
Abstract
Intrinsic apoptosis is principally governed by the BCL-2 family of proteins, but some non-BCL-2 proteins are also critical to control this process. To identify novel apoptosis regulators, we performed a genome-wide CRISPR-Cas9 library screen, and it identified the mitochondrial E3 ubiquitin ligase MARCHF5/MITOL/RNF153 as an important regulator of BAK apoptotic function. Deleting MARCHF5 in diverse cell lines dependent on BAK conferred profound resistance to BH3-mimetic drugs. The loss of MARCHF5 or its E3 ubiquitin ligase activity surprisingly drove BAK to adopt an activated conformation, with resistance to BH3-mimetics afforded by the formation of inhibitory complexes with pro-survival proteins MCL-1 and BCL-XL. Importantly, these changes to BAK conformation and pro-survival association occurred independently of BH3-only proteins and influence on pro-survival proteins. This study identifies a new mechanism by which MARCHF5 regulates apoptotic cell death by restraining BAK activating conformation change and provides new insight into how cancer cells respond to BH3-mimetic drugs. These data also highlight the emerging role of ubiquitin signalling in apoptosis that may be exploited therapeutically.
Similar content being viewed by others


Introduction
Intrinsic apoptosis plays a pivotal role in tissue homeostasis, development, and immunity. Dysregulated apoptosis contributes to a variety of human pathologies including neurodegeneration [1], autoimmune diseases [2], and cancers [3]. Therefore, targeting apoptosis is an emerging strategy to either block cell death in neurodegenerative disease or to induce cell death to treat cancer. In vertebrates, intrinsic apoptosis is principally regulated by BCL-2 proteins, which comprise three subgroups: pro-survival proteins, pro-apoptotic effector proteins, and initiator BH3-only proteins. These proteins control cellular fate through protein-protein interactions [4,5,6], with some associations promoting death and others favouring survival.
Although apoptosis is mainly controlled by BCL-2 proteins, some non-BCL-2 proteins also serve as important regulators [7,8,9]. These include voltage-dependent anion channel 2 (VDAC2), which supports the integration of the cell death effector proteins BAK and BAX into the mitochondrial outer membrane (MOM) [10,11,12,13].
In healthy cells, BAK and BAX that reside on the MOM associate with VDAC2 [13]. However, interactions with VDAC2 have seemingly opposing effects on BAX and BAK apoptotic function. Whilst deleting Vdac2 inhibits BAX apoptotic activity by impairing BAX MOM localisation [14], VDAC2 deficiency elevates BAK apoptotic activity [15], even though BAK levels at mitochondria are reduced in Vdac2-deficient cells [11, 13, 14]. These findings emphasise that although BAX and BAK are typically regarded as redundant apoptosis effector proteins, they are regulated distinctly from one another.
To understand why BAK exhibits heightened activity in the absence of VDAC2, we performed a CRISPR-Cas9 whole genome library screen that identified the mitochondrial E3 ubiquitin ligase MARCHF5 as a novel regulator of BAK-dependent apoptosis. Loss of MARCHF5, or abolishing its ubiquitin ligase activity, rendered BAX-deficient cells highly resistant to prolonged treatment with BH3 mimetics. This involved BAK adopting an active conformation and forming inhibitory Mode 2 complexes with the pro-survival proteins MCL-1 and BCL-XL. Surprisingly, BAK conformation change did not require BH3-only proteins. Whilst the in vivo consequence of MARCHF5-mediated control of BAK needs to be resolved, our data suggest that MARCHF5 can dictate a cell’s response to BH3 mimetic drugs through controlling BAK conformation and this represents a new opportunity to control apoptosis in a broad spectrum of cells.
Methods and materials
Cell culture and stable cell lines
SV40-transformed Mouse Embryonic Fibroblasts (MEFs) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% (v/v) Fetal Bovine Serum (FBS), 50 μM 2-Mercaptoethanol (2-ME) and 100 μM asparagine. HeLa cells (ATCC #CCL-2) and Human Embryonic Kidney 293 T (HEK293T) cells (ATCC #CRK-3216) were cultured in DMEM supplemented with 10% (v/v) FCS. HCT116 (gift from Xu Luo [16]), KM12 and KMS-12-PE (DSMZ #ACC606) cells were cultured in RPMI1640 supplemented with 10% (v/v) FCS. Cells were cultured in humidified incubators maintained at 37 °C and 10% CO2 for MEFs, HeLa and 293T cells or 5% CO2 for KMS-12-PE and HCT116 cells. Cells used were authenticated by STR profiling (https://www.garvan.org.au/research/capabilities/molecular-genetics/cell-line-identification) and were routinely (every two months) screened for Mycoplasma contamination by MycoAlert Kit (Cat#LT07218, Lonza) as the manufacturer’s instructions. Only Mycoplasma negative cells were analysed.
Retroviral and lentiviral infection
3xMyc (3xMEQKLISEEDLNE)-tagged wild-type MARCHF5 or MARCHF5C65/68S were synthesised by Integrated DNA Technologies (IDT) and subcloned into pMSCV-IRES-puro (pMIP) vector [17] or pMSCV-IRES-hygro (pMIH) vector. HEK293T cells were used as packaging cells. The retroviral or lentiviral constructs were first introduced into packaging cells by FuGENE6 (Cat#E2691, Promega) transfection according to the manufacturer’s instructions. Viral supernatants were filtered and used to infect MEF, HeLa, and KMS-12-PE cells by spin-infection (2500 rpm centrifugation at 25 °C for 1 h) in the presence of 4 µg/mL polybrene (Cat#S2667, Sigma-Aldrich). Stably transduced cells were selected either by antibiotic selection (2 mg/mL hygromycin (Cat# 10687010, Thermo Fisher) or 2 µg/mL puromycin (Cat#P9620, Sigma-Aldrich)) or expression of fluorescent protein markers (GFP, BFP or CFP) using a BD FACSAria Fusion cell sorter.
Generating knockout cell lines by CRISPR/Cas9 gene editing
Gene-deletions were engineered using CRISPR-Cas9 gene editing technology either by transient transfection or lentiviral transduction. The sgRNA sequences used for targeting specific genes (Supplementary Table 1) were taken from available whole genome libraries Gecko [18] or Brunello [19]. Oligonucleotides encoding these sgRNA sequences were ordered from IDT and annealed as previously described [20]. The annealed oligos were cloned into pSpCas9(BB)-2A-GFP/PX458 (for transient co-expression of sgRNA and Cas9) [21], pKLV-U6RNA(BbsI)-PGKpuro2ABFP (for constitutive lentiviral sgRNA expression) [22], or FgH1tUTG-CFP or FgH1tUTG-GFP vectors (for inducible lentiviral sgRNA expression) [23]. FUCas9Cherry lentiviral vector was used to generate stable Cas9 expressing HeLa, HCT116, and KMS-12-PE cells [23]. sgRNA were expressed for 48 h before cells were sorted into single cells based on fluorescent marker protein expression using a BD FACSAria Fusion cell sorter. Individual clones were expanded, and successful knock-out clones were identified by sequencing PCR amplicons encompassing the sgRNA target sites. The primers for amplifying the sgRNA target sites are listed in Supplementary Table 2.
Immunoblotting and immunoprecipitation
Cell pellets were lysed in ONYX lysis buffer (20 mM Tris-pH 7.4, 135 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% (v/v) glycerol, supplemented with 1% (v/v) Triton X-100 or 1% digitonin, 0.5 μg/ml complete protease inhibitor (Sigma-Aldrich)) for 30 min on ice. Following lysis, samples were centrifuged at 13,000 rpm at 4 °C for 10 min and supernatants were collected. Protein concentration was determined using the Bradford protein assay. For immunoprecipitation, bait proteins were captured with antibodies to BAK (Clone# G317-2, BD Pharmingen), MCL-1 (Clone#19C4-15, Walter and Eliza Hall Institute), or BCL-XL (Clone#E18, Abcam) and Protein G Sepharose beads. 25 μg of whole cell lysate was loaded per well and resolved by SDS-PAGE. Gels were transferred onto nitrocellulose membrane (Invitrogen) using an iBlot Gel Transfer Device (Invitrogen). Non-specific binding was blocked with 5% w/v non-fat milk in TBS-T (20 mM Tris-HCL pH 7.6, 137 mM NaCl, 0.1% Tween-20) for 1 h at room temperature before immunoblotting with primary antibodies overnight at 4 °C. The membranes were washed with TBS-T three times, followed by incubating with appropriate HRP-conjugated secondary antibodies for 1 h in room temperature. The membranes were washed with TBS-Tween (0.01% v/v) prior to detection using enhanced chemiluminescent (ECL) reagent (Millipore) and imaged using a ChemiDoc Imaging System (BioRad). Monoclonal antibodies to MCL-1 (Clone#19C4-15), BAK (Clone#7D10), BCL-2 (Clone#BCL-2-100), BCL-W, BIM (Clone#3C5), BID (Clone#2D1-3), MYC (Clone#7E10), VDAC2 (gift of M. Ryan) and HSP70 (Clone#N6, gift of W. Welch and R. Anderson) were produced with the WEHI antibody facility. The following antibodies were purchased from commercial sources: BAK (Cat#B5897, Sigma-Aldrich), BCL-XL (Clone#44/BCL-X, BD Bioscience), BCL-XL (Clone#E18, Abcam), BCL-2 (Clone#7/BCL-2, BD Bioscience), MCL-1 (Cat#600-401-394, Rockland), NOXA (Clone#114C307.1, Novus Biologicals), BIM (Cat#ADIAAP330, Stressgen), BAD (Cat#ADIAAP020, Stressgen), PUMA (Cat#3043, ProSci), HA (Clone#3F10, Roche Applied Science), Cytochrome c (Clone#7H8.2C12, BD Pharmingen), Cytochrome c (Clone#6H2.B4, BD Pharmingen), DRP1 (611113, BD Transduction), MiD49 (16413-1-AP, Proteintech) and FAR1 (NBP1-89847, Novus Biologicals). Secondary antibodies: anti-rabbit IgG (Cat#403005), anti-mouse IgG (Cat#103005), anti-rat IgG (Cat#303005) were obtained from Southern Biotech.
