
Abstract
Mutation of the TP53 tumor suppressor gene is the most common genetic alteration in cancer, and almost 1000 alleles have been identified in human tumors. While virtually all TP53 mutations are thought to compromise wild type p53 activity, the prevalence and recurrence of missense TP53 alleles has motivated countless research studies aimed at understanding the function of the resulting mutant p53 protein. The data from these studies support three distinct, but perhaps not necessarily mutually exclusive, mechanisms for how different p53 mutants impact cancer: first, they lose the ability to execute wild type p53 functions to varying degrees; second, they act as a dominant negative (DN) inhibitor of wild type p53 tumor-suppressive programs; and third, they may gain oncogenic functions that go beyond mere p53 inactivation. Of these possibilities, the gain of function (GOF) hypothesis is the most controversial, in part due to the dizzying array of biological functions that have been attributed to different mutant p53 proteins. Herein we discuss the current state of understanding of TP53 allele variation in cancer and recent reports that both support and challenge the p53 GOF model. In these studies and others, researchers are turning to more systematic approaches to profile TP53 mutations, which may ultimately determine once and for all how different TP53 mutations act as cancer drivers and whether tumors harboring distinct mutations are phenotypically unique. From a clinical perspective, such information could lead to new therapeutic approaches targeting the effects of different TP53 alleles and/or better sub-stratification of patients harboring TP53 mutant cancers.
Similar content being viewed by others


A long history
p53 was first discovered over 40 years ago in complex with the SV40 large T antigen in virally transformed cells [1, 2]. Although it was first classified as an oncogene, possibly because the initial studies inadvertently utilized a mutant p53 cDNA, it was later reclassified as a tumor suppressor gene after additional studies demonstrated that it could suppress growth and oncogenic transformation of cultured cells [3, 4]. In vivo studies of p53 null mice corroborated the in vitro data: while p53 null mice are developmentally normal, they ultimately develop tumors with nearly 100% penetrance [5].
In its essence, p53 is a stress-responsive transcription factor. Upon activation in response to a diverse array of stressors, the tetrameric form of the protein binds to DNA in a sequence-specific manner [6]. Once bound to DNA, p53 activates a range of antiproliferative programs, as well as the E3 ligase MDM2 to create a negative feedback loop that ultimately leads to degradation of p53 (refs. [7,8,9,10,11,12,13]). Virtually all p53 mutants studied to date have lost the ability to bind to DNA, thereby impairing its function as a transcription factor, and it seems likely that loss of this molecular function largely explains its role in tumor formation [14]. Owing to disruption of the p53-MDM2 negative feedback loop, many p53 mutant proteins are stabilized, allowing them to engage in aberrant interactions with other cellular factors, potentially altering their function and leading to GOF phenotypes [15,16,17].
The spectrum of TP53 mutations is unique among cancer genes
The spectrum of TP53 mutations in human tumors is remarkable for its diversity and tissue specificity. There is strong enrichment for mutations in the DNA binding domain (DBD). DBD mutations are predominantly missense (~80%), including six “hotspot” codons (R175, R213, G245, R248, R273, and R282), which account for ~25% of all TP53 mutations. In contrast, mutations that occur outside of the DBD are more likely to be nonsense or truncating mutations (~67%) than missense mutations [18, 19]. In addition, beyond acquiring a TP53 mutation in one allele, most tumors lose the second allele by deletion or copy neutral loss of heterozygosity [20, 21]. The extent to which this allelic variation is a consequence of the underlying mutational mechanisms or biological selection for functionally meaningful mutations—or both—remains incompletely understood. Most of the hotspot residues contain methylated CpGs, which are five times more likely than unmethylated cytosines to undergo spontaneous deamination producing the observed C > T mutation [19, 22]. Since the majority of TP53 mutations are not encoded in the germline, the observed frequency of TP53 mutations may also be influenced by the immunogenicity of the mutant protein, with those least likely to be surveilled producing a greater advantage [23, 24]. Finally, different mutants may have different biological potencies, with more recurrent alleles having more potent oncogenic effects.
