
Abstract
Kinase signaling in the tiered activation of inflammasomes and associated pyroptosis is a prime therapeutic target for inflammatory diseases. While MAPKs subsume pivotal roles during inflammasome priming, specifically the MAP3K7/JNK1/NLRP3 licensing axis, their involvement in successive steps of inflammasome activation is poorly defined. Using live-cell MAPK biosensors to focus on the inflammasome triggering event allowed us to identify a subsequent process of biphasic JNK activation. We find that this biphasic post-trigger JNK signaling initially facilitates the mitochondrial reactive oxygen species generation needed to support core inflammasome formation, then supports the gasdermin-mediated cell permeation required for release of active IL-1β from human macrophages. We further identify and characterize a xanthine oxidase-ROS activated MAP3K5/JNK2 substrate licensing complex as a novel regulator of the GSDMD mobilization which precedes pyroptosis. We show that inhibitors targeting this MAP3K5 cascade alleviate morbidity in mouse models of colitis and dampen both augmented IL-1β release and cell permeation in monocytes derived from patients with gain-of-function inflammasomopathies.
Similar content being viewed by others


Introduction
High-order inflammasome complexes assemble following pathogenic infection or sterile stress to activate and secrete the inflammatory IL-1β, IL-1α, and IL-18 [1,2,3,4]. Here, three stepwise regulatory gates restrict pathological activation of the canonical inflammasome: (1) Priming: TLR or cytokine-induced innate immune signaling, transcriptionally upregulates limiting inflammasome components (2) Assembly and Activation: ASC oligomerizes into a platform to support cleavage of caspase 1 and its zymogen substrates (3) Release: Active inflammatory cytokines disperse following gasdermin-mediated exocytosis, cell permeation, and pyroptosis [5, 6]. Additional regulatory and licensing events that refine checkpoint function by altering biochemical activities and localizations of select inflammasome components remain an active area of study [7].
Mitogen-activated protein kinases (MAPKs) serve as central stress and immune integration hubs during inflammatory responses [8, 9]. Apart from their well-established role facilitating TLR priming, the contribution of each major MAPK branch (ERK, p38, and JNK) during inflammasome triggering has yet to be clarified [10,11,12,13]. Dynamic MAPK nuclear-to-cytosol kinase translocation reporter (KTR) biosensors overcome many of the technical limitations associated with traditional approaches to study MAPKs in inflammasome activation [14]. First, the dynamic nature of KTRs affords high temporal resolution allowing analysis of MAPK activities downstream of prerequisite priming, a key factor that traditional genetic approaches cannot overcome. Second, their single-celled nature facilitates signaling investigation proximal to cell death, an event obscured when using population-level MAPK readouts. Lastly, multiplexing MAPK KTRs with second-messenger biosensors yields real-time correlations, which coupled with inhibitors can illuminate causal signaling cascades. Therefore, MAPK KTRs are valuable tools for studying MAPK dynamics in inflammasome activation models.
MAPK responses are the composite integration of environmental response signals from diverse upstream MAP3Ks [15]. Receptors and proximal kinases activate MAP3Ks to both regulate specific cellular processes and contribute to downstream MAP2K/MAPK signaling cascades, often with the aid of scaffolding proteins [16]. While MAP3K involvement in innate immune signaling and apoptotic cell death is widely recognized, prior studies supporting MAPK contributions to inflammasome activation and pyroptotic cell death remain controversial [13, 17,18,19]. Since numerous chemical inhibitors of MAPK cascade components have been developed, identification and characterization of the role of these pathways during inflammasome triggering may provide new therapeutic pathways to treat inflammatory morbidities.
Diverse sensory-proximal gain-of-function mutations that sensitize inflammasome seeding underlie many inborn inflammatory disorders. Neonatal-onset multisystem inflammatory disease (NOMID) and macrophage activation syndrome (MAS) manifest from gain-of-function mutations in cryopyrin (NLRP3) and ipaf (NLRC4) respectively [20]. While molecularly distinct, hyper-secreted IL-1β and IL-18 both drive inflammatory flares and persistent inflammation. Although neutralizing biologics (anakinra, rilonacept, and canakinumab) alleviate symptoms, small molecules targeting gasdermin D, the shared executor of pyroptosis required to release both IL-1β and IL-18, represent an attractive therapeutic alternative for these diseases [6, 21,22,23].
Here, we have used a combinatorial approach harnessing multiplexed biosensors, acute pharmacological screening, and traditional genetic and biochemical approaches to interrogate MAPK cascades during inflammasome activation. We delineate both an unappreciated role for JNK in supporting trigger-phase mitochondrial ROS (mtROS) production, and a novel MAP3K substrate licensing complex that facilitates subcellular mobilization of GSDMD. Further investigation reveals that mtROS supports inflammasome formation while xanthine oxidase-induced ROS production promotes distinct MAP3K activation to facilitate pyroptosis. Genetics and inhibitors targeting this GSDMD licensing cascade mitigate disease progression in a whole animal model of inflammatory colitis and limit IL-1β release from molecularly distinct inflammasomopathy peripheral blood mononuclear cells (PBMCs).
Results
JNK is the primary MAPK supporting inflammasome-mediated IL-1β secretion and pyroptosis in human macrophages
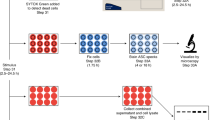
To limit priming-associated interference with interpretation of MAPK perturbation during triggering, we established an extended priming regimen with the active signaling component of LPS (KDO-Lipid A (KLA)) in primary human monocyte-derived macrophages (hMDM), whereby MAPK prime-induced activity returned close to baseline levels after 18 h (Fig. S1A). Specific MAPK inhibitors were then applied 1 h prior to NLRP3 inflammasome triggering with nigericin to investigate trigger-induced MAPK dependencies required for downstream inflammasome outputs (Fig. 1A). Kinetic analysis of ASC speck formation (Fig. 1B, D), IL-1β release (Fig. 1E), and cell permeation through propidium iodide (PI) uptake (Fig. 1C, F) revealed specific MAPK involvement during the inflammasome trigger step in hMDM. While ERK pathway inhibition resulted in little to no delay in ASC speck formation, IL-1β release, or cell permeation (Fig. 1D–F), p38 inhibition augmented ASC specking without impacting IL-1β release or cell permeability kinetics, implicating p38 as a negative regulator of core-inflammasome ASC speck formation (Fig. 1D–F). In contrast, JNK inhibition led to broad defects in ASC speck formation, diminished IL-1β release, and delayed PI uptake (Fig. 1D–F).

A hMDM were subjected to an 18 h 100 nM KLA extended prime regimen prior to inflammasome triggering with 10 µM nigericin. B, C Inflammasome formation was evaluated by endogenous ASC speck imaging (B; White arrows) while pyroptotic cell permeation was evaluated by propidium iodide (PI) uptake (C; Red stain) to analyze kinetic differences between the two events. Blue indicates Hoechst-stained nuclei. D–F Following 18 h 100 nM KLA priming, individual MAPK inhibitors (ERK [10 µM U0126], JNK [30 µM JNK-IN-8], p38α [10 µM SB203580], and pan-p38 [30 µM doramapimod]) were applied for 1 h before inflammasome triggering with 10 µM nigericin to examine roles for MAPKs in ASC specking (D), IL-1β release (E), and cell permeation (F). D–F Shading represents standard deviation from the mean. Two-way ANOVA followed by Tukey’s multiple comparison test; *p < 0.05, **p < 0.01. Comparable results were obtained in parallel studies with U937 cells (see Figs. S1 and S2). B–F Data shown are representative of at least three independent experiments.
Extended-prime NLRP3 inflammasome activation in the U937 human monocyte line exhibited JNK-supported ASC specking kinetics comparable to primary hMDM (Fig. S1B–F). Using this system, we developed a higher-throughput, coupled PI uptake/IL-1β release assay in 384-well format to permit testing of kinase inhibitors at a broader concentration range flanking the reported on-target dose (Fig. S2A, B). This revealed a clear dose-dependent requirement for JNK, but not p38 or ERK, in both PI uptake (Fig. S2C) and IL-1β release (Fig. S2D). Additional JNK inhibitors also dampened ASC speck formation, IL-1β release, and DRAQ7 uptake, supporting inhibitor specificity (Fig. S2E–G). This JNK requirement persisted irrespective of the NLRP3 activator as evidenced by diminished IL-1β release from U937 cells treated with the lysosome damaging dipeptide LeuLeuOMe (LLoM) or the mitochondrial reactive oxygen species (mtROS) stimulator menadione (Fig. S2H, I). These findings allude to critical JNK signaling dependencies for inflammasome activation and cell lysis following the inflammasome trigger step in human macrophages beyond the established NLRP3-licensing role for JNK1 during priming [10, 13].