Cell viability assays
Cells were cultured with the indicated concentrations of BH3 mimetics (ABT-737 (Cat#A-1002, Active Biochem), A-1331852 (WEHI Chemical Biology Division) and S-63845 (Cat#A-6044, Active Biochem)) or with mDivi1(M0199, Sigma-Aldrich). After 24 h, both viable and dead cells were harvested and re-suspended in KDS-BSS buffer supplemented with 2 µg/ml propidium iodide (PI) (Sigma Aldrich). Cell viability (PI negative) was assessed using an LSR-II flow cytometer (BD Biosciences).
Colony formation assay
Cells were seeded in 6-well plate at a density of 1,000 cells per well and cultured with titrated concentrations of BH3 mimetics for 5 days. After treatment, cells were stained with 0.5% crystal violet (Cat#C0775, Sigma-Aldrich) for 20 min at room temperature and photographed using a ChemiDoc Imaging System (BioRad).
BAK and BAX activation, cytochrome c release and caspase-3 activation by Flow Cytometry
Cells were harvested following 0.5 or 2 h of culture with BH3 mimetic drugs. 20 µM caspase inhibitor QVD-OPh (Cat#OPH109, MP Biomedicals) was included when assays were performed for BAK and BAX activation or cytochrome c release, but not when caspase-3 activation was measured. Harvested cells were washed once with ice-cold PBS.
To assess BAK and BAX conformation change and caspase-3 activation, cell pellets were fixed and then permeabilised using the eBioscience cell fixation and permeabilisation kit (Cat#88882400, Thermo Fischer) according to the manufacturer’s instructions. Fixed cells were incubated with either conformation-specific BAK antibody (Clone#G317-2, 1:100, Cat# 556382, BD Pharmingen), BAX antibody (Clone#3, 1:100, Cat#610982, BD Pharmingen), or anti-caspase-3-phycoerythrin antibody (Clone#C92-605, 1:25, Cat#550821, BD Pharmingen), followed by phycoerythrin (PE)-conjugated anti-mouse antibody (1:200, Cat#1031309, SouthernBiotech). To quantify % cells with activated caspase-3, markers were set to <5% caspase-3 positive in untreated cells of each genotype.
Cytochrome c release was assayed as described [24]. Briefly, cells were permeabilised with 0.025% w/v digitonin in permeabilisation buffer (20 mM HEPES pH 7.5, 100 mM sucrose, 2.5 mM MgCl2, 100 mM KCl) for 10 min on ice before fixation. Fixed cells were incubated with anti-cytochrome c-APC antibody (Clone#REA702, 1:50) (Cat#130111180, Miltenyi Biotec). Samples were analysed on an LSR-II flow cytometer (BD Biosciences). To quantify % cells with released cytochrome c, markers were set to <5% cytochrome c negative in untreated BAX−/−BAK−/− cells.
BAK and BAX subcellular redistribution assay
Cells were permeabilised with 0.025% w/v digitonin in permeabilisation buffer for 10 min on ice and cytosol and heavy membrane fractions separated by centrifugation at 13000 g for 5 min at 4 °C. Membrane fractions were resuspended in permeabilisation buffer and incubated at 30 °C. At specific times membrane and supernatant were separated by centrifugation at 13,000 g for 5 min at 4 °C prior to SDS-PAGE and immunoblotting.
Mass Spectrometric analysis
MEF cells were harvested, and mitochondria were isolated and enriched using a Mitochondria isolation kit as the manufacturer’s introductions (Cat#130096946, Miltenyi Biotec). HeLa cells were harvested and permeabilised by 0.025% w/v digitonin (Biosynth) in permeabilisation buffer for 10 min on ice, and then mitochondria were isolated and enriched using gradient centrifugation [25]. As previously described [26], membrane fractions were normalised for protein content using the Pierce BCA Protein Assay Kit (Cat#23227, ThermoFisher Scientific), subjected to trypsinisation, and peptides were labelled with Tandem Mass Tags (Cat#TMT; 90061, ThermoFisher Scientific).
For TMT labelling, protein pellets were solubilised and digested into tryptic peptides for mass spectrometry using the iST-NHS kit (PreOmics GmbH, Planegg, Germany) as per manufacturer instructions. Peptides were labelled with 6plex Tandem Mass Tags (TMT) (Thermo Fisher Scientific) in 8:1 label:protein ratio as per manufacturer instructions. Pooled samples were fractionated using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Fisher Scientific) as per manufacturer’s instructions. Individual fractions were dried using a CentriVap Benchtop Vacuum Concentrator (Labconco) and reconstituted in 2% (v/v) acetonitrile (ACN) and 0.1% (v/v) trifluoroacetic acid (TFA). Liquid chromatography (LC) coupled MS/MS was carried out on an Orbitrap Q Exactive Plus mass spectrometer (Thermo Fisher Scientific) with a nanoESI interface in conjunction with an Ultimate 3000 RSLC nanoHPLC (Dionex Ultimate 3000). The LC system was equipped with an Acclaim Pepmap nano-trap column (Dionex-C18, 100 Å, 75 μm × 2 cm) and an Acclaim Pepmap RSLC analytical column (Dionex-C18, 100 Å, 75 μm × 50 cm). The mass spectrometer was operated in a centroid data spectrum mode. The tryptic peptides were injected to the trap column at an isocratic flow of 5 μl/min of 2% (v/v) CH3CN containing 0.1% (v/v) formic acid for 5 min applied before the trap column was switched in-line with the analytical column. The eluents were 5% DMSO in 0.1% v/v formic acid (solvent A) and 5% DMSO in 100% v/v CH3CN and 0.1% v/v formic acid (solvent B). The flow gradient was (1) 0–6 min at 3% B, (2) 6–95 min, 3–22% B (3) 95–105 min 22–40% B (4) 105–110 min, 40–80% B (5) 110–115 min, 80–80% B (6) 115–125 min, 80–3% and equilibrated at 3% B for 5 min before the next sample injection.
For TMT and LFQ analysis, in brief data were collected in positive mode using Data Dependent Acquisition using m/z 375–1450 as MS scan range, HCD for MS/MS of the 12 most intense ions with charge ≥2. Other instrument parameters were: MS1 scan at 70,000 resolution (at 200 m/z), MS maximum injection time 50 ms, AGC target 3E6, normalised collision energy was at 27% energy, isolation window of 1.2 Da, MS/MS resolution 17,500, MS/MS AGC target of 5E4, MS/MS maximum injection time 50 ms, minimum intensity was set at 2E3 and dynamic exclusion was set to 30 s. Raw files were processed using the MaxQuant platform (version 1.6.5.0) and searched against UniProt human or mouse database using default settings for a TMT 6plex or LFQ experiment. In brief: oxidation of methionine and N-terminal acetylation were specified as variable modifications. Trypsin/P cleavage specificity (cleaves after lysine or arginine, even when proline is present) was used with a maximum of 2 missed cleavages. Carbamidomethylation of cysteine was set as a fixed modification. A search tolerance of 4.5 ppm was used for MS1 and 20 ppm for MS2 matching. False discovery rates (FDR) were determined through the target-decoy approach set to 1% for both peptides and proteins.
Log2-transformed TMT reporter intensity corrected values were grouped and fit to a moderated t-test to identify significant differences in protein abundance between samples (limma Bioconductor package—version 3.46.0). For the MEF data, as these data were derived from independent experiments analysed on separate mass spectrometry runs, the Log2-transformed reporter intensities were normalised and batch corrected prior to fitting the linear model with limma. For the HeLa data, the label-free samples were run through the mass spectrometer in a single experiment, so batch correction was not necessary, with the LFQ intensity data otherwise treated similarly to TMT reporter intensity data described above.
