
Abstract
Apoptosis is a regulated cellular pathway that ensures that a cell dies in a structured fashion to prevent negative consequences for the tissue or the organism. Dysfunctional apoptosis is a hallmark of numerous pathologies, and treatments for various diseases are successful based on the induction of apoptosis. Under homeostatic conditions, apoptosis is a non-inflammatory event, as the activation of caspases ensures that inflammatory pathways are disabled. However, there is an increasing understanding that under specific conditions, such as caspase inhibition, apoptosis and the apoptotic machinery can be re-wired into a process which is inflammatory. In this review we discuss how the death receptor and mitochondrial pathways of apoptosis can activate inflammation. Furthermore, we will highlight how cell death due to mitotic stress might be a special case when it comes to cell death and the induction of inflammation.
Similar content being viewed by others

Facts
-
Death receptors can signal both cell death and inflammation.
-
Through release of mtDNA, mitochondrial cell death can be inflammatory.
-
Problems during mitosis lead to cell death.
-
Mitotic stress can also induce inflammation.
Open Questions
-
Under which circumstances is physiological apoptosis inflammatory?
-
What are the differences between death receptors with regards to inflammation?
-
Why is mitotic stress inflammatory?
Introduction
Every day billions of cells die in our bodies. In order to maintain a healthy, well-functioning organism, these cells must be rapidly removed to prevent any unwanted immune responses. To cope with this constant turnover, cells have developed elegant pathways of programmed cell death which execute the cell’s demise [1, 2]. The best studied of these is apoptosis, a complex but highly regulated form of cell death which serves to activate caspase proteases resulting in the demolition of the cell. Apoptosis is executed through two main pathways: the extrinsic pathway, which is activated by death receptors on the cell membrane or in the cytosol, and the intrinsic pathway, which harnesses unique features of the mitochondria to initiate cell death. Other forms of regulated cell death, such as necroptosis and pyroptosis, are highly inflammatory, serving as powerful barriers against bacterial, viral, protozoan, and fungal infection [3]. In contrast, given the large number of cells which undergo apoptosis, apoptosis does not elicit an immune response. However, work in the last number of years has contradicted this, and in certain and specific situations apoptosis (or rather activation of the apoptotic machinery) can be highly inflammatory leading to a potent induction of various inflammatory pathways, such as NF-κB and type I interferons [4,5,6]. In this review we will summarise the recent evidence that apoptosis and activation of the extrinsic, intrinsic and mitotic apoptotic machinery can be pro-inflammatory.
The apoptotic machinery: a brief primer
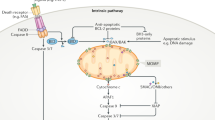
Broadly, apoptosis is activated through one of two mechanisms. The first of these is the death receptor, or extrinsic pathway. Binding of a cognate ligand to a plasma membrane-bound death receptor (such as death domain (DD)-containing TRAIL-R1, TRAIL-R2 or Fas/CD95) stimulates the trimerisation of the receptor which is then capable of recruiting FADD through homotypic DD interactions on the cytoplasmic portion of the death receptor forming the death-inducing signalling complex (DISC) [7]. FADD in turn recruits death effector domain (DED)-containing proteins such as caspase-8, which is activated and cleaves a number of substrates [8, 9]. These substrates include the effector caspases-3 and -7 (in type I cells) or BID, a pro-apoptotic BH3-only protein which permeabilises the mitochondrial outer membrane (in type II cells) [10]. In addition, other DED proteins, notably c-FLIP, also form part of the DISC and depending on levels of expression can promote or inhibit apoptosis [11,12,13,14,15] (Fig. 1). Furthermore, DISCs can form intracellularly on autophagosomal membranes, not only at the plasma membrane [16].

Ligation of the TRAIL or Fas death receptor stimulates its trimerisation. FADD binds to the cytoplasmic death domain (DD) portion of the death receptor, which in turn recruits death effector domain-containing proteins such as caspase-8, cFLIP, and caspase-10 to form the death-inducing signalling complex (DISC). Activation of caspase-8 at the DISC forms a catalytically active dimer, which can cleave a limited number of substrates, but crucially including caspases −3 and −7 and BID. Cleavage and activation of the executioner caspases −3 and −7 drives cellular demolition.
The other pathway of apoptosis is the intrinsic, or mitochondrial pathway. Upon encountering a cellular stress, such as ER stress, DNA damage, or most anti-cancer drugs, cells activate BH3-only proteins. They bind and inhibit members of the pro-survival BCL-2 family, such as BCL-2, BCL-xL and MCL-1, whose main function is to safeguard the integrity of the mitochondrial outer membrane by binding and inhibiting BAX and BAK [17] (Fig. 1). When pro-survival BCL-2 proteins are inhibited BAX and BAK are free to be activated by BH3-only proteins, and form pores in the mitochondrial outer membrane. This process is known as mitochondrial outer membrane permeabilisation, or MOMP, and is the defining hallmark of mitochondrial apoptosis. BAX/BAK pores allow for the efflux of a number of different intermembrane space proteins, such as cytochrome c, SMAC, and Omi [18]. Upon entering the cytoplasm, cytochrome c binds and facilitates the formation of the apoptosome, comprised of multiple copies of APAF-1, which acts as an activation platform for caspase-9 [19]. Active caspase-9 cleaves a number of proteins, but most importantly it cleaves and activates caspases −3 and −7, which together execute the demolition phase of apoptosis [20, 21].
Inflammation arising from death receptors
Although most studies of death receptors focus on death as the outcome, death is not always inevitable from death receptor activation. Amongst the many studies which focus on the death of a cell from death receptor activation, there are reports that both TRAIL and Fas receptors promote the production of pro-inflammatory chemokines and cytokines [22,23,24,25] and that cells can survive TRAIL treatment [26]. Moreover, ligation of TRAIL and Fas death receptors can have many other outcomes, such as increased cell invasiveness [27], cancer metastasis [28,29,30], proliferation [31], activation of dendritic cells [32], and entosis [33]. Indeed, the founding member of the tumour necrosis factor (TNF) superfamily, TNF, despite its name rarely kills cells, and requires the blockade of NF-κB-mediated transcription to do so (see Box 1). Together, the observations that TRAIL or Fas ligation can have pro-survival roles in part explains the relative lack of success of TRAIL receptor agonists in the clinic, particularly given that TRAIL can actually promote tumourigenesis [34].
At the molecular level, complexes formed following TRAIL and Fas receptor ligation are more complicated than originally understood. Initially, TRAIL receptor activation leads to the formation of a plasma membrane-bound DISC complex, comprised of the adaptor protein FADD, caspase-8, the long and short isoforms of c-FLIP, TRAF2, and RIPK1, among others, termed complex I. Here, ubiquitination events determine the signalling outcomes, whether that be cell death or gene expression. For example, caspase-8 can be modified with K63 ubiquitin chains by the E3 ligase cullin-3 [35], promoting its activation, whereas TRAF2 can modify caspase-8 with K48 ubiquitin chains, which trigger the proteasomal destruction of caspase-8 [36]. In this way, temporal changes in ubiquitination can act as a molecular timer of caspase-8 activation. More recently, the linear ubiquitin chain assembly complex (LUBAC) has been shown to ubiquitinate caspase-8 and RIPK1 with linear chains whilst they are in complex I [37]. LUBAC-mediated ubiquitination of caspase-8 serves to limit caspase-8 activation and, through NEMO binding to linear ubiquitin chains, recruits the IKK complex. LUBAC also ubiquitinates caspase-8 in complex II, a complex which forms after complex I previously thought to be mainly responsible for gene activation [38]. However, it is now understood that both TRAIL complexes I and II can mediate apoptosis and cytokine production [37].