Single-macrophage MAPK biosensors illuminate biphasic JNK signaling following inflammasome triggering
Adoption of the MAPK KTR biosensor system permitted simultaneous single-cell investigation in human macrophages [14]. We first generated a single lentiviral polycistronic multi-KTR (mKTR) construct encoding distinct fluorophore reporters for each major MAPK branch (ERK, JNK, and p38) separated by 2A autoproteolysis sites (Fig. 2A). Stable expression of this reporter in human THP1 macrophages (mKTR B6 clone), provided dynamic single cell readouts of MAPK activation (cytosolic fluorescence) vs inactivation (nuclear fluorescence) (Fig. 2B). We confirmed the specificity of each KTR in anisomycin-activated cells (Fig. S3A–D) and established that the mKTR-B6 cells showed expected kinetics of ASC speck formation (Fig. S3E, F) and IL-1β cleavage (Fig. S3G) in response to nigericin. We then examined parallel, single-cell MAPK dynamics in real-time following NLRP3 inflammasome triggering (Fig. 2C, D). We observed a pattern of biphasic activation for both JNK and p38 kinases in individual cells, whereas ERK showed a slight inactivation prior to cell lysis (Fig. 2C, D). These MAPK activity patterns were confirmed in population-level western blots from hMDMs (Fig. S3H, I) and U937 cells (Fig. 2E, F), which also uncovered a temporally-distinct, isoform-specific activation of JNK with initial activation of p44 phospho-JNK1 in the first hour followed by delayed activation of p52 phospho-JNK2 peaking around 2 h after nigericin stimulation (Fig. 2F). These data together allude to molecularly distinct, phasic JNK activation steps following inflammasome triggering that may drive specific intracellular signaling events. We therefore sought to investigate whether this might lead to disparate downstream functionality.

A Construct design of lentivirus and THP1 stable clones expressing multiple kinase translocation reporters (mKTRs) representing each major MAPK branch. B Key for interpreting augmented or depressed MAPK activity using KTRs. C Live imaging of THP1 mKTR B6 line following differentiation for one day in 20 ng/mL PMA, treatment with 100 nM KLA for 18 h, and 10 µM nigericin trigger prior to imaging every 5 min for 4 h. Yellow arrows indicate relative activation and purple arrows indicate relative inactivation of KTRs over time. Images are representative of three independent experiments consisting of four fields per condition. D Nuclear KTR signal was segmented and quantitated in Imaris followed by statistical graphing on an inverted y-axis in seaborn. Swarm violins representing at least 200 cells for each timepoint were plotted. Internal violin breaks represent the mean and 95% CI. E Western blot analysis of population-level MAPK dynamics in PMA differentiated U937 cells treated with 100 nM KLA for 18 h preceding the 10 µM nigericin trigger. F Blot densitometry quantitation of population-level MAPK activation from (E). F Dual y-axes with distinct scales were used to plot more robust p-p38 activation (dashed y-axis) alongside ERK inactivation and JNK activation (solid y-axis). C–F Data are representative of three independent experiments.
Trigger-induced JNK activity spurs mitochondrial ROS production to tune NLRP3 inflammasome formation
While mtROS influences inflammasome responses [24,25,26], signaling cascades promoting and responding to ROS dynamics during the triggering step have not been thoroughly examined. We employed multiplex live-cell mKTR analysis coupled with chemical far-red ROS reporters to examine MAPK activity and ROS generation in parallel. We found that early-phase JNK activation correlated closely with, and was necessary for, a compartmentalized ROS spike (Fig. 3A, B) that largely colocalized with mitoTrackerRed (Fig. S4A). The mtROS scavenger mitoTEMPO correspondingly reduced early ROS production further implying a mitochondrial origin (Fig. 3C). MitoTEMPO treatment also significantly delayed ASC speck formation (Fig. 3D), but failed to impact PI uptake (Fig. 3E), implying a distinct mechanism controlling cell permeation downstream of inflammasome formation.

A, B Live confocal imaging of ROS production via ROSBrite670 in the stable THP1 mKTR B6 line triggered with 10 µM nigericin compared to cells pretreated with 30 µM JNK-IN-8 (A) and accompanying quantitation (B). C, E, F, H, I Live epifluorescent imaging quantitation of hMDM labeled with green-shifted chemical biosensor (DCFDA) to measure ROS production (C, F), PI (E, H), or Draq7 (I) to assess cell permeation following a 10 µM nigericin trigger ±30 µM JNK-IN-8 (JNKi), 10 µM SB203580 (p38αi), 30 µM doramapimod (p38i), 5 mM N-acetyl cysteine (NAC), or 300 µM mitoTEMPO. D, G Endogenous hMDM ASC speck quantitation performed by high-content imaging under inhibitor conditions described above. B Line plots of data summarized from at least 200 cells, as in Fig. 2D, represent gated mean cellular fluorescence for each biosensor while shading represents one standard deviation from the mean. Two-way ANOVA followed by Tukey’s multiple comparison test; **p < 0.01, ***p < 0.001, ****p < 0.0001. A–I Imaging is representative of two (C, D, F, G) or three (A, B, E, H, I) independent experiments consisting of four (A, B), ten (C, E, F, H, I), or 20 (D, G) independent fields per condition.
We then tested MAPK dependencies of post-trigger ROS induction in DCFDA loaded hMDM. We found that multiple JNK inhibitors, but not p38 inhibitors, strongly suppressed ROS production throughout the 5 h time course, at a level comparable to the global ROS scavenger N-acetyl cysteine (NAC) (Figs. 3F, S4B). JNK inhibition similarly dampened compartmentalized mtROS production comparable to the pan-ROS inhibitor DPI (Fig. S4C). Consistent with our prior findings, JNK inhibition strongly reduced ASC specking (Fig. 3G), but unlike mitoTEMPO, it prevented pyroptotic cell permeation measured by either PI uptake (Fig. 3H) or DRAQ7 imaging (Fig. 3I). While acute, JNK-dependent mtROS responses support core inflammasome formation, the contrasting requirement in lytic cell death suggests that a distinct JNK signal facilitates cell permeation and pyroptosis.
MAP3K inhibitor screening identifies MAP3K5 as a prominent, specific regulator of cellular pyroptosis
Activation of MAP3K signaling hubs provide functional specifity to well characterized MAP2K-MAPK activation cascades (Fig. S5A) [15, 16]. Since JNK facilitates early mtROS/ASC specking, and a distinct JNK-dependent activity promotes cell lysis, we reasoned that independent MAP3K signaling scaffolds may explain this dichotomy. We coupled pan-family specific MAP3K inhibitors (to overcome known intra-familial paralog redundancies) with the 384-well PI uptake assay described previously (Fig. S2A, B) to broadly assess MAP3K involvement in inflammasome-induced cell permeation (Fig. 4A, B). Inhibition of insulin signaling-related ALK family kinases, Syk kinase, or stress-response signaling protein IRE1α resulted in significant delays in PI uptake following nigericin treatment (Fig. 4A), consistent with existing literature [27,28,29,30]. Although few MAP3K inhibitors impaired PI uptake, significant delays in cell permeation were observed with inhibitors targeting MAP3K5, at levels comparable to PI uptake defects observed following JNK inhibition (Fig. 4A, B).

A, B Propidium iodide uptake kinetics in PMA differentiated, 18 h 100 nM KLA primed U937 cells triggered with 10 µM nigericin in the presence of of MAP3K (A) or MAPK (B) inhibitors at multiples of indicated target-concentrations (Table S1). Data were reduced as in Fig S2B. C, D Kinetics of PI uptake, IL-1β release, and ASC speck formation in PMA differentiated U937 cells after extended priming followed by triggering with 10 µM nigericin ±30 µM MAP3K5 inhibitor selonsertib (M3K5i) (C) or 10 µM MAP3K7 inhibitor 7-oxozeanol (M3K7i) (D). E Differentiated U937 cells following extended priming were triggered with 10 µM nigericin to assess kinetic biochemical cleavage of caspase 1 and GSDMD ±30 µM selonsertib (M3K5i) (E) or 10 µM 7-oxozeanol (M3K7i) (F). A, B Error bars represent standard deviation while shading represents one and two standard deviations from the cumulative mean of 16 DMSO control samples. C, D Shading represents one standard deviation from the mean. A, B Pair-wise two-tailed student’s T test of unequal variance; *p < 0.05, **p < 0.01, ***p < 0.001. C, D Two-way ANOVA followed by Tukey’s multiple comparison test; ***p < 0.001, ****p < 0.0001. A–F Data are representative of at least two (A, B) or three (C, D) experiments containing four replicates per group (A–D).
