apoptosis; cells receiving lethal doses of ionizing radiation (blue: affected cells) undergo apoptosis and release death-promoting (red) signals to unirradiated neighbors (pink: bystander cells), thereby causing them to undergo apoptosis. b Apoptosis-induced compensatory proliferation; cells stimulated by death-promoting signals (blue: affected cells) undergo apoptosis and release growth-promoting (yellow) signals to non-stimulated neighbors (pink; bystander cells), thereby causing them to proliferate. c Non-autonomous induction of differentiation; cells stimulated by differentiation-promoting signals (blue; affected cells) undergo changes in cell fate and release differentiation-promoting (green) signals to non-stimulated neighbors (pink; bystander cells), thereby inducing differentiation of these cells to the same fate.
Upon elimination of aberrant cells, neighboring cells can take over the available space through “compensatory” proliferation, a process discovered in the wing disc of Drosophila [109]. This was found to occur in response to mitogenic cues such as Wnt and TGF-β homologs, which are secreted by dying loser cells and stimulate proliferation of adjacent cells in the environment [110,111,112]. This phenomenon was also observed in the Drosophila follicular epithelium, where competitive elimination of aberrant cells triggered local cellular hypertrophy for repair of tissue loss in an insulin growth factor (IGF)-dependent manner [113]. Interestingly, as these processes occurred in post-mitotic rather than stem or progenitor cells, this highlights how competition can stimulate non-autonomous tissue plasticity and homeostasis. Further illustrating this, in tumors arising from Rab5-mutation in the Drosophila wing disc, establishment of a protective environment was required to shield mutant cells from competitive elimination [114]. Notably, formation of proliferative zones could be observed adjacent to tumor borders, where JNK-induced apoptotic elimination of loser Rab5-mutants by WT cells occurred [114]. Consistent with the established role of JNK activity in stimulating release of Dpp and Wg, tumor growth was found to be contingent on these pro-proliferative factors, exemplifying how the tumor-suppressive role of cell competition can be exploited to instead promote tumorigenesis [114]. Similarly, in intestinal organoids comprised of tumor and WT cells, mutants not only engaged in super competition but additionally exhibited greater proliferation in the presence of WT cells [84].
Contrasting their role in stimulating cellular expansion, dying cells have also been shown to secrete signals that promote death of other cells within the tissue environment. This was demonstrated when apoptotic induction in one compartment of the Drosophila imaginal wing disc stimulated apoptosis in the other compartment through long-ranged release of the death ligand Eiger, the tumor necrosis factor-alpha (TNFα) homolog, from dying cells and subsequent JNK activation in recipient cells [108]. Furthermore, this mechanism of apoptosis-induced-apoptosis was also observed in mice, where hair follicle cells undergoing coordinated death were found to release TNFα [108]. It is tempting to speculate that winner cells mediating competition not only thrive from the direct elimination of loser cells, but also through the release of dying signals that further prime surrounding loser cells for elimination in certain contexts. Characterizing the environmental footprint of cell competition may reveal yet another means of abrogating tumorigenesis and promoting tissue remodeling.
Stem cell competition in transplantation
Competitive transplantation assays have long been established as a method for investigating SC competition for niche occupancy post-grafting in mammals, especially within the mouse hematopoietic system and testes [115, 116]. Moreover, such assays can prove to be highly consequential in harnessing the therapeutic potential of SCs [48, 49, 117,118,119,120,121,122]. Co-transplantation of WT and p27−/− spermatogonial SCs (SSCs) into mouse testes revealed that while cells deficient in p27, a cyclin-dependent kinase inhibitor critical for self-renewal in SSCs, can successfully engraft and give rise to progeny under non-competitive conditions, they are outcompeted by WT SSCs for access to the niche [117]. Furthermore, although testes from p27−/− mice exhibited an increased number of spermatogonial progenitor cells, likely due to aberrant self-renewal, this ultimately caused defects in spermatogenesis and germline transmission [117].