What is the biological significance of cytokine and chemokine production in response to TRAIL and Fas receptor signalling? This has perhaps been best studied in the context of cancer, especially since cytokines and chemokines have been shown to be important in the tumour microenvironment [39]. Cancer cells treated with TRAIL secrete a vast array of proteins, including the cytokines and chemokines CXCL1, CXCL4, CCL2, IL-8, and NAMPT [40]. Importantly, these cytokines and chemokines are secreted by cells which survive TRAIL treatment, rather than those which die, and strictly require the presence of FADD and caspase-8 (although studies differ as to whether TRADD is required) [40,41,42]. However, it does not require the caspase activity of caspase-8, as treatment with the pan-caspase inhibitor QVD does not alter the inflammatory outcome [42]. In line with this, Henry & Martin show that TRAIL treatment results in the production of cytokines and chemokines in HeLa cells, which can be uncoupled from cell death [42]. While caspase-8 activity is dispensable for this cytokine production, it requires FADD, RIPK1 and caspase-8 protein expression. Cytokine and chemokine secretion also occurs when cells are treated with doses of TRAIL insufficient to cause cell death. In this scenario, procaspase-8 aids the assembly of a cytoplasmic “FADDosome” complex consisting of FADD, RIPK1 and caspase-8, also known as complex II to draw parallels to TNF signalling (Fig. 2). Formation of the “FADDosome”/complex II drives NF-κB-dependent inflammatory gene expression. Additionally, LUBAC is recruited to both TRAIL complex I and II promoting gene expression and preventing apoptosis [37]. What is the function of this in cancer models? Non-small cell lung cancer (NSCLC) patients with high levels of FADD mRNA have poorer survival than patients with low FADD mRNA [43], leading to the tempting suggestion that the FADDosome might form downstream of TRAIL receptor ligation in cancers, driving inflammatory gene expression. Accordingly, deletion of FADD in a mouse model of NSCLC significantly decreased tumour burden as well as levels of IL-8, CXCL1 and CCL2 [40]. Furthermore, secretion of TRAIL/TRAIL receptor-dependent CCL2 produces a tumour-supportive microenvironment in which cancers can thrive. The FADDosome also forms in vivo following activation of the Fas receptor in mice lacking MLKL (crucial for necroptosis) and harbouring a non-cleavable caspase-8 (Casp8DA/DAMlkl-/-), and deletion of one allele of Fasl, Fadd or Ripk1 was sufficient to abrogate FADDosome-dependent inflammation, highlighting the important role for this complex in vivo [44]. Interestingly, selective expression of different head and neck cancer associated-caspase-8 mutations show different capacities to mediate inflammation [45]. For example, the D303G mutation appears to increase chemokine and cytokine production in cells and in xenograft in vivo models. It will be interesting to see if these findings can be confirmed in more sophisticated mouse models of cancer.

Ligation of the TRAIL receptor stimulates the formation of the death-inducing signalling complex (DISC). FADD is the first to be recruited, which allows the recruitment of multiple copies of caspase-8 through death-effector domain (DED) interactions. RIPK1 is also recruited, and can be modified with ubiquitin chains by cIAP1/2. This complex can then dissociate from the plasma membrane, forming the FADDosome which can activate NF-κB-dependent gene transcription and cytokine and chemokine production.
Taxanes are a group of microtubule poisons that induce mitotic arrest and cell death via various cell death modalities [46] and are commonly used in the clinic for the treatment of lung, breast, and ovarian cancers. There is extensive evidence in the literature showing that taxanes invoke inflammatory responses in cancers, facilitating cancer progression and resistance [47,48,49,50,51]. However, in addition to being microtubule poisons that induce cell death, they also robustly induce endoplasmic reticulum (ER) stress, which is known to cause ligand-independent TRAIL receptor activation, although this remains the topic of some debate [52,53,54,55]. How TRAIL receptors can activate and signal without a ligand was a mystery. Whilst others have reported that misfolded proteins can directly bind and activate TRAIL-R2 to invoke cell death [56], Sullivan et al showed that ER stress leads to the transcriptional upregulation of TRAIL receptors, which can then stimulate the formation of the FADDosome complex, activating NF-κB and expression of cytokines and chemokines [57] (Fig. 2). This induction of inflammation does not fit within the usual pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) paradigm [58], and so a new term has been coined: stress-associated molecular patterns (SAMPs), which could potentially form in response to a wide variety of stressors.
Similar to TRAIL/TRAIL-R signalling, binding of Fas ligand to the Fas receptor stimulates the formation of a FADD/caspase-8/c-FLIP DISC which is capable of activating caspase-8 and inducing rapid cell death. Until recently, apoptosis was thought to be an immune-silent event, which does not invoke an immune response. However, work in the last decade has shown that dying cells secrete a number of “find-me” signals that attract phagocytic cells to the dying cell to aid in the removal of the corpse. These “find-me” signals include ATP [59], lysophosphatidylcholine (LPC) [60], and IL-8, among others [61]. More recently, it was found that upon stimulation with Fas ligand, multiple cell types can secrete more conventional cytokines and chemokines, such as IL-6, CCL2, CXCL1, and sICAM-1 in a manner that can be uncoupled from cell death [62]. Importantly, this appears to be dependent on inhibitor of apoptosis (IAP) proteins, as treatment with BV6 (an IAP antagonist) blocks the production of cytokines as well as RIPK1 [62].
Inflammation arising from the mitochondrial cell death machinery
The large majority of apoptosis is executed via the mitochondrial cell death pathway. In a healthy cell, the mitochondrial outer membrane is kept under constant guard by a delicate balance of pro- and anti-apoptotic proteins, which together maintain membrane integrity. Upon encountering a cellular insult, the balance of these proteins is disrupted in such a way that it tips in favour of pro-apoptotic proteins, allowing BAX/BAK to form pores on the mitochondrial outer membrane [17] (Fig. 1). Through these pores flow a number of different proteins, such as cytochrome c, SMAC and Omi, and when they enter the cytoplasm they activate caspases [17]. For example, when cytochrome c enters the cytoplasm it triggers the formation of the apoptosome, a multimeric complex of APAF-1 at which the initiator procaspase-9 is activated. This triggers a caspase cascade which activates caspases −3 and −7 which ultimately demolish the cell by cleaving a large array of substrates. Similarly, when SMAC enters the cytoplasm it binds and neutralizes members of the IAP family, cellular inhibitors of caspases. Importantly, mitochondrial permeabilisation is a point-of-no-return, and once it has occurred, the cell will die regardless of caspase activation.
