
Abstract
Human mesenchymal stem cells (hMSCs) can be differentiated into adipocytes and osteoblasts. The processes are driven by the rewiring of chromatin architectures and transcriptomic/epigenomic changes. Here, we induced hMSCs to adipogenic and osteogenic differentiation, and performed 2 kb resolution Hi-C experiments for chromatin loops detection. We also generated matched RNA-seq, ChIP-seq and ATAC-seq data for integrative analysis. After comprehensively comparing adipogenesis and osteogenesis, we quantitatively identified lineage-specific loops and screened out lineage-specific enhancers and open chromatin. We reveal that lineage-specific loops can activate gene expression and facilitate cell commitment through combining enhancers and accessible chromatin in a lineage-specific manner. We finally proposed loop-mediated regulatory networks and identified the controlling factors for adipocytes and osteoblasts determination. Functional experiments validated the lineage-specific regulation networks towards IRS2 and RUNX2 that are associated with adipogenesis and osteogenesis, respectively. These results are expected to help better understand the chromatin conformation determinants of hMSCs fate commitment.
Similar content being viewed by others


Introduction
Human mesenchymal stem cells (hMSCs) are multipotential cells and capable of differentiating into adipocytes (AC) and osteoblasts (OB). Studies have characterized key factors that manipulate hMSCs differentiation. For example, the transcriptome profiling during adipogenesis [1] and osteogenesis [2] have identified several signature genes. In particular, the investigation of core transcription factors (TFs) has uncovered master regulators for adipogenesis [3, 4] and osteogenesis [5, 6]. Besides, epigenomic programming indicates dynamic histone modifications during mouse MSCs differentiation [7, 8]. Recently, studies focusing on open chromatin have attracted attention by showing the rewiring of chromatin accessibility during adipogenesis and osteogenesis [9]. These observations highlight the distinct genetic regulatory characteristics between AC and OB commitment.
Taking advantage of high-throughput chromosome conformation capture (Hi-C) technology, the spatial organization of human genome has been revealed at unprecedented resolution, and the structures of chromatin loops can be detected. Chromatin loops directly bring distal elements into close proximity with target promoters [10], which further contributes to gene regulation [10, 11]. Studies have reported that the reorganization of chromatin loops can facilitate cell differentiation [12, 13]. In terms of hMSCs differentiation, by utilizing enhancer capture Hi-C techniques, the enhancer interactome at early stage (day 1) of adipogenesis and osteogenesis was revealed [14]. Although they have identified lineage-specific interactions, only those with highly connected enhancers were considered. Moreover, the transcriptomic and epigenomic characterization of these interactions is rather limited. Up to now, a genome-wide view of how chromatin loops rewire during hMSCs lineage determination, and the affiliated transcriptome and epigenome alternation is still blank.
Here we performed high resolution Hi-C experiments before and after inducing hMSCs differentiation into AC and OB, and simultaneously included RNA-seq, ChIP-seq and ATAC-seq at each stage to provide a comprehensive insight of gene regulation patterns. By integrating these data, we identified lineage-specific chromatin loops, and showed the associated reprograming of gene expression, enhancer mapping and TF binding. We also constructed lineage-specific long-range regulation networks and identified novel controlling factors for hMSCs fate determination.
Results
High-resolution Hi-C analyses identify lineage-specific loops during adipocytes and osteoblasts commitment
We first carried out high-resolution Hi-C experiments before and after hMSCs differentiation (Fig. 1A and S1). At least six replicates were generated for each cell type. The Hi-C samples were well stratified by cell types (Fig. S2A) and highly correlated between replicates (Fig. S2B). Hi-C data were then combined to produce an average of ~2.5 billion valid read pairs after quality control (Supplementary Tables S1 and S2). The intrachromosomal contact probability curves were similar across cells (Fig. S2C) and consistent with previous reports [15]. As shown in Fig. 1B, local chromatin interactions were able to be observed in chromatin contact maps at 2 kb resolution.

A Schematic representation of this study. hMSCs were differentiated into adipocytes and osteoblasts. 15 days of adipogenic differentiation and 21 days of osteogenic differentiation was conducted by supplying with specific differentiation media. The collections of cells were subjected to Hi-C, RNA-seq, ChIP-seq and ATAC-seq measurement. B Normalized Hi-C interaction frequencies displayed as two-dimensional heatmaps at 50 kb, 10 kb and 2 kb/1 kb resolutions for different cells. An expected OB-specific interaction is circled, which also shows mesenchymal-lineage specificity. C Differential contacted loops during adipogenesis and osteogenesis were identified by performing statistical analyses. The overlap represents the counts of loops that were not differentially contacted. The number of lineage-specific loops are noted in bold. D The enrichment of enhancers and open chromatin at lineage-specific loop anchors and control regions. Enhancers were identified by combining H3K27ac and H3K4me1 ChIP-seq data, and open chromatin was recognized from ATAC-seq peaks. Enrichment Z-scores are plotted. E ChromHMM annotation enrichment at lineage-specific loop anchors and control regions. Enrichment Z-scores are plotted. The more deviation from zero indicates the more significant enrichment or depletion. Cells are separated by different shapes. F The enrichment of CTCF binding sites at lineage-specific loop anchors, control regions and the 2 kb interval neighbors to anchors. Enrichment Z-scores are plotted. G The enrichment of lineage-specific enhancer-enhancer interactions in lineage-specific loops. Boxplots showing the distribution of fold change ratios by comparing the number of lineage-specific loops with control pairs that were overlapped with enhancer-enhancer interactions. Each point represents a fold change ratio of overlapping lineage-specific loops to the overlapping control pairs. 1000 sets of control pairs were generated in total. Empirical P values are indicated (***P < 0.001).
The genome is hierarchically organized at different genomic scales, including A/B compartments and topologically associated domains (TADs) [16]. By exploring our Hi-C data, we observed lineage-specific A/B compartment switch after differentiation (Fig. S3A, B). Among 4303 and 5329 TADs identified in AC and OB (Supplementary Table S3), 10% and 23% were adipogenesis and osteogenesis specific, respectively, although TADs were generally consistent (Fig. S3C). Moreover, over 60% of TAD boundaries can be found in other cells [16] (Fig. S3D).
Taking advantage of the high-resolution Hi-C data, we called chromatin loops in each cell (Supplementary Table S4; Methods). Loop reproducibility was verified between replicates (Fig. S3E; Methods). In contrast with TADs, loop structures changed more dramatically that only ~33% of those were shared across cells. As a loop shown in Fig. 1B, when comparing with hMSCs, the contact frequency was elevated in OB but weakened in AC. It was also a mesenchymal-lineage-specific loop when comparing with Hi-C data from GM12878 [16]. This loop harbors the IQCJ-SCHIP1 readthrough gene, whose defect leads to abnormal skeleton morphology in mouse [17].
We then identified differentially contacted loops during adipogenesis or osteogenesis by conducting quantitative comparison (Methods). We found comparable proportions of remodeled and stable TADs that harbored differentially contacted loops (the proportions were 80.9% (remodeled) and 79.0% (stable) in AC; 69.4% (remodeled) and 76.1% (stable) in OB), indicating that loop structures are able to capture more detailed chromatin conformation alternation. Among differentially contacted loops, we further identified lineage-specific loops (Methods). Finally, we found 4899 AC-specific loops, 5062 OB-specific loops and 5685 hMSCs-specific loops, respectively (Fig. 1C).
Active regulatory elements are enriched in lineage-specific loops
Previous studies have highlighted the enhancer-involving interactions facilitated by chromatin loops. To see whether this is confirmed by lineage-specific loops, we initially generated ChIP-seq data of two enhancer-associated histone modifications, H3K4me1 and H3K27ac in each cell (Fig. S4A). Both marks were correlated between replicates but more lineage-specific across cells (Fig. S4B), and were successfully recognized at the upstream of representative genes’ TSSes (Fig. S4C, D). After peak calling and integrating, we identified 128,179, 224,322 and 167,451 putative enhancers in hMSCs, AC and OB, respectively (Methods). Chromatin accessibility is another critical condition for enhancer-centered regulation. Here, we detected accessible chromatin using ATAC-seq (Fig. S4E) and identified 138,820 and 120,209 confidential peaks in AC and OB, respectively. We also retrieved accessible regions in hMSCs from ENCODE [18].
Enrichment analysis suggested that the lineage-specific loop anchors were significantly enriched for both enhancers and ATAC-seq peaks comparing to control regions (Fig. 1D; Methods). We also obtained chromatin states of regulatory elements from ChromHMM annotations. Likewise, lineage-specific loop anchors were significantly enriched for active elements, while less occupied by transcription signals (Fig. 1E and Supplementary Table S5; Methods). We next obtained ChIP-seq data of the architectural protein CTCF from ENCODE [18] and Mikkelsen et al. [19], and found that lineage-specific loop anchors contained more CTCF binding sites than their 2 kb interval neighbors or control regions (Fig. 1F).