CRISPR/Cas9 library screens
Cells were first engineered to stably express Cas9 by transduction with FUCas9Cherry lentivirus [23]. mCherryhigh cells were sorted as population using a BD FACSAria Fusion cell sorter. The Cas9-expressing cells were seeded in 10 cm2 dishes at a density of 1 ×106 one day prior to transduction with lentiviral sgRNA libraries. A genome-wide library was used for the MEF screen in Fig. 1c; [22] and a custom library was used for the HeLa screen in Fig. 6c–e (synthesised by Synbio Technologies), which included 3326 sgRNAs (Supplementary Table 5) targeting 514 human protein-coding genes (258 potential MARCHF5 substrates identified by our mass spectrometry experiments along with other previously described MARCHF5 substrates, known mitochondrial and peroxisomal proteins, and BCL-2 family members). Transduced cells were cultured with 2 μg/ml puromycin for 48 h to enrich sgRNA-expressing cells. Each transduced pool of cells was then split into two groups: one was treated with DMSO, and the other was subjected to a lethal dose of BH3 mimetics (10 µM ABT-737 or 500 nM A1331852 for MEFs and 2 µM S63845 + 1 µM A1131852 for HeLa cells) for 48 h. Surviving cells were expanded for 5 days, their DNA was extracted, and sgRNA sequences were amplified by PCR and sequenced as described [22]. For the MEF experiments, the screen was performed with two independently derived cell lines, each transduced 5-6 times with library sgRNAs, then divided and selected with either ABT-737 or A1331852 in parallel, making >20 biological replicate samples. Likewise, the Hela screen was performed with triplicate viral transductions in two independent cell clones, for a total of six replicate samples. Raw counts of each sgRNA were normalised to counts per million (CPM) reads and Log2 transformed following the addition of a 1 CPM offset to each sgRNA. The log2 transformed values were fit to a linear model and a moderated t-test was performed to identify sgRNA whose relative abundance differed significantly between the BH3 mimetic and DMSO treated samples (and also between Cas9 expressing and non-expressing cells in HeLa screen) (limma Bioconductor package—version 3.46.0). The sgRNAs were then ranked in order of their t-statistic. An iterative hypergeometric scoring algorithm [27, 28] was applied to identify genes whose sgRNA were enriched among the most significantly depleted sgRNA in the library.

a Bax−/− Mcl-1−/−Vdac2−/− MEFs were used for the CRISPR/Cas9 library screen. On this genetic background, apoptosis is driven exclusively by the effector protein BAK (because BAX is absent) and BAK is controlled primarily by the pro-survival protein BCL-XL (due to the absence of MCL1—its other major regulator [5]). Apoptosis can be provoked in these cells with BH3 mimetics that target BCL-XL, such as A1331852, which selectively inhibits BCL-XL, or ABT-737, which also inhibits BCL-W and BCL-2. b Outline of the genome-wide CRISPR/Cas9 library screen. c sgRNAs targeting Bak, Ubqln1, and Marchf5 were significantly enriched following selection with ABT-737. Bax−/−Mcl-1−/−Vdac2−/− MEFs expressing Cas9 and a whole-genome sgRNA library were cultured with 10 µM ABT-737 or 500 nM A1331852 for 48 h. Surviving cells were expanded for 5 days and sgRNA abundance was quantified by deep-sequencing. Plot shows the abundance of independent sgRNAs in selected versus untreated cells. Values represent the mean of >20 biological replicate samples (2 cell lines x 5-6 independent library transductions subjected to selection with ABT-737 or A1331852 in parallel). Dashed line represents a LOWESS regression curve fit to the data. d MEFs of the indicated genotypes (Mcl1−/− genetic background) were treated with escalating does of the BCL-XL inhibitor A1331852 for 48 h. Cell death was assessed by PI uptake and flow cytometry. Data are mean ± SD of three independent experiments. e MEFs of the indicated genotypes (Mcl1+/+ genetic background) were treated with (10 µM MCL-1 inhibitor S63845 and escalating does of A1331852) for 24 h. Cell death was assessed by PI uptake and flow cytometry. Data are mean ± SD of three independent experiments.
Results
MARCHF5 regulates BAK-dependent apoptosis
To identify genes that regulate BAK-driven apoptosis independently of (or redundantly with) VDAC2, a CRISPR-Cas9 whole genome library screen was performed in Bax−/− Mcl1−/− Vdac2−/− MEFs. Removing MCL-1 enabled apoptosis to be driven in these cells with the BH3-mimetic compounds ABT-737 or A1331852 (Fig. 1a), while the absence of BAX and VDAC2 ensured that apoptosis was mediated by BAK and controlled independently of VDAC2. Bax−/− Mcl1−/− Vdac2−/− MEFs were transduced with genome-wide lentiviral single guide RNA (sgRNA) library [22], treated with ABT-737 or A1331852 to induce apoptosis, and surviving cells were expanded to identify enriched sgRNA (Fig. 1b). As expected, sgRNA targeting Bak were the most enriched (Fig. 1c, Supplementary Fig. 1a), as this yielded BAK/BAX-deficient cells that are refractory to intrinsic apoptosis [29]. Clear enrichment was also observed for multiple sgRNAs targeting two genes involved in ubiquitin signalling: the mitochondrial E3 ubiquitin ligase Membrane Associated RING-CH-type Finger 5 (Marchf5) and the ubiquitin receptor ubiquilin-1 (Ubqln1) (Fig. 1c, Supplementary Fig. 1a). sgRNAs targeting these genes were not enriched in the equivalent screen performed in Bax−/−Mcl1−/−Vdac2+/+ MEFs [14], suggesting that their influence on BAK in these cells becomes evident when VDAC2 is absent.
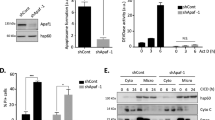
To validate the candidates, Marchf5 or Ubqln1 were deleted in Bax−/−Vdac2−/− MEFs on both an Mcl1−/− and an Mcl1+/+ genetic background (Supplementary Fig. 1b–g, j). In both contexts, the Marchf5 or Ubqln1 knockout cells were less sensitive to BH3 mimetics compared with empty vector control cells (Fig. 1d, e, Supplementary Fig. 1h, k). As deleting Marchf5 produced the strongest phenotype in these experiments, subsequent studies focused on elucidating the mechanism by which MARCHF5 influenced BAK.
Deleting Marchf5 and Vdac2 in Bax −/− MEFs impairs MOMP
Given that Bax−/−Vdac2−/−Marchf5−/− MEFs were highly resistant to treatment with BH3 mimetic drugs in short-term cell death assays, we explored whether this protective effect preserved clonogenic potential. After 5 days treatment with combined BH3 mimetics, significantly more colonies formed by Bax−/−Vdac2−/−Marchf5−/− MEFs compared with MEFs of other genotypes (Bax−/− or Bax−/−Vdac2−/− or Bax−/−Marchf5−/−) (Fig. 2a). Mitochondrial outer membrane permeabilisation (MOMP) is the point of no return in apoptosis as cells eventually die irrespective of caspase activity following MOMP [30, 31]. Hence, that MARCHF5-deficent cells retained clonogenic potential suggests that the blockade is upstream of MOMP.

a MEFs of the indicated genotypes were cultured continuously with BH3 mimetic drugs (10 µM S63845 and escalating concentration of A1331852) for 5 days. Colonies were stained with crystal violet and photographed. Representative images of three independent experiments are shown. b Schematic of the intrinsic apoptosis pathway. c Cells were cultured with BH3 mimetics (10 µM S63845 + 1 µM A1331852) for 0.5 h or 2 h in the presence of 20 µM caspase inhibitor QVD-OPh. Cytochrome c release was assessed by immunostaining and flow cytometry. Histograms are representative of three independent experiments and the proportion of cells with low cytochrome c collated from three independent experiments, data are mean ± SD. d Cells were cultured with BH3 mimetics (10 µM S63845 + 1 µM A1331852) for 0.5 h or 2 h. Caspase-3 activation was assessed by immunostaining and flow cytometry. Histograms are representative of three independent experiments with the proportion of cells exhibiting active caspase-3 collated from three independent experiments, data are mean ± SD. ****p ≤ 0.0001 based on unpaired Student’s t-test.
To explore this, we assessed cytochrome c release and caspase-3 activation in cells exposed to BH3 mimetic drugs, both of which follow MOMP (Fig. 2b). Bax−/−Vdac2−/− and Bax−/−Marchf5−/− MEFs exhibited cytochrome c release following treatment with BH3 mimetics, whereas cytochrome c release was limited in Bax−/−Vdac2−/−Marchf5-/- MEFs (Fig. 2c). These data indicate that MARCHF5 loss impairs BAK-driven MOMP induced by BH3 mimetics in MEFs, but only when VDAC2 is absent.
When assessed by subcellular fractionation, BH3 mimetics caused complete cytochrome c release in Bax−/−Vdac2−/− and Bax−/−Marchf5−/− MEFs, but incomplete and more gradual loss from Bax−/−Vdac2−/−Marchf5−/− MEFs (Supplementary Fig. 2). Interestingly, cytochrome c only accumulated to appreciable levels in the cytosolic fractions of the Bax−/−Vdac2−/− and Bax−/−Marchf5−/− MEFs, with only very low levels of cytochrome c observed in the cytosol of Bax−/−Vdac2−/−Marchf5−/− MEFs. In all genotypes, a proteasome-dependent (MG-132-inhibited) degradation pathway acted to eliminate cytochrome c once it reached the cytosol [32]. We hypothesised that the slower kinetics of release, together with this proteasomal turnover, limited cytochrome c from activating caspases in Bax−/−Vdac2−/−Marchf5−/− MEFs.