MAP3K5 is a large ROS-responsive kinase that promotes p38 and JNK signaling during apoptotic cell death following DNA damage, an event also linked to pyroptotic cell death [31,32,33]. While MAP3K5 has recently been implicated in sustained activation of p38 and JNK during late-phase toll-like receptor responses, infection, and autoinflammation [34,35,36], our extended priming regimen implies a specific role for MAP3K5 proximal to inflammasome-induced cell death. Consistent with this idea, post-prime inhibition of MAP3K5 failed to prevent either ASC specking or caspase 1 activation, however PI uptake, IL-1β release, and the activatory cleavage of pore-forming GSDMD were all impaired (Fig. 4C, E). These data allude to a pyroptosome-centric role for MAP3K5 signaling that operates either parallel to or downstream of inflammasome formation, but precedes and facilitates GSDMD processing, IL-1β release, and cellular pyroptosis.
MAP3K7 and MAP3K5 regulate distinct JNK activities to license inflammasome formation and pyroptosis respectively
Since MAP3K7 has been causally linked to the prime-associated JNK1 activation necessary for NLRP3 phosphorylation [10, 13, 37], we further investigated the mechanistic impacts of MAP3K7 inhibition specifically during the inflammasome triggering step. Following extended priming, inhibition of MAP3K7, but not MAP3K5, dampened trigger-induced mtROS production (Fig. S5B) and abrogated ASC specking, IL-1β release, and caspase 1 cleavage without exerting significant effects on lytic cell death (Figs. 4A, D, F, S5C, D); findings consistent with our earlier mitoTEMPO observations (Fig. 3C–E) and prior studies [38,39,40,41,42,43]. Furthermore, phosphorylation of the JNK substrate c-Jun was diminished by both MAP3K5 and MAP3K7 inhibition, corroborating downstream JNK activation by both MAP3Ks (Fig. 4E, F). Our findings agree with previously reported, prime-independent roles for MAP3K7 in core inflammasome formation [13, 37], but further stress a mechanistic separation between inflammasome formation and lytic cell death, where MAP3K5 appears to be a key regulator.
Intracellular mobilization of GSDMD is regulated by a MAP3K5:JNK signaling axis
Although ASC speck formation has been used extensively as an endpoint to study core inflammasome formation [44], specific regulators of the subsequent GSDMD pore formation are just being defined [40, 45, 46]. While proteolysis and assembly of N-terminal GSDMD oligomers into membrane pores represent well established bottlenecks in the late pyroptotic response, early factors that mobilize GSDMD to inflammasomes and further deliver GSDMD to the plasma membrane are less well understood. Confocal imaging of endogenous GSDMD in primary hMDM revealed a KLA-prime associated shift to the perinuclear region, while inflammasome triggering with nigericin further redistributed GSDMD to the plasma membrane (Fig. 5A). We found that nigericin triggering increased staining of pJNK and its colocalization with GSDMD, alluding to potential GSDMD regulation by active JNK (Fig. 5A). Biophysical fluorescence lifetime imaging (FLIM) also suggested close proximity (<10 nm) of active pJNK and GSDMD proteins, as shown by GSDMD-induced reduction of the 488-pJNK donor lifetime (Fig. 5B). Inhibition or knockout of upstream MAP3K5 reduced pMAP3K5 staining (Fig. S6A, B, C), cytosolic pJNK staining (Figs. 5C, D, S6H, J), and associated pJNK/GSDMD colocalization (Fig. 5C, D) while downstream JNK inhibition did not substantially reduce upstream MAP3K5 or JNK phosphorylation (Figs. 5D, S6B). Importantly, both MAP3K5/JNK inhibition and MAP3K5 deficiency (but not MAP3K7 inhibition), dramatically reduced GSDMD plasma-membrane mobilization following nigericin triggering (Figs. 5C, E, S6H, I, S7A, B), supporting a role for the MAP3K5/JNK signaling axis in subcellular mobilization of GSDMD .

A Coimmunostaining of pJNK and GSDMD in hMDM following extended priming and 2 h of 10 µM nigericin triggering. Yellow arrows indicate plasma-membrane localization. B Fluorescence lifetime (FLIM) of 488-pJNK in extended-primed hMDM triggered with 10 µM nigericin for 2 h. FLIM was quantitated as the reduction of Alexa 488-anti-pJNK fluorescence lifetime in the presence of Alexa 560-anti-GSDMD. C Extended-primed hMDM were treated with MAP3K5 and JNK inhibitors for one hour before triggering with 10 µM nigericin for 2 h. Cells were stained for pJNK and GSDMD to assess JNK activation and GSDMD localization. D Enumeration of hMDM pJNK mean cytosolic immunofluorescence following a 10 µM nigericin trigger ±30 µM M3K5i (selonsertib) or 30 µM JNKi (JNK-IN-8). E Quantitation of plasma-membrane localized GSDMD in hMDM treated as in (C). F CoIPs were performed on WT iBMDM or iBMDM stably expressing GSDMD-HA in naïve state (−) 100 nM KLA primed for 18 h (P) or additionally treated with 10 µM nigericin for 1 h (N) by immunoprecipitating HA and immunoblotting for indicated targets. G Protein interaction mapping was performed by co-expression in 293T17 cells with MAP3K5-FLAG fragments and either JNK2-HA or GSDMD-HA followed by IP of FLAG and immunoblotting of HA. Yellow arrows represent upshifted band migration. H SDS-resistant GSDMD super-shifting was performed by co-expressing MAP3K5, JNK2, and GSDMD in 293T17 cells in the presence or absence of MAP3K5 inhibitor [30 µM selonsertib]. A minimum of six confocal fields were acquired per sample condition per experiment (A–C) while quantitation represents five confocal (B, E) or 30 epifluorescent (D) fields in each of four replicates per group. B, D Error bars represent standard deviation from the mean. A, C Pearson’s correlation for pJNK/GSDMD colocalization was calculated by applying identical thresholding to experimental samples using Imaris. B, E Pair-wise two-tailed student’s T test of unequal variance; *p < 0.05, ***p < 0.001. D Two-way ANOVA followed by Tukey’s multiple comparison test; *p < 0.05, ****p < 0.0001. A–H Data shown are representative of at two (B, H) or three (A, C–H) experiments.
MAP3K5 and JNK2 regulate FL-GSDMD complexes
In light of the data suggesting close proximity of JNK and GSDMD, and the dependency of intracellular GSDMD mobilization on the MAP3K5/JNK signaling axis, we sought to investigate whether these components form a biochemical complex capable of regulating GSDMD. Co-immunoprecipitation (Co-IP) studies from macrophages stably expressing low levels of HA-tagged GSDMD demonstrated that GSDMD constitutively associates with MAP3K5 and the p52 isoform of JNK2, but not JNK1 (Fig. 5F), while evidence of active pJNK2 association was enriched in primed and triggered lysates (Fig. 5F, red asterisk).
We then mapped the MAP3K5 domain requirements for JNK2 and GSDMD interaction through fragment co-expression in inflammasome devoid 293T17 cells. This revealed robust association between full-length MAP3K5 and GSDMD, while JNK2 most strongly associated with fragments containing the C-terminal coiled-coil domain of MAP3K5 (Fig. 5G). Closer inspection of input lysates suggested partial band-shifting of GSDMD only in the presence of full-length MAP3K5 (Fig. 5G, lowest panel, red asterisk), which may be indicative of GSDMD phosphorylation. Co-expression of both MAP3K5 and JNK2 with GSDMD-HA in inflammasome deficient 293T17 cells promoted high molecular weight GSDMD super-shifting in denaturing, reducing SDS-PAGE gels (Fig. 5H) which was substantially reduced by lysate treatment with calf intestinal phosphatase or cell pretreatment with MAP3K5 inhibitor, further indicating regulatory importance of MAP3K5 kinase activity (Fig. 5H, S7C). Together, these data suggest that MAP3K5 and JNK2 can together promote stable, full-length GSDMD super-shifting independent of caspase-1 mediated cleavage.