Recently, enhancer-enhancer interactions were identified during the early stage of adipogenesis and osteogenesis [14]. We wondered whether they can be well captured by lineage-specific loops. We identified lineage-specific enhancer-enhancer interactions (Methods) and found that 45–53% of those were located within lineage-specific loops. Statistical analysis confirmed the significant enrichment for lineage-specific enhancer-enhancer interactions in lineage-specific loops comparing to null distributions (empirical Penrichment < 0.001; Fig. 1G). Together, these results indicate the overall enrichment of active elements at lineage-specific loops during hMSCs fate commitment.
Lineage-specific loops are accompanied by the activation of lineage-determining genes
We generated RNA-seq data to explore the relationship between chromatin conformation and gene expression during hMSCs differentiation. After gene expression quantification and differentially expressed gene identification, we found that active genes were associated with A compartment (Fig. S5A), prone to entirely resided in TADs (Fig. S5B), generally observed around the loop anchors of AC and OB (Fig. S5C). And gene expression increased as the distance to loop anchors decreased (Fig. 2A).

A Comparison of gene expression levels with respect to different distances to loop anchors. P values were calculated by Wilcoxon signed-rank test (***P < 0.001). B The expression levels of genes located within AC- or OB-specific loops were compared between hMSCs, AC and OB. P values were calculated by paired sample Wilcoxon test (**P < 0.01, ***P < 0.001). C All expressed genes were grouped by comparing the number of surrounding lineage-specific loops (±1 Mb around TSS). The expression of genes having more surrounding AC-/OB-specific loops were compared between cells. P values were calculated by paired sample Wilcoxon test (**P < 0.01, ***P < 0.001). D Upregulated genes during adipogenesis or osteogenesis were mapped to lineage-specific or common loops under different distances to loop anchors. The similarity of each gene set between AC and OB was indicated by Jaccard similarity coefficient. E Differentiation associated GO pathway enrichment analyses using lineage-specific genes located within or beyond 40 kb to lineage-specific loops. F An illustration of the AC-specific loop near APOE. The lineage-specific interaction (purple red arc, right) was confirmed by enhanced contact frequency (circled in heatmap, left) in AC comparing with hMSCs and OB, which was accompanied by elevated APOE expression (middle) in AC.
We next investigated whether lineage-specific loop alternation was associated with gene activation. We found that genes located within AC/OB-specific loops had higher expression in respective cell than that in the other cells (Fig. 2B; paired samples Wilcoxon test). Considering some genes located outside but near lineage-specific loops, we wondered if genes were lineage-specific activated when there were more surrounding lineage-specific loops. After counting surrounding lineage-specific loops (±1 Mb) for each gene, we found significantly higher expression of genes with more surrounding AC/OB-specific loops (Fig. 2C; paired samples Wilcoxon test).
We questioned if lineage-specific loops were selectively contacted lineage-determining genes that are supposed to be cell-specific activated. Up-regulated genes were grouped by different distance to lineage-specific/common loops, and compared for gene set similarity between AC and OB. We found that, as distance decreased, gene similarity decreased for lineage-specific loops while increased for common loops (Fig. 2D; Jaccard similarity coefficient), suggesting that active genes tend to be more distinct near lineage-specific loops. We then identified AC/OB-specific genes as up-regulated only during adipogenesis or osteogenesis. Lineage-specific genes close to lineage-specific anchors (≤40 kb) were significantly enriched for adipogenesis/osteogenesis-related pathways (Fig. 2E). As an example, the AC-specific activation was observed for adipogenesis-associated gene APOE that located within an AC-specific loop (Fig. 2F). Together, our results suggest a close distance between lineage-determining genes and lineage-specific loops.
Cell fate determination is achieved by lineage-specific loops collaborating with lineage-specific enhancers
Long-range enhancer-promoter interaction is usually facilitated by chromatin loops [20, 21]. We then investigated whether gene activation during hMSCs differentiation was caused by rearrangement of enhancer-promoter interactions through lineage-specific loops. Putative enhancers were identified as mentioned above. We found that the contact frequency of loops mapping with enhancers was significantly higher than those without enhancers mapping (Fig. 3A). By selecting enhancers located within lineage-specific loops, we observed that the ChIP-seq signals were also likely to be lineage-specific (Fig. 3B). Lineage-specific enhancers were then screened out, and were found significantly enriched in lineage-specific loops (AC: P = 2.44 × 10−68, OR = 1.08; hMSCs: P = 1.77 × 10−11, OR = 1.03; OB: P < 1 × 10−100, OR = 1.39; fisher exact test) (Fig. 3C). However, for shared enhancers across cells, they were found depleted in AC/OB-specific loops (AC: P = 3.45 × 10−8, OR = 0.94; OB: P < 1 × 10−100, OR = 0.67) but enriched in hMSCs-specific loops (P = 8.92 × 10−22, OR = 1.07), suggesting that some preestablished enhancers in hMSCs-specific loops are likely to be retained after differentiation.

A Comparison of Hi-C contact frequency for loops with or without enhancer mapping in each cell. Statistical significance was indicated by Wilcoxon signed-rank test (***P < 0.001). B The H3K27ac and H3K4me1 ChIP-seq profiles of different cells around the enhancers (±2 kb around enhancer midpoint) that located within lineage-specific loops. ChIP-seq signals were normalized for input and log2 transformed. C The enrichment/depletion of lineage-specific or common enhancers at lineage-specific loops. Fisher exact test was used to determine enrichment/depletion status. D The schematic diagram showing 4 enhancer-mediated regulation patterns proposed by mapping lineage-specific enhancers to lineage-specific activated genes under different circumstances (left panel). The first was regulated by local lineage-specific enhancers located near gene promoters (5 kb upstream of TSS). The second and third were both related to long-range enhancer-promoter interactions but in lineage-specific loops or common loops, respectively. The other situations were considered as the fourth pattern. The pathway enrichment for lineage-specific activated genes in each pattern (right panel). GO pathways related to adipogenesis or osteogenesis are coded by different colors. E Venn diagrams displaying the overlapping gene counts between different patterns in (D). The overlap between genes from the first and second/third patterns indicated that they were possibly regulated by both local and distal enhancers. F Comparison of gene co-expression coefficients between lineage-specific loops and background groups. The co-expression of genes located within lineage-specific loops or backgrounds was estimated by Pearson correlation. The comparison was conducted by Wilcoxon signed-rank test. n represents the group sizes.
We next hypothesized four enhancer-mediated regulation patterns by mapping lineage-specific enhancers to lineage-specific genes under different circumstances (Fig. 3D). Genes interacting with distal enhancers through lineage-specific loops were significantly enriched for adipogenesis/osteogenesis-related pathways, emphasizing the importance of lineage-specific long-range regulation (Fig. 3D). We found that 9% and 14% of OB-specific and AC-specific genes interacted with distal enhancers through both lineage-specific and common loops, respectively. Pathway enrichment analysis with genes exclusively mapping to lineage-specific loops showed consistent and stronger associations with adipogenesis/osteogenesis-related pathways comparing to those exclusively mapping to common loops (Fig. S6A). These results indicate that lineage-specific loops collaborating with lineage-specific enhancers is essential to AC and OB commitment.
Up to 57% of genes were possibly regulated by both local and distal enhancers (Fig. 3E). We then wondered whether the distal enhancers were functional when local enhancers existed. We hypothesized that the functional distal enhancers could synchronously regulate genes locating in the same loop through chromatin looping, which led to co-expression. The co-expression was then detected using population-based gene expression data (Methods). Comparing to backgrounds, genes located within the same loops were more likely to be co-expressed when both distal and local enhancers existed. (Fig. 3F and S6B; Wilcoxon signed-rank test). Additionally, by performing soft clustering with time-course gene expression data during adipogenesis and osteogenesis, we found a higher tendency of sharing expression profiles for genes within same loops (Fig. S6C; Methods), also indicating functional distal enhancers.
Chromatin accessibility reveals lineage-specific TFs activation during cell fate determination
Chromatin accessibility is another critical condition for enhancer-centered gene regulation. As expected, we found ATAC-seq signal was highly correlated with H3K27ac modification (Fig. S7). Comparing to GC and length-match control regions, ATAC-seq peaks were more colocalized with H3K27ac marks (Fig. 4A). We also found that 84.71% and 68.82% of up-regulated gene promoters in AC and OB were mapped with reliable ATAC-seq peaks. These open promoters were significantly associated with higher Hi-C contact density and gene expression levels (Fig. 4B).