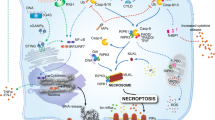
In contrast to other forms of cell death, such as necrosis, apoptosis has been considered a largely regulated and “immune-silent” form of cell death. However, work from the past number of years has shown that mitochondrial apoptosis in particular is far from immune-silent and in fact has the potential to trigger potent immune responses under specific conditions, such as when caspases are inhibited. One of the major drivers of this is due to the origins of mitochondria, which arrived into a host cell related to Asgard archaea as an endosymbiotic alphaproteobacterium, and over time have become adapted to the host through significant proteome rewiring [63, 64]. However, due to this bacterial origin they have maintained their own independent genome, mitochondrial DNA (mtDNA). Usually mtDNA is kept inside the mitochondria and away from innate immune sensors in the cytoplasm. But what happens if the mitochondrial membranes are permeabilised? A number of studies in 2014 were the first to show that pharmacological or genetic blockade of caspases was sufficient to drive a cGAS-STING type I interferon response in cells [5, 6]. Further investigation revealed that mtDNA was being released from the mitochondria into the cytoplasm. Importantly, the activity of apoptotic caspases (such as caspase-9 and caspase-3/7) was enough to block this inflammatory response, rendering apoptosis immunologically silent. Using super-resolution microscopy, we and others have shown that BAX/BAK pores during the initial stage of MOMP are large enough to only permit the release of small proteins, such as cytochrome c. However, these initial pores rapidly unite to form large BAX/BAK “macropores” through which the mitochondrial inner membrane herniates, ruptures, and allows the release of mtDNA into the cytoplasm [65,66,67]. Once in the cytoplasm, it is detected by cGAS, which catalyses production of a second messenger cGAMP, which then activates STING. STING subsequently activates TBK1, which phosphorylates IRF3 allowing the expression of type I interferon genes [68] (Fig. 3). Importantly, it should be noted that for MOMP-induced inflammation to proceed, caspases must be inhibited. One of the reasons for this is that proteins in innate immune pathways, such as cGAS, IRF3 and MAVS are rapidly cleaved and inactivated by caspases, helping to maintain the immuno-silent nature of apoptosis [69].

Permeabilisation of the mitochondrial outer membrane by BAX/BAK can trigger inflammation in a number of different ways. Firstly, following MOMP, the inner membrane herniates and permeabilises, allowing the efflux of mtDNA into the cytoplasm where it activates cGAS-STING-dependent inflammatory responses, such as activation of type I interferon responses and NF-κB. SMAC release during MOMP triggers IAP degradation and NIK activation, leading to NF-κB activation. During infection with viruses, such as SFTSV, BAX/BAK pores facilitate the release of oxidized mtDNA, which can bind and activate the NLRP3 inflammasome, triggering NF-κB responses. Finally, mitochondrial RNA can be released via BAX/BAK pores, which are detected by MDA5 and MAVS in the cytoplasm, triggering a type I interferon response.
The release of mtDNA during cell death has now been noted in a variety of different pathological settings [70]. In cancer, inducing mitochondrial cell death with BH3 mimetics while simultaneously inhibiting caspase activity may seem counterintuitive, but has actually been shown to be a more effective means of controlling tumour growth. Using a variety of in vitro and in vivo models of cancer, Giampazolias et al. showed that deleting APAF-1 or pharmacological caspase inhibition not only activated NF-κB-dependent inflammation (through the release of SMAC and activation of NIK), but also cGAS-STING-dependent type I interferon responses (through the release of mtDNA into the cytoplasm) [4]. In a mouse model of breast cancer, irradiation causes MOMP, leading to the release of mtDNA through BAX/BAK pores [71]. In this scenario, if autophagy is inhibited mtDNA accumulates in the cytoplasm, triggering cGAS-STING-dependent type I interferon responses, a process that bears resemblance to mtDNA in heart failure [72]. In mouse models of colorectal cancer, inhibiting apoptotic caspase activity using FDA-approved emricasan/IDN-6556 combined with radiation enhances anti-tumour immunity, resulting in better tumour control [73]. Together, this strongly suggests that targeting MOMP while simultaneously inhibiting caspases could be a new strategic approach to treating a variety of different cancers.
The cGAS-STING axis is not the only immune pathway activated by mtDNA release into the cytoplasm. Some viruses, such as severe fever with thrombocytopenia syndrome virus (SFTSV) also release mtDNA in a BAX/BAK-dependent mechanism (though the precise mechanism is unclear); however the mtDNA is not recognised by cGAS but rather by the NLRP3 inflammasome, a multiprotein cytosolic sensor of DNA [74] (Fig. 3). Caspase-1 is recruited to, and activated at, the NLRP3 inflammasome, which enables the cleavage and secretion of IL-1β. IL-1β is secreted by cells as part of an innate anti-microbial defence system. Interestingly, cells exposed to IL-1β are known to activate cGAS-STING signalling, which is now understood to be due to mtDNA release [75]. While not formally tested, the authors speculate that this might be due to BAX/BAK pore formation, since FOXO3 is activated following IL-1β stimulation, and there is evidence, albeit limited, suggesting that FOXO3 is linked to a “transient MOMP” [76, 77]. Similarly, TNF treatment induces the release of mtDNA into the cytoplasm in a model of arthritis, and again, although not formally tested, it seems plausible that this could be due to BAX/BAK pore formation [78]. Although this observation may be completely unrelated to apoptosis since there appears to be no loss of cell viability, it is intriguing that pharmacological caspase inhibition with zVAD-fmk enhances interferon production. Thus, it is tempting to speculate that there may be some degree of non-lethal “minority MOMP” (see below), although this should be formally tested.
In another example of inflammatory signals flowing from permeabilised mitochondria, SMAC release via BAX/BAK pores can trigger NLRP3 inflammasome activation in macrophages. Here, released SMAC binds and facilitates the degradation of IAPs, licensing formation of the Ripoptosome and caspase-8 activation which enables IL-1β secretion [79] (Fig. 3). Alternatively, SMAC release can also lead to IAP depletion allowing NLRP3 inflammasome formation which can likewise lead to IL-1β secretion, in a manner independent of gasdermin proteins [80].
It should be noted that not all mtDNA release occurs as a result of BAX/BAK-mediated permeabilisation of the mitochondrial outer membrane. Amyotrophic lateral sclerosis (ALS) is characterised by cytoplasmic accumulation of TDP-43, a nuclear DNA/RNA binding protein, which can enter the mitochondria [81,82,83]. TDP-43 expression in cells triggers the release of mtDNA into the cytoplasm through the mitochondrial permeability transition pore (mPTP) where it is detected by cGAS, activating a STING-dependent inflammatory response [84]. In a mouse model of the autoimmune disease lupus, voltage-dependent anion channels (VDACs) can form pores in the mitochondrial outer membrane in response to oxidative stress, leading to mtDNA expulsion and cGAS-STING-dependent inflammation [85]. VDAC channels have also been implicated in mtDNA release observed in skin and lung samples from patients infected with SARS-CoV-2 [86]. Given the intimate relationship between BAX/BAK and VDACs, it is possible BAX/BAK may have a role [87], though the requirement for VDACs in mitochondrial permeabilisation is still a subject of debate [88, 89]. Finally, gasdermins D and E (GSDMD, GSDME) are pore forming proteins more recognised for their roles in permeabilising the plasma membrane during pyroptosis. However, a number of laboratories have shown that GSDMD and GSDME can both permeabilise the mitochondrial outer membrane [90, 91], and in the case of NLRP3 inflammasome activation, GSDMD can permeabilise the mitochondria to allow the efflux of mtDNA [91].