To investigate whether MAP3K5/JNK2 regulation of GSDMD mobilization extends to inflammasome-independent processes; we assessed IFNγ-induced GSDMD PIT formation previously described in HeLa cells [47,48,49]. We observed substantial reduction of GSDMD PIT mobilization following treatment with either MAP3K5 or JNK inhibitors (Fig. S7D, E). Together these data suggest that MAP3K5/JNK2 may act broadly as a substrate licensing complex that redistributes GSDMD to specific subcellular locations where it can be appropriately deployed in response to a variety of activation stimuli.
Xanthine oxidase inhibition prevents MAP3K5 activation and GSDMD mobilization
Since MAP3K5 is a ROS-responsive kinase [32], we evaluated trigger-induced ROS sourcing in hMDM loaded with DCFDA by employing general and specific ROS scavengers and inhibitors. While the global ROS scavenger N-Acetylcysteine (NAC) robustly prevented DCFDA fluorescence accumulation (Fig. 6A), the mtROS scavenger mitoTEMPO specifically attenuated early ROS production, consistent with prior work [24, 41, 42]. In contrast, a late phase of ROS production was specifically dampened by the xanthine oxidase (XO) inhibitor febuxostat (Fig. 6A). This supported our earlier observation of acute-phase mtROS supporting ASC speck formation and led us to speculate whether the later phase of XO-dependent ROS may be required for MAP3K5 activity and downstream pyroptosis.

A ROS production following extended priming and 10 µM nigericin triggering was assessed in DCFDA labeled hMDM treated with indicated ROS inhibitors (XOi [300 µM febuxostat], 300 µM mitoTEMPO, or NAC [5 mM N-acetyl cysteine]). B PI-uptake kinetics in extended-primed, PMA differentiated U937 cells triggered with 10 µM nigericin in the presence of indicated target-concentrations of ROS inhibitors (Table S1). Data were reduced as in Fig S2B. C CoIP was performed using iBMDM stably expressing GSDMD-HA as described in Fig. 5F while the presence of xanthine oxidase (XO) was assessed by immunoblot. D, G Confocal imaging of GSDMD, pJNK, and pMAP3K5 immunofluorescence was assessed in extended-primed hMDM treated for 1 h with indicated ROS inhibitors (5 mM NAC, XOi: 300 µM Febuxostat, 300 µM mitoTEMPO) followed by 4 h of 10 µM nigericin triggering. E, F Enumeration of pMAP3K5 and cytosolic pJNK immunofluorescence of samples treated as in (D). H Quantitation of plasma membrane localization of GSDMD in hMDM following treatment with ROS inhibitors treated as in (G). A Data are represented as the mean of four replicates per group while error bars represent standard deviation. B Shading represents one and two standard deviations from the cumulative mean of 16 DMSO control samples. E, F Quantitation was performed by epifluorescent high-content imaging parallel to the confocal imaging for (D). H GSDMD plasma membrane localization was enumerated from a minimum of 200 cells via confocal microscopy. B, H Pair-wise two-tailed student’s T test of unequal variance; **p < 0.01, ****p < 0.0001. A, E,F Two-way ANOVA followed by Tukey’s multiple comparison test; *p < 0.05, **p < 0.01, ****p < 0.0001. A–H Data shown are representative of three experiments.
We found that enzymatic inhibition of XO dampened pyroptotic cell permeation and IL-1β release, without affecting early-phase ASC specking in response to nigericin (Figs. 6B, S8A, B) implicating the later-phase of XO-induced ROS in cellular pyroptosis. Likewise, Co-IP of GSDMD-HA from immortalized bone marrow-derived macrophages (iBMDM) revealed a conditional association of GSDMD with XO only after priming and during inflammasome activation (Fig. 6C) providing a biochemical basis for tuning GSDMD-dependent pyroptosis.
We then questioned whether XO supports signaling in the MAP3K5/JNK2/GSDMD pyroptotic licensing axis. Fixed immunofluorescence imaging revealed an increase in active phospho-MAP3K5 (pM3K5) and phospho-JNK in nigericin triggered hMDM with pM3K5 subcellular localization mirroring that of GSDMD (Fig. 6D–G). We find that XO inhibitor pretreatment significantly reduced trigger-induced pM3K5 and cytosolic pJNK staining and delayed cell permeation in hMDM to levels comparable to those achieved with NAC-mediated global ROS scavenging (Fig. 6B, D–F). Inhibition of XO also attenuated GSDMD redistribution to the plasma membrane, supporting a role for XO-generated ROS in regulating GSDMD through the MAP3K5/JNK signaling axis (Fig. 6D, G, H). In contrast, specific inhibition of acute-phase mtROS failed to impact pMAP3K5, cytosolic pJNK, or GSDMD mobilization to the plasma membrane (Fig. 6B, D–H), nor did MAP3K5 or XO inhibition affect early ROS generation (Fig. S5B, S8C), suggesting specificity of XO-induced late ROS in the MAP3K5/JNK2 pyroptotic cascade.
XO:MAP3K5:JNK2 pyroptotic signaling is required for DSS-induced colitis in mice
Since the mouse model of dextran sodium sulfate (DSS) colitis relies on GSDMD-induced pyroptosis [43, 50, 51], we reasoned that selective inhibitors of the XO/MAP3K5/JNK2/GSDMD cascade may affect DSS-induced morbidity. Accordingly, intraperitoneal injection of inhibitors for XO (febuxostat), MAP3K5 (selonsertib), and JNK (SP600125) all reduced DSS-induced colon shortening, weight loss, and morbidity scores when administered after the start of DSS treatment (Fig. 7A–D). While these chemical inhibitors partially alleviated morbidity, Jnk2−/−, but not Jnk1−/− nor Nlrp3−/− mice, exhibited both complete protection from colon shortening and reduced weight loss comparable to Gsdmd−/− and caspase1/11 double knockout mice (Fig. 7B–D, S9A). These effects are unlikely to be driven by priming defects in PRR-driven innate immune transcription since Jnk2−/− bone marrow-derived macrophages (BMDM) exhibited no defects in Il6, Tnf, or Cxcl10 transcriptional responses to KDO-lipid A (Fig. S9B–D). Nor are these observations likely to be caused by defective core inflammasome formation since endogenous ASC specking kinetics and caspase-1 cleavage in Jnk2−/− BMDM were comparable to wildtype controls (Fig. 7E, H, S9E, F). Meanwhile, Jnk2−/− BMDM exhibited diminished IL-1β release, PI uptake, and GSDMD cleavage following nigericin triggering (Fig. 7F–H), results that largely phenocopy our earlier findings for MAP3K5 inhibition (Fig. 4C, E). In contrast, Jnk1−/− BMDM exhibit defective mtROS production and ASC specking, consistent with a role in the early trigger-phase JNK response, but fail to limit cell lysis (Fig. S9F–I). Together, these findings provide in vivo support for the importance of the XO/MAP3K5/JNK2 signaling cascade in regulating the pyroptosis contributing to DSS-induced colitis, distinct from JNK1 involvement in canonical inflammasome formation.

A–D Mice injected intraperitoneally with inhibitors (XO [febuxostat 30 mg/kg], JNK [SP600125 30 mg/kg], or MAP3K5 [selonsertib 30 mg/kg]) according to the treatment regimen (A), or the indicated knockouts, were assessed for susceptibility to 2% DSS-induced colitis via monitoring colon shortening (B), weight loss (C), and morbidity score from stool condition (D). E–G BMDM were assessed for ASC specking (E), IL-1β release (F), and PI uptake (G), following 18 h 100 nM KLA priming and a 10 µM nigericin trigger. H Jnk2−/− BMDM were primed for 18 h with 100 nM KLA and triggered with 10 µM nigericin to assess activatory cleavage of Caspase 1 (p20) and Gsdmd (cl-Gsdmd) via western blot. I–J Human PBMC from patients harboring gain-of-function mutations in various inflammasomes (NOMID: NLRP3 and MAS: NLRC4) were assessed for IL-1β release (I) and PI uptake (J) following extended priming −/+ 1 h 30 µM M3K5 inhibitor selonsertib and 10 µM nigericin triggering. A, B, D Data are represented as the mean of five mice (B–D), sample quadruplicates (E–G), or duplicate wells (I, J) per group while error bars (B–D, I) and shading (E–G, J) represent standard deviation from the mean. B, I Pair-wise two-tailed student’s T test of unequal variance; *p < 0.05, ****p < 0.0001. C–G, J Two-way ANOVA followed by Tukey’s multiple comparison test; ****p < 0.0001. A–J Data shown are representative of two (A–D, I, J) or three (E–H) experiments.