As successful cell transplantation and integration post-grafting inherently relies on the responsiveness and relative fitness of grafted cells within the host environment, transplantation assays have highlighted several factors that affect the ability of grafted SCs to compete with host cells [49, 117, 118, 120,121,122]. An important role was established for the axonal guidance receptor Robo4, which has been shown to play a role in HSC adhesion within the niche [118]. Robo4 mutant cells were not able to effectively compete for niche occupancy upon transplantation into the bone marrow and also exhibited compensatory increase in Cxcr4, a chemokine receptor required for HSC mobilization and self-renewal [118, 119]. Manipulation of Robo4 may therefore serve as a potential therapeutic target to promote the competitive ability of HSCs during transplantation [118, 119]. Another factor functioning in cellular adhesion, the cell surface integrin αvβ3, has been shown to promote HSC competition post-grafting as regulators of its expression including thrombopoietin, STAT5, and JAK-STAT signaling have been implicated in enabling HSCs to compete for niche occupancy [120,121,122]. The role of cellular adhesion in shaping cellular fitness during grafting is further depicted, as HSC transplantation assays revealed that cells depleted for p53 display increased expression of cytokines and adhesion molecules that promote competitiveness post-transplantation [49, 118].
Furthermore, during liver transplantation, fetal liver stem/progenitor cells (FLSPC) have been shown to repopulate recipient livers through cell competition [123]. Examining the contribution of age in cellular fitness, a 3-fold higher occurrence of apoptosis was observed in host hepatocytes surrounding transplanted FLSPC clusters in older versus younger livers, with up to 5-fold greater liver repopulation when FLSPCs were transplanted into older livers [124]. This was coupled with an increase in Activin A in host hepatocytes [124], which can induce apoptosis, downregulate anti-apoptotic genes, and inhibit proliferation [125, 126]. Moreover, as FLSPCs lack Activin receptor expression, this enabled them to efficiently outcompete host hepatocytes in older livers [124]. Further examining the application of cell competition for replacement of aberrant host hepatocytes, healthy liver cells were transplanted into transgenic mice bearing a mutation in α1-Antitrypsin (AAT) [127]. This plasma glycoprotein is typically secreted by hepatocytes but accumulates within cells in its mutant form, causing stress and abnormal cellular functions [127]. Post-transplantation, efficient cellular engraftment and proliferation was observed, with 20-98% of mutant host hepatocytes replaced over time [127]. This is likely to be due to a combination of increased host cell apoptosis resulting from cell competition as well as growth signals sent by these cells [127].
Recent work has further implicated SC competition as a barrier to human-animal chimeras, which are being investigated as a therapeutic approach for transplantation and tissue engineering [128]. To unveil factors rendering donor cells less fit in host environments, human and mouse pluripotent SCs (PSCs) co-cultures were established, after which competitive elimination of human PSCs was observed [128]. This occurred via NFκB activation in loser cells, as inhibition enabled human cells to overcome elimination both in vitro and post-transplantation into mouse embryos [128].
These results unveil important functional roles of cell competition during transplantation, while enriching our understanding of the intrinsic and environmental factors that underlie cellular fitness and foster competition. Deeper understanding of the underlying molecular mechanisms can therefore hold tremendous promise for catalyzing regenerative medicine and therapeutic innovation.
Future applications and perspectives
Insights into the molecular and genetic contributions of cell competition in mammalian systems have already unlocked tremendous potential that can be harnessed for a variety of therapeutic applications [129]. Illustrating this, manipulation in Myc level expression in a subset of cardiomyocytes enabled these cells to contribute to mouse cardiac replenishment through competition with WT cardiomyocytes [129]. Strikingly, this did not result in any atypical morphological or functional phenotypes [129]. As the heart tissue does not exhibit the capacity for endogenous regeneration observed in other tissues, such findings have important implications for heart disease therapeutics and can be applied to other systems.
Targeting cell competition-dependent tumorigenesis, a model was designed in which tetracycline-inducible RasV12-GFP loser epithelial cells were co-cultured with WT winner cells and subjugated to high-throughput drug screening [130]. Subsequent measurement of GFP intensity enabled identification of compounds that promote mutant cell elimination by WT cells, while individual cultures assessed which of these compounds exhibited preferentially toxicity to loser cells without compromising WT cells [130]. As tumors typically exhibit interfaces of transformed and WT cells, this provides an alternative screening approach that better accounts for cell competition and the heterogenous cellular context in which tumors arise. Expanding upon this, cell competition screens can enable more customized modeling of clonal expansion and drug resistance in patient tumors, and potentially predict how heterogenous cell populations may dynamically respond to various treatments.