A Colocalization of chromatin accessible regions and H3K27ac modification sites. The colocalization was compared between ATAC-seq peaks (left column) and control regions (right column) in AC and OB. The ATAC-seq peak summits occurred at H3K27ac depletion sites, which was identified as the available regions for TFs binding [70]. B Comparison of normalized Hi-C contact frequency (left panel) and gene expression levels (right panel) for promoters mapping with ATAC-seq peaks or control regions. Statistical significance was estimated by Wilcoxon signed-rank test. C The enrichment profiles of 413 known TF motifs at ATAC-seq peaks in each cell. TF motifs were clustered according to their lineage-specific activation manner. Known regulatory TFs related to adipogenesis and osteogenesis are listed on the right and marked by different colors. D The enrichment of AC- or OB-specific motif peaks in lineage-specific or common loops. The loops were trimmed to be non-overlapping. Fisher exact test was used to determine statistical significance. The odds ratios are plotted to indicate enrichment (OR > 1) or depletion (OR < 1). E The normalized Hi-C contact frequency at lineage-specific motif peaks, which overlapped with lineage-specific genes and enhancers, was compared between AC and OB. Statistical significance was estimated by paired samples Wilcoxon test. F The subtractive interaction heatmaps and genome browser screenshots showing the lineage-specific loop structures, differential distal enhancer signals and lineage-specific open chromatin around IRS2 and RUNX2 loci.
We further interrogated whether loop formation is coupled with activating TF binding affinity during hMSCs differentiation. Known TF motifs were discovered within open chromatin and classified into 6 clusters according to their lineage-specific accessible manner (Fig. 4C; Methods). Known adipogenesis-related TFs were successfully found in AC-specific motifs (cluster (3)). Interestingly, some osteogenesis-related motifs were accessible in both hMSCs and OB (Fig. 4C, clusters (4) and (6)), supporting previous observation that activation of hMSCs TFs was required for osteogenesis [9]. The motif of osteogenic regulator RUNX2 was found more accessible in hMSCs than in OB (cluster (2)), which is consistent with the observation of fewer RUNX2 binding sites at the late stage of osteogenesis compared to the beginning [22, 23]. We finally collected AC-specific motifs (cluster (3)) and OB-specific motifs (clusters (4), (5) and (6)). We detected significant enrichment of ATAC-seq peaks annotated with AC/OB-specific motifs in AC/OB-specific loops, respectively (Fig. 4D; Fisher test). In contrast, we observed significant depletion of both AC- and OB-specific motif peaks in hMSCs-specific loops, yet a mild enrichment in common loops (Fig. 4D). Besides, we found that AC/OB-specific motif peaks annotated with lineage-specific genes and enhancers were associated with higher Hi-C contact frequency in respective cell (Fig. 4E; paired samples Wilcoxon test). These results link chromatin remodeling with lineage-specific TFs activation that might involve in hMSCs fate determination.
Next, we selected lineage-specific loops connecting lineage-specific enhancers and promoters that were mapped individually with lineage-specific motif peaks. 22 AC-specific and 40 OB-specific genes were screened out (Supplementary Table S6), including adipocytes functional genes PDK4 [24], IRS2 [25] etc. and osteoblasts functional genes RUNX2 [26], SIGLEC15 [27] etc. Multi-level regulatory data accounting for IRS2 and RUNX2 activation are presented in Fig. 4F.
Comprehensive loop-mediated regulatory networks indicate key regulators for adipogenesis and osteogenesis
So far, we have emphasized the association between chromatin loops and gene regulation by mapping different regulatory elements. We next aimed to tie multi-omics data together and find out the prospective regulatory networks for adipogenesis and osteogenesis. We assumed that lineage-specific loops shortened the spatial distance between distal enhancers and target genes during differentiation, which was conducive to gene-TFs binding (Fig. 5A; Methods). We finally identified 20 and 40 genes involved in AC-specific and OB-specific regulatory networks, respectively (Fig. 5B). The putative “gene-TFs” pairs were summarized in Supplementary Table S7. Among 12 and 19 genes in AC and OB regulatory networks whose defect were linked with abnormal mouse phenotypes [17], 7 and 6 genes were associated with adipose and skeleton disfunctions, respectively (Supplementary Table S8).

A The illustration of regulatory network construction. The lineage-specific gene activation is expected to be regulated through shortening the spatial distance between open promoter and distal lineage-specific enhancer by lineage-specific loops, and also facilitated by TFs binding to promoters and enhancers. B The regulatory networks targeting lineage-specific activated genes in AC (upper panel) and OB (lower panel). The network nodes represent genes and TFs, and the network edges stand for the lineage-specific loops that linking open promoters with distal active enhancers. TFs are linked with genes through binding with either promoters or distal enhancers. TF nodes are marked by different colors according to their binding properties. C Representative networks for adipogenesis related gene IRS2 and osteogenesis related gene RUNX2. Only TFs binding both enhancers and promoters of same genes are displayed. TFs subjected to subsequent functional experiments are marked in red. D Genome browser screenshots presenting the lineage-specific transcription profiles and lineage-specific loop structures of the AC-specific activated gene IRS2 (upper panel) and OB-specific activated gene RUNX2 (lower panel), respectively. The ChIP-seq data of putative TFs CTCF STAT3 are presented to show the expected binding sites around loop anchors.
Evidence showed that some TFs facilitating gene transcription by binding to both promoters and distal enhancers through chromatin interaction [28]. Here, during hMSCs differentiation, we reported a considerable proportion of TFs synchronously binding to promoters and enhancers (Fig. 5B). These TFs linked with more genes in the networks, especially for those involving in adipogenesis and osteogenesis (Fig. S8). We still took IRS2 and RUNX2, two essential genes to AC [25] and OB [26] commitment, as examples (Fig. 5C). We have detected an AC-specific loop anchored at IRS2 promoter and interacted with a distal lineage-specific enhancer (Figs. 4F and 5D, upper panel). We found 18 TFs possibly binding at both IRS2 promoter and distal enhancer (Fig. 5C, upper panel), including architectural proteins CTCF and YY1. CTCF ChIP-seq data from human adipose [18] confirmed its binding at both anchors (Fig. 5D, upper panel). For RUNX2, we also established an OB-specific regulatory network specifically connecting the promoter of RUNX2 transcript with an upstream distal enhancer (Fig. 5D, lower panel). 9 binding events were observed at both promoter and enhancer (Fig. 5C, lower panel), including the osteogenic regulator STAT3 [29]. STAT3 binding at both anchors was also confirmed by ChIP-seq data from human fetal osteoblasts [30] (Fig. 5D, lower panel). Overall, we proposed the loop-mediated regulatory networks for both adipogenesis and osteogenesis. Particularly, we have pointed out the underlying long-range regulation mechanisms for representative genes IRS2 and RUNX2.
Functional experiments validate the lineage-specific long-range regulatory networks towards IRS2 and RUNX2
We next performed functional experiments to validate the representative regulatory networks towards IRS2 and RUNX2. Firstly, chromosome conformation capture (3 C) assay was used to validate the lineage-specific loops in hMSCs, AC, OB, and corresponding cell lines. As expected, the loop linking IRS2 was only observed in AC and preadipocytes, and the loop linking RUNX2 was only detected in OB and human osteoblast-like U2OS (Fig. 6A, B and S9A, B). 3D-FISH experiments also confirmed the OB-specific loop by showing anchor colocalization only in U2OS (Fig. S9C), while the detection of AC-specific loop was restricted by limited genomic distance. To further investigate the regulatory role of lineage-specific loops, we knocked-out the distal loop anchors using CRISPR/Cas9 system. The efficient abrogation was verified by PCR sequencing (Fig. S9D). The loop structures were successfully interrupted in anchor-deleted cells (Fig. 6C, D). The expression of IRS2 and RUNX2 were significantly decreased after deletion (Fig. 6E, F). Additionally, no relevant effect was found on nearby genes (Fig. S9E), and both in-house and public data [14, 31] indicated no other genes targeted by the deleted anchor (Fig. S9F), which can eliminate the concern of side-effects.

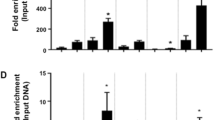
A, B Chromosome conformation capture (3 C) assay verified chromatin interactions towards IRS2 gene in AC and preadipocytes (A), and towards RUNX2 gene in OB and U2OS (B), respectively. The normalized percentages of chromatin interaction frequencies between implicated loop anchors (N3 and N7) or between baits (N3) and other neighboring EcoRI sites are shown. C, D Distal loop anchors were deleted by CRISPR/Cas9. The effect of distal anchor deletion on corresponding loop formation was detected by 3 C in preadipocytes (C) and U2OS (D). The 3 C products were not detectable after anchor deletion in U2OS. E, F The effect of distal anchor deletion on the expression of IRS2 (E) in preadipocytes or RUNX2 (F) in U2OS cells. Gene expression was estimated by qRT-PCR. G, H The effect of CTCF or EP300 knockdown on IRS2 expression in preadipocytes (G) and EP300 or STAT3 knockdown on RUNX2 expression in U2OS (H). Two independent shRNAs (shRNA-1 and shRNA-2) and shNC (negative control) were used. Gene expression was estimated by qRT-PCR. I The chromatin interaction frequency between implicated AC-specific loop anchors (N3 and N7 presented in A) before and after CTCF/EP300 knockdown in preadipocytes. J The chromatin interaction frequency between implicated OB-specific loop anchors (N3 and N7 presented in B) before and after EP300/STAT3 knockdown in U2OS. The 3 C products were not detectable after STAT3 knockdown. K Schematics representation of the underlying three-dimensional regulation mechanisms for IRS2 and RUNX2 expression activation during adipogenesis and osteogenesis, respectively. The data are plotted as mean ± SD (n = 3 or 4). t test was implemented to evaluate significant differences (*P < 0.05, **P < 0.01, ***P < 0.001).