Consistent with this hypothesis, robust caspase-3 activation was observed in Bax−/−Vdac2−/− and Bax−/−Marchf5−/− MEFs exposed to BH3 mimetic drugs, but almost no caspase-3 activation occurred in Bax−/−Vdac2−/−Marchf5−/− cells (Fig. 2d). Hence, we infer that these cells contain enough intact mitochondria to support clonogenic growth [33].
MARCHF5 deletion alone can be sufficient to regulate BAK-dependent apoptosis
Given our findings in MEFs, we sought to explore if the role of MARCHF5 in regulating BAK was conserved in human cells. We deleted MARCHF5 and/or VDAC2 in BAX−/− HeLa and KMS-12-PE human multiple myeloma cells using CRISPR/Cas9 gene editing (Supplementary Fig. 3a, b). Interestingly, deleting MARCHF5 alone (i.e independent of VDAC2 deletion) was sufficient to inhibit BAK-dependent apoptosis in HeLa and KMS-12-PE cells induced by BH3 mimetics in short-term (Fig. 3a and Supplementary Fig. 3c) and long-term clonogenic assays (Fig. 3b), as well as impair cytochrome c release (Fig. 3c and Supplementary Fig. 3d) and caspase-3 activation (Fig. 3d and Supplementary Fig. 3e). KM-12 colorectal cancer cells do not express BAX [34] (Supplementary Fig. 3f). As in cells with engineered BAX deletion, MARCHF5 loss likewise rendered KM-12 cells highly resistant to BH3-mimetic treatment (Supplementary Fig. 3g, h). A previous report indicated that MARCHF5 deletion renders cells that expressed both BAX and BAK more sensitive to BH3-mimetic treatment [35]. To determine if this implied a differential effect on BAX and BAK apoptotic activity, we deleted MARCHF5 in wildtype KMS-12-PE cells that express both BAK and BAX (Supplementary Fig. 3i, j). Interestingly, MARCHF5 deletion rendered these cells highly resistant to MCL-1 inhibition that is largely BAK-dependent (Fig. 3e). In contrast, MARCHF5 deletion rendered these cells more sensitive to BCL-2 inhibition with ABT-199 that can be mediated by either BAX or BAK (Fig. 3f), suggesting that MARCHF5 indeed differentially controls BAK and BAX apoptotic activity.

a HeLa cells of the indicated genotypes were treated with BH3 mimetic drugs (2 µM S63845 and escalating concentration of A1331852) for 24 h. Cell death was assessed by PI uptake and flow cytometry. Data are represented as mean ± SD of three independent experiments. b Cells were cultured with BH3 mimetic drugs (2 µM S63845 and escalating concentration of A1331852) for 5 days, colonies were stained with crystal violet and photographed. Representative images of three independent experiments. c Cells were cultured with BH3 mimetic drugs (2 µM S63845 and escalating concentration of A1331852) for 0.5 h or 2 h in the presence of caspase inhibitor QVD-OPh (20 µM). Cytochrome c release was assessed by immunostaining and flow cytometry. Representative histograms shown from 3 independent experiments and proportion of cells with low cytochrome c collated from three independent experiments, data are mean ± SD. d Cells were cultured with BH3 mimetic drugs (2 µM S63845 and escalating concentration of A1331852) for 0.5 h or 2 h. Caspase-3 activation was assessed by immunostaining and flow cytometry. Representative histograms are from 3 independent experiments with proportion of cells exhibiting active caspase-3 collated from three independent experiments, data are mean ± SD. KMS-12-PE cells of the indicated genotypes were treated with escalating concentration of S63845 (e) or the BCL-2 inhibitor ABT-199 (f) for 24 h. Cell death was assessed by PI uptake and flow cytometry. Data are mean ± SD of three independent experiments. **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 based on unpaired Student’s t-test.
MARCHF5 deletion causes BAK activation and sequestration by pro-survival proteins
MARCHF5 is an E3 ubiquitin ligase that resides in the MOM and targets various protein substrates for proteasomal turnover by adding K48-linked ubiquitin chains [17, 35,36,37,38,39,40,41,42,43,44]. MARCHF5 also modulates substrate localisation through K63-linked ubiquitin modifications [45] and also regulates mitochondrial protein import [46].
As MARCHF5 can target MCL-1 proteasomal turnover [17, 35,36,37,38], we first investigated whether MARCHF5 regulates the level and/or mitochondrial localisation of other BCL-2 family proteins. Deleting MARCHF5 stabilised MCL-1 and NOXA protein levels in HeLa and KMS-12-PE cells, but not in MEFs, which express low levels of NOXA (Fig. 4a and Supplementary Fig. 4a, b) [17]. There were no significant changes in expression level among the BCL-2 family proteins examined (BAK, BCL-XL, BCL-2, PUMA, BAD, BIM, BID), or changes in cleavage of BID to its p15 activated form (Fig. 4a), that were consistent across cell types that would likely confer the resistance phenotype observed (Fig. 4a, Supplementary Fig. 4a, b).

a BCL-2 family protein abundance was assessed by Western blotting whole cell lysates prepared from HeLa cells of the indicated genotype. Data are representative of two independent experiments. *indicates cross-reactive non-specific protein. b Cells were cultured with BH3 mimetic drugs (2 µM S63845 and 1 µM A1331852) for 2 h. BAK conformation was assessed by immunostaining and flow cytometry using an antibody that recognises BAK in its active conformation (clone G317-2). Histograms are representative of three independent experiments. c Schematic representation of inhibitory modes of pro-survival proteins. Cell protection is afforded by sequestering BH3-only proteins (Mode 1) or sequestering activated BAK and BAX (Mode 2). BAK (d), MCL-1 (e), or BCL-XL (f) were immunoprecipitated from whole cell lysates. Bound proteins were resolved by SDS-PAGE and immunoblotted for BAK, MCL-1, or BCL-XL. * indicates cross-reactive non-specific protein. Data are representative of three independent experiments. g MCL-1 was immunoprecipitated from whole cell lysates prepared from untreated cells or cells treated with BH3 mimetic drugs (2 µM S63845 and 1 µM A1331852) for 4 h. Bound proteins were resolved by SDS-PAGE and immunoblotted for BAK, NOXA, and MCL-1. Data are representative of three independent experiments.
Although MCL-1 level was elevated following MARCHF5 loss, this was unlikely to be the sole factor impairing BAK as similar apoptosis resistance was observed in Mcl1-deficient MEFs (Fig. 1d). Nevertheless, as MCL-1 was the most significantly altered BCL2 protein in MARCHF5−/− HeLa cells, we determined whether this served to impair BAK. To test this, we assessed the response of MTCH2-deficient cells to BH3 mimetics because MTCH2 is necessary for MARCHF5 to degrade MCL-1 [17]. Deleting MTCH2 in BAX−/− HeLa stabilised MCL-1 to similar levels as MARCHF5 deletion (Supplementary Fig. 4c). However, in contrast to MARCHF5-deficient cells, MTCH2 loss did not confer resistance to BH3-mimetic drugs (Supplementary Fig. 4d), indicating that MCL-1 stabilisation was not sufficient for the protection afforded by MARCHF5 loss.
To drive apoptosis, BAK undergoes significant conformational rearrangement [47,48,49,50,51,52,53,54,55,56,57,58]. We hypothesised that BAK conformation change was inhibited in the absence of MARCHF5. BAK activation was assessed by intracellular flow cytometry with the conformation-specific N-terminal antibody G317-2. As expected, limited activated BAK was observed in untreated MARCHF5 wild-type HeLa (BAX-/-), MEFs (Bax−/−Vdac2−/−), and KMS-12-PE cells (wild-type and BAX-/-), whilst BAK became activated following treatment with BH3 mimetics (Fig. 4b, Supplementary Fig. 4e, f). Surprisingly, in MARCHF5-deficient cells (MEFs, HeLa, KM12, and KMS-12-PE), BAK adopted an activated conformation even in the absence of BH3 mimetics (Fig. 4b, Supplementary Fig. 4e–h) and underwent little further activation when exposed to BH3 mimetics (Fig. 4b, Supplementary Fig. 4e, f), suggesting that most of BAK in untreated MARCHF5-deficient cells was in an activated conformation.