Directing efforts to preventative intervention of cell competition-dependent tumorigenesis has also shown tremendous promise [131]. As p53 mutants in human and mouse esophageal epithelium exhibit resistance to low-dose ionizing radiation, which typically causes oxidative stress that elicits DNA repair, they can outcompete WT neighbors upon stimulation. However, pretreatment of irradiated mice with the antioxidant N-acetyl cysteine remarkably prevented this displacement of WT cells by mutants post-irradiation [131]. In an additional example, enhancement of SC competition through caloric restriction resulted in an increased SC pool in the intestine, coupled with slower but more efficient cell competition and diminished retention of neutral and Apc−/− mutant SCs over time [132].
Building upon these and other studies, identifying the critical drivers of SC competition will yield profound consequences on our understanding of fundamental biological processes and ability to innovate novel therapeutic strategies. Through induction of transient genetic changes, supplementation of appropriate extrinsic signals, and employment of cellular-based strategies, cell competition can enable preferential manipulation of aberrant cells and environmental remodeling. Ultimately, these approaches hold tremendous promise for targeting of developmental abnormalities, tumorigenesis, and aging, facilitating transplantation, and engineering tissue regeneration.
References
Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. 2015;16:329–44.
Soteriou D, Fuchs Y. A matter of life and death: stem cell survival in tissue regeneration and tumour formation. Nat Rev Cancer. 2018;18:187–201.
Koren E, Fuchs Y. Modes of regulated cell death in cancer. Cancer Discov. 2021;11:245–65.
Morata G. Cell competition: a historical perspective. Dev Biol. 2021;476:33–40.
Levayer R, Moreno E. Mechanisms of cell competition: themes and variations. J Cell Biol. 2013;200:689–98.
Cosentino K, García-Sáez AJ. Bax and Bak pores: are we closing the circle? Trends Cell Biol. 2017;27:266–75.
Walczak H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol. 2013;5:a008698.
Morata G, Ripoll P. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev Biol. 1975;42:211–21.
Simpson P, Morata G. Differential mitotic rates and patterns of growth in compartments in the Drosophila wing. Dev Biol. 1981;85:299–308.
Cohen B, Simcox AA, Cohen SM. Allocation of the thoracic imaginal primordia in the Drosophila embryo. Development. 1993;117:597–608.
Marygold SJ, Roote J, Reuter G, Lambertsson A, Ashburner M, Millburn GH, et al. The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol. 2007;8:R216.
Lindsley DL, Grell EH. Genetic variations of Drosophila melanogaster. Science 1968;162:993–993.
Moreno E, Basler K, Morata G. Cells compete for Decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature. 2002;416:755–9.
Moreno E, Basler K. DMyc transforms cells into super-competitors. Cell. 2004;117:117–29.
de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila Myc regulates organ size by inducing cell competition. Cell 2004;117:107–16.
Tolwinski NS. Introduction: Drosophila-a model system for developmental biology. J Dev Biol. 2017;5:9.
Baker NE. Emerging mechanisms of cell competition. Nat Rev Genet. 2020;21:683–97.
Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010;143:508–25.
Evans M, Kaufman M. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–6.
Sionov RV, Haupt Y. The cellular response to p53: the decision between life and death. Oncogene. 1999;18:6145–57.
Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604.
Tarkowski AK, Witkowska A, Opas J. Development of cytochalasin B-induced tetraploid and diploid/tetraploid mosaic mouse embryos. J Embryol Exp Morphol. 1977;41:47–64.
Nagy A, Gocza E, Merentes Diaz E, Prideaux VR, Ivanyi E, Markkl M, et al. Embryonic stem cells alone are able to support fetal development in the mouse. Development. 1990;110:815–21.
Horii T, Yamamoto M, Morita S, Kimura M, Nagao Y, Hatada I. P53 suppresses tetraploid development in mice. Sci Rep. 2015;5:8907.