We next validated the role of implicated TFs in regulating target gene expression. We each selected two candidate TFs (CTCF and EP300 for IRS2, STAT3 and EP300 for RUNX2). Even though CTCF is crucial in genome-wide transcriptional regulation, here we found the binding of CTCF at IRS2 locus was AC-specific (Fig. S9G). After suppressing TFs using short hairpin RNAs (shRNAs), the IRS2 expression was significantly declined in CTCF/EP300 knockdown preadipocytes (Fig. 6G). Likewise, knockdown of STAT3/EP300 in U2OS significantly affected RUNX2 expression (Fig. 6H). 3C experiments further validated that suppressing CTCF/EP300 dismissed the AC-specific loop around IRS2 (Fig. 6I), and knockdown of STAT3/EP300 in U2OS observably disrupted the OB-specific interaction to RUNX2 (Fig. 6J). Together, we validated the three-dimensional regulation mechanisms for IRS2 and RUNX2 (Fig. 6K), which required lineage-specific loop integrality and specific TF binding.
eQTL variants are linked with target genes through chromatin loops
We next wondered whether eQTL and target genes were linked through chromatin loops. We included eQTL data from GTEx project and mapped SNP-gene pairs to lineage-specific loops. The Q-Q plots of adipose eQTL indicated more significant associations recognized by AC-specific loops (Fig. 7A). Given the large overlap in the transcriptomic effects of genetic variation between osteoblasts and lymphocytes [32, 33], we used lymphocytes eQTL data and observed predominant enrichment at OB-specific loops (Fig. 7A). The associations at lineage-specific loops were even more dominant than those without loop mapping (P < 2.2 × 10−16; Kolmogorov-Smirnov test). In light of eQTL dominance at lineage-specific loops, we added eQTL information to the regulatory networks. SNPs locating at TF binding sites in loop anchors or in LD with intra-anchor SNPs and impacting target gene expression were added to the networks. Eventually, we suggested 13 and 15 genes that were possibly affected by distal genomic variations in AC and OB, respectively (Supplementary Table S9). Particularly, we identified two distal SNPs significantly associated with IRS2 expression (Fig. 7B). This information is expected to go forward and reveal the underlying “SNP-TFs-gene” regulation cascade.

A Q-Q plots of eQTL associations by mapping “gene-SNP” pairs to lineage-specific loops. eQTL data were derived from adipose tissues and lymphocytes. Kolmogorov-Smirnov test was implemented to evaluate significant enrichment of eQTL associations at AC-/OB-specific loops. B The regulation network identified for IRS2 through jointly analyzing multi-omics data. The original network was extended by adding eQTL associations (P < 0.05). SNPs located at TF binding sites within anchors are marked in grey, while the eQTL SNPs located in 20 kb flanking regions around anchors and in high LD (R2 > 0.8) with intra-anchor SNPs are marked in pale red. TFs binding to both enhancer and promoter are colored in green. Network edges indicating different connections are marked in different colors.
Discussion
Here, we performed the comprehensive assessment of chromatin loops, mRNA transcription, H3K27ac and H3K4me1 histone modifications as well as chromatin accessibility before and after inducing hMSCs to adipogenic/osteogenic differentiation. After leveraging these data, we revealed the lineage-specific rearrangement of chromatin loops and epigenomic features during adipogenesis and osteogenesis, and suggest close relevance with AC/OB determination, which is expected to provide better understanding of the controlling factors during hMSCs differentiation.
We reported an osteogenesis-specific loop linking RUNX2 with an upstream enhancer, which was verified by various experiments including 3 C assay, 3D-FISH and CRISPR/Cas9 system. The chromatin structure at RUNX2 locus has raised a lot attention. Barutcu et al. observed intra-gene interactions during early stage of mouse MC3T3-E1 osteogenic differentiation, which controlled Runx2 transcription [34]. Sancisi et al. reported interactions between RUNX2 and distal downstream enhancers, which facilitated aberrant expression of RUNX2 in thyroid and breast cancer cells [35]. Additionally, Kawane et al. identified an upstream enhancer, 30 kb to Runx2 promoter, that regulated Runx2 expression in mouse primary osteoblasts [36]. Although different cellular contexts were investigated, the higher-order organization at RUNX2 locus was successfully detected in these studies. Different distal enhancers were discovered here and in previous studies, which hints that, in different biological conditions, the eventual RUNX2 expression is regulated by forming cell/tissue-specific chromatin structures.
It’s also worth noting that we found the impact of STAT3 on both RUNX2 expression and OB-specific loop formation. The gene activation effect is concordance with previous observation that STAT3 binds to RUNX2 promoter and activates transcription through histone demethylation [37]. Here, we reported the chromatin interactions facilitating by STAT3 for the first time. Though this is novel, study on another STAT family member, STAT5, has revealed the special function of promoting chromatin looping [38]. Both STAT3 and STAT5 are involved in JAK–STAT pathway and responsible for important cellular processes such as cell growth and apoptosis [39]. Nevertheless, how STAT3 controls RUNX2 expression through chromatin folding remains further investigation.
We reported AC-specific regulatory networks for IRS2 and validated that IRS2 expression is significantly affected after disrupting loop structure. Experimental evidence also suggests the function of CTCF on regulating IRS2 expression and chromatin interaction. CTCF is an essential TF and famous for its predominant role on 3D chromatin organization. Moreover, CTCF binding is required during adipogenesis [40]. We have retrieved CTCF binding events at IRS2 promoter, but the question remains whether this is one of the triggers for adipocytes differentiation. Here, based on our results, we proposed an underlying regulation cascade linking CTCF with IRS2 activation through chromatin loop formation. These observations can provide novel insights to understand CTCF-dependent adipogenesis mechanisms.
Overall, our study provides a holistic comparison of chromatin conformation changes between adipogenesis and osteogenesis, which is useful to understand cell fate determination during hMSCs differentiation.
Materials and methods
Cell culture and hMSCs differentiation
Primary human umbilical cord derived hMSCs were obtained frozen from Shaanxi Stem Cell Engineering Co., Ltd from 1 donor who have signed the informed consent for this study. Cells were thawed and expanded for an additional passage for all the subsequent experiments. hMSC cells were seeded at a density of 1 × 104 cells/cm2 and cultured at 37 °C, 5% CO2 in Dulbecco modified Eagle medium (DMEM; GE, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and 1% antibiotics (penicillin 100 U/ml, streptomycin 100 μg/ml; Solarbio, China). When 80% confluence was reached, part of the cells was harvested, and the left were switched to differentiation culture medium to induce adipogenesis and osteogenesis.
For osteogenic differentiation, hMSC cells were grown in DMEM medium supplemented with 10% FBS, 1% antibiotics, 10 mM glycerol-2-phosphate, 50 μM L-ascorbic acid, and 100 nM dexamethasone for 21 days. Medium was replaced every three days.
Adipogenic differentiation was induced in hMSC cells cultured by alternately supplying treatment of solution A and B. Solution A: DMEM medium containing 10% FBS, 1% antibiotics, 10 mg/L insulin (Novo Nordisk), 1 μM dexamethasone, 0.5 mM IBMX, 100 μM indometacin. Solution B: DMEM medium containing 10% FBS, 1% antibiotics, 10 mg/L insulin. Cells were firstly cultured in solution A for three days and were additionally supplied with solution B for another day. Cells were harvested after adipogenic induction for 15 days. All reagents were purchased from Sigma (USA).
Cell differentiation status were further verified at 4 time points (0d, 5d, 10d, 15d for adipogenic differentiation; 0d, 7d, 14d, 21d for osteogenic differentiation) through microscopic identification, Oil Red O/Alizarin Red S staining and qRT-PCR quantification of marker genes (fig. S1). The staining areas were counted by ImageJ [41] software at each time point, and the statistical significance was indicated by t-test.
Human embryonic kidney 293 T cells (HEK293T) and human bone osteosarcoma epithelial cells (U2OS) were purchased from ATCC (USA), and human preadipocytes were obtained from Bluefbio (China). U2OS cells were cultured in RPMI-1640 medium, while HEK293T and preadipocyte cells were cultured in DMEM medium. Both were further supplemented with 10% FBS, 1% antibiotics, and were cultured at 37 °C, 5% CO2.
