
Abstract
Gliomas are the most aggressive primary brain tumors. However, no significant improvement in survival has been achieved with the addition of temozolomide (TMZ) or radiation as initial therapy, although many clinical efforts have been carried out to target various signaling pathways or putative driver mutations. Here, we report that glycosyltransferase 8 domain containing 1 (GLT8D1), induced by HIF-1α under a hypoxic niche, significantly correlates with a higher grade of glioma, and a worse clinical outcome. Depletion of GLT8D1 inhibits self-renewal of glioma stem cell (GSC) in vitro and represses tumor growth in glioma mouse models. GLT8D1 knockdown promotes cell cycle arrest at G2/M phase and cellular apoptosis with or without TMZ treatment. We reveal that GLT8D1 impedes CD133 degradation through the endosomal-lysosomal pathway by N-linked glycosylation and protein-protein interaction. Directly blocking the GLT8D1/CD133 complex formation by CD133N1~108 (referred to as FECD133), or inhibiting GLT8D1 expression by lercanidipine, suppresses Wnt/β-catenin signaling dependent tumorigenesis both in vitro and in patient-derived xenografts mouse model. Collectively, these findings offer mechanistic insights into how hypoxia promotes GLT8D1/CD133/Wnt/β-catenin signaling during glioma progression, and identify GLT8D1 as a potential therapeutic target in the future.
Similar content being viewed by others


Introduction
Malignant brain tumors are mostly intractable, and gliomas are the most aggressive type. Temozolomide (TMZ) is an alkylating agent exerting antitumor action through the methylation of DNA, which is used as standard therapy for gliomas [1, 2]. Based on the status of isocitrate dehydrogenase (IDH) mutation, gliomas are mainly categorized into wild-type (WT) and mutant IDH groups, and patients with IDH mutant gliomas have a more favorable clinical outcome compared to patients with wild-type (WT) IDH tumors [3, 4]. Limited by the treatment option, the overall survival time remains measured in months [5, 6]. During the past decades, many studies have been focused on the increased glycolytic or hypoxic metabolic environments in human cancers, and both in vitro and in vivo evidence have shown that altered glycosylation is a universal feature of cancer cells, and certain glycans are well known biomarkers for various types of tumors [7, 8]. Based on the evidence that high glucose energy demand favors glioma stem cell (GSC) survival, targeting glycosylation process related to GSC represents attractive therapeutic strategies in the future for gliomas [9, 10].
Increasing evidence has demonstrated that GSC often resides in a hypoxic niche, and hypoxia has been documented to promote expansion