Bowling S, di Gregorio A, Sancho M, Pozzi S, Aarts M, Signore M, et al. P53 and mTOR signalling determine fitness selection through cell competition during early mouse embryonic development. Nat Commun. 2018;9:1763.
Zhang G, Xiea Y, Zhou Y, Xiang C, Chen L, Zhang C, et al. P53 pathway is involved in cell competition during mouse embryogenesis. Proc Natl Acad Sci USA. 2017;114:498–503.
Dejosez M, Ura H, Brandt VL, Zwaka TP. Safeguards for cell cooperation in mouse embryogenesis shown by genome-wide cheater screen. Science. 2013;341:1511–4.
Sancho M, Di-Gregorio A, George N, Pozzi S, Sánchez JM, Pernaute B, et al. Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev Cell. 2013;26:19–30.
Clavería C, Giovinazzo G, Sierra R, Torres M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature. 2013;500:39–44.
Hashimoto M, Sasaki H. Epiblast formation by TEAD-YAP-dependent expression of pluripotency factors and competitive elimination of unspecified cells. Dev Cell. 2019;50:139–54.
Díaz-Díaz C, Fernandez de Manuel L, Jimenez-Carretero D, Montoya MC, Clavería C, Torres M. Pluripotency surveillance by Myc-driven competitive elimination of differentiating cells. Dev Cell. 2017;4:585–99.
Ellis SJ, Gomez NC, Levorse J, Mertz AF, Ge Y, Fuchs E. Distinct modes of cell competition shape mammalian tissue morphogenesis. Nature. 2019;569:497–502.
Mesa KR, Rompolas P, Zito G, Myung P, Sun TY, Brown S, et al. Niche-induced cell death and epithelial phagocytosis regulate hair follicle stem cell pool. Nature. 2015;522:94–97.
Lima A, Lubatti G, Burgstaller J, Hu D, Green AP, di Gregorio A, et al. Cell competition acts as a purifying selection to eliminate cells with mitochondrial defects during early mouse development. Nat Metab. 2021;3:1091–108.
Telang S, Lane AN, Nelson KK, Arumugam S, Chesney J. The oncoprotein H-RasV12 increases mitochondrial metabolism. Mol Cancer. 2007;6:77.
Jam FA, Morimune T, Tsukamura A, Tano A, Tanaka Y, Mori Y, et al. Neuroepithelial cell competition triggers loss of cellular juvenescence. Sci Rep. 2020;10:18044.
Kucinski I, Dinan M, Kolahgar G, Piddini E. Chronic activation of JNK JAK/STAT and oxidative stress signalling causes the loser cell status. Nat Commun. 2017;8:136.
Nagata R, Nakamura M, Sanaki Y, Igaki T. Cell competition is driven by autophagy. Dev Cell. 2019;51:99–112.
Baumgartner ME, Dinan MP, Langton PF, Kucinski I, Piddini E. Proteotoxic stress is a driver of the loser status and cell competition. Nat Cell Biol. 2021;23:136–46.
Recasens-Alvarez C, Alexandre C, Kirkpatrick J, Nojima H, Huels DJ, Snijders AP, et al. Ribosomopathy-associated mutations cause proteotoxic stress that is alleviated by TOR inhibition. Nat Cell Biol. 2021;23:127–35.
Langton PF, Baumgartner ME, Logeay R, Piddini E. Xrp1 and Irbp18 trigger a feed-forward loop of proteotoxic stress to induce the loser status. PLoS Genet. 2021;17:e1009946.
Lee CH, Kiparaki M, Blanco J, Folgado V, Ji Z, Kumar A, et al. A regulatory response to ribosomal protein mutations controls translation, growth, and cell competition. Dev Cell. 2018;46:456–69.
Baillon L, Germani F, Rockel C, Hilchenbach J, Basler K. Xrp1 is a transcription factor required for cell competition-driven elimination of loser cells. Sci Rep. 2018;8:17712.
Ochi N, Nakamura M, Nagata R, Wakasa N, Nakano R, Igaki T. Cell competition is driven by Xrp1-mediated phosphorylation of eukaryotic initiation factor 2α. PLoS Genet. 2021;17:e1009958.
Kiparaki M, Khan C, Folgado-Marco V, Chuen J, Moulos P, Baker NE. The transcription factor Xrp1 orchestrates both reduced translation and cell competition upon defective ribosome assembly or function. Elife 2022;11:e71705.