Given that BAK failed to drive apoptosis in MARCHF5-deficient cells despite adopting an activated conformation, we hypothesised that the activated BAK was sequestered by pro-survival BCL-2 proteins thereby limiting its ability to form the homo-oligomers that kill cells. Pro-survival proteins prevent cell death by interacting with BH3-only proteins (Mode 1) or binding to activated BAK/BAX (Mode 2) (Fig. 4c) [59]. Mode 2 interactions are proposed to be less easily disrupted compared to Mode 1 interactions [59, 60] and may confer resistance to anti-cancer treatments [59, 61,62,63]. We evaluated the interactions between BAK and the pro-survival proteins MCL-1 or BCL-XL by co-immunoprecipitation. Using a conformation-specific antibody to pull down the active BAK (clone G317-2) [51], greater amounts of both MCL-1 and BCL-XL were co-precipitated from MARCHF5-deficient HeLa than from control cells (Fig. 4d). Likewise, when either MCL-1 or BCL-XL were immunoprecipitated, more BAK co-precipitated in cells lacking MARCHF5 (Fig. 4e, f). Similar profiles were also observed when MARCHF5 was deleted in KMS-12-PE (Supplementary Fig. 4l, m) and Vdac2−/− MEFs (Supplementary Fig. 4i–k). Treatment of MARCHF5-deficient HeLa cells with BH3 mimetics disrupted the Mode 1 interaction between MCL-1 and NOXA, but MCL-1-BAK Mode 2 complexes were retained (Fig. 4g). Stabilisation of MCL-1 was not sufficient to drive this interaction with BAK in MTCH2-deficient HeLa cells (Supplementary Fig. 4n). These data are consistent with the need for BAK to be in an activated conformation with its BH3 domain exposed to interact with pro-survival proteins and suggest that in the absence of MARCHF5, BAK changes conformation and engages in Mode 2 complexes with pro-survival proteins that are not readily disrupted by BH3 mimetics.
To determine if MARCHF5 loss similarly provoked BAX to adopt an activated conformation, we deleted MARCHF5 in BAK−/− HeLa cells and assessed BAX conformation with the conformation-specific antibody [64] (Supplementary Fig. 5a). Limited BAX activation was detected in untreated MARCHF5−/− HeLa cells, and BAX activation was significantly induced by BH3-mimetics irrespective of MARCHF5 expression (Supplementary Fig. 5b).
MARCHF5 loss does not provoke BAK retrotranslocation
Interaction with pro-survival BCL-2 proteins retrotranslocates BAX and BAK from the MOM thereby determining their apoptotic potential [65,66,67]. We tested whether BAK interaction with pro-survival proteins due to MARCHF5-deficiency provoked BAK retrotranslocation (Fig. 4d–f). Consistent with previous reports, BAX retrotranslocated from mitochondrial fractions over time (Supplementary Fig. 6) [66], whilst BAK retrotranslocation was limited in the absence of ectopic expression of pro-survival BCL-2 proteins (Supplementary Fig. 6) [67]. Although MARCHF5 loss promoted BAK Mode 2 interactions (Fig. 4d–f), it did not cause BAK to dissociate from mitochondria (Supplementary Fig. 6). We cannot exclude that pro-survival interactions driven by MARCHF5 loss may influence BAK subcellular distribution, but as the majority of BAK remained stably associated with mitochondria, dysregulated BAK retrotranslocation was unlikely responsible for the impaired MOMP in MARCHF5-deficient cells.
MARCHF5 regulates BAK apoptotic activity dependent on its E3 ubiquitin ligase activity
We tested whether MARCHF5’s E3 ubiquitin ligase activity was necessary to regulate BAK by engineering HeLa cells expressing MYC-tagged wild-type MARCHF5 or a mutant form that lacks ubiquitin ligase activity (MARCHF5ΔCys) [39, 42]. Whilst wild-type MARCHF5 rescued the apoptotic response of MARCHF5-deficient cells, cells expressing MARCHF5ΔCys remained highly resistant to BH3 mimetic drugs (Fig. 5a, b). Consistent with this, only ligase-active MARCHF5 restored cytochrome c release (Fig. 5c) and caspase-3 activation (Fig. 5d) in response to BH3 mimetics, and reverted BAK to an inactive conformation (Fig. 5e) and disengaged Mode 2 interactions (Fig. 5f, g) in HeLa, MEFs and KMS-12-PE cells (Supplementary Fig. 7). These data indicate that MARCHF5 restrains BAK conformation and apoptotic function via its E3 ubiquitin ligase activity.

a MYC-tagged wild-type MARCHF5 or catalytically inactive mutant MARCFH5ΔCys (C65S, C68S) were expressed in BAX−/−MARCHF5−/− HeLa cells. Exogenous MARCHF5 protein expression was confirmed by immunoblotting for the Myc-tag. *indicates cross-reactive non-specific protein. Data are representative of three independent experiments. b The indicated cells were cultured with BH3 mimetic drugs (2 µM S63845 and escalating concentration of A1331852) for 24 h. Cell death was assessed by PI uptake and flow cytometry. Data are mean ± SD of three independent experiments. c Cells were cultured with BH3 mimetic drugs (2 µM S63845 with 1 µM A1331852) for 0.5 h or 2 h in the presence of caspase inhibitor QVD-OPh (20 µM). Cytochrome c release was assessed by immunostaining and flow cytometry. Proportion of cells with low cytochrome c collated from three independent experiments. Data are presented as mean ± SD. d Cells were cultured with BH3 mimetic drugs (2 µM S63845 and 1 µM A1331852) for 0.5 h or 2 h. Caspase-3 activation was assessed by immunostaining and flow cytometry. Proportion of cells exhibiting active caspase-3 collated from three independent experiments. Data are presented as mean ± SD. e BAK conformation was assessed in the indicated cells by immunostaining and flow cytometry using a BAK conformation-specific antibody (clone G317-2). Histograms are representative of three independent experiments. MCL-1 (f) or BCL-XL (g) were immunoprecipitated from whole cell lysates prepared from the indicated cells. Bound proteins were resolved by SDS-PAGE and immunoblotted for BAK, MCL-1, or BCL-XL. Data are representative of three independent experiments. **p ≤ 0.01, ***p ≤ 0.001 based on unpaired Student’s t-test.
MARCHF5 regulation of BAK conformation is independent of DRP1
MARCHF5 regulates mitochondrial dynamics through its ubiquitination of DRP1 or the DRP1 receptor MiD49 [39, 44]. Recently, DRP1 has been shown to interact with BAX to provoke its activation [68]. Hence, we tested whether BAK activation in MARCHF5-deficient cells was due to DRP1. Consistent with previous findings, MARCHF5−/− HeLa had elevated mitochondrial MiD49, although DRP1 levels remained unchanged (Supplementary Fig. 8a) [44]. Interestingly, treatment with the DRP1 inhibitor, mDivi1 [69], in combination with BH3 mimetics, induced some cell death of the otherwise resistant MARCHF5−/− HeLa cells (Supplementary Fig. 8b). However, DRP1 inhibition did not alter BAK conformation (Supplementary Fig. 8c), suggesting that MARCHF5 loss provokes BAK activation independent of DRP1. The induction of limited cell death by mDivi1 may be due to DRP1-independent inhibition of mitochondrial complex I [70].
Identification of MARCHF5 substrates that contribute to BAK activation
BAK has a sole lysine that is exposed to the cytosol (K113) that we have shown is a target for the E3 ubiquitin ligase Parkin [71], which might be a target for MARCHF5-mediated ubiquitination. However, we did not observe changes in BAK levels following MARCHF5 loss, nor was BAK constitutively ubiquitinated that might implicate BAK as a target of MARCHF5 (Fig. 4a and Supplementary Fig. 4a). Moreover, mutation of this lysine does not influence BAK expression, apoptotic activity, and regulation. These data suggest that BAK is unlikely to be a substrate of MARCHF5. Instead, we hypothesised that MARCHF5 regulates BAK indirectly. MARCHF5 targets many substrates for protein degradation through the addition of K48-linked ubiquitin [17, 35, 39,40,41,42,43,44, 72]. To identify MARCHF5 substrates that are responsible for controlling BAK conformation change, we used two complementary approaches. Firstly, we compared the proteome of MARCHF5 wild-type and MARCHF5-deficient HeLa and MEFs by mass spectrometry. We identified 32 proteins that were significantly elevated in BAX−/−MARCHF5−/− HeLa (Log2FC > 1; FDR < 0.05) (Fig. 6a, Supplementary Fig. 9a and Supplementary Table 3). The known MARCHF5 substrate MFN1 was significantly enriched in this dataset using a slightly less stringent Log2FC cut-off (Log2FC = 0.81; FDR = 0.008) [39,40,41] (Fig. 6a and Supplementary Table 3). Sample preparation was more variable for the MEF proteomics dataset, resulting in fewer proteins reaching a statistically significant threshold, but three proteins were elevated in Vdac2−/−Marchf5−/− relative to Vdac2−/−Marchf5+/+ MEFs (Log2FC > 1; FDR = 0.05) (Supplementary Figs. 9b, c and Supplementary Table 4). Interestingly, one of those proteins was fatty acyl CoA reductase 1 (FAR1), which was also the most significantly elevated protein in MARCHF5−/− HeLa (Log2FC = 3.0; FDR = 6.3 × 10−7) (Fig. 6a, Supplementary Fig. 9a and Supplementary Table 3).