MAP3K5 signaling contributes to IL-1β production in gain-of-function NLRP3 (NOMID) and NLRC4 (MAS) patient PBMCs
We then investigated the impact of the MAP3K5:GSDMD substrate licensing complex in monocytes from patients carrying gain-of-function alleles in cryopyrin (NLRP3: NOMID) or nlrc4 (MAS). Mutations in these distinct inflammasome seeding components lead to augmented IL-1β release and manifestation of inflammatory flares in a largely GSDMD-dependent manner [20, 52]. Inhibition of MAP3K5 in these cells both dampened IL-1β release and significantly delayed PI uptake (Fig. 7I, J). These data suggest that MAP3K5 inhibitors may provide an alternative to expensive biologics for management of inflammasome-associated autoinflammatory flares.
Discussion
We demonstrate that JNK kinases support IL-1 family cytokine release beyond their reported roles in inflammasome priming [10]. Here, a trigger-induced early role for JNK1 mobilizes mtROS to facilitate ASC speck formation in hMDM. A second JNK2 activation driven by xanthine-oxidase produced ROS and MAP3K5 promoted GSDMD mobilization and cell lysis suggesting a signaling pathway parallel to core inflammasome formation. Pharmacological inhibition of this parallel cascade suppresses inflammatory morbidities in a whole animal colitis model and limits IL-1 release in cellular models of distinct gain-of-function inflammasomopathies.
Leveraging extended TLR priming, which better mimics conditions at PAMP-laden mucosal surfaces, allowed us to investigate trigger-induced MAPK signaling [13]. This approach identified reciprocal roles for JNK in supporting, and p38 in regulating, core inflammasome ASC speck formation and pyroptosis, a finding consistent with MAPK pro and anti-regulation observed in other settings [53]. Multi-MAPK-KTR macrophage lines (mKTR) revealed trigger-induced, biphasic activation of both JNK and p38 correlated with early-phase ASC speck formation and late-phase pyroptotic cell death, prompting us to investigate up and downstream factors associated with each phase of JNK activation.
Diverse MAP3Ks promote specific MAPK activities in addition to contributing to canonical MAPK-AP1 responses. Inhibition of distinct MAP3Ks during the inflammasome trigger step uncoupled discrete MAPK signaling origins underlying both inflammasome formation and pyroptosis. Inhibition of MAP3K7 or knockout of Jnk1 diminished inflammasome ASC speck formation, but failed to limit lytic cell death, consistent with literature linking MAP3K7 inhibition to necrolytic cell death and panoptosis [13, 37, 39, 41,42,43]. In contrast, MAP3K5 inhibition or knockout of Jnk2 limited the subcellular mobilization and cleavage of GSDMD necessary for pyroptotic cell permeation, without impairing inflammasome formation or activation of caspase 1 . We therefore questioned if signaling intermediates coordinate MAPK activation in inflammasome formation and pyroptosis.
Both inflammasome formation and pyroptotic cell death rely on ROS generation [24, 41]; however, ROS sourcing and associated signaling during these events is not well understood. We demonstrate that early JNK, but not p38, promotes trigger-induced mtROS production, consistent with studies linking JNK1 signaling to mtROS in other settings [54, 55]. Meanwhile mtROS scavenging with mitoTEMPO limits ASC specking to levels observed following JNK inhibition, or deletion of the Jnk1 gene, and complements prior findings for mtROS during inflammasome activation [24, 26, 56]. Unexpectedly, scavenging mtROS failed to reduce GSDMD-mediated cell permeation, a finding consistent with cell-permeation sufficiency following upstream MAP3K7 inhibition or Jnk1 deletion.
These observations supported distinct regulation of the pyroptosis-specific MAP3K5/JNK2 signaling node. Since MAP3K5 is normally inactivated by covalent cysteine bridging with thioredoxin or periredoxin [31, 32], we reasoned that other ROS sources following inflammasome triggering may facilitate MAP3K5 activation and downstream pyroptosis. Here, XO inhibition, but not mtROS scavenging, specifically diminished late phase ROS production, reduced phosphorylation of both MAP3K5 and JNK, and limited GSDMD mobilization and cell permeation, an observation accordant with FDA-approved therapies targeting XO-induced IL-1 release in inflammatory gout [42]. These data demonstrate that distinct ROS sources differentially contribute to inflammasome formation and pyroptosis; however, context-specific variation in ROS sourcing cannot fully be ruled out [40].
While GSDMD regulation has primarily focused on late-phase pore formation and plasma membrane repair [40, 45, 57,58,59,60], we questioned whether the XO-ROS:MAP3K5:JNK2 signaling chain functioned as a licensing complex to mobilize GSDMD for pyroptosis. Immunoprecipitation of GSDMD from macrophages established constitutive interaction with MAP3K5 and the p52 isoform of JNK2, whereas XO recruitment was conditional following the nigericin trigger. These interactions, combined with inhibitor data, support a model where a subpopulation of GSDMD associates with inactive MAP3K5 and JNK2 which is conditionally activated by XO-ROS. This activation promotes full-length GSDMD super-shifting reminiscent of N-terminal oligomerization [61]; however, more studies are needed to clarify how the exact mode of action relates to cell permeation.
The existence of a new GSDMD-centric regulatory node could impact multiple inflammasomes. Indeed, PBMCs harboring gain-of-function mutations in either NLRP3 (NOMID) or NLRC4 (MAS) inflammasome seeding components secreted less IL-1β and delayed cell permeation after MAP3K5 inhibition. Similarly, MAP3K5 cascade inhibitors and genetic deletion of JNK2, GSDMD, or caspase 1/11, but not NLRP3 or JNK1, alleviated colon shortening in a pyroptosis-dependent, canonical inflammasome independent, mouse model of DSS-induced colitis. We additionally find that MAP3K5 inhibition disassembles IFNγ-induced GSDMD PITs in HeLa cells extending its functional significance beyond macrophage inflammasomes [49, 62,63,64,65] . Similarly, co-expression of MAP3K5, JNK2, and GSDMD in inflammasome-deficient 293T17 cells promotes MAP3K5 dependent full-length GSDMD super-shifting in denaturing, reducing PAGE gels, suggesting that MAP3K5 operates broadly in GSDMD regulation that is distinct from, but operates parallel to, inflammasome-mediated cleavage of GSDMD and associated pore formation. GSDMD embodies a major bottleneck in inflammatory cytokine release and its licensing complex may be a suitable therapeutic target to limit recurrent morbidities of heightened, chronic secretion of IL-1 family cytokines.
Materials and methods
Mice and cell lines
Mice were maintained in specific-pathogen-free conditions. All procedures were approved by the NIAID Animal Care and Use Committee (National Institutes of Health, Bethesda, MD). Mice were euthanized either before the development of clinical disease or at the defined humane endpoint. Wild-type B57Bl6N mice were obtained from Taconic. GSDMD KO, NLRP3 KO, Caspase 1/11 DKO, JNK1 KO, and JNK2 KO mice were obtained from The Jackson Laboratory. ASC knockout mice were a generous gift from the Flavell lab. Mice were backcrossed where needed and were allowed to establish stable colonies in the same facility for two generations prior to use in experiments. Murine BMDM were isolated by aseptic bone flushing and were differentiated in complete DMEM containing 50 ng/mL mM-CSF for 6 days. Primary hMDM were obtained as elutriated monocytes from screened, healthy donors under the NIH Clinical Center IRB-approved protocol 99-CC-0168 from the NIH Department of Transfusion Medicine and were differentiated by culturing in complete RPMI containing 50 ng/mL hGM-CSF for 8 days. U937 (CRL-1593.2) and THP1 (TIB-202) immortal monocytes were obtained from ATCC, were cultured below a density of 106 cells/mL, and were differentiated into macrophages by a 24 h incubation in 20 ng/mL PMA. HeLa (CCL-2) were obtained from ATCC.
Isolation of human blood cells
Peripheral blood was collected in tubes containing sodium heparin. Erythrocytes were removed using ACK lysis buffer (8.3 g/L NH4Cl, KHCO3 1 g/L, EDTA 0.0372 g/L) (Quality Biological, Inc. Gaithersburg, MD) in 0.01 M Tris–HCl buffer pH 7.4. Blood cells were pelleted and mixed with 10 ml of ACK lysis buffer for 1 min. Cells were washed and resuspended in RPMI. Cell viability was assessed using trypan blue exclusion (Invitrogen, Frederick MD).