Ji Z, Chuen J, Kiparaki M, Baker N. Cell competition removes segmental aneuploid cells from drosophila imaginal disc-derived tissues based on ribosomal protein gene dose. Elife. 2021;10:e61172.
Tseng CY, Burel M, Cammer M, Harsh S, Flaherty MS, Baumgartner S, et al. chinmo-mutant spermatogonial stem cells cause mitotic drive by evicting non-mutant neighbors from the niche. Dev Cell. 2022;57:80–94.
Marusyk A, Porter CC, Zaberezhnyy V, DeGregori J. Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol. 2010;8:e1000324.
Bondar T, Medzhitov R. p53-Mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–22.
Watanabe H, Ishibashi K, Mano H, Kitamoto S, Sato N, Hoshiba K, et al. Mutant p53-expressing cells undergo necroptosis via cell competition with the neighboring normal epithelial cells. Cell Rep. 2018;23:3721–9.
Norman M, Wisniewska KA, Lawrenson K, Pablo GM, Tada M, Kajita M, et al. Loss of scribble causes cell competition in mammalian cells. J Cell Sci. 2012;125:59–66.
Wagstaff L, Goschorska M, Kozyrska K, Duclos G, Kucinski I, Chessel A, et al. Mechanical cell competition kills cells via induction of lethal p53 levels. Nat Commun. 2016;7:11373.
Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10:207–17.
Mascré G, Dekoninck S, Drogat B, Youssef KK, Brohée S, Sotiropoulou PA, et al. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature. 2012;489:257–62.
Kato T, Liu N, Morinaga H, Asakawa K, Muraguchi T, Muroyama Y, et al. Dynamic stem cell selection safeguards the genomic integrity of the epidermis. Dev Cell. 2021;56:3309–20.
Liu N, Matsumura H, Kato T, Ichinose S, Takada A, Namiki T, et al. Stem cell competition orchestrates skin homeostasis and ageing. Nature. 2019;568:344–50.
Penzo-Méndez AI, Chen YJ, Li J, Witze ES, Stanger BZ. Spontaneous cell competition in immortalized mammalian cell lines. PLoS ONE. 2015;10:e0132437.
Colom B, Herms A, Hall MWJ, Dentro SC, King C, Sood RK, et al. Mutant clones in normal epithelium outcompete and eliminate emerging tumours. Nature. 2021;598:510–4.
Brown S, Pineda CM, Xin T, Boucher J, Suozzi KC, Park S, et al. Correction of aberrant growth preserves tissue homeostasis. Nature 2017;548:334–7.
Moya IM, Castaldo SA, van den Mooter L, Soheily S, Sansores-Garcia L, Jacobs J, et al. Peritumoral activation of the Hippo pathway effectors YAP and TAZ suppresses liver cancer in mice. Science. 2019;336:1029–34.
Martins VC, Busch K, Juraeva D, Blum C, Ludwig C, Rasche V, et al. Cell competition is a tumour suppressor mechanism in the thymus. Nature. 2014;509:465–70.
Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16.
Martincorena I, Campbell P. Somatic mutation in cancer and normal cells. Science. 2015;349:1478–83.
Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018;362:911–7.
Yizhak K, Aguet F, Kim J, Hess JM, Kübler K, Grimsby J, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;364:eaaw0726.
Kajita M, Fujita Y. EDAC: Epithelial defence against cancer - cell competition between normal and transformed epithelial cells in mammals. J Biochem. 2015;158:15–23.
Kajita M, Sugimura K, Ohoka A, Burden J, Suganuma H, Ikegawa M, et al. Filamin acts as a key regulator in epithelial defence against transformed cells. Nat Commun. 2014;5:4428.
Cho M, Thompson D, Cramer C, Vidmar T, Scieszka J. The Madin Darby canine kidney (MDCK) epithelial cell monolayer as a model cellular transport barrier. Pharm Res. 1989;6:71–7.
Hogan C, Dupré-Crochet S, Norman M, Kajita M, Zimmermann C, Pelling AE, et al. Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol. 2009;11:460–7.