a Volcano plot depicting differences in protein abundance between BAX-/-MARCHF5+/+ and BAX−/−MARCHF5−/− HeLa cells. See Supplementary Table 3 for additional details. b Schematic of the rationale for the drop-out CRISPR/Cas9 library screen. If BAK is driven into inhibitory Mode 2 complexes by a substrate protein that accumulates upon MARCHF5 deletion, then deleting the substrate should restore BAK-mediated apoptosis in MARCHF5 knockout cells. As such, sgRNA targeting the substrate should be depleted following BH3 mimetic treatment. (c, d) BAX−/−MARCHF5−/− HeLa cells with or without Cas9 expression were transduced with a custom lentiviral sgRNA library (see Supplementary Table 5 for additional details). Transduced cells cultured for 5 days. sgRNA were amplified from surviving cells and quantified by sequencing. The significance (FDR-corrected) of sgRNA enriched (c) or depleted (d) was calculated using the ranked order of the Log2FC for individual sgRNA targeting each gene [28]. See Supplementary Table 6A and 6B for additional detail. (e) BAX−/−MARCHF5−/− HeLa cells with Cas9 expression from (c) were treated with DMSO or BH3 mimetic drugs (2 µM S63845 and 1 µM A1331852) for 48 h. sgRNA were amplified from surviving cells and quantified by sequencing. Gene-level significance values were calculated as above. See Supplementary Table 6C for additional detail. f The cells were cultured with BH3 mimetic drugs (2 µM S63845 and escalating concentration of A1331852) for 24 h. Cell death was assessed by PI uptake and flow cytometry. Data are presented as mean ± SD of three independent experiments. g BAX−/−MARCHF5−/−C8ORF82−/− and control HeLa cells were cultured with BH3 mimetic drugs (2 µM S63845 and 1 µM A1331852) in the presence or absence of caspase inhibitor (QVD-OPh, 50 µM) for 24 h. Cell death was assessed by PI uptake and flow cytometry. Data are presented as mean ± SD of two independent experiments. h BAK conformation was assessed by immunostaining and flow cytometry in BAX−/−MARCHF5−/−C8ORF82−/− and control HeLa cells using a BAK conformation-specific antibody (clone G317-2). Histograms are representative of two independent experiments.
To explore whether any of these elevated proteins was responsible for the resistance of MARCHF5-deficient cells, we generated a custom sgRNA library to perform a CRISPR/Cas9 drop-out screen. The sgRNA library comprised 3325 sgRNAs targeting 461 genes (6 or 7 sgRNAs for each gene) identified in our proteomic analyses, previously reported MARCHF5 substrates, peroxisomal proteins and other known components of the MOM, together with 150 non-targeting control sgRNAs (Supplementary Table 5).
The sgRNA library was introduced into BAX−/−MARCHF5−/− HeLa that either did or did not express Cas9. To identify genes that influenced the steady-state growth and survival of BAX-/-MARCHF5−/− HeLa, we compared changes in sgRNA abundance between Cas9+ve cells to Cas9−ve cells. As expected, sgRNAs targeting BAK were enriched in Cas9+ve cells (Fig. 6c and Supplementary Fig. 9d). We also observed a significant depletion of sgRNA targeting BCL2L1/BCLXL and MCL1 in Cas9+ve cells (Fig. 6d and Supplementary Fig. 9d), supporting that these were necessary to maintain survival of BAX−/−MARCHF5−/− cells through their Mode 2 engagement of BAK.
Next, we sought to identify sgRNA that would render MARCHF5-deficient cells sensitive to BH3 mimetics. BAX−/−MARCHF5−/− cells expressing Cas9 and the sgRNA library were treated with BH3-mimetics (2 µM S63845 + 1 µM A1331852) for 48 h. sgRNAs targeting 3 genes were significantly depleted in the treated cells (FDR < 0.05): FAR1, C8orf82, and MYOF (Fig. 6e and Supplementary Fig. 9e), suggesting that expression of these candidates may provoke BAK conformation change in BAX−/−MARCHF5−/− cells.
FAR1 and C8orf82 were of particular interest given that these were among the most upregulated proteins following MARCHF5 deletion (Fig. 6a and Supplementary Fig. 9a–c). To validate if these proteins regulate BAK apoptotic activity, each was deleted in BAX-/-MARCHF5−/− HeLa (Supplementary Fig. 9f–h). Deleting FAR1 or MYOF alone did not markedly affect the response of BAX-/-MARCHF5−/− cells to BH3 mimetics (Fig. 6f). Intriguingly, deleting C8orf82 partially restored caspase-mediated apoptosis in BAX-/-MARCHF5−/− MEFs in response to BH3 mimetics (Fig. 6f, g). However, C8orf82 loss did not reverse the constitutive BAK activation (Fig. 6h), suggesting that C8orf82 was not responsible for this aspect of the MARCHF5-deficient phenotype and might instead influence BAK-mediated apoptosis through a different mechanism.
MARCHF5 controls BAK independently of BH3-only proteins
Our proteomics and genetic screening, which were predicated on the hypothesis that the MARCHF5 substrate would be degraded, and so would accumulate in its absence, did not reveal a strong candidate for the MARCHF5 substrate regulating BAK. Engagement with BH3-only proteins, such as BIM or BID, is typically regarded as necessary to initiate BAK activating conformation change. Therefore, we sought to test whether BH3-only proteins were critical for driving BAK conformation change following MARCHF5 deletion. We utilised HCT116 cells engineered to lack all known BCL-2 proteins, including the eight BH3-only family members (“All Bcl-2 ko”) [16]. BAK and MCL-1 (Cer-hMCL-1) were stably expressed in these cells (Fig. 7b), and MARCHF5 was subsequently deleted (Supplementary Fig. 10). HCT116 All Bcl-2 ko cells expressing Cer-hMCL-1 and hBAK underwent cell death with MCL-1 inhibitor treatment (Fig. 7c) and MARCHF5 deletion rendered these cells insensitive (Fig. 7c). Flow cytometry revealed that MARCHF5 deletion provoked constitutive activated conformation of BAK similar to MARCHF5-expressing cells treated with MCL-1 inhibitor (Fig. 7d). Moreover, MARCHF5 loss also promoted constitutive BAK: MCL-1 Mode 2 interaction (Fig. 7e). These data suggest that MARCHF5 controls BAK conformation and Mode 2 interactions independent of known BH3-only proteins.

a, b hBAK and Cerulean-hMCL-1 were stably expressed in HCT116 cells lacking all BCL-2 family genes [16]. Protein expression was confirmed by immunoblotting. Two independent MARCHF5−/− clones were generated (see Supplementary Fig. 10 for indel confirmation). Data are representative of two independent experiments. c Cells in b were treated with MCL-1 inhibitor S63845 at the indicated concentrations for 24 h and cell death was assessed by PI uptake. Data are presented as mean ± SD of two independent experiments. d BAK conformation was assessed by immunostaining and flow cytometry using a BAK conformation-specific antibody (G317-2) either in untreated cells or cells treated with S63845 (2 μM) for 2 h. Histograms are representative of two independent experiments. e BAK or MCL-1 were immunoprecipitated from whole cell lysates prepared from MARCHF5+/+ or MARCHF5−/− All BCL-2 knockout HCT116 cells re-expressing Cer-hMCL-1 and hBAK. Bound proteins were resolved by SDS-PAGE and immunoblotted for BAK or MCL-1. *indicates cross-reactive non-specific protein. Data are representative of two independent experiments.
Discussion
MARCHF5 has been linked to apoptosis because it can ubiquitinate the pro-survival protein MCL-1 [17, 35,36,37,38], and proteins that regulate mitochondrial morphology [39,40,41,42,43,44,45, 44, 73, 74]. MARCHF5-deficient cells exhibit a hyper-fused mitochondrial network and diminished sensitivity to death stimuli in some cellular contexts [44, 73, 74]. In other cases, MARCHF5 loss can promote apoptosis even though MCL-1 is elevated [17, 35, 38]. Here, we have identified that MARCHF5 ubiquitin ligase activity restrains BAK conformation change, such that MARCHF5 loss triggers BAK activation and formation of Mode 2 complexes that block BAK-mediated apoptosis.
Although the target, or targets, of MARCHF5’s ubiquitin ligase activity that limit BAK conformation change remain unresolved, that this is independent of all BH3-only proteins suggests an unexpected new level of control. Intriguingly, whilst it was necessary to delete Vdac2 in MEFs to reveal the BAK phenotype caused by MARCHF5-deficiency (Figs. 1d, e, and 2a), deleting MARCHF5 alone was sufficient in HeLa, KMS-12-PE, and KM-12 cells (Fig. 3a, b, and Supplementary Fig. 3c, h). This difference may relate to disparities in how mouse and human BAK interact with VDAC2, as noted from the divergent impact of small molecules on these interactions [75]. However, other factors may also be involved, and follow-up studies in additional contexts would help to determine whether species-differences in BAK: VDAC2 interaction contribute to the observed phenotypes.