In addition to mtDNA, mitochondria also contain mtRNA. Under homeostatic conditions, this mtRNA is degraded by the degradosome, principally comprised of the mitochondrial RNA helicase SUV3 and polynucleotide phosphorylase PNPase. When this is disrupted (for example in patients with mutations in PNPase) mtRNA accumulates. In such pathogenic conditions, mtRNA is released through BAX/BAK pores on the mitochondrial outer membrane and into the cytoplasm where it is detected by the RNA sensor MDA5, which through MAVS elicits a type I interferon response [92] (Fig. 3). Similarly, mitochondrial-targeted transcription activator-like effector nuclease (TALEN)-mediated induction of mtDNA damage or radiation activates BAX/BAK pore formation on the mitochondrial outer membrane, facilitating the release of mtRNA which is detected by RIG-I (and presumably MDA5), triggering a type I interferon response [93] (Fig. 3). Notably, BAX/BAK pore formation following mtDNA damage induced by TALENs is not apoptotic, and thus does not cause caspase activation. How BAX/BAK can form pores on the mitochondrial outer membrane that does not result in apoptotic signalling is intriguing, but as of yet unclear.
Mitochondrial permeabilisation as an immune alarm
Permeabilisation of the mitochondrial outer membrane has always been thought of as an all-or-nothing event, in that within a single cell all mitochondria undergo MOMP and the cell rapidly dies [94, 95]. However, the general idea that all cells treated with a cell death-inducing stimulus die has been challenged: in 2010, Lovric & Hawkins noted that in a given population of cells treated with TRAIL some cells die, but not all [26]. Although they did not test a role for mitochondria, the cells which survive often have mutations dependent on caspase-8 activation, showing that surviving cell death creates mutations in the cells that do not die. Subsequently, we and others showed that treating cells with sub-lethal doses of drugs which invoke cellular stresses or anti-cancer drugs was able to permeabilise only a few mitochondria within a cell, challenging the notion that MOMP occurs in all mitochondria in a cell. Importantly, in cells which undergo “minority MOMP”, we were able to detect a small, but significant, degree of caspase activation, enough to cleave the inhibitor of caspase-activated DNase (ICAD), releasing active CAD which induces DNA damage and genomic instability [96, 97]. We also recently defined the underlying basis of this process, showing that minority MOMP induced by BCL-2 antagonism occurs preferentially on dysfunctional, fragmented mitochondria, which possess higher levels of anti-apoptotic BCL-2 expression and higher BAX due to reduced retrotranslocation, rendering them more primed towards minority MOMP [98].
Since mtDNA can be extruded from mitochondria during cell death, and that it is possible that only a limited subset of mitochondria can undergo MOMP in a given cell, Brokatzky et al set out to test the possibility that pathogens could trigger minority MOMP. Indeed, they were able to show that sub-lethal doses of BH3 mimetics was sufficient to trigger IL-6 release in a BAX/BAK-, BCL-xL- and STING-dependent manner [99]. Furthermore, when screening various of bacterial, viral and protozoan pathogens they were able to show that infection also causes minority MOMP and DNA damage. In cells, the function of this is likely to induce MOMP to trigger mtDNA release (or release of other proteins) to activate cGAS-STING and elicit a type I interferon response. In this manner, the infected cell becomes an “alarm” for the surrounding cells, perhaps to warn them of a potential threat. Similarly, infection with Helicobacter pylori also induces sub-lethal caspase activation and inflammatory signalling. In contrast to other pathogens however, Hp infection seems to only permit the release of SMAC from BAX/BAK pores, and not cytochrome c. How SMAC can be preferentially released from BAX/BAK pores remains to be elucidated, but it is in line with other data [100, 101]. One tempting hypothesis may be drawn from recent data from the Garcia-Saéz laboratory, who showed that BAX and BAK form different-sized pores at divergent rates. It has been known for a number of years that BAX and BAK can form pores that resemble lines, arcs and rings [102, 103]; however we now know that BAK can oligomerise and form pores faster, but at a smaller size than BAX [104]. Crucially, this means cells which predominantly express BAK release mtDNA faster than cells which only express BAX. The mitochondrial protein ERA G-protein-like 1 (ERAL1) is also released from the mitochondrial matrix through BAX/BAK pores following infection with RNA viruses such as VSV and Sendai virus [105]. Translocation of ERAL1 into the cytoplasm appears to promote ubiquitination of RIG-I and MDA5, which promote oligomerisation of MAVS on the mitochondrial outer membrane, resulting in anti-viral responses [105]. However, it is not clear from this study how cells remain alive, as formation of the BAX/BAK pore would usually result in a rapid and complete cell death, so it is possible that other processes, such as minority MOMP and/or caspase inhibition could play a role.
Inflammation arising from mitotic cell death
The correct and error-free execution of cell division is essential for an organism to prevent the introduction or propagation of damaged or dangerous cells [106]. Errors introduced during mitosis can have a profoundly negative impact if passed on to daughter cells. Cells have therefore developed several strict checkpoints during the cell cycle that have to be passed before they can progress [106]. Failure to fulfill these checkpoint requirements can lead to a stall in mitosis to remedy the underlying reason of the failure to pass the checkpoint. If this stall cannot be overcome, e.g., if the sustained damage is too severe, the cell undergoes cell death. While cell death due to errors in mitosis often uses much of the same machinery as the intrinsic pathway of apoptosis described previously, particularly in the execution of the death, how it is specifically induced is still a matter of active research and not yet understood in detail.
Recent publications suggest that activation of caspase-2 in a complex called the PIDDosome promotes cleavage and inactivation of MDM2, which under basal conditions promotes degradation of p53 [107,108,109,110]. Activation of p53 can then push cells towards cell cycle arrest, senescence, or apoptosis, the distinction between those different outcomes believed to be due to the dynamics and sustained levels of increased p53 [111,112,113]. In some cancer cells, active nuclear caspase-8 cleaves the deubiquitinase USP28 in response to DNA damage. The cleaved USP28 is therefore incapable of stabilizing p53, enabling the cell to progress in mitosis instead of undergoing p53-mediated apoptosis [114].
Like apoptosis triggered by other stresses, mitotic cell death relies on activation of pro-apoptotic BH3-only proteins and concomitant inactivation of pro-survival BCL-2 family members [115]. Several of these pro- and anti-apoptotic proteins are modified by components of the cell cycle machinery. While in reality the picture might be more complicated and is still to be elucidated, current models suggest that the interplay between mitotic progression and induction of apoptosis ultimately decides the fate of a cell encountering problems during mitosis.