Kajita M, Hogan C, Harris AR, Dupre-Crochet S, Itasaki N, Kawakami K, et al. Interaction with surrounding normal epithelial cells influences signalling pathways and behaviour of Src-transformed cells. J Cell Sci. 2010;123:171–80.
Leung CT, Brugge JS. Outgrowth of single oncogene-expressing cells from suppressive epithelial environments. Nature. 2012;482:410–3.
Chiba T, Ishihara E, Miyamura N, Narumi R, Kajita M, Fujita Y, et al. MDCK cells expressing constitutively active Yes-associated protein (YAP) undergo apical extrusion depending on neighboring cell status. Sci Rep. 2016;6:28383.
Tamori Y, Bialucha CU, Tian AG, Kajita M, Huang YC, Norman M, et al. Involvement of Lgl and mahjong/VprBP in cell competition. PLoS Biol. 2010;8:e1000422.
Ohoka A, Kajita M, Ikenouchi J, Yako Y, Kitamoto S, Kon S, et al. EPLIN is a crucial regulator for extrusion of RasV12- transformed cells. J Cell Sci. 2015;128:781–9.
Tanimura N, Fujita Y. Epithelial defense against cancer (EDAC). Semin Cancer Biol. 2020;63:44–48.
Kon S, Ishibashi K, Katoh H, Kitamoto S, Shirai T, Tanaka S, et al. Cell Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol. 2017;19:530–41.
Akter E, Tasaki Y, Mori Y, Nakai K, Hachiya K, Lin H, et al. Non Non-degradable autophagic vacuoles are indispensable for cell competition. Cell Rep. 2022;40:111292.
Menéndez J, Pérez-Garijo A, Calleja M, Morata G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc Natl Acad Sci USA. 2010;107:14651–6.
Mamada H, Sato T, Ota M, Sasaki H. Cell competition in mouse NIH3T3 embryonic fibroblasts is controlled by the activity of Tead family proteins and Myc. J Cell Sci. 2015;128:790–803.
di Giacomo S, Sollazzo M, de Biase D, Ragazzi M, Bellosta P, Pession A, et al. Human cancer cells signal their competitive fitness through MYC activity. Sci Rep. 2017;7:12568.
Price CJ, Stavish D, Gokhale PJ, Stevenson BA, Sargeant S, Lacey J, et al. Genetically variant human pluripotent stem cells selectively eliminate wild-type counterparts through YAP-mediated cell competition. Dev Cell. 2021;56:2455–70.
van Neerven SM, de Groot NE, Nijman LE, Scicluna BP, van Driel MS, Lecca MC, et al. Apc-mutant cells act as supercompetitors in intestinal tumour initiation. Nature. 2021;594:436–41.
Pronobis MI, Rusan NM, Peifer M. A novel GSK3-regulated APC:Axin interaction regulates Wnt signaling by driving a catalytic cycle of efficient βcatenin destruction. Elife. 2015;4:e08022.
Krotenberg Garcia A, Fumagalli A, Le HQ, Jackstadt R, Lannagan TRM, Sansom OJ, et al. Active elimination of intestinal cells drives oncogenic growth in organoids. Cell Rep. 2021;36:109307.
Sasaki A, Nagatake T, Egami R, Gu G, Takigawa I, Ikeda W, et al. Obesity suppresses cell-competition-mediated apical elimination of RasV12-transformed cells from epithelial tissues. Cell Rep. 2018;23:974–82.
Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–8.
Braakhuis BJM, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1723–30.
Aster JC, Pear WS, Blacklow SC. The varied roles of Notch in cancer. Annu Rev Pathol. 2017;12:245–75.
Alcolea MP, Greulich P, Wabik A, Frede J, Simons BD, Jones PH. Differentiation imbalance in single oesophageal progenitor cells causes clonal immortalization and field change. Nat Cell Biol. 2014;16:612–9.
Vermeulen L, Morrissey E, van der Heijden M, Nicholson AM, Sottoriva A, Buczacki S, et al. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 2013;342:995–8.
Snippert HJ, Haegebarth A, Kasper M, Jaks V, van Es JH, Barker N, et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science. 2010;327:1385–9.
Snippert HJ, Schepers AG, van Es JH, Simons BD, Clevers H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep. 2014;15:62–69.