Hi-C library preparation and sequencing
6 technical replicates of adipocytes and osteoblasts, and 7 technical replicates of hMSCs were generated after cell differentiation with each replicate containing about 1 × 107 cells. In situ Hi-C was next performed on each replicate using methods as previously described [16]. Briefly, after harvesting from plates, cells were crosslinked with 1 ml of freshly made 1% formaldehyde solution and incubated for 10 min at room temperature. The reaction was quenched by adding glycine solution to a final concentration of 0.2 M. Cells were lysed and chromatin was next digested with 200 U of MboI restriction enzyme for 16 h at 37 °C. Digested DNA ends were labeled using biotinylated nucleotides and incubated at 37 °C for 90 min. Fragments were proximity ligated by adding T4 DNA ligase and were incubated at 4 °C for 1 h, followed by 4 h at room temperature. Samples were supplemented with SDS, Proteinase K, and NaCl to reverse crosslinking, and incubated overnight at 65 °C. After that, DNA fragments were purified and dissolved. Purified DNA fragments were sheared to a size of 300–500 bp. Ligation junctions labeled with biotin were subsequently pulled down using streptavidin C1 beads. After adding A-tails to the fragment ends and following ligation by the Illumina paired-end sequencing adapters, Hi-C libraries were sequenced on an Illumina HiSeq X Ten system. Hi-C libraries were prepared and sequenced by Novogene Co. Ltd. (Beijing, China) as previously described [42].
RNA-seq data generation
Two technical replicates were generated for each cell type. Total RNA was extracted using the TRIzol (Invitrogen, USA) method [43]. RNA concentration and purity were evaluated with a NanoDrop spectrophotometer (Thermo Fisher Scientific). RNA-seq libraries were generated using NEBNext Ultra RNA Library Prep Kit for Illumina (New England BioLabs, USA) following manufacturer’s instructions and were then sequenced on the Illumina HiSeq X Ten platform using the 150-bp pair-end sequencing strategy. Finally, an average of 47 million pair-end reads were obtained per sample.
Chromatin immunoprecipitation assay
ChIP assay was performed using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, USA) as previously described [44]. Briefly, cells were crosslinked with 1% formaldehyde. After quenching with glycine solution, cells were rinsed, pelleted and resuspended in cold PBS, and then resuspended and pelleted twice with buffer A and B, respectively. Nuclei were digested with Micrococcal Nuclease. The digestion reaction was deactivated with 0.5 M EDTA. The nuclei were then pelleted, and sediment resuspended in ChIP buffer using protease inhibitor cocktail. The lysate was sonicated with the VirTis Virsonic 100 Ultrasonic Homogenizer/Sonicator for 3 pulses. After centrifuging, the supernatant was collected and immunoprecipitated with H3K4me1 (Abcam Cat. #ab176877) and H3K27ac (Abcam Cat. #ab177178) antibodies or normal immunoglobulin G (IgG) as a negative control and precleared with agarose beads. DNA protein complex was then precipitated with agarose beads, eluted from the beads, and reversely cross-linked by 5 M NaCl and Proteinase K. Libraries for ChIP-seq were prepared by Novogene Co., Ltd. (Beijing, China) using custom protocol as previously described [45]. Libraries were next sequenced on the Illumina HiSeq X Ten platform configured for 150-bp pair-end reads.
ATAC-seq data generation
ATAC-seq libraries were constructed for adipocytes and osteoblasts following the original protocol [46]. In brief, 20,000 cells were lysed with cold lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.03% Tween-20), and centrifuged at 500 g for 8 min at 4 °C. The supernatant was carefully removed, and the nuclei was resuspended with Tn5 transposase reaction mix (25 μl 2 × TD buffer, 2.5 μl Tn5 transposase and 22.5 μl nuclease-free water) (Illumina) at 37 °C for 30 min. Immediately after the transposition reaction, DNA was purified using a Qiagen MinElute kit. Libraries were constructed using TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme, China) under manufacturer’s instructions and were then sequenced on an Illumina HiSeq X Ten sequencer to an average read depth of 52 million pair-end reads per sample. The ATAC-seq experiment and library sequencing were performed by Frasergen Bioinformatics Co., Ltd, Wuhan, China.
qRT-PCR
Cells at each differentiation time point were partially collected to detect marker gene expression. Total RNA was isolated with TRIzol reagent (Invitrogen, USA), and was converted to cDNA with reagents purchased from Vazyme Biotech (China). PCR procedure was performed using Qigen SYBR Green PCR Kit (Qiagen, German) and was operated with Bio-Rad System (CFX Connect™, Bio-Rad). The specific qRT-PCR primers used in this study were summarized in Supplementary Table S10. All the experiments were conducted following the manufacturer’s instructions.
Computational analysis
Hi-C data processing
Hi-C reads from each replicate were aligned (hg19), filtered and paired using HiCUP pipeline [47] with parameters (–longest 800 –shortest 150). In summary, ~0.53 billion (~81% of total read pairs) paired reads uniquely mapped to the genome. After removing self-ligation and invalid pairs, ~0.46 billion (~61% of total read pairs) valid pairs were remained for subsequent analysis (Supplementary Tables S1 and S2). Valid pairs for replicates of each cell were combined to generate raw contact matrices at different binning resolutions. We next normalized the raw contact matrices using ICE normalization [48] with parameters (-filter_low_counts_perc 0.02 -eps 0.1 -remove-all-zeros-loci). PCA analysis was executed to indicate the good quality of Hi-C libraries. Hi-C data reproducibility was indicated according to both stratum adjusted correlation [49] and Pearson correlation analyses.
TAD calling and TAD signal calculation
TADs were called with Domaincalling pipeline as described by Dixon et al. [50]. The ICE-normalized matrix was subjected to calculate Directionality Index values. The results were applied with Hidden Markov Model to call TADs. We executed this TAD calling procedure at 10 kb binning resolution in this study. In total, 4556, 4303 and 5329 TADs were identified for hMSCs, adipocytes and osteoblasts, respectively.
In order to compare TAD stabilization, we evaluate TAD signals indicating the strength of TAD contact. We used the method described by Ke et al. [51] to calculate TAD signals. Intra-chromosomal maps were first prepared, and the TAD signal for each bin was next calculated as the log2 ratio of the number of normalized upstream-to-downstream interactions within a 2 Mb region. Bins with less than 10 interactions at either upstream or downstream region were filtered. TAD signals were then used to perform Pearson correlation test between cells.
Identification of A/B compartments
Hi-C output from HiCUP was transformed to compatible file format to work with HOMER software [52]. The PC1 values were determined using the “runHiCpca.pl” function with parameters (-res 25000 -window 25000 -pc 1). The signs of PC1 values were used to assign the chromatin into A/B compartment. To detect either positive or negative PC1 represents open A compartment, we compared the PC1 values with backup data from Barutcu et al. [53]. They assigned positive PC1 to open A-type and negative to B-type. The generally consistent positive and negative values between ours and the reference suggest that the genomic regions with positive PC1 here should be identified as A compartment, while the negative regions should be assigned to B compartment.
Identification of chromatin loops
Loops were called by two computational strategies. The first is “findTADsAndLoops.pl” function packaged in HOMER. It was utilized to call loops at 2 kb resolution with parameters (-res 2000 -window 2000 -minDist 6000 -maxDist 1000000). The other software, HiCCUPS [16], was applied separately to identify loops at 5 and 10 kb resolutions with default parameters. Finally, loops from two methods were pooled together, which yielded a list of 21,738, 12,460 and 16,930 loops in hMSCs, adipocytes and osteoblasts, respectively. The loop calls identified by two software are noted in Supplementary Table S4. To test loop reproducibility between replicates, we performed loop calling again with leaving one replicate out at a time. The similarity between new loops and original loops was next indicated by Jaccard similarity coefficient. We observed high similarities (Jaccard similarity coefficient ≥ 0.85) for replicates of the same cell type, while significantly decreased when comparing different cells (Fig. S3E).
Statistical identification of differential contact loops and lineage-specific loops
To find differential contact loops in adipocytes and osteoblasts comparing with hMSCs, we first merged loops in chosen cells into a union set by “merge2Dbed.pl” function in HOMER with default parameters. Next, we counted raw contact frequencies from filtered Hi-C read pairs of each cell replicate and built a contact frequency matrix with respect to loop sets and replicates. The contact frequency matrix was then inputted into edgeR [54]. After normalizing by the trimmed mean of M values (TMM), differential contact loops between hMSCs and adipocytes or osteoblasts were identified using a generalized linear model likelihood ratio test. The significance was determined by P < 0.01. After filtering out the loops that were failed to pass significant threshold, we further identified “AC-specific” and “OB-specific” loops as those were only up-regulated in AC and OB, and “hMSCs-specific” loops as those were down-regulated during both adipogenesis and osteogenesis. We also recognized “common” loops as those were simultaneously up-regulated in AC and OB.