Chemicals and antibodies
Nigericin, blasticidin, menadione, doramapimod, SB203580, JNK-IN-VIII, U0126, selonsertib, febuxostat, CC401, JNK-IN-V, takinib, and deabeitic acid were purchased from Cayman Chemical. DCFDA and ROSBrite670 were purchased from AAT Bioquest. Hoechst 33342 and Draq7 were purchased from ThermoFisher. LeuLeuOMe was purchased from ChemImpex. Propidium iodide and mitoTEMPO were purchased from Sigma Aldrich. Human IFNγ was purchased from R&D Systems. 16% PFA was purchased from Electron Microscopy Services. See Supplemental Tables S1 and S2 for additional chemical and antibody information.
Inflammasome activation
Classical inflammasome activation was stimulated with 10 µM nigericin, 1 mM LeuLeuOMe, or 250 µM menadione. Each agonist was lot validated to confirm induction of caspase-1 dependent cell-permeation and IL-1β release.
Fixed immunofluorescence imaging
hMDM were differentiated for 5 days followed by seeding at 1 × 104 cells/well in a 384-well imaging plates while U937 were seeded at 2 × 104 cells/well in a 384-well imaging plate and were differentiated with 50 ng/mL PMA for 1 day. After differentiation, cells were treated with 100 nM KLA for 18 h. Respective inhibitors were added 1 h prior to addition of 10 µM nigericin. Nigericin was added to wells at indicated times before adding paraformaldehyde to a final concentration of 2%. Cells were fixed for 10 min, permeabilized in cold 70% ethanol, and stored at −20 °C until use. Wells were warmed to room-temperature and rehydrated for 5 min in TBS followed by blocking with 2% BSA TBS for 1 h. Primary antibodies were applied at indicated concentrations (Table S2) in 2% BSA TBS for 1 h followed by two washes in TBS. Fluor conjugated secondary antibodies were applied at a concentration of 1:2000 in 2% BSA TBS for 30 min followed by four TBS washes. Cells were counterstained in 1 ug/mL Hoechst 33342 in TBS. 384-well plates were imaged on both a ThermoScientific CX7 epifluorescent high-content imager for quantitations as well as a Leica SP8 confocal microscope for high-resolution imaging and quantification of colocalization. Colocalization analysis was performed across parallelly acquired samples with identical thresholding in Imaris.
ASC Puncta quantitation
Endogenous ASC puncta quantitation was performed by subtracting basal cytosolic fluorescence from images of unstimulated cells. An ROI was set by applying a sigmoidal filter to the nuclear Hoechst stain followed by expansion of 15 pixels to capture the cell cytoplasm. A spot-detection algorithm was applied using a box smoothing of four and a sigmoidal threshold to assure stringent counting of objects with strong signal to noise ratios. Nuclear ASC was filtered out of the ASC speck count by applying an area size filter on detected spots. Mean, total, and binary-thresholded positivity measures were acquired.
ELISA
Elisa for both human and murine IL-1β were purchased from R&D Systems and were used according to manufacturer protocols. Briefly, 10 µL of capture antibody diluted in PBS was bound to 384-well NUNC immunosorp plates for 2 h. Antibody was removed and plates were blocked in PBS containing 2% BSA for 1 h followed by sample binding for 2 h. Sample was removed and a mix of biotin-conjugated detection antibody and streptavidin HRP was applied for 1 h. Plates were washed four times with PBS containing 0.02% Tween-20. Plates were developed by adding 10 µL of TMB-Plus substrate for 20 min then quenched with 10 µL 2 M sulfuric acid. Plates were read at 450 nm with baseline subtraction from 570 nm on a BMG-Omegastar. A standard curve was calculated and samples in the quantitative linear range were interpolated.
PI uptake imaging
hMDM were differentiated with 50 ng/mL GM-CSF for 5 days followed by seeding at 1 × 104 cells/well in a 384-well imaging plate while U937 were seeded at 2 × 104 cells/well in a 384-well imaging plate and were differentiated with 50 ng/mL PMA for 2 days. Cells were then treated with 100 nM KLA for 18 h. Respective inhibitors were added 1 h prior to addition of 10 µM nigericin. Media was exchanged to label cells in 1 µg/mL Hoechst 33342 in 5% FBS RPMI lacking phenol-red supplemented with 5 µg/mL PI. A final concentration of 10 µM nigericin was added to wells and kinetic imaging was performed on a CX7 high-content imager equipped with an environmental control unit and laser autofocus control. Images were acquired from ten non-adjacent fields in each well at five-minute intervals for 12 h. ROI were calculated by nuclear stain and expanded 15 pixels to encompass the cytoplasm. Basal cytosolic fluorescence from images of living cells was subtracted and a fixed threshold was applied to gauge PI positivity. Mean, total, and binary-thresholded measures were acquired for each cell and were compared to wells fully permeabilized by Triton-X 100.
PI uptake screen
U937 were seeded at 5 × 104 cells/well in a 384-well imaging plate and were differentiated with 50 ng/mL PMA for 2 days. Cells were then treated with 100 nM KLA for 18 h. Respective inhibitors were added 1 h prior to addition of 10 µM nigericin. Media was exchanged to label cells in 1 µg/mL Hoechst 33342 in 5% FBS RPMI lacking phenol-red supplemented with 5 μg/mL PI. A final concentration of 10 µM nigericin was added to wells and kinetic imaging was performed on a ClarioSTAR plate reader equipped with an environmental control unit. Hoechst was quantified prior to kinetic reads. PI scans were acquired every 15 min using bottom-read orbital averaging for 24 h. PI measurements were ratioed to Hoechst readings to normalize PI to total cell number. Kinetics of PI uptake in control samples were calculated in R and further used to gate the kinetic window used for AUC calculation for parallelly treated samples.
Multiplex MAPK reporter generation
Individual MAPK translocation reporters were a generous gift from Drs. Markus Covert (Stanford University) and Sergi Regot (Johns Hopkins University). Individual reporters were amplified with primers containing sequence-specific 2A cleavage sites (Table S3). Fragments were assembled with NEB builder into a third generation pLenti-Puro backbone cut with EcoRI-PacI. The construct was further subcloned into a second-generation pLenti-Blast at AgeI-BamHI cut sites to allow higher efficiency virus production.
Lentivirus and stable cell line generation
293T17 cells were grown to log phase and were transfected with 3:1:0.1:0.1 construct:psPAX2:MD.G:pRSV-Rev using Mirus TransIT Lenti. After 48 h, viral supernatant was harvested and filtered through 0.45 µM cellulose. Crude viral supernatant was concentrated with LentiX Concentrator and was stored at −80 °C. Transduction of 1/10 total virus was performed on log-phase target cells. After 48 h, 10 µg/mL blasticidin or 2 µg/mL puromycin selection medium was added, and clonal populations were generated by single-cell limiting dilution. Immortalized bone marrow-derived macrophages (iBMDMs) were generated by adding supernatant from J2Cre oncogenic virus producing cells to differentiated murine BMDM. Cells were gradually, but fully weaned off their M-CSF growth dependency before use in experiments.
Biosensor live imaging
5 × 104 of THP1 MKTR clones B6 and K14 were differentiated in complete RPMI lacking phenol-red containing 50 ng/mL PMA on 12-chamber #1.5 coverglass for 1 day. Cells were then primed with 100 nM KLA for 18 h. Inhibitor was applied to cells 1 h before imaging. Hoechst 33342 was applied at 500 ng/mL for 30 min prior to imaging. DCFDA labeling was performed at 5 µM for 30 min prior to media washout where indicated. ROSbright670 was added at 10 µM 15 min prior to imaging without washout to maintain high signal to noise ratios. Following labeling, 2 μg/mL Propidium Iodide or 1 µM Draq7 was added in fresh imaging media supplemented with indicated inhibitors for the duration of inflammasome activation. Chambers were mounted on a Leica SP8 confocal microscope equipped with an environmental control chamber. Multiplexed imaging was performed with sequential scanning to prevent spectral overlap at five-minute intervals over 3–12 h. Analysis was performed with Imaris or KNIME software wherein nuclei were identified from cells and ROI were expanded to encompass the cytosol. Cell boundaries were further defined using the GFP channel as Erk-mClover regularly displayed detectable levels of cytosolic signal. Nuclear and cytosol average and total intensities were calculated, and cell tracks were exported and graphed using seaborn.