Rhiner C, López-Gay JM, Soldini D, Casas-Tinto S, Martín FA, Lombardía L, et al. Flower forms an extracellular code that reveals the fitness of a cell to its neighbors in Drosophila. Dev Cell. 2010;18:985–98.
Merino MM, Rhiner C, Portela M, Moreno E. “Fitness fingerprints” mediate physiological culling of unwanted neurons in drosophila. Curr Biol. 2013;23:1300–9.
Merino MM, Rhiner C, Lopez-Gay JM, Buechel D, Hauert B, Moreno E. Elimination of unfit cells maintains tissue health and prolongs lifespan. Cell. 2015;160:461–76.
Moreno E, Fernandez-Marrero Y, Meyer P, Rhiner C. Brain regeneration in Drosophila involves comparison of neuronal fitness. Curr Biol. 2015;25:955–63.
Madan E, Pelham CJ, Nagane M, Parker TM, Canas-Marques R, Fazio K, et al. Flower isoforms promote competitive growth in cancer. Nature. 2019;572:260–4.
Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131:966–79.
Li W, Baker NE. Engulfment is required for cell competition. Cell. 2007;129:1215–25.
Lolo FN, Casas-Tintó S, Moreno E. Cell competition time line: winners kill losers, which are extruded and engulfed by hemocytes. Cell Rep. 2012;2:526–39.
Bozkurt E, Düssmann H, Salvucci M, Cavanagh BL, van Schaeybroeck S, Longley D, et al. Trail signaling promotes entosis in colorectal cancer. J Cell Biol. 2021;220:e202010030.
Fadok PM, Voelker DR, Campbell PA, Cohen JJ. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–16.
Sun Q, Cibas ES, Huang H, Hodgson L, Overholtzer M. Induction of entosis by epithelial cadherin expression. Cell Res. 2014;24:1288–98.
Rizzotto D, Villunger A. P53 clears aneuploid cells by entosis. Cell Death Differ. 2021;28:818–20.
Mackay HL, Moore D, Hall C, Birkbak NJ, Jamal-Hanjani M, Karim SA, et al. Genomic instability in mutant p53 cancer cells upon entotic engulfment. Nat Commun. 2018;9:3070.
Lyng FM, Seymour CB, Mothersill C. Initiation of apoptosis in cells exposed to medium from the progeny of irradiated cells: a possible mechanism for bystander-induced genomic instability? Radiat Res. 2002;157:365–70.
Seymour CB, Mothersill C. Relative contribution of bystander and targeted cell killing to the low-dose region of the radiation dose-response curve. Radiat Res. 2000;153:508–11.
Pérez-Garijo A, Fuchs Y, Steller H. Apoptotic cells can induce non-autonomous apoptosis through the TNF pathway. Elife. 2013;2013:e01004.
Haynie JL, Bryant PJ. The effects of X-rays on the proliferation dynamics of cells in the imaginal wing disc of Drosophila melanogaster. Wilehm Roux’s Arch Dev Biol. 1977;183:85–100.
Pérez-Garijo A, Martín FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004;131:5591–8.
Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the wingless signaling pathways. Dev Cell. 2004;7:491–501.
Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol. 2004;14:1262–6.
Tamori Y, Deng WM. Tissue repair through cell competition and compensatory cellular hypertrophy in postmitotic epithelia. Dev Cell. 2013;25:350–63.
Ballesteros-Arias L, Saavedra V, Morata G. Cell competition may function either as tumour-suppressing or as tumour-stimulating factor in Drosophila. Oncogene. 2014;33:4377–84.
Harrison DE. Competitive repopulation: a new assay for long-term stem cell functional capacity. Blood. 1980;55:77–81.
Shinohara T, Orwig KE, Avarbock MR, Brinster RL. Germ line stem cell competition in postnatal mouse testes 1. Biol Reprod. 2002;66:1491–7.
Kanatsu-Shinohara M, Takashima S, Shinohara T. Transmission distortion by loss of p21 or p27 cyclin-dependent kinase inhibitors following competitive spermatogonial transplantation. Proc Natl Acad Sci USA. 2010;107:6210–5.