ChIP-seq data processing and enhancer analysis
ChIP-seq reads were aligned to the human genome assembly (hg19) using Bowtie2 [55] with default settings. Duplicate reads and reads with MAPQ < 30 were discarded. MACS2 [56] was used to perform peak calling with the following parameters (-g hs -p 0.01 -nomodel -extsize 147 -keep-dup all). Peaks of H3K4me1 and H3K27ac were found for each cell replicate separately. Replicated peaks were identified by at least 50% overlap. Putative enhancers were further defined by merging replicated peaks of H3K4me1 and H3K27ac in each cell.
ATAC-seq data processing and peak calling
Adaptors were trimmed from ATAC-seq reads sequences using custom python scripts. Pair-end reads were aligned to hg19 using Bowtie2. Duplicate reads and reads with MAPQ < 30 were discarded. Reads mapping to the mitochondria and chromosome Y were removed. After filtering, the qualified reads were subjected to MACS2 to call peaks with parameters (-q 0.05 -nomodel -shift −100 -extsize 200 -keep-dup all). Peaks mapped to the consensus excludable ENCODE blacklist (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/) were filtered out. Correlation analysis validated replicates concordance (fig. S7A). The peaks between replicates of the same cell were merged using BEDTools [57]. In total, we identified 138,820 and 120,209 peaks from adipocytes and osteoblasts, respectively. In order to compare TFs footprints in adipocytes/osteoblasts with hMSCs and another unrelative cell, we obtained ATAC-seq peaks of hMSCs from Rauch et al. (GSE113253) [9] and GM12878 cell line from Buenrostro et al. (GSE47753) [46].
Genomic elements and enhancer-enhancer interactions enrichment
We compared the chromatin elements enrichment between loop anchors and control regions that were generated by collecting genomic fragments with the same distance to anchors as the detected interactions but in the opposite direction. The enrichment of regulatory elements at loop anchors were estimated by XGR package [58] implemented in R. Enrichment Z-scores resulting from XGR were plotted to show different enrichment preference. Chromatin states from an imputed 25-state model of bone marrow derived MSCs (E026), MSCs derived adipocytes (E023) and osteoblasts (E129) were obtained from the Roadmap Epigenomics project (https://personal.broadinstitute.org/jernst/MODEL_IMPUTED12MARKS/). 16 annotations associated with transcription and gene regulation were selected to test enrichment. Enrichment P values are indicated in Supplementary Table S5. CTCF ChIP-seq data derived from ENCODE [18] and Mikkelsen et al. (GSE21366) [19] were used to compare CTCF binding sites enrichment between loop anchors, control regions and the disjoint 2 kb apart neighbors to loop anchors.
The enhancer-enhancer interactions identified in 1-day differentiated adipocytes and osteoblasts were obtained from GEO (GSE140782) [14]. Differential interactions during either adipogenesis or osteogenesis were estimated by edgeR with 10% FDR. Lineage-specific enhancer-enhancer interactions were found in the same way as identifying lineage-specific loops. To assess the enrichment of lineage-specific enhancer-enhancer interactions in lineage-specific loops, we generated a null distribution by randomly pairing two loop anchors at the same chromosome (distance <2 Mb) to transfer the loop interactions from the true anchors to select control anchors. 1000 sets of control pairs (with the same number of control pairs as that of the lineage-specific loops in each set) were built. We next mapped both the lineage-specific loops and the control pairs to lineage-specific enhancer-enhancer interactions and quantified the number of overlapping pairs. We estimated the fold enrichment by the ratio of the overlapping number of loops to the mean of null distribution and computed the empirical P values.
RNA-seq data processing and differential expression analysis
RNA-seq reads were aligned to human genome (hg19) using STAR [59] with default parameters. Duplicates were marked by PicardTools (v2.18.9) [60]. Low mapping quality reads (MAPQ < 30) were removed for subsequent analyses. The transcript and gene-based expression levels were quantified and normalized to transcript per million (TPM) using RSEM (v1.3.0) [61]. The expected counts of genes/transcripts from RSEM were next normalized by the TMM method. Genes/transcripts that had TMM count >1 in at least 50% of the samples were selected, and were transformed to estimate the mean-variance relationship by voom function implemented in limma package [62] from R. The data were then tested for differential expression by linear model.
Co-expression and soft clustering analyses
In order to reveal the synchronous expression of genes located within same loops when both local and distal enhancers existed, we performed co-expression analysis using population-based gene expression data from adipose tissues [63] (GTEx, phs000424.v8.p2) and human primary osteoblasts [64] (GSE15678). We also implemented soft clustering analysis with time-series gene expression data during adipogenesis and osteogenesis derived from Rauch et al. (GSE113253) [9] using Mfuzz software [65]. The background set was comprised of pairwise genes that matched expression levels (with ±20% variation) and genomic distances (±2 kb) to intra-loop gene pairs but didn’t locate within same loops. Time-course gene expression matrices were input into Mfuzz with default parameters. Genes having similar time-series expression profiles were clustered together. The soft clustering analysis was performed with intra-loop gene pairs and backgrounds separately. The pairwise genes within same loops or backgrounds were further counted if they were grouped into same cluster. The proportion of intra-loop gene pairs within same cluster was then compared with that from backgrounds through chi-square test.
Colocalization between ATAC-seq and H3K27ac ChIP-seq
Complementary genomic regions to ATAC-seq peaks were selected for adipocytes and osteoblasts, from which length matching regions were randomly generated. The GC contents of random regions were calculated by BEDtools. Regions with GC content matching with peaks were integrated to construct the matching region set. ATAC-seq reads and H3K27ac ChIP-seq reads mapped to ATAC-seq peaks or matching regions were counted and normalized by RPKM using deepTools software [66]. Colocalization profiles were plotted at a 10 kb region flanking the ATAC-seq peak summits/region midpoints.
TF motifs discovery in ATAC-seq peaks
The HOMER motif finding function “findMotifsGenome.pl” was used to detect enriched TF motifs in ATAC-seq peaks with parameters (-size 200 -mask). We found motifs within ATAC-seq peaks identified in hMSCs, adipocytes, osteoblasts and GM12878 using the union peak set as background. 413 known motifs available in HOMER were used to test for enrichment. The enrichment Z-scores were used to find lineage-specific motifs across cells.
Regulatory networks construction
We constructed regulatory networks using multi-omic data, including loop structures, gene expression levels, enhancers and chromatin accessible regions, as well as TFs binding sites collecting by Yevshin et al. [67]. The loop anchors were first rescaled to 10 kb, and then searched for ATAC-seq peaks. Lineage-specific loops with both anchors mapping with ATAC-seq peaks were kept. The lineage-specific activated gene promoters were consisted of −2 kb to +1 kb regions around TSS of gene transcript. Next, one side of loop anchors was mapped with promoters while the other side was mapped with lineage-specific enhancers. Both promoters and enhancers were then searched for TFs binding sites separately. By this way, target genes and TFs were connected. The connections were further categorized into 3 groups according to TFs binding properties, including “both” as TFs binding to both distal enhancers and promoters of target genes simultaneously, “only enhancer” as only binding to enhancers, and “only promoter” as only binding to gene promoters. The edge weight was defined as:
where Gfc and TFfc refer to expression fold change of target gene and TFs after differentiation. N refers to the peak caller counts that were able to recognize the binding events at either promoters or enhancers, or the sum of peak callers for binding to both promoters and enhancers. The node weight was defined as the expression fold change. The TFs binding sites and target genes were next utilized to search for eQTL associations. The eQTL data (P < 0.05) derived from subcutaneous and visceral omentum adipose and lymphocytes were obtained from GTEx database (v8) [63]. SNPs located at the TFs binding sites on distal enhancers and effecting the same genes as the loop interacting with were subsequentially added to the networks. We also extended the candidate eQTL to locating within 20 kb flanking regions around anchors and having high LD (R2 > 0.8) with intra-loop SNPs. These significant SNPs (association P < 0.05) were also added to the final networks.
Functional experiments
3C assay
The chromosome conformation capture (3 C) assay was performed following previous methods [68]. The experiment procedures are briefly described below.
3C library preparation
Cells were first fixed with 1% formaldehyde for 10 min and quenched with 2.5 M glycine. The cross-linked cells were then lysed and homogenized. Following a centrifugation step, the nuclear pellets were washed with 1 × NEB Buffer 2 (New England BioLabs, USA) and resuspended with 1 × CutSmart Buffer (New England BioLabs, USA). 1% SDS was added, and the samples were next incubated at 65 °C for 10 min to remove any proteins that were not cross-linked to DNA. Then, quenched with 10% Triton X-100. The EcoRI restriction enzyme (New England BioLabs, USA) was chosen to digest the cross-linked DNA overnight at 37 °C, then religated the digested DNA with T4 DNA ligase (New England BioLabs, USA) at 16 °C for 2 h. The ligated samples were treated with proteinase K, and DNA was extracted with phenol-chloroform.
Control library preparation
The control library was constructed to correct primer efficiency. The control library contained several random EcoRI cut site-enriched genomic loci (NCs), either spanning or near the two anchor regions. Amplified the NCs fragments from human genomic DNA by PCR, mixed each purified NC fragment equimolarly, digested by EcoRI and ligated by T4 DNA ligase. The random ligation products were finally extracted by phenol-chloroform to form the control library.