Mitochondrial live imaging
5 × 104 U937 cells were differentiated in complete RPMI lacking phenol-red containing 50 ng/mL PMA on clear-bottom black 384-well plates for 2 days. Cells were then primed with 200 nM KLA for 18 h. Hoechst 33342 (500 ng/mL), MitoTrackerRed (5 µM), ROSBrite670 (5 µM) were applied 30 min prior to imaging on a CX7 high-content imager. Pearson’s correlation analysis was performed with Imaris.
Immunoblotting (IB)
2 × 106 U937 were differentiated in 50 ng/mL PMA for 2 days in each well of 6-well plates. Cells were treated with 100 nM KLA for 18 h where indicated. Inhibitors were added 1 h before addition of nigericin to 10 µM final concentration. Cells were incubated for the indicated times followed by cell scraping at 4 °C. Cells were washed in PBS followed by lysis in 30 µL 2% Triton-X 100 TBS 2 mM MgCl +2 u/mL Benzonase + 2x protease inhibitor + 2x phos-stop for 10 min. Samples were stored at −80 °C until use. Upon thawing, samples were mixed by pipetting and were added to 2x LDS sample buffer at a 1:1 ratio. Samples were heated at 95 °C for 10 min and were vortexed and centrifuged prior to loading on 12% Bis-Tris gels. Gels were run at 100 V for 2.5 h in MES running buffer followed by wet tank transfer (Towbin) at 70 V for 2 h. Ladder was marked with xylene marker and blots were blocked in 2% BSA 0.05% Tween-20 TBS for 1 h. Primary antibodies were applied overnight at 4 °C in blocking buffer at indicated concentrations. Blots were washed twice with TBS-T and secondary HRP antibody was applied at a concentration of 1:50,000 in TBS-T for 30 min. Blots were washed four times in TBS-T for 5 min each followed by developing in Dura-ECL or ECL Lightning on a chemidoc. HRP was inactivated in 0.02% azide for 1 h and blots for alternative species antibodies were performed. For caspase 1 and GSDMD blots, cells were exchanged to Optimem supplemented with inhibitors 1 h prior to commencing nigericin treatment. Supernatants were collected and protein was precipitated in a final concentration of 80% acetone. Pellets were resuspended in 2x LDS loading buffer and were sonicated for 1 h to generate secreted protein samples which were then mixed with respective cell lysates.
Fluorescence lifetime imaging (FLIM)
Fluorescence lifetime imaging was performed on hMDM cultured on #1.5 glass coverslips using a Leica SP8 WLL Falcon inverted confocal microscope with a 63X objective (Leica Microsystems, Buffalo Grove, IL). Cells were stained with pJNK and GSDMD primaries (Invitrogen 700031 and SCBT (64-Y)) and Alexa-488 and Alexa-555 secondaries, respectively. Analysis of fluorescence time decays were resolved by time-correlated single-photon counting using an SP8 FLIM FALCON system equipped with a tunable WLL system set at 488 nm excitation wavelength and at 80mhz frequency. Images were acquired at 512-512-pixel format, collecting in excess of 2000 photons per pixel. FRET efficiency transients and FRET-FLIM Images were analyzed and processed using LASX single molecule detection analysis software.
Immunoprecipitation (IP)
Lentiviral C-terminally HA-tagged murine GSDMD was generated through amplification of the ORF from iBMDM cDNA (Table S3). The amplicon was then assembled into second-generation pLenti-BLAST at AgeI-BamHI cut sites. Stable iBMDMs expressing low levels of tagged construct were generated to use for semi-physiologic IP. iBMDMs either expressing or not expressing tagged GSDMD were plated at 6 × 107 cells in three 15 cm TC plates for each IP sample. Cells were either left untreated, were treated with 100 nM KLA for 18 h, or were treated with KLA followed by stimulation with 10 µM nigericin for 1 h. Samples were cooled on ice in a cold room where cells were scraped and pelleted followed by lysis in cold 0.2% Triton-X 100 TBS containing protease inhibitor cocktail and phos-stop. Insoluble material was removed by centrifugation, input samples were taken. IP proceeded by adding 20 µL of 50% protein A/G magnetic beads to samples along with 5 µg HA antibody. Samples were rotated end-over-end for 2 h at 4 °C. Beads were washed six times in 500 µL lysis buffer followed by elution in 50 µL 2× LDS sample buffer. Samples were subject to western blotting as described above.
Biochemical mapping
C-terminally tagged Human GSDMD (HA) and MAP3K5 (FLAG) were amplified and cloned from hMDM cDNA. Fragments were generated by subcloning with designated primers (Table S3). Fragments and ORFs were cloned into pLenti-BLAST at AgeI and BamHI cut-sites. Minimalist interaction was performed by co-expressing fragments along with full-length human GSDMD-HA or JNK2-HA in 293T17 cells followed by IP and western blotting.
CRISPR RNP knockout cell line generation
spCas9x2NLS was purchased from Synthego. sgRNA targeting MAP3K5 were designed in-house and ordered through Genscript. 1 µL of three sgRNA (50 μM each) was added to 30 pmol spCas9x2NLS and incubated at 4 °C for 10 min. 4 × 106 PBS washed U937 cells were resuspended in Neon buffer R added for a final volume of 12 μL. RNP-cell mix was electroporated in 10 μL format tips using electrode buffer E (1500 V 10 ms x5). Cells were immediately single-cell plated, amplified, and were screened for effective gene editing by genomic PCR and western blot.
GSDMD PIT induction
5 × 103 HeLa were plated in 96-well plates. 200 u/mL IFNγ was administered for 48 h. Inhibitors were applied to cells at indicated concentrations 1 h prior to cell fixation as described above. Cells were stained with anti-GSDMD antibody (H-6) and Alexa 660 secondary (Table S3). Cells were imaged on a CX7 high content imager as described above and GSDMD PITs were quantitated in accordance with ASC speck quantitation described above.
DSS colitis
2% 40kD DSS water was administered to age and sex-matched mice for 10 days. Inhibitors were administered intraperitoneally as 200 μL doses where indicated. Mice were weighed and observed for morbidity phenotypes each morning while inhibitors were administered in the afternoon. Mice incurring lethality were excluded from colon-length analyses. All mouse procedures were performed according to NIAID Animal Care and Use Committee approved protocols.
qRT-PCR
RNA was extracted from wild-type and JNK2−/− BMDM following treatment with 100 nM KLA for the indicated times and was further purified with a Qiagen RNAeasy kit. 500 ng of RNA was used for cDNA synthesis using Biorad iScript master mix with oligo dT primers according to manufacturer protocols. For each gene quantitated, ΔΔCT values were calculated by normalization to the HPRT housekeeping gene once similar cDNA load was validated. Log2 normalization was performed followed by plotting in seaborn.
Data availability
Please direct material requests and correspondence to fraseri@niaid.nih.gov.
References
Martinon F, Burns K, Tschopp J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol Cell. 2002;10:417–26.
Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281:8–27.
Lamkanfi M, Dixit VM. Mechanisms and Functions of Inflammasomes. Cell 2014;157:1013–22.
Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013;341:1250–3.
Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–89.
Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018;48:35–44.e6.
Song N, Li T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front Immunol [Internet]. 2018 [cited 2021 Apr 28];9. https://www.frontiersin.org/articles/10.3389/fimmu.2018.02305/full
Darling NJ, Cook SJ. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim Biophys Acta BBA. Mol Cell Res. 2014;1843:2150–63.
Arthur JSC, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13:679–92.
Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol Cell. 2017;68:185–197.e6.
Ali SR, Timmer AM, Bilgrami S, Park EJ, Eckmann L, Nizet V, et al. Anthrax Toxin Induces Macrophage Death by p38 MAPK Inhibition but Leads to Inflammasome Activation via ATP Leakage. Immunity 2011;35:34–44.
Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411.
Okada M, Matsuzawa A, Yoshimura A, Ichijo H. The Lysosome Rupture-activated TAK1-JNK Pathway Regulates NLRP3 Inflammasome Activation. J Biol Chem. 2014;289:32926–36.
Regot S, Hughey JJ, Bajar BT, Carrasco S, Covert MW. High-sensitivity measurements of multiple kinase activities in live single cells. Cell 2014;157:1724–34.
MAP kinase kinase kinases and innate immunity: Trends in Immunology [Internet]. [cited 2021 Apr 28]. https://www.cell.com/trends/immunology/comments/S1471-4906(05)00290-5
Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP. Scaffold proteins of MAP-kinase modules. Oncogene 2007;26:3185–202.