Smith-Berdan S, Nguyen A, Hassanein D, Zimmer M, Ugarte F, Ciriza J, et al. Robo4 cooperates with Cxcr4 to specify hematopoietic stem cell localization to bone marrow niches. Cell Stem Cell. 2011;8:72–83.
Peled A, Petit I, Kollet O, Magid M, Ponomaryov T, Byk T, et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283:845–8.
Umemoto T, Yamato M, Ishihara J, Shiratsuchi Y, Utsumi M, Morita Y, et al. Integrin-v3 regulates thrombopoietin-mediated maintenance of hematopoietic stem cells. Blood. 2012;119:83–94.
Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007;1:685–97.
Wang Z, Li G, Tse W, Bunting KD. Conditional deletion of STAT5 in adult mouse hematopoietic stem cells causes loss of quiescence and permits efficient nonablative stem cell replacement. Blood. 2008;113:4856–65.
Oertel M, Menthena A, Dabeva MD, Shafritz DA. Cell competition leads to a high level of normal liver reconstitution by transplanted fetal liver stem/progenitor cells. Gastroenterology. 2006;130:507–20.
Menthena A, Koehler CI, Sandhu JS, Yovchev MI, Hurston E, Shafritz DA, et al. Activin A, p15INK4b signaling, and cell competition promote stem/progenitor cell repopulation of livers in aging rats. Gastroenterology. 2011;140:1009–1020.
Schwall R, Robbins K, Jardieu P, Chang L, Lai C, Terrell T. Activin induces cell death in hepatocytes in vivo and in vitro. Hepatology. 1993;18:347–56.
Hully JR, Chang L, Schwall RH, Widmer RH, Terrell TG. Induction of apoptosis in the murine liver with recombinant human activin A. Hepatology. 1994;4:854–62.
Ding J, Yannam GR, Roy-Chowdhury N, Hidvegi T, Basma H, Rennard SI, et al. Spontaneous hepatic repopulation in transgenic mice expressing mutant human α1-antitrypsin by wild-type donor hepatocytes. J Clin Investig. 2011;121:1930–4.
Zheng C, Hu Y, Sakurai M, Pinzon-Arteaga CA, Li J, Wei Y, et al. Cell competition constitutes a barrier for interspecies chimerism. Nature. 2021;592:272–6.
Villa del Campo C, Clavería C, Sierra R, Torres M. Cell competition promotes phenotypically silent cardiomyocyte replacement in the mammalian heart. Cell Rep. 2014;8:1741–51.
Yamauchi H, Matsumaru T, Morita T, Ishikawa S, Maenaka K, Takigawa I, et al. The cell competition-based high-throughput screening identifies small compounds that promote the elimination of RasV12-transformed cells from epithelia. Sci Rep. 2015;5:15336.
Fernandez-Antoran D, Piedrafita G, Murai K, Ong SH, Herms A, Frezza C, et al. Outcompeting p53-mutant cells in the normal esophagus by redox manipulation. Cell Stem Cell. 2019;25:329–41.
Bruens L, Ellenbroek SIJ, Suijkerbuijk SJE, Azkanaz M, Hale AJ, Toonen P, et al. Calorie restriction increases the number of competing stem cells and decreases mutation retention in the intestine. Cell Rep. 2020;32:107937.
Acknowledgements
We apologize to colleagues whose contributions we could not adequately cite due to space constraints. We thank Fuchs lab members for helpful discussion and input. Figures and graphical abstract were generated using BioRender.
Funding
YF was supported by the EMBO Young Investigator program, ICRF (15-771-RCDA) grants, ISF individual 2124/19, and IPMP 1019045 2029637 grants, and ICRF acceleration AG-17-917.
Author information
Authors and Affiliations
Contributions
MY and YF wrote and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yusupova, M., Fuchs, Y. To not love thy neighbor: mechanisms of cell competition in stem cells and beyond. Cell Death Differ 30, 979–991 (2023). https://doi.org/10.1038/s41418-023-01114-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-023-01114-3
This article is cited by
-
Dying in self-defense: cell death signaling in animals and plants
Cell Death & Differentiation (2024)
-
Apoptotic dysregulation mediates stem cell competition and tissue regeneration
Nature Communications (2023)