In addition to the intrinsic pathway, components of the extrinsic pathway interface with the regulation of mitosis. A non-apoptotic and non-inflammatory “Ripoptosome” (see Box 1) forms during mitosis to ensure that chromosomes are segregated correctly [116]. Furthermore, phosphorylation of procaspase-8 by Cdk1/cyclin B1 and polo-like kinase 1 (Plk1) prevents its activation during mitosis [117], while phosphorylation of FADD by casein kinase 1α is required for the progression of mitosis [118].
While canonical apoptosis is considered immunologically silent and not able to trigger an inflammatory response, does the same hold true for mitotic cell death? While clear evidence that mitotic cell death itself is inflammatory is unavailable, some features leading to its induction make it more likely to induce an inflammatory response. One trigger of mitotic cell death is DNA damage, induced by radiation or genotoxic drugs. Additionally, in a pathway reminiscent of minority MOMP (see above), prolonged mitotic arrest can lead to limited activation of caspases. After caspase-mediated cleavage of ICAD, activated CAD can then cleave genomic DNA and thereby induce DNA damage [119]. Even though it has been known for a while that DNA damage can induce inflammation, this activation seems to be independent of mitosis and rather mediated by the canonical DNA damage response [120, 121]. DNA damage occurred during mitosis can lead to DNA double-strand breaks and the formation of micronuclei, fragments of chromosomes encapsulated by nuclear membranes outside the nucleus. These micronuclei can then be detected by cGAS, leading to activation of an interferon response and inflammation [122, 123]. A similar effect has also been described during senescence: here cytoplasmic chromatin fragments (CCF) translocate from the nucleus to the cytoplasm, where they are subsequently detected by cGAS and induce inflammation [124,125,126]. These data are in line with the general perception that cGAS is a sensor of “out of place” dsDNA, for example from pathogens or mitochondria. Chromatin fragments, micronuclei or other sources of nuclear DNA in the cytoplasm are detected by cGAS and constitute signs that necessitate an inflammatory response. Several mechanisms exist to prevent accidental activation of cGAS, which can also be found in the nucleus, such as its tight tethering to nucleosomes [127]. Inhibition of cGAS in this way also prevents its activation during mitosis, when the nuclear envelope breaks down and nuclear DNA is exposed to cGAS.
Surprisingly, activation of the cGAS-STING pathway itself can eventually lead to the induction of mitotic cell death [128]. Here, prolonged mitotic arrest leads to low-level activation of IRF3 by cGAS. Interestingly, while this IRF3 activation is insufficient to induce an inflammatory response, by a yet undefined mechanism this cGAS-STING-IRF3 axis promotes MOMP via inactivation of BCL-xL [128] (Fig. 4).

DNA damage or other mitotic stresses can lead to defects in mitosis, including chromosomal instability and micronuclei. These issues are detected by various surveillance mechanisms to prevent the propagation of potentially mutated or otherwise harmful cells. Micronuclei or cytoplasmic chromatin fragments are detected by the cGAS-STING pathway, leading to the activation of type I interferon, NF-κB signaling, and subsequently inflammation, but also metastasis or cell death. Supernumerary centrosomes promote the activation of the PIDDosome, which cleaves and inhibits MDM2. Therefore, p53 is stabilized and can promote apoptosis, cell cycle arrest or senescence. Mitotic stress also shifts the balance of pro-survival BCL-2 proteins and proapoptotic BH3-only proteins to promote apoptosis.
In cancer, activation of cGAS due to chromosomal instability and micronuclei can also have negative effects. Mediated by STING, tumor cells with chromosomal instability induce NF-κB signaling and inflammation, promoting metastasis [129]. These results are in accordance with previous research showing heightened carcinogenesis due to STING-dependent inflammation [130]. Remarkably, tumour cells have even developed mechanisms to counteract anti-tumour immunity evoked by STING in chromosomally instable cells. By expressing the ectonucleotidase ENPP1, they degrade the cGAMP that is produced by cGAS and excreted to the extracellular space [131]. Therefore, cGAMP cannot be recognised by surrounding immune cells and activate an anti-tumour response in them. In this way, the tumor can become resistant to elimination by the host immune system.
Concluding remarks
It is now evident that in addition to being immuno-silent, apoptosis can be, under certain circumstances, an inflammatory event. Active areas of current research include uncovering in which physiological settings apoptosis can be inflammatory, independent of pharmacological or genetic inhibition of caspases, as well as how it can be exploited therapeutically. For example, cardiomyocytes show a decreased expression of APAF-1 [132, 133], rendering them functionally deficient in the activation of caspase-9 and thus intrinsic apoptosis. Whether this deficiency results in increased inflammation of the heart when cardiomyocytes undergo mitochondrial permeabilisation is an interesting avenue for future research, particularly since heart disease is the leading cause of death in the developed world.
Another interesting potential role of apoptosis-induced inflammation mediated by caspase inhibition is during cellular infection with viruses, which often express proteins that inhibit caspases [134]. These inhibitors, therefore, prevent apoptosis of the infected cell and give the virus more time to replicate. One can speculate that inflammation induced by the induction of apoptosis triggered by viral infection, in combination with viral caspase inhibition, is an additional cellular mechanism to boost inflammation to fight the virus.
Clinically, some chemotherapeutic drugs like doxorubicin have been shown to induce an inflammatory response even under caspase-proficient conditions [135]. However, it is believed that these and some other drugs initiate a specific form of cell death termed “immunogenic cell death” [136], characterised by the release of several DAMPs which activate the immune system.
While there is an increasing number of reports suggesting an inflammatory outcome of apoptosis, so far they are limited to fairly specific situations. It remains to be seen whether apoptosis is potentially inflammatory, or if this controversial role is merely a byproduct of the circumstances.
References
Green DR, Victor B. The pantheon of the fallen: why are there so many forms of cell death? Trends Cell Biol. 2012;22:555–6.
Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. 2020;21:398–414.
Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17:151–64.
Giampazolias E, Zunino B, Dhayade S, Bock F, Cloix C, Cao K, et al. Mitochondrial permeabilization engages NF-κB-dependent anti-tumour activity under caspase deficiency. Nat Cell Biol. 2017;19:1116–29.
Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014;159:1563–77.
White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014;159:1549–62.
Riley JS, Malik A, Holohan C, Longley DB. DED or alive: assembly and regulation of the death effector domain complexes. Cell Death Dis. 2015;6:e1866.
Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J, et al. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000;12:599–609.
Scott FL, Stec B, Pop C, Dobaczewska MK, Lee JJ, Monosov E, et al. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009;457:1019–22.
Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 2009;460:1035–9.
Micheau O, Thome M, Schneider P, Holler N, Tschopp J, Nicholson DW, et al. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem. 2002;277:45162–71.
Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, et al. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J. 2011;433:447–57.
Safa AR. c-FLIP, a master anti-apoptotic regulator. Exp Oncol. 2012;34:176–84.
Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, et al. Co-operative and hierarchical binding of c-FLIP and Caspase-8: A unified model defines how c-FLIP isoforms differentially control cell fate. Mol Cell. 2016;61:834–49.
Majkut J, Sgobba M, Holohan C, Crawford N, Logan AE, Kerr E, et al. Differential affinity of FLIP and procaspase 8 for FADD’s DED binding surfaces regulates DISC assembly. Nat Commun. 2014;5:3350.
Hattori T, Takahashi Y, Chen L, Tang Z, Wills CA, Liang X, et al. Targeting the ESCRT-III component CHMP2A for noncanonical Caspase-8 activation on autophagosomal membranes. Cell Death Differ. 2021;28:657–70.
Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85–100.
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 1997;275:1132–6.
Dorstyn L, Akey CW, Kumar S. New insights into apoptosome structure and function. Cell Death Differ. 2018;25:1194–208.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997;91:479–89.
Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–57.
Berg D, Stuhmer T, Siegmund D, Muller N, Giner T, Dittrich-Breiholz O, et al. Oligomerized tumor necrosis factor-related apoptosis inducing ligand strongly induces cell death in myeloma cells, but also activates proinflammatory signaling pathways. FEBS J. 2009;276:6912–27.
Altemeier WA, Zhu X, Berrington WR, Harlan JM, Liles WC. Fas (CD95) induces macrophage proinflammatory chemokine production via a MyD88-dependent, caspase-independent pathway. J Leukoc Biol. 2007;82:721–8.
Farley SM, Dotson AD, Purdy DE, Sundholm AJ, Schneider P, Magun BE, et al. Fas ligand elicits a caspase-independent proinflammatory response in human keratinocytes: implications for dermatitis. J Invest Dermatol. 2006;126:2438–51.
Park DR, Thomsen AR, Frevert CW, Pham U, Skerrett SJ, Kiener PA, et al. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J Immunol. 2003;170:6209–16.
Lovric MM, Hawkins CJ. TRAIL treatment provokes mutations in surviving cells. Oncogene 2010;29:5048–60.
Barnhart BC, Legembre P, Pietras E, Bubici C, Franzoso G, Peter ME. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. EMBO J. 2004;23:3175–85.
Trauzold A, Siegmund D, Schniewind B, Sipos B, Egberts J, Zorenkov D, et al. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 2006;25:7434–9.
von Karstedt S, Conti A, Nobis M, Montinaro A, Hartwig T, Lemke J, et al. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell. 2015;27:561–73.
Hoogwater FJ, Nijkamp MW, Smakman N, Steller EJ, Emmink BL, Westendorp BF, et al. Oncogenic K-Ras turns death receptors into metastasis-promoting receptors in human and mouse colorectal cancer cells. Gastroenterology 2010;138:2357–67.
Chen L, Park SM, Tumanov AV, Hau A, Sawada K, Feig C, et al. CD95 promotes tumour growth. Nature 2010;465:492–6.
Rescigno M, Piguet V, Valzasina B, Lens S, Zubler R, French L, et al. Fas engagement induces the maturation of dendritic cells (DCs), the release of interleukin (IL)-1beta, and the production of interferon gamma in the absence of IL-12 during DC-T cell cognate interaction: a new role for Fas ligand in inflammatory responses. J Exp Med. 2000;192:1661–8.
Bozkurt E, Dussmann H, Salvucci M, Cavanagh BL, Van Schaeybroeck S, Longley DB, et al. TRAIL signaling promotes entosis in colorectal cancer. J Cell Biol. 2021;220:e202010030.
von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer. 2017;17:352–66.
Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009;137:721–35.
Gonzalvez F, Lawrence D, Yang B, Yee S, Pitti R, Marsters S, et al. TRAF2 Sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol Cell. 2012;48:888–99.
Lafont E, Kantari-Mimoun C, Draber P, De Miguel D, Hartwig T, Reichert M, et al. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 2017;36:1147–66.
Varfolomeev E, Maecker H, Sharp D, Lawrence D, Renz M, Vucic D, et al. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem. 2005;280:40599–608.
Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17:559–72.
Hartwig T, Montinaro A, von Karstedt S, Sevko A, Surinova S, Chakravarthy A, et al. The TRAIL-induced cancer secretome promotes a tumor-supportive immune microenvironment via CCR2. Mol Cell. 2017;65:730–42 e5.
Grunert M, Gottschalk K, Kapahnke J, Gundisch S, Kieser A, Jeremias I. The adaptor protein FADD and the initiator caspase-8 mediate activation of NF-kappaB by TRAIL. Cell Death Dis. 2012;3:e414.
Henry CM, Martin SJ. Caspase-8 acts in a non-enzymatic role as a scaffold for assembly of a pro-inflammatory “FADDosome” complex upon TRAIL stimulation. Mol Cell. 2017;65:715–29 e5.
Chen G, Bhojani MS, Heaford AC, Chang DC, Laxman B, Thomas DG, et al. Phosphorylated FADD induces NF-kappaB, perturbs cell cycle, and is associated with poor outcome in lung adenocarcinomas. Proc Natl Acad Sci USA. 2005;102:12507–12.
Tummers B, Mari L, Guy CS, Heckmann BL, Rodriguez DA, Ruhl S, et al. Caspase-8-dependent inflammatory responses are controlled by its adaptor, FADD, and Necroptosis. Immunity. 2020;52:994–1006.
Cui Z, Dabas H, Leonard BC, Shiah JV, Grandis JR, Johnson DE. Caspase-8 mutations associated with head and neck cancer differentially retain functional properties related to TRAIL-induced apoptosis and cytokine induction. Cell Death Dis. 2021;12:775.
Sazonova EV, Kopeina GS, Imyanitov EN, Zhivotovsky B. Platinum drugs and taxanes: can we overcome resistance? Cell Death Disco. 2021;7:155.
Collins TS, Lee LF, Ting JP. Paclitaxel up-regulates interleukin-8 synthesis in human lung carcinoma through an NF-kappaB- and AP-1-dependent mechanism. Cancer Immunol Immunother. 2000;49:78–84.
Ding AH, Porteu F, Sanchez E, Nathan CF. Shared actions of endotoxin and taxol on TNF receptors and TNF release. Science 1990;248:370–2.
Wang TH, Chan YH, Chen CW, Kung WH, Lee YS, Wang ST, et al. Paclitaxel (Taxol) upregulates expression of functional interleukin-6 in human ovarian cancer cells through multiple signaling pathways. Oncogene 2006;25:4857–66.
Dong QG, Sclabas GM, Fujioka S, Schmidt C, Peng B, Wu T, et al. The function of multiple IkappaB: NF-kappaB complexes in the resistance of cancer cells to Taxol-induced apoptosis. Oncogene 2002;21:6510–9.
Olson OC, Kim H, Quail DF, Foley EA, Joyce JA. Tumor-associated macrophages suppress the cytotoxic activity of antimitotic agents. Cell Rep. 2017;19:101–13.