Fischer FA, Mies LFM, Nizami S, Pantazi E, Danielli S, Demarco B, et al. TBK1 and IKKε act like an OFF switch to limit NLRP3 inflammasome pathway activation. Proc Natl Acad Sci [Internet]. 2021 Sep [cited 2021 Dec 20];118. https://www.pnas.org/content/118/38/e2009309118
Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, et al. NF-κB Is a Negative Regulator of IL-1β Secretion as Revealed by Genetic and Pharmacological Inhibition of IKKβ. Cell 2007;130:918–31.
Karin M, Delhase M. The IκB kinase (IKK) and NF-κB: key elements of proinflammatory signalling. Semin Immunol. 2000;12:85–98.
de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular Mechanisms in Genetically Defined Autoinflammatory Diseases: Disorders of Amplified Danger Signaling. Annu Rev Immunol. 2015 ;33:823–74.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015;526:660–5.
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015;526:666–71.
Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020;369:1633–7.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011;469:221–5.
Ernst O, Sun J, Lin B, Banoth B, Dorrington MG, Liang J, et al. A genome-wide screen uncovers multiple roles for mitochondrial nucleoside diphosphate kinase D in inflammasome activation. Sci Signal. 2021;14:eabe0387.
Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018;560:198–203.
Gross O, Poeck H, Bscheider M, Dostert C, Hannesschläger N, Endres S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 2009;459:433–6.
Zhang B, Wei W, Qiu J. ALK is required for NLRP3 inflammasome activation in macrophages. Biochem Biophys Res Commun. 2018;501:246–52.
Lerner AG, Upton JP, Praveen PVK, Ghosh R, Nakagawa Y, Igbaria A, et al. IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 2012;16:250–64.
Pereira M, Tourlomousis P, Wright JP, Monie T, Bryant CE. CARD9 negatively regulates NLRP3-induced IL-1β production on Salmonella infection of macrophages. Nat Commun. 2016;7:12874.
Ichijo H, Nishida E, Irie K, Dijke P, ten, Saitoh M, Moriguchi T, et al. Induction of Apoptosis by ASK1, a Mammalian MAPKKK That Activates SAPK/JNK and p38 Signaling Pathways. Science 1997;275:90–4.
Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–606.
Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8:1812–25.
Tartey S, Gurung P, Dasari TK, Burton A, Kanneganti TD. ASK1/2 signaling promotes inflammation in a mouse model of neutrophilic dermatosis. J Clin Investig. 2018;128:2042–7.
Place DE, Samir P, Karki R, Briard B, Vogel P, Kanneganti TD. ASK Family Kinases Are Required for Optimal NLRP3 Inflammasome Priming. Am J Pathol. 2018;188:1021–30.
Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–92.
Gong YN, Wang X, Wang J, Yang Z, Li S, Yang J, et al. Chemical probing reveals insights into the signaling mechanism of inflammasome activation. Cell Res. 2010;20:1289–305.
Malireddi RKS, Gurung P, Mavuluri J, Dasari TK, Klco JM, Chi H, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med. 2018;215:1023–34.
Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science 2018;362:1064–9.
Evavold CL, Hafner-Bratkovič I, Devant P, D’Andrea JM, Ngwa EM, Boršić E, et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell. 2021 Jul;S0092-8674 00796-0.
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–40.
Ives A, Nomura J, Martinon F, Roger T, LeRoy D, Miner JN, et al. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat Commun. 2015;6:1–11.
Bulek K, Zhao J, Liao Y, Rana N, Corridoni D, Antanaviciute A, et al. Epithelial-derived gasdermin D mediates nonlytic IL-1β release during experimental colitis [Internet]. Am Soc Clin Investig. 2020 [cited 2021 Apr 29]. https://www.jci.org/articles/view/138103/figure/1
Stutz A, Horvath GL, Monks BG, Latz E. ASC Speck Formation as a Readout for Inflammasome Activation. In: De Nardo CM, Latz E, editors. The Inflammasome: Methods and Protocols [Internet]. Totowa, NJ: Humana Press; 2013 [cited 2021 Apr 29]. p. 91–101. (Methods in Molecular Biology). https://doi.org/10.1007/978-1-62703-523-1_8
Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21:736–45.
de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019;26:146–61.
Wandel MP, Kim BH, Park ES, Boyle KB, Nayak K, Lagrange B, et al. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol. 2020;21:880–91.
Gaudet RG, Zhu S, Halder A, Kim BH, Bradfield CJ, Huang S, et al. A human apolipoprotein L with detergent-like activity kills intracellular pathogens. Science 2021;373:eabf8113.
Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. 2016;213:2113–28.
Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1β-converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci. 2001;98:13249–54.
Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol [Internet]. 2018 Aug [cited 2020 Mar 22];3. https://immunology.sciencemag.org/content/3/26/eaat2738
Xiao J, Wang C, Yao JC, Alippe Y, Xu C, Kress D, et al. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLOS Biol. 2018;16:e3000047.
Fey D, Croucher D, Kolch W, Kholodenko B. Crosstalk and Signaling Switches in Mitogen-Activated Protein Kinase Cascades. Front Physiol. 2012;3:355
Chambers JW, LoGrasso PV. Mitochondrial c-Jun N-terminal Kinase (JNK) Signaling Initiates Physiological Changes Resulting in Amplification of Reactive Oxygen Species Generation. J Biol Chem. 2011;286:16052–62.
Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK Translocation to Mitochondria Leading to Inhibition of Mitochondria Bioenergetics in Acetaminophen-induced Liver Injury*. J Biol Chem. 2008;283:13565–77.
Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V. Cutting Edge: Reactive Oxygen Species Inhibitors Block Priming, but Not Activation, of the NLRP3 Inflammasome. J Immunol. 2011;187:613–7.
Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018;362:956–60.
Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016;535:111–6.
Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–78.
Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021;591:131–6.
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016;535:153–8.
Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat Commun. 2020;11:2212.
Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10:1689.
Zanoni I, Tan Y, Gioia MD, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 2016;352:1232–6.
Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016;44:833–46.
Acknowledgements
We thank the NIH Department of Transfusion Medicine for providing human blood-derived monocytes, Richard Flavell (Yale University) for generously providing ASC knockout mice, Sergi Regot (Johns Hopkins University) and Markus Covert (Stanford University) for providing KTR constructs and analysis help, colleagues in the Laboratory of Immune System Biology, and Lara Kohler for helpful discussions and critical reading of the paper.
Funding
This work was supported by the Intramural Research Program of NIAID, NIH.
Author information
Authors and Affiliations
Contributions
Conceptualization, CJB, OE, JJL, and IDCF; Methodology, CJB, SPJ, JS, SG, AAJ, CEB; Software, CJB, and SG; Validation, JS, OE, SPJ, CJB and JJL; Formal Analysis, CJB and IDCF; Investigation, CJB, OE, SPJ, JS, JL, and SG; Resources, AAJ, RGM, and IDCF; Data Curation, CJB, OE, SPJ, JS, JJL, SG, and IDCF; Writing—Original Draft, CJB and IDCF; Writing—Review & Editing, CJB, JJL, OE, SPJ, JS, CEB, RGM and IDCF; Visualization, CJB, SG and IDCF, Supervision, CEB, RGM, and IDCF; Project Administration, CJB and IDCF; Funding Acquisition, CEB, RGM and IDCF.
Corresponding author
Ethics declarations
Competing interests
CEB is on the scientific advisory board of Nodthera, Related Sciences and Lightcast, and is a consultant for Janssen. All other authors declare no competing interests.
Ethics approval
Human peripheral blood from screened, healthy donors was obtained under the NIH Clinical Center IRB-approved protocol 99-CC-0168 from the NIH Department of Transfusion Medicine. Mice were maintained in specific-pathogen-free conditions and all procedures were approved by the NIAID Animal Care and Use Committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by H Ichijo
Rights and permissions
About this article

Cite this article
Bradfield, C.J., Liang, J.J., Ernst, O. et al. Biphasic JNK signaling reveals distinct MAP3K complexes licensing inflammasome formation and pyroptosis. Cell Death Differ 30, 589–604 (2023). https://doi.org/10.1038/s41418-022-01106-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-022-01106-9
This article is cited by
-
(Pro)renin receptor mediates tubular epithelial cell pyroptosis in diabetic kidney disease via DPP4-JNK pathway
Journal of Translational Medicine (2024)
-
Molecular mechanisms of gasdermin D pore-forming activity
Nature Immunology (2023)