The frequency of TP53 mutation varies greatly between different tumor types. TP53 is mutated in more than 90% of ovarian cancers whereas less than 15% of acute myeloid leukemias (AML) have TP53 mutations, suggesting that there may be some tissue-specific requirements for loss of wild type or gain of mutant p53 functions [25]. Further support for the tissue specificity of mutant p53 function comes from studies of Li Fraumeni Syndrome patients. Li Fraumeni patients have inherited a germline TP53 mutation and therefore have a high risk of developing cancer [26,27,28]. Intriguingly, a Li Fraumeni cohort from southern Brazil harbors a founder mutation, R337H, that gives rise to pediatric adrenal cortical carcinoma at a much higher frequency than other mutations. These observations hint that the mutant R337H protein may have gained a function that specifically promotes tumor formation in the kidney [29]. That said, some aspects of genetic background may also contribute to this specificity, since an analysis of the MSK-IMPACT dataset indicates that the one patient with a germline TP53R337H allele presented with prostate and stomach cancer, and none of the 15 patients with acquired TP53R337H mutations develop adrenal carcinoma [30, 31].
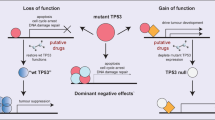
If TP53 mutations across tumor types produce a heterogeneous functional output, it is possible that mutants differ in the extent to which they inactivate wild type p53, serve as a DN, and/or produce oncogenic GOF activities that promote cancer beyond inactivating p53. An overhwhelming body of evidence suggests that p53 must be inactivated to promote tumorigenesis [32]. There is also no question that certain missense mutant proteins can have dominant negative activities [33]. Still, the fact that the vast majority of tumors ultimately inactivate the remaining wild type p53 allele implies that the DN effect might not be able to completely inactivate wild type p53 tumor suppressive function. Indeed, the composition of tetramers in a heterozygous cell range from fully wild type to fully mutant, and the strength of the DN correlates with increasing numbers of mutant subunits in the tetramer [34]. Alternatively, there may be selection for additional pro-oncogenic effects. Whether any or all TP53 mutations have GOF activities, and whether these functions are biologically relevant, remains the subject of intense research. Below we discuss recent studies refuting or supporting the p53 mutant GOF hypothesis and refer the reader to other excellent reviews for a comprehensive summary of the data [7, 11, 35,36,37].
Evidence for gain of function
The potential GOF effects of p53 mutant proteins have been the topic of debate for nearly 30 years. The first hint that p53 mutant proteins could have GOF activity came from Levine and colleagues, who showed that ectopic expression of certain TP53 mutant alleles could activate the expression of a multi-drug resistance gene reporter while the wild type p53 could not. Different TP53 mutants showed a range of activities, providing an early hint that not all TP53 mutations function equivalently [38]. Later, two other groups found that although mice engineered to have germline missense mutations (R175H and R273H) succumbed to cancer at a similar rate as p53 null, they displayed a broader tumor spectrum and a higher incidence of metastasis [39, 40]. These data were viewed as decisive evidence of a GOF effect.
Since these early studies, thousands of more recent papers provide additional evidence that mutant p53 can influence biological functions such as metastasis, stemness, epithelial to mesenchymal transition, and many others [35,36,37]. Despite this, the field has yet to converge on a cohesive model to explain these provocative results. Although many of the proposed GOF mechanisms involve mutant p53 interacting with other transcriptional regulators to induce changes in gene expression, a staggering number of effectors have been identified [41]. Not only do different TP53 mutations appear to have different GOF capabilities, but even the same mutant appears to act through distinct mechanisms depending on the context (see for example [42,43,44]), suggesting that genetic background may be responsible for some of the inconsistencies. This complexity is even more remarkable given that most studies only characterize one or a few mutants. It is therefore not surprising that some investigators have begun to question whether p53 GOF plays a meaningful role in cancer biology, and they are beginning to take a comprehensive approach to address this question.
A more systematic approach
Recently, two groups conducted saturation mutagenesis screens by exogenously expressing a library of mutant p53 cDNAs in cancer cell lines. Giacomelli and colleagues conducted their screen in isogenic TP53+/+ and TP53−/− human A549 lung adenocarcinoma cells while Boettcher et al. did the same in a K562-TP53wild type CML cell line which expressed a GFP reporter at the CDKN1A locus [33, 45]. Both groups found that DBD mutants showed a dampened senescence response following treatment with the MDM2 inhibitor Nutlin-3a. In contrast, cells harboring N- or C-terminal mutations in p53 maintained an intact senescence response. Together, these reports provide strong evidence that, at least in the in vitro assay used, p53 DBD mutants, but not other mutants, can act as DN proteins.