An interesting aspect of the phenotype that results from MARCHF5 loss is that BAK appears to be driven into its active conformation, but paradoxically this is associated with diminished sensitivity to BH3-mimetics. In most cases, BAK activation and conformation change would drive cell death through the formation of BAK dimers and higher-order oligomers that damage the mitochondrial outer membrane [48, 52, 54, 76]. In cells lacking MARCHF5, constitutively activated BAK engages with pro-survival proteins in inhibitory Mode 2 complexes thereby limiting its apoptotic capacity (Fig. 4d–f).
Our studies focused on BAK-driven apoptosis by using cells that did not express BAX. In this context, BAK function was markedly reduced upon loss of MARCHF5. This contrasts with studies in cells expressing both BAX and BAK, where MARCHF5 deletion resulted in greater sensitivity to apoptosis triggers [17, 35, 37]. One hypothesis to reconcile these findings is that deleting MARCHF5 could also promote BAX-activating conformation change, but that there may be insufficient free pro-survival proteins to sequester the activated populations of both BAK and BAX in inhibitory Mode 2 complexes. Indeed, we observed that MARCHF5 loss provoked, albeit limited, BAX conformation change (Supplementary Fig. 5b). Why the constitutive BAX conformation change is limited compared to the near complete BAK conformation change is unclear. However, given that MARCHF5 is an integral mitochondrial ligase, it is intriguing to speculate that it is the mitochondrial population of BAX that is activated in response to MARCHF5 loss.
The E3 ubiquitin ligase activity of MARCHF5 ligase activity restricts BAK from becoming activated and being locked into inactive Mode 2 complexes with pro-survival proteins. This suggests that the ubiquitination of a MARCHF5 substrate controls the observed phenotype. Our proteomics and genetic screens failed to identify a compelling candidate suggesting possible redundancy or even a non-proteinaceous substrate. The substrate may directly influence BAK and/or its pro-survival counterparts, but could equally influence these proteins indirectly, perhaps by altering the mitochondrial lipid environment in which they reside. Mitochondrial lipids can actively participate in the regulation of BAX/BAK-driven apoptosis [77,78,79], and MARCHF5 is known to influence lipid and fatty acid metabolism as well as mitochondrial lipid modifications [80, 81]. There could also be interplay in this regard with the dominant influence of VDAC2 over the phenotype in certain contexts (Fig. 1e), given the propensity for VDAC2 loss to disrupt peroxisomes [82], which are major sites of lipid metabolism.
It is noteworthy that BAK was driven to its active form in cells lacking MARCHF5 independently of its canonical activators - the BH3-only proteins (Fig. 7d). This finding aligns with observations that BAK and BAX can be driven to their active conformation potentially through interaction with mitochondrial lipids [16]. Changes in the nature or composition of mitochondrial lipids following MARCHF5 deletion [80, 81] could conceivably impact the propensity for BAK to become activated as well as influence whether activated BAK molecules form homodimers or engage with pro-survival proteins.
Altogether, these findings uncover a novel pathway for BAK to transition to its activated conformation that is controlled by MARCHF5 ubiquitin ligase activity. Our studies raise several interesting questions about the mechanisms underlying this process that warrant further exploration, including the identity of the MARCHF5 substrate responsible, and why BAK molecules activated in this way engage with pro-survival proteins rather than homo-oligomerise. Furthermore, although we show that MARCHF5 controls BAK conformation and apoptotic activity in a conserved manner in a variety of cell types, how this mechanism governs cellular fate in vivo during development, tissue homeostasis, and possibly tumour progression remains unclear. Answering these questions will provide important new insights into the molecular control of apoptosis signalling and may provide a new opportunity to intervene.
References
Ferriero DM. Neonatal brain injury. N. Engl J Med. 2004;351:1985–95.
Oliveira JB, Gupta S. Disorders of apoptosis: mechanisms for autoimmunity in primary immunodeficiency diseases. J Clin Immunol. 2008;28:20–8.
Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56.
Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63.
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–305.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403.
Lee Y-J, Jeong S-Y, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–11.
Yu T, Fox RJ, Burwell LS, Yoon Y. Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein hFis1. J Cell Sci. 2005;118:4141–51.
Weaver D, Eisner V, Liu X, Várnai P, Hunyady L, Gross A, et al. Distribution and apoptotic function of outer membrane proteins depend on mitochondrial fusion. Mol Cell. 2014;54:870–8.
Roy SS, Ehrlich AM, Craigen WJ, Hajnóczky G. VDAC2 is required for truncated BID‐induced mitochondrial apoptosis by recruiting BAK to the mitochondria. EMBO Rep. 2009;10:1341–7.
Naghdi S, Várnai P, Hajnóczky G. Motifs of VDAC2 required for mitochondrial Bak import and tBid-induced apoptosis. Proc Natl Acad Sci. 2015;112:E5590–E5599.
Yamagata H, Shimizu S, Nishida Y, Watanabe Y, Craigen W, Tsujimoto Y. Requirement of voltage-dependent anion channel 2 for pro-apoptotic activity of Bax. Oncogene 2009;28:3563–72.
Ma S, Nguyen T, Tan I, Ninnis R, Iyer S, Stroud D, et al. Bax targets mitochondria by distinct mechanisms before or during apoptotic cell death: a requirement for VDAC2 or Bak for efficient Bax apoptotic function. Cell Death Differ. 2014;21:1925–35.
Chin HS, Li MX, Tan IK, Ninnis RL, Reljic B, Scicluna K, et al. VDAC2 enables BAX to mediate apoptosis and limit tumor development. Nat Commun. 2018;9:4976.
Cheng EH-Y, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003;301:513–7.
O’Neill KL, Huang K, Zhang J, Chen Y, Luo X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016;30:973–88.
Djajawi TM, Liu L, Gong J-N, Huang AS, Luo M-J, Xu Z, et al. MARCH5 requires MTCH2 to coordinate proteasomal turnover of the MCL1: NOXA complex. Cell Death Differ. 2020;27:2484–99.
Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783.
Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–91.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–23.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308.
Koike-Yusa H, Li Y, Tan E-P, Velasco-Herrera MDC, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32:267–73.
Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L, et al. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 2015;10:1422–32.
Waterhouse NJ, Steel R, Kluck R, Trapani JA, Assaying cytochrome C translocation during apoptosis. Signal Transduction Protocols. Springer, 2004, pp 307–13.
Johnston AJ, Hoogenraad J, Dougan DA, Truscott KN, Yano M, Mori M, et al. Insertion and assembly of human tom7 into the preprotein translocase complex of the outer mitochondrial membrane. J Biol Chem. 2002;277:42197–204.
Hock DH, Reljic B, Ang C-S, Muellner-Wong L, Mountford HS, Compton AG, et al. HIGD2A is required for assembly of the COX3 module of human mitochondrial complex IV. Mol Cell Proteom. 2020;19:1145–60.
Eden E, Lipson D, Yogev S, Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol. 2007;3:e39.
König R, Chiang C-Y, Tu BP, Yan SF, DeJesus PD, Romero A, et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat Methods. 2007;4:847–9.
Wei MC, Zong W-X, Cheng EH-Y, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001;292:727–30.
Haraguchi M, Torii S, Matsuzawa S-I, Xie Z, Kitada S, Krajewski S, et al. Apoptotic protease activating factor 1 (Apaf-1)–independent cell death suppression by Bcl-2. J Exp Med. 2000;191:1709–20.
Tait SW, Green DR. Caspase-independent cell death: leaving the set without the final cut. Oncogene 2008;27:6452–61.
Gama V, Swahari V, Schafer J, Kole AJ, Evans A, Huang Y, et al. The E3 ligase PARC mediates the degradation of cytosolic cytochrome c to promote survival in neurons and cancer cells. Sci Signal. 2014;7:ra67.
Tait SW, Parsons MJ, Llambi F, Bouchier-Hayes L, Connell S, Muñoz-Pinedo C, et al. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell. 2010;18:802–13.
Hostein I, Robertson D, DiStefano F, Workman P, Clarke PA. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 2001;61:4003–9.
Subramanian A, Andronache A, Li YC, Wade M. Inhibition of MARCH5 ubiquitin ligase abrogates MCL1-dependent resistance to BH3 mimetics via NOXA. Oncotarget 2016;7:15986–6002.
Cherok E, Xu S, Li S, Das S, Meltzer WA, Zalzman M, et al. Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5–dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol Biol Cell. 2017;28:396–410.
Arai S, Varkaris A, Nouri M, Chen S, Xie L, Balk SP. MARCH5 mediates NOXA-dependent MCL1 degradation driven by kinase inhibitors and integrated stress response activation. Elife. 2020;9:e54954.
Haschka MD, Karbon G, Soratroi C, O’Neill KL, Luo X, Villunger A, MARCH5-dependent degradation of MCL1/NOXA complexes defines susceptibility to antimitotic drug treatment. Cell Death Differ. 2020;27:2297–312.
Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007;178:71–84.
Park Y-Y, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J Cell Sci. 2010;123:619–26.
Park Y-Y, Cho H, Mitofusin 1 is degraded at G 2/M phase through ubiquitylation by MARCH 5. Cell Div. 2012;7:25.
Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006;25:3618–26.
Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH‐V is a novel mitofusin 2‐and Drp1‐binding protein able to change mitochondrial morphology. EMBO Rep. 2006;7:1019–22.
Xu S, Cherok E, Das S, Li S, Roelofs BA, Ge SX, et al. Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol Biol Cell. 2016;27:349–59.
Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, et al. MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol Cell. 2013;51:20–34.
Phu L, Rose CM, Tea JS, Wall CE, Verschueren E, Cheung TK, et al. Dynamic regulation of mitochondrial import by the ubiquitin system. Mol Cell. 2020;77:1107–23.e1110.
Westphal D, Kluck R, Dewson G. Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014;21:196–205.
Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, et al. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3: groove interactions. Mol Cell. 2008;30:369–80.
Griffiths GJ, Corfe BM, Savory P, Leech S, Degli Esposti M, Hickman JA, et al. Cellular damage signals promote sequential changes at the N-terminus and BH-1 domain of the pro-apoptotic protein Bak. Oncogene 2001;20:7668–76.
Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, et al. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell. 2014;55:938–46.
Alsop AE, Fennell SC, Bartolo RC, Tan IK, Dewson G, Kluck RM. Dissociation of Bak α1 helix from the core and latch domains is required for apoptosis. Nat Commun. 2015;6:1–13.
Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, Kluck RM. Bak activation for apoptosis involves oligomerization of dimers via their α6 helices. Mol Cell. 2009;36:696–703.
Iyer S, Bell F, Westphal D, Anwari K, Gulbis J, Smith B, et al. Bak apoptotic pores involve a flexible C-terminal region and juxtaposition of the C-terminal transmembrane domains. Cell Death Differ. 2015;22:1665.
Pang Y-P, Dai H, Smith A, Meng XW, Schneider PA, Kaufmann SH. Bak conformational changes induced by ligand binding: insight into BH3 domain binding and Bak homo-oligomerization. Sci Rep. 2012;2:257.
Li MX, Tan IK, Ma SB, Hockings C, Kratina T, Dengler MA, et al. BAK α6 permits activation by BH3-only proteins and homooligomerization via the canonical hydrophobic groove. Proc Natl Acad Sci. 2017;114:7629–34.
Uren RT, O’Hely M, Iyer S, Bartolo R, Shi MX, Brouwer JM, et al. Disordered clusters of Bak dimers rupture mitochondria during apoptosis. Elife 2017;6:e19944.
Bleicken S, Classen M, Padmavathi PV, Ishikawa T, Zeth K, Steinhoff H-J, et al. Molecular details of Bax activation, oligomerization, and membrane insertion. J Biol Chem. 2010;285:6636–47.
Subburaj Y, Cosentino K, Axmann M, Pedrueza-Villalmanzo E, Hermann E, Bleicken S, et al. Bax monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nat Commun. 2015;6:1–11.
Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44:517–31.
Hockings C, Alsop AE, Fennell SC, Lee EF, Fairlie WD, Dewson G, et al. Mcl-1 and Bcl-x L sequestration of Bak confers differential resistance to BH3-only proteins. Cell Death Differ. 2018;25:721–34.
Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88.
Roberts AW, Stilgenbauer S, Seymour JF, Huang DC. Venetoclax in patients with previously treated chronic lymphocytic leukemia. Clin Cancer Res. 2017;23:4527–33.
Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016;538:477–82.
Dewson G, Snowden R, Almond J, Dyer M, Cohen G. Conformational change and mitochondrial translocation of Bax accompany proteasome inhibitor-induced apoptosis of chronic lymphocytic leukemic cells. Oncogene 2003;22:2643–54.
Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, et al. Bcl-x(L) Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 2011;145:104–16.
Lauterwasser J, Todt F, Zerbes RM, Nguyen TN, Craigen W, Lazarou M, et al. The porin VDAC2 is the mitochondrial platform for Bax retrotranslocation. Sci Rep. 2016;6:1–11.
Todt F, Cakir Z, Reichenbach F, Emschermann F, Lauterwasser J, Kaiser A, et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2015;34:67–80.
Jenner A, Pena-Blanco A, Salvador-Gallego R, Ugarte-Uribe B, Zollo C, Ganief T, et al. DRP1 interacts directly with BAX to induce its activation and apoptosis. EMBO J. 2022;41:e108587.
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204.
Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, et al. The Putative Drp1 Inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell. 2017;40:583–94.e586
Bernardini JP, Brouwer JM, Tan IK, Sandow JJ, Huang S, Stafford CA, et al. Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J. 2019;38:e99916.
Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005;121:1085–95.
Fang L, Hemion C, Goldblum D, Meyer P, Orgül S, Frank S, et al. Inactivation of MARCH5 prevents mitochondrial fragmentation and interferes with cell death in a neuronal cell model. PLoS One. 2012;7:e52637.
Wang J, Aung LH, Prabhakar BS, Li P, The mitochondrial ubiquitin ligase plays an anti‐apoptotic role in cardiomyocytes by regulating mitochondrial fission. J Cell Mol Med. 2016, 20: 2278–88.
van Delft MF, Chappaz S, Khakham Y, Bui CT, Debrincat MA, Lowes KN, et al. A small molecule interacts with VDAC2 to block mouse BAK-driven apoptosis. Nat Chem Biol. 2019;15:1057–66.
Aluvila S, Mandal T, Hustedt E, Fajer P, Choe JY, Oh KJ. Organization of the mitochondrial apoptotic BAK pore oligomerization of the bak homodimers. J Biol Chem. 2014;289:2537–51.
Cowan AD, Smith NA, Sandow JJ, Kapp EA, Rustam YH, Murphy JM, et al. BAK core dimers bind lipids and can be bridged by them. Nat Struct Mol Biol. 2020;27:1024–31.
Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, et al. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012;148:988–1000.
Vasquez-Montes V, Rodnin MV, Kyrychenko A, Ladokhin AS, Lipids modulate the BH3-independent membrane targeting and activation of BAX and Bcl-xL. Proc Natl Acad Sci. 2021;118:e2025834118.
Bond ST, Moody SC, Liu Y, Civelek M, Villanueva CJ, Gregorevic P, et al. Mitochondria dysfunction in aging and metabolic diseases: The E3 ligase MARCH5 is a PPARγ target gene that regulates mitochondria and metabolism in adipocytes. Am J Physiol-Endocrinol Metab. 2019;316:e293.
Kitakata H, Endo J, Matsushima H, Yamamoto S, Ikura H, Hirai A, et al. MITOL/MARCH5 determines the susceptibility of cardiomyocytes to doxorubicin-induced ferroptosis by regulating GSH homeostasis. J Mol Cell Cardiol. 2021;161:116–29.
Hosoi K-I, Miyata N, Mukai S, Furuki S, Okumoto K, Cheng EH, et al. The VDAC2–BAK axis regulates peroxisomal membrane permeability. J Cell Biol. 2017;216:709–22.
Deutsch EW, Bandeira N, Sharma V, Perez-Riverol Y, Carver JJ, Kundu DJ, et al. The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020;48:D1145–D1152.
Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450.
Acknowledgements
We would like to thank Ruth Kluck, Peter Czabotar, Daniel Gray, and Andreas Strasser for helpful discussions and suggestions; Melissa Shi for technical assistance; Stephen Wilcox for DNA sequencing; Simon Cobbold for assistance with proteomic analysis; Prof Xu Luo for providing all BCL-2 genes knockout HCT116 cells; Christine White for technical support. We thank the Bio21 Mass Spectrometry and Proteomics Facility (MMSPF) for the provision of instrumentation, training, and technical support, and the Mito Foundation for the provision of instrumentation through the large equipment grant support scheme. This work was supported by scholarship fellowship and grants from the Australian National Health and Medical Research Council (NHMRC) to DAS (#1140851, #1140906); Melbourne University (MIRS and MIFRS scholarships to ASH and TMD); the Walter and Eliza Hall Institute of Medical Research (to ASH); the Bodhi Foundation and the McPhee Charitable Trust (to GD). Research was supported by an NHMRC Independent Research Institutes Infrastructure Support Scheme grant (361646) and Victorian State Government Operational Infrastructure Support grant.
Author information
Authors and Affiliations
Contributions
DCSH, MvD, and GD devised the study and interpreted data. ASH and HSC planned, performed experiments, and interpreted data. BR and DAS performed the proteomics and interpreted data. TMD, JNG, and IKLT generated reagents; ASH and MvD collated and analysed data. ASH, MvD, GD wrote the manuscript. All authors contributed to manuscript review.
Corresponding authors
Ethics declarations
Competing interests
GD, DCSH and MvD are employees of the Walter and Eliza Hall Institute which receives royalty payments relating to the use of Venclexta/venetoclax.
Ethics approval
This study did not include human participants or animal research and so did not require ethics approval.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by C Borner
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, A.S., Chin, H.S., Reljic, B. et al. Mitochondrial E3 ubiquitin ligase MARCHF5 controls BAK apoptotic activity independently of BH3-only proteins. Cell Death Differ 30, 632–646 (2023). https://doi.org/10.1038/s41418-022-01067-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-022-01067-z

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}