Iurlaro R, Puschel F, Leon-Annicchiarico CL, O’Connor H, Martin SJ, Palou-Gramon D, et al. Glucose deprivation induces ATF4-mediated apoptosis through TRAIL death receptors. Mol Cell Biol. 2017;37:e00479–16.
Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS, et al. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014;345:98–101.
Glab JA, Doerflinger M, Nedeva C, Jose I, Mbogo GW, Paton JC, et al. DR5 and caspase-8 are dispensable in ER stress-induced apoptosis. Cell Death Differ. 2017;24:944–50.
Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–97.
Lam M, Marsters SA, Ashkenazi A, Walter P Misfolded proteins bind and activate death receptor 5 to trigger apoptosis during unresolved endoplasmic reticulum stress. Elife. 2020;9:e52291.
Sullivan GP, O’Connor H, Henry CM, Davidovich P, Clancy DM, Albert ML, et al. TRAIL Receptors Serve as Stress-Associated Molecular Patterns to Promote ER-Stress-Induced Inflammation. Dev Cell. 2020;52:714–730.
Matzinger P. The danger model: a renewed sense of self. Science 2002;296:301–5.
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009;461:282–6.
Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003;113:717–30.
Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity 2011;35:445–55.
Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, Tynan GA, et al. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol Cell. 2013;49:1034–48.
Roger AJ, Munoz-Gomez SA, Kamikawa R. The origin and diversification of Mitochondria. Curr Biol. 2017;27:R1177–R92.
Gogoi J, Bhatnagar A, Ann KJ, Pottabathini S, Singh R, Mazeed M, et al. Switching a conflicted bacterial DTD-tRNA code is essential for the emergence of mitochondria. Sci Adv. 2022;8:eabj7307.
Riley JS, Quarato G, Cloix C, Lopez J, O’Prey J, Pearson M, et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018;37:e99238.
McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018;359:eaao6047.
Ader NR, Hoffmann PC, Ganeva I, Borgeaud AC, Wang C, Youle RJ, et al. Molecular and topological reorganizations in mitochondrial architecture interplay during Bax-mediated steps of apoptosis. Elife 2019;8:e40712.
Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17:1142–9.
Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, et al. Apoptotic caspases suppress Type I Interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol Cell. 2019;74:19–31.
Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020;21:e49799.
Yamazaki T, Kirchmair A, Sato A, Buque A, Rybstein M, Petroni G, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol. 2020;21:1160–71.
Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012;485:251–5.
Han C, Liu Z, Zhang Y, Shen A, Dong C, Zhang A, et al. Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nat Immunol. 2020;21:546–54.
Li S, Li H, Zhang YL, Xin QL, Guan ZQ, Chen X, et al. SFTSV infection induces BAK/BAX-dependent mitochondrial DNA release to trigger NLRP3 inflammasome activation. Cell Rep. 2020;30:4370–85.
Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M Jr. Interleukin-1beta Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol Cell. 2019;74:801–15.
Obexer P, Geiger K, Ambros PF, Meister B, Ausserlechner MJ. FKHRL1-mediated expression of Noxa and Bim induces apoptosis via the mitochondria in neuroblastoma cells. Cell Death Differ. 2007;14:534–47.
Hagenbuchner J, Kuznetsov A, Hermann M, Hausott B, Obexer P, Ausserlechner MJ. FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3. J Cell Sci. 2012;125:1191–203.
Willemsen J, Neuhoff MT, Hoyler T, Noir E, Tessier C, Sarret S, et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep. 2021;37:109977.
Chauhan D, Bartok E, Gaidt MM, Bock FJ, Herrmann J, Seeger JM, et al. BAX/BAK-induced apoptosis results in Caspase-8-dependent IL-1β maturation in macrophages. Cell Rep. 2018;25:2354–68.
Vince JE, De Nardo D, Gao W, Vince AJ, Hall C, McArthur K, et al. The Mitochondrial apoptotic effectors BAX/BAK activate Caspase-3 and -7 to Trigger NLRP3 Inflammasome and Caspase-8 driven IL-1beta activation. Cell Rep. 2018;25:2339–53 e4.
Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2014;23:1413–24.
Wang W, Li L, Lin WL, Dickson DW, Petrucelli L, Zhang T, et al. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet. 2013;22:4706–19.
Wang W, Arakawa H, Wang L, Okolo O, Siedlak SL, Jiang Y, et al. Motor-coordinative and cognitive dysfunction caused by mutant TDP-43 could be reversed by inhibiting its mitochondrial localization. Mol Ther. 2017;25:127–39.
Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 triggers Mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. 2020;183:636–49.
Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019;366:1531–6.
Domizio JD, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature 2022;603:145–51.
Ma SB, Nguyen TN, Tan I, Ninnis R, Iyer S, Stroud DA, et al. Bax targets mitochondria by distinct mechanisms before or during apoptotic cell death: a requirement for VDAC2 or Bak for efficient Bax apoptotic function. Cell Death Differ. 2014;21:1925–35.
Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–5.
Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003;301:513–7.
Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10:1689.
de Torre-Minguela C, Gomez AI, Couillin I, Pelegrin P. Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB J. 2021;35:e21757.
Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rotig A, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018;560:238–42.
Tigano M, Vargas DC, Tremblay-Belzile S, Fu Y, Sfeir A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 2021;591:477–81.
Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–62.
Goldstein JC, Munoz-Pinedo C, Ricci JE, Adams SR, Kelekar A, Schuler M, et al. Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 2005;12:453–62.
Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell. 2015;57:860–72.
Liu X, He Y, Li F, Huang Q, Kato TA, Hall RP, et al. Caspase-3 promotes genetic instability and carcinogenesis. Mol Cell. 2015;58:284–96.
Cao K, Riley JS, Heilig R, Montes-Gomez AE, Vringer E, Berthenet K, et al. Mitochondrial dynamics regulate genome stability via control of caspase-dependent DNA damage. Dev Cell. 2022;57:1211–25 e6.
Brokatzky D, Dorflinger B, Haimovici A, Weber A, Kirschnek S, Vier J, et al. A non-death function of the mitochondrial apoptosis apparatus in immunity. EMBO J. 2019;38:e100907.
Andree M, Seeger JM, Schull S, Coutelle O, Wagner-Stippich D, Wiegmann K, et al. BID-dependent release of mitochondrial SMAC dampens XIAP-mediated immunity against Shigella. EMBO J. 2014;33:2171–87.
Flores-Romero H, Hohorst L, John M, Albert MC, King LE, Beckmann L. et al. BCL-2-family protein tBID can act as a BAX-like effector of apoptosis. EMBO J. 2022;41:e108690.
Grosse L, Wurm CA, Bruser C, Neumann D, Jans DC, Jakobs S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016;35:402–13.
Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, et al. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016;35:389–401.
Cosentino K, Hertlein V, Jenner A, Dellmann T, Gojkovic M, Pena-Blanco A, et al. The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol Cell. 2022;82:933–49 e9.
Li S, Kuang M, Chen L, Li Y, Liu S, Du H, et al. The mitochondrial protein ERAL1 suppresses RNA virus infection by facilitating RIG-I-like receptor signaling. Cell Rep. 2021;34:108631.