Boettcher et al. also conducted RNA sequencing of K562 cells that express wild type p53 or a hotspot mutation in the absence of the wild type [45]. They found that there was little variation between the genes that were downregulated by different mutants, leading the authors to conclude that the primary function of mutant p53 is to inactivate wild type transcriptional programs. However, these data were also consistent with a potential GOF role for the mutants in this system: there is a subset of genes that were activated by the mutants to a greater extent than the wild type, though the specific genes varied between mutants.
To assess the potential of mutant p53 to function as a DN in vivo, Aubrey et al. conducted competitive transplantation assays with hematopoietic stem and progenitor cells derived from three different mouse models (Trp53+/−, Trp53−/−, and Eμ-Myc;Trp53+/+) that express p53 cDNAs harboring select DBD mutations [32]. They found that mutant p53 cDNAs could dramatically accelerate tumorigenesis in the Trp53+/+ background but had no impact on the already rapid tumorigenesis that occurs in the Trp53−/− background. In the Trp53+/− background, the Trp53 mutant cDNAs apparently confer a selective advantage not by accelerating tumorigenesis but rather by biasing the tumors to a myeloid differentiation state.
The above results reiterate the potential role for a DN effect at early stages of tumorigenesis prior to the ‘second hit’ but did not support a GOF activity. Still, the latter conclusion is based largely on the lack of tumor acceleration when p53 mutant cDNAs are introduced into the p53 null background, an observation that is potentially confounded by the potential presence of pre-existing tumor cells in the transduced Trp53−/− population. Nonetheless, the same group has provided orthogonal support for this notion by showing that CRISPR-mediated disruption of mutant TP53 in a range of human cancer cell lines has no effect on proliferation or survival in vitro or following transplantation into immunocompromised animals [46].
Irreconcilable differences
Collectively, the above studies elegantly confirm what we have known for some time—that TP53 mutations both impair wild-type p53 functions and can act, to varying degrees, as dominant negative proteins. However, their impact relates less to what was observed compared to what was not: any meaningful oncogenic GOF for mutant p53. How then, can we reconcile these systematic studies with the countless peer-reviewed studies showing p53 can have GOF oncogenic effects, often in well-controlled isogenic settings?
In principle, there could be some underlying feature about mutant p53 that produces real biology but may not be selected for during tumorigenesis. Another possibility is that the assays used do not fully capture the range of settings where p53 GOF plays a role. Most in vitro studies relied heavily on profiling the proliferation of cancer cells, which is a too limited readout to assess the function of a protein that is known to regulate many different cellular processes. In support of the need to systematically conduct assays that capture the breadth of p53 functions, one group found that hotspot mutations did not proliferate any better than other mutants in 2D or 3D cell culture, yet they were robustly selected for upon transplantation into nude mice, implying a GOF effect [47].
Although some of the studies described above did look at tumor initiation in vivo, many of the GOF activities affect aspects of cancer that may not be as relevant in the hematologic cancer models used. For example, the best characterized GOF activity of mutant p53 is its role in promoting metastasis [43, 48]. Furthermore, the tumor-promoting functions of mutant p53 may be influenced by the microenvironment. Indeed, a recent study showed that mutant p53 promotes tumor formation in the distal gut due to an interaction with gut microbiota, whereas the mutant was tumor suppressive in the proximal gut which has much lower levels of bacteria [49]. Moreover, recent reports have uncovered a role for mutant p53 in modulating immune surveillance. Both the R249S and the R175H mutants suppress the recruitment of T cells and NK cells while promoting the recruitment of pro-angiogenic M2 macrophages and neutrophils [23, 24].
It is hard to refute the plethora of technically sound studies that identify a GOF role for mutant p53. Because many of these studies have focused on only a few mutants, it has been extraordinarily difficult to sort out to what extent there is a shared GOF mechanism between different mutants in the same setting, and to what extent a mechanism is conserved between the same mutant in different settings. To that end, one solution to the DN vs GOF debate is to continue to systematically study a range of mutants, but expand the readout to a range of phenotypes, including ones in vivo, to capture both potential DN and GOF properties and any allelic variation. Thus, the saga continues.