3C interaction frequency evaluation
Both 3C and control library products were measured simultaneously by qPCR. The “bait” primer was designed targeting the EcoRI enzyme cut site at one loop anchor and other primers targeting the EcoRI cut sites on either NCs fragments or another anchor. Then, an additional primer next to the “bait” primer was designed to measure the PCR products of “bait fragment” as baseline. The percentage of interaction frequencies of both 3 C and control library were calculated by dividing the amount of PCR products between bait and distal fragments by the amount of local PCR products of bait fragment. The 3 C interaction frequency was then normalized by control library. The primers used in 3 C assay are summarized in Supplementary Table S10.
Interaction frequency fold change assessment
In order to make 3 C results comparable between different cells, we introduced interaction frequency fold change to evaluate relative chromatin interaction strength. Briefly, the mean value of interaction frequencies between bait and NCs in respective cells were first calculated, and then divided into the interaction frequency between anchor fragments to get the relative interaction frequency fold change.
3D-FISH
Two sets of oligonucleotide probes were designed and listed in Supplementary Table S10. The primary probes were complementary to target anchors and contained a particular region with fixed sequence. The secondary probes were designed targeting the particular region of primary probes and labeled with biotin (distal anchor) or digoxin (proximal anchor) at 5′ end. 3D-FISH was performed according to previous method with modifications [69]. Briefly, U2OS, hMSCs and preadipocytes were seeded on poly-L-lysine coated coverslips and cultured at 37 °C overnight for attachment. Cells were then fixed with 4% paraformaldehyde for 10 min. After washing with 0.01% Triton X-100, cells were permeabilized with 0.5% Triton X-100 for 15 min, then immerged in 20% glycerol. Next, coverslips were frozen in liquid nitrogen (for ~30 s) and thawed for three times. Cells were washed and incubated in 0.1 M HCl for 5 min at room temperature, and then immerged twice in 2 × SSC and put in formamide-SSC solution overnight at 4 °C. After that, coverslips were incubated in each of 2 × SSC and 70%, 85%, and 100% ethanol sequentially. Hybridization mixture containing hybridization buffer (Focobio, China) and primary probes was added to the coverslips then sealed with rubber cement. Slides were then heated at 75 °C for 2 min and incubated at 37 °C overnight. Coverslips were washed and incubated in 3% BSA (Beyotime, China) for 1 h. Next, coverslips were again treated with gradient alcohol then supplied with secondary probes and incubated for 2 h. After 1 h treatment with 3% BSA, the coverslips were firstly incubated with horseradish peroxidase (HRP) anti-streptavidin antibody (Focobio Cat. #F-2001) for 1 h and followed by signal amplification using tyramide signal amplification (TSA) kit (Focobio, China). Then HRP anti-digoxin antibody (Focobio Cat. #F-2008) was supplied and followed by signal amplification. Coverslips were finally stained with DAPI (Beyotime, China). Images of 3D-FISH were captured using a fluorescence confocal microscope (Leica TCS SP8).
Anchor deletion by CRISPR/Cas9
Distal loop anchors were deleted in corresponding cells using CRISPR/Cas9 system. Briefly, three pairs of single-guided RNAs (sgRNAs) were designed for target anchor fragments (https://zlab.bio/guide-design-resources), and cloned into lentiCRISPR v2 transfer plasmid (Addgene, plasmid no. 52961). The transfer plasmid was then co-transfected with packaging plasmid (psPAX2) and envelope plasmid (pCMV-VSV-G) into HEK 293 T cells for 48 h to produce lentivirus. The supernatant medium containing target lentivirus was collected and filtrated by 0.22 μm aperture PES membranes (Millipore, Germany). U2OS and human preadipocytes were next infected with lentivirus suspension mixed with 8 μg/ml polybrene (Santa Cruz Biotechnology, USA). Then, cells were treated with 2 μg/ml puromycin for two weeks, and the remaining positive cells were harvested and verified by PCR sequencing. Total RNA was subsequently extracted from wildtype and anchor-deletion cells, and qRT-PCR was performed to evaluate and compare target gene expression levels. The results were obtained from four independent experiments. sgRNA primers used here are listed in Supplementary Table S10.
shRNA knockdown of selected TFs
To validate the function of selected TFs in regulating target gene expression and loop formation, we performed short hairpin RNA (shRNA) knockdown experiments targeting CTCF, EP300 in preadipocytes and targeting STAT3, EP300 in U2OS. Two independent shRNAs (shRNA-1 and shRNA-2) were designed for each TF as listed in Supplementary Table S10. The shRNA and negative control (shNC) fragments were then cloned into lenti-CMV-MCS-EF1α-PuroR plasmid separately. The resulted plasmids were then co-transfected with packaging plasmid (psPAX2) and envelope plasmid (pCMV-VSV-G) into HEK 293 T cells for 48 h to produce lentivirus. U2OS and preadipocytes were next infected with lentivirus suspension mixed with 8 μg/ml polybrene. 2 μg/ml puromycin was finally used to treat the cells for two weeks. Positive cells were further collected and subjected to subsequent analysis.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and the Supplementary data files. The raw and processed data generated in this study are deposited in the Gene Expression Omnibus under accession number GSE151324.
References
Brunmeir R, Wu J, Peng X, Kim SY, Julien SG, Zhang Q, et al. Comparative transcriptomic and epigenomic analyses reveal new regulators of murine brown adipogenesis. PLoS Genet. 2016;12:e1006474.
Piek E, Sleumer LS, van Someren EP, Heuver L, de Haan JR, de Grijs I, et al. Osteo-transcriptomics of human mesenchymal stem cells: accelerated gene expression and osteoblast differentiation induced by vitamin D reveals c-MYC as an enhancer of BMP2-induced osteogenesis. Bone 2010;46:613–27.
Lehrke M, Lazar MA. The many faces of PPARγ. Cell 2005;123:993–9.
Rosen ED, Hsu CH, Wang X, Sakai S, Freeman MW, Gonzalez FJ, et al. C/EBPα induces adipogenesis through PPARγ: a unified pathway. Genes Dev. 2002;16:22–6.
Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 1997;89:747–54.
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002;108:17–29.
Meyer MB, Benkusky NA, Sen B, Rubin J, Pike JW. Epigenetic plasticity drives adipogenic and osteogenic differentiation of marrow-derived mesenchymal stem cells. J Biol Chem. 2016;291:17829–47.
Wang L, Niu N, Li L, Shao R, Ouyang H, Zou W. H3K36 trimethylation mediated by SETD2 regulates the fate of bone marrow mesenchymal stem cells. PLoS Biol. 2018;16:e2006522.
Rauch A, Haakonsson AK, Madsen JGS, Larsen M, Forss I, Madsen MR, et al. Osteogenesis depends on commissioning of a network of stem cell transcription factors that act as repressors of adipogenesis. Nat Genet. 2019;51:716–27.
Sexton T, Cavalli G. The role of chromosome domains in shaping the functional genome. Cell 2015;160:1049–59.
Greenwald WW, Chiou J, Yan J, Qiu Y, Dai N, Wang A, et al. Pancreatic islet chromatin accessibility and conformation reveals distal enhancer networks of type 2 diabetes risk. Nat Commun. 2019;10:2078.
Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, et al. Multiscale 3D genome rewiring during mouse neural development. Cell 2017;171:557–72.e24.
Greenwald WW, Li H, Benaglio P, Jakubosky D, Matsui H, Schmitt A, et al. Subtle changes in chromatin loop contact propensity are associated with differential gene regulation and expression. Nat Commun. 2019;10:1054.
Madsen JGS, Madsen MS, Rauch A, Traynor S, Van Hauwaert EL, Haakonsson AK, et al. Highly interconnected enhancer communities control lineage-determining genes in human mesenchymal stem cells. Nat Genet. 2020;52:1227–38.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009;326:289–93.
Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159:1665–80.
Bult CJ, Blake JA, Smith CL, Kadin JA, Richardson JE. Mouse Genome Database G. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019;47:D801–6.
The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74.
Mikkelsen TS, Xu Z, Zhang X, Wang L, Gimble JM, Lander ES, et al. Comparative epigenomic analysis of murine and human adipogenesis. Cell 2010;143:156–69.
Siersbaek R, Madsen JGS, Javierre BM, Nielsen R, Bagge EK, Cairns J, et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Mol Cell. 2017;66:420–35.e5.
Rubin AJ, Barajas BC, Furlan-Magaril M, Lopez-Pajares V, Mumbach MR, Howard I, et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nat Genet. 2017;49:1522–8.
Wu H, Whitfield TW, Gordon JA, Dobson JR, Tai PW, van Wijnen AJ, et al. Genomic occupancy of Runx2 with global expression profiling identifies a novel dimension to control of osteoblastogenesis. Genome Biol. 2014;15:R52.