Matthews HK, Bertoli C, de Bruin RAM. Cell cycle control in cancer. Nat Rev Mol Cell Biol. 2022;23:74–88.
Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ, et al. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol Cell. 2011;43:57–71.
Burigotto M, Mattivi A, Migliorati D, Magnani G, Valentini C, Roccuzzo M, et al. Centriolar distal appendages activate the centrosome-PIDDosome-p53 signalling axis via ANKRD26. EMBO J. 2020;40:e104844.
Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 2017;31:34–45.
Evans LT, Anglen T, Scott P, Lukasik K, Loncarek J, Holland AJ. ANKRD26 recruits PIDD1 to centriolar distal appendages to activate the PIDDosome following centrosome amplification. EMBO J. 2020;40:e105106.
Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell 2016;165:631–42.
Reyes J, Chen JY, Stewart-Ornstein J, Karhohs KW, Mock CS, Lahav G. Fluctuations in p53 signaling allow escape from cell-cycle arrest. Mol Cell. 2018;71:581–91 e5.
Tsabar M, Mock CS, Venkatachalam V, Reyes J, Karhohs KW, Oliver TG, et al. A Switch in p53 dynamics marks cells that escape from DSB-induced cell cycle arrest. Cell Rep. 2020;32:107995.
Muller I, Strozyk E, Schindler S, Beissert S, Oo HZ, Sauter T, et al. Cancer cells employ nuclear Caspase-8 to overcome the p53-dependent G2/M checkpoint through cleavage of USP28. Mol Cell. 2020;77:970–84.
Haschka M, Karbon G, Fava LL, Villunger A. Perturbing mitosis for anti-cancer therapy: is cell death the only answer? EMBO Rep. 2018;19:e45440.
Liccardi G, Ramos Garcia L, Tenev T, Annibaldi A, Legrand AJ, Robertson D, et al. RIPK1 and Caspase-8 ensure chromosome stability independently of their role in cell death and inflammation. Mol Cell. 2019;73:413–28 e7.
Matthess Y, Raab M, Knecht R, Becker S, Strebhardt K. Sequential Cdk1 and Plk1 phosphorylation of caspase-8 triggers apoptotic cell death during mitosis. Mol Oncol. 2014;8:596–608.
Alappat EC, Feig C, Boyerinas B, Volkland J, Samuels M, Murmann AE, et al. Phosphorylation of FADD at serine 194 by CKIalpha regulates its nonapoptotic activities. Mol Cell. 2005;19:321–32.
Orth JD, Loewer A, Lahav G, Mitchison TJ. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell. 2012;23:567–76.
Cytlak UM, Dyer DP, Honeychurch J, Williams KJ, Travis MA, Illidge TM. Immunomodulation by radiotherapy in tumour control and normal tissue toxicity. Nat Rev Immunol. 2022;22:124–38.
Bock FJ, Krumschnabel G, Manzl C, Peintner L, Tanzer MC, Hermann-Kleiter N, et al. Loss of PIDD limits NF-kappaB activation and cytokine production but not cell survival or transformation after DNA damage. Cell Death Differ. 2013;20:546–57.
Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017;548:461–5.
Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017;548:466–70.
Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017;550:402–6.
Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA. 2017;114:E4612–E20.
Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19:1061–70.
Wischnewski M, Ablasser A. Interplay of cGAS with chromatin. Trends Biochem Sci. 2021;46:822–31.
Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki H. The Cytoplasmic DNA Sensor cGAS promotes mitotic cell death. Cell 2019;178:302–15 e23.
Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018;553:467–72.
Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun. 2014;5:5166.
Li J, Duran MA, Dhanota N, Chatila WK, Bettigole SE, Kwon J, et al. Metastasis and immune evasion from extracellular cGAMP hydrolysis. Cancer Discov. 2021;11:1212–27.
Potts MB, Vaughn AE, McDonough H, Patterson C, Deshmukh M. Reduced Apaf-1 levels in cardiomyocytes engage strict regulation of apoptosis by endogenous XIAP. J Cell Biol. 2005;171:925–30.
Sanchis D, Mayorga M, Ballester M, Comella JX. Lack of Apaf-1 expression confers resistance to cytochrome c-driven apoptosis in cardiomyocytes. Cell Death Differ. 2003;10:977–86.
Callus BA, Vaux DL. Caspase inhibitors: viral, cellular and chemical. Cell Death Differ. 2007;14:73–8.
Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–701.
Kroemer G, Galassi C, Zitvogel L, Galluzzi L. Immunogenic cell stress and death. Nat Immunol. 2022;23:487–500.
Nauts HC, Swift WE, Coley BE. The treatment of malignant tumors by bacterial toxins as developed by the late William B. Coley, M.D., reviewed in the light of modern research Cancer Res. 1946;6:205–16.
Tracey KJ, Lowry SF, Cerami A. Cachetin/TNF-alpha in septic shock and septic adult respiratory distress syndrome. Am Rev Respir Dis. 1988;138:1377–9.
Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23:1565–76.
Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 1998;8:297–303.
Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 2009;136:1098–109.
Ori D, Kato H, Sanjo H, Tartey S, Mino T, Akira S, et al. Essential roles of K63-linked polyubiquitin-binding proteins TAB2 and TAB3 in B cell activation via MAPKs. J Immunol. 2013;190:4037–45.
Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol. 2018;20:1389–99.
Micheau O, Tschopp J. Induction of TNF Receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003;114:181–90.
Vince JE, Pantaki D, Feltham R, Mace PD, Cordier SM, Schmukle AC, et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-{kappa}b and to prevent tnf-induced apoptosis. J Biol Chem. 2009;284:35906–15.
Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem. 2006;281:13636–43.
Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008;133:693–703.
Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–48.
Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449–63.
Brusilovsky M, Rochman M, Rochman Y, Caldwell JM, Mack LE, Felton JM, et al. Environmental allergens trigger type 2 inflammation through ripoptosome activation. Nat Immunol. 2021;22:1316–26.
Acknowledgements
We are indebted to our colleagues and mentors, past and present, for continuous discussions. We would like to apologise to the many authors whose work we could not cite due to space restrictions. Figures were created with Biorender.com.
Author information
Authors and Affiliations
Contributions
FJB and JSR wrote the manuscript and drew the figures. Both authors approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by F Pentimalli
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bock, F.J., Riley, J.S. When cell death goes wrong: inflammatory outcomes of failed apoptosis and mitotic cell death. Cell Death Differ 30, 293–303 (2023). https://doi.org/10.1038/s41418-022-01082-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-022-01082-0
This article is cited by
-
Mechanism study of oleanolic acid derivative, K73-03, inducing cell apoptosis in hepatocellular carcinoma
Cancer Cell International (2024)
-
Mcl-1 mediates intrinsic resistance to RAF inhibitors in mutant BRAF papillary thyroid carcinoma
Cell Death Discovery (2024)
-
Dying in self-defense: cell death signaling in animals and plants
Cell Death & Differentiation (2024)
-
Gold nanoparticles (AuNPs) decrease the viability of cervical cancer cells by inducing the BAX gene and activating antioxidant enzymes
Molecular Biology Reports (2024)