An alternative hypothesis
Whether one or more of the above hypotheses prove to be biologically relevant, it nonetheless remains difficult to conceptualize how a range of distinct mutant proteins could acquire such potent biological activities without some basis in normal physiology. As such, it is attractive to consider the possibility that some TP53 mutants acquire pro-oncogenic activities through a ‘separation-of-function’ (SOF) rather than GOF mechanism. In principle, mutations that selectively retain the pro-proliferative or survival functions of wild type p53 (e.g. adaptation to metabolic stress) while disrupting the canonical tumor suppressive activities (e.g. apoptosis, senescence) might produce phenotypes that appear similar to a GOF (Table 1) (refs. [7, 37, 41]). As one example, cancer-associated recurrent exon 6 TP53 truncating mutations mimic a naturally occurring, pro-proliferative splice variant, p53-psi, and can produce a metastatic phenotype that non-exon 6 truncations cannot [20]. Other reports hint at SOF in what might classically have been labelled GOF: for example, R248W, but not R175H, is able to promote cell survival in serine depleted conditions by activating the serine synthesis pathway and antioxidant response; other mutants retain wild type p53’s ability to suppress autophagy which promotes survival in hypoxic conditions [50, 51]. RNA sequencing data generated by Aubrey et al, though analyzed through the lens of the DN hypothesis, supports the existence of mutant p53 SOF at the transcriptional level: while many well-known p53 target genes were downregulated by expression of a mutant, about 40% of canonical targets were not. Furthermore, the hotspot mutations had higher expression of target genes that may be advantageous for tumor development than the other mutants [32]. While evidence for the SOF hypothesis remains sparse, if confirmed, could lead to a more predictable and unified model explaining TP53 mutational variation and thus is worthy of further exploration.
An important question to resolve
Achieving clarity in our understanding of how mutant p53 proteins influence cancer phenotypes would be of great benefit to the field and to the treatment of patients with tumors that harbor mutant p53. Currently, patients are stratified into wild type and mutant p53 during diagnosis; however, if there is substantial allelic variation, a better classification system would be to stratify patients by functional class of the mutant. One can imagine that this type of classification of mutant p53 may also reveal novel dependencies and therapeutic vulnerabilities such that in the future, patients may receive targeted therapy based on the functional class of the TP53 mutation.
References
Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–3.
Linzer DIH, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52.
Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–93.
Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Wild-type p53 can inhibit oncogene-mediated focus formation. PNAS. 1989;86:8763–7.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature. 1991;356:215–21.
El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49.
Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–31.
Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20:199–210.
Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170:1062–78.
Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2:a000935.
Boutelle AM, Attardi LD. p53 and tumor suppression: it takes a network. Trends Cell Biol. 2021;31:298–310.
Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45.
Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275:8945–51.
Kato S, Han S-Y, Liu W, Otsuka K, Shibata H, Kanamaru R, et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. PNAS. 2003;100:8424–9.
Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9.
Midgley CA, Lane DP. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene. 1997;15:1179–89.
Wiech M, Olszewski MB, Tracz-Gaszewska Z, Wawrzynow B, Zylicz M, Zylicz A. Molecular mechanism of mutant p53 stabilization: the role of HSP70 and MDM2. PLoS One. 2012;7:e51426.
Hainaut P, Pfeifer GP. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb Perspect Med. 2016;6:a026179.
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008.
Shirole NH, Pal D, Kastenhuber ER, Senturk S, Boroda J, Pisterzi P, et al. TP53 exon-6 truncating mutations produce separation of function isoforms with pro-tumorigenic functions. Elife. 2016;5:e17929.
Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature. 2016;531:471–5.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Ghosh M, Saha S, Bettke J, Nagar R, Parrales A, Iwakuma T, et al. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell. 2021;39:494–508.
Siolas D, Vucic E, Kurz E, Hajdu C, Bar-Sagi D. Gain-of-function p53 R172H mutation drives accumulation of neutrophils in pancreatic tumors, promoting resistance to immunotherapy. Cell Rep. 2021;36:109578.
The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93.
Li FP, Fraumeni JF. Rhabdomyosarcoma in children: Epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst. 1969;43:1365–73.
Malkin D, Li FP, Strong LC, Fraumeni JF, Nelson CE, Kim DH, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–8.
Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, et al. Revisiting Li-Fraumeni Syndrome from TP53 mutation carriers. J Clin Oncol. 2015;33:2345–52.
Achatz MI, Zambetti GP. The inherited p53 mutation in the Brazilian population. Cold Spring Harb Perspect Med. 2016;6:a026195.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disco. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Aubrey B, Janic A, Chen Y, Chang C, Lieschke E, Diepstraten S, et al. Mutant TRP53 exerts a target gene-selective dominant-negative effect to drive tumor development. Genes Dev. 2018;32:1420–9.
Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet. 2018;50:1381–7.
Chan WM, Siu WY, Lau A, Poon RYC. How many mutant p53 molecules are needed to inactivate a tetramer? Mol Cell Biol. 2004;24:3536–51.
Aschauer L, Muller PAJ. Novel targets and interaction partners of mutant p53 Gain-Of-Function. Biochem Soc Trans. 2016;44:460–6.
Bargonetti J, Prives C. Gain-of-function mutant p53: History and speculation. J Mol Cell Biol. 2019;11:605–9.
Muller PAJ, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–17.
Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, et al. Gain of function mutations in p53. Nat Genet. 1993;4:42–46.
Lang GA, Iwakuma T, Suh Y-A, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–72.
Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni Syndrome. Cell. 2004;119:847–60.
Pfister NT, Prives C. Transcriptional regulation by wild-type and cancer-related mutant forms of p53. Cold Spring Harb Perspect Med. 2017;7:a026054.
Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–58.
Weissmueller S, Manchado E, Saborowski M, Morris JP IV, Wagenblast E, Davis CA, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell. 2014;157:382–94.
Muller PAJ, Trinidad AG, Timpson P, Morton JP, Zanivan S, van den Berghe PVE, et al. Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene. 2012;32:1252–65.
Boettcher S, Miller PG, Sharma R, McConkey M, Leventhal M, Krivtsov AV, et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science. 2019;365:599–604.
Wang Z, Vaillant F, Chang C, Riffkin C, Lieschke E, Diepstraten S, et al. Loss of mutant TP53 does not impair the sustained proliferation, survival or metastasis of diverse cancer cells. Preprint at https://www.biorxiv.org/node/1218384 (2020).
Kotler E, Shani O, Goldfeld G, Lotan-Pompan M, Tarcic O, Gershoni A, et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell. 2018;71:178–90.
Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. PNAS. 2010;107:246–51.
Kadosh E, Snir-Alkalay I, Venkatachalam A, May S, Lasry A, Elyada E, et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature. 2020;586:133–8.
Humpton TJ, Hock AK, Maddocks ODK, Vousden KH. p53-mediated adaptation to serine starvation is retained by a common tumour-derived mutant. Cancer Metab. 2018;6:18.
Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, et al. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle. 2008;7:3056–61.
Pfister NT, Fomin V, Regunath K, Zhou JY, Zhou W, Silwal-Pandit L, et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 2015;29:1298–315.
Klimovich B, Merle N, Neumann M, Elmshäuser S, Nist A, Mernberger M, et al. p53 partial loss-of-function mutations sensitize to chemotherapy. Oncogene. 2022;41:1011–23.
Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10:191–202.
Acknowledgements
We thank all members of the Lowe lab, especially Francisco Sánchez-Rivera, Varun Narendra, and Francisco Barriga, as well as Arnie Levine and Karen Vousden for their helpful discussions and comments. We apologize to the many investigators whose work could not be included in this manuscript owing to space constraints.
Funding
SWL is an Investigator of the Howard Hughes Medical Institute and the Geoffrey Beene Chair for Cancer Biology.
Author information
Authors and Affiliations
Contributions
MCK and SWL conceived of the idea and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
SWL is on the scientific advisory board and holds equity in PMV Pharmaceuticals, a biotech company interested in targeting p53 mutants for cancer therapy.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Melino
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kennedy, M.C., Lowe, S.W. Mutant p53: it’s not all one and the same. Cell Death Differ 29, 983–987 (2022). https://doi.org/10.1038/s41418-022-00989-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-022-00989-y
This article is cited by
-
TP53 gain-of-function mutations promote osimertinib resistance via TNF-α–NF-κB signaling in EGFR-mutated lung cancer
npj Precision Oncology (2024)
-
A guardian turned rogue: TP53 promoter translocations rewire stress responses to oncogenic effectors in osteosarcoma
Cancer Gene Therapy (2024)
-
Decoding p53 tumor suppression: a crosstalk between genomic stability and epigenetic control?
Cell Death & Differentiation (2024)
-
TP53 missense mutation reveals gain-of-function properties in small-sized KRAS transformed pancreatic ductal adenocarcinoma
Journal of Translational Medicine (2023)
-
Nanomedicine for autophagy modulation in cancer therapy: a clinical perspective
Cell & Bioscience (2023)