Meyer MB, Benkusky NA, Pike JW. The RUNX2 cistrome in osteoblasts: characterization, down-regulation following differentiation, and relationship to gene expression. J Biol Chem. 2014;289:16016–31.
Cadoudal T, Distel E, Durant S, Fouque F, Blouin JM, Collinet M, et al. Pyruvate dehydrogenase kinase 4: regulation by thiazolidinediones and implication in glyceroneogenesis in adipose tissue. Diabetes 2008;57:2272–9.
Miki H, Yamauchi T, Suzuki R, Komeda K, Tsuchida A, Kubota N, et al. Essential role of insulin receptor substrate 1 (IRS-1) and IRS-2 in adipocyte differentiation. Mol Cell Biol. 2001;21:2521–32.
Komori T. Molecular mechanism of Runx2-dependent bone development. Mol Cells. 2020;43:168–75.
Hiruma Y, Tsuda E, Maeda N, Okada A, Kabasawa N, Miyamoto M, et al. Impaired osteoclast differentiation and function and mild osteopetrosis development in Siglec-15-deficient mice. Bone 2013;53:87–93.
Zhou Q, Yu M, Tirado-Magallanes R, Li B, Kong L, Guo M, et al. ZNF143 mediates CTCF-bound promoter-enhancer loops required for murine hematopoietic stem and progenitor cell function. Nat Commun. 2021;12:43.
Itoh S, Udagawa N, Takahashi N, Yoshitake F, Narita H, Ebisu S, et al. A critical role for interleukin-6 family-mediated Stat3 activation in osteoblast differentiation and bone formation. Bone 2006;39:505–12.
Najafova Z, Tirado-Magallanes R, Subramaniam M, Hossan T, Schmidt G, Nagarajan S, et al. BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Res. 2017;45:127–41.
Chesi A, Wagley Y, Johnson ME, Manduchi E, Su C, Lu S, et al. Genome-scale Capture C promoter interactions implicate effector genes at GWAS loci for bone mineral density. Nat Commun. 2019;10:1260.
Kwan T, Grundberg E, Koka V, Ge B, Lam KC, Dias C, et al. Tissue effect on genetic control of transcript isoform variation. PLoS Genet. 2009;5:e1000608.
Hsu YH, Zillikens MC, Wilson SG, Farber CR, Demissie S, Soranzo N, et al. An integration of genome-wide association study and gene expression profiling to prioritize the discovery of novel susceptibility loci for osteoporosis-related traits. PLoS Genet. 2010;6:e1000977.
Barutcu AR, Tai PW, Wu H, Gordon JA, Whitfield TW, Dobson JR, et al. The bone-specific Runx2-P1 promoter displays conserved three-dimensional chromatin structure with the syntenic Supt3h promoter. Nucleic Acids Res. 2014;42:10360–72.
Sancisi V, Manzotti G, Gugnoni M, Rossi T, Gandolfi G, Gobbi G, et al. RUNX2 expression in thyroid and breast cancer requires the cooperation of three non-redundant enhancers under the control of BRD4 and c-JUN. Nucleic Acids Res. 2017;45:11249–67.
Kawane T, Komori H, Liu W, Moriishi T, Miyazaki T, Mori M, et al. Dlx5 and mef2 regulate a novel runx2 enhancer for osteoblast-specific expression. J Bone Min Res. 2014;29:1960–9.
Kurozumi A, Nakano K, Yamagata K, Okada Y, Nakayamada S, Tanaka Y. IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone 2019;124:53–61.
Li P, Mitra S, Spolski R, Oh J, Liao W, Tang Z, et al. STAT5-mediated chromatin interactions in superenhancers activate IL-2 highly inducible genes: Functional dissection of the Il2ra gene locus. Proc Natl Acad Sci USA. 2017;114:12111–9.
Wingelhofer B, Neubauer HA, Valent P, Han X, Constantinescu SN, Gunning PT, et al. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018;32:1713–26.
Dubois-Chevalier J, Oger F, Dehondt H, Firmin FF, Gheeraert C, Staels B, et al. A dynamic CTCF chromatin binding landscape promotes DNA hydroxymethylation and transcriptional induction of adipocyte differentiation. Nucleic Acids Res. 2014;42:10943–59.
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5.
Li D, Ning C, Zhang J, Wang Y, Tang Q, Kui H, et al. Dynamic transcriptome and chromatin architecture in granulosa cells during chicken folliculogenesis. Nat Commun. 2022;13:131.
Gayral P, Weinert L, Chiari Y, Tsagkogeorga G, Ballenghien M, Galtier N. Next-generation sequencing of transcriptomes: a guide to RNA isolation in nonmodel animals. Mol Ecol Resour. 2011;11:650–61.
Chen XF, Zhu DL, Yang M, Hu WX, Duan YY, Lu BJ, et al. An osteoporosis risk SNP at 1p36.12 acts as an allele-specific enhancer to modulate LINC00339 expression via long-range loop formation. Am J Hum Genet. 2018;102:776–93.
Chen X, Loo JX, Shi X, Xiong W, Guo Y, Ke H, et al. E6 protein expressed by high-risk HPV activates super-enhancers of the EGFR and c-MET oncogenes by destabilizing the histone demethylase KDM5C. Cancer Res. 2018;78:1418–30.
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–8.
Wingett S, Ewels P, Furlan-Magaril M, Nagano T, Schoenfelder S, Fraser P, et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res. 2015;4:1310.
Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods. 2012;9:999–1003.
Yang T, Zhang F, Yardimci GG, Song F, Hardison RC, Noble WS, et al. HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res. 2017;27:1939–49.
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012;485:376–80.
Ke Y, Xu Y, Chen X, Feng S, Liu Z, Sun Y, et al. 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell 2017;170:367–81.e20.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89.
Barutcu AR, Lajoie BR, McCord RP, Tye CE, Hong D, Messier TL, et al. Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells. Genome Biol. 2015;16:214.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2.
Fang H, Knezevic B, Burnham KL, Knight JC. XGR software for enhanced interpretation of genomic summary data, illustrated by application to immunological traits. Genome Med. 2016;8:129.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 2011;12:323.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648–60.
Grundberg E, Kwan T, Ge B, Lam KC, Koka V, Kindmark A, et al. Population genomics in a disease targeted primary cell model. Genome Res. 2009;19:1942–52.
Futschik ME, Carlisle B. Noise-robust soft clustering of gene expression time-course data. J Bioinform Comput Biol. 2005;3:965–88.
Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–5.
Yevshin I, Sharipov R, Kolmykov S, Kondrakhin Y, Kolpakov F. GTRD: a database on gene transcription regulation-2019 update. Nucleic Acids Res. 2019;47:D100–5.
Thynn HN, Chen XF, Hu WX, Duan YY, Zhu DL, Chen H, et al. An allele-specific functional SNP associated with two systemic autoimmune diseases modulates IRF5 expression by long-range chromatin loop formation. J Invest Dermatol. 2020;140:348–60.e11.
Cremer M, Muller S, Kohler D, Brero A, Solovei I. Cell preparation and multicolor FISH in 3D preserved cultured mammalian cells. CSH Protoc 2007;2007:pdb prot4723.
Ott CJ, Federation AJ, Schwartz LS, Kasar S, Klitgaard JL, Lenci R, et al. Enhancer architecture and essential core regulatory circuitry of chronic lymphocytic leukemia. Cancer Cell. 2018;34:982–95.e7.
Acknowledgements
We would like to thank the GTEx Consortium. We obtained GTEx data through dbGaP authorized access at https://dbgap.ncbi.nlm.nih.gov/aa/wga.cgi?page=login with the accession number of phs000424.v8.p2. This study is also supported by the High-Performance Computing Platform and Instrument Analysis Center of Xi’an Jiaotong University. This work was supported by grants from the National Natural Science Foundation of China (82170896, 32170616, 31970569, 32100416, 31871264), the Natural Science Basic Research Program of Shaanxi Province (2021JC-02), Innovation Capability Support Program of Shaanxi Province (2022TD-44), China Postdoctoral Science Foundation (2021M702618), and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
TLY and YG conceived and supervised this project. RHH conducted the computational work. CW, CXD and FC performed the functional experiments. JG, RHH and QLC performed the cell culture experiments. YR performed visualization. SSD built the pipeline for Hi-C data analysis. HC and DLZ carried out the library construction experiments. SY and YXC participated in data analysis. RHH and YG wrote the manuscript with the assistance of other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The research was approved by the Ethics Committee of Xi’an Jiaotong University (Shaanxi, China). This study was performed in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by D Aberdam
Rights and permissions
About this article

Cite this article
Hao, RH., Guo, Y., Wang, C. et al. Lineage-specific rearrangement of chromatin loops and epigenomic features during adipocytes and osteoblasts commitment. Cell Death Differ 29, 2503–2518 (2022). https://doi.org/10.1038/s41418-022-01035-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-022-01035-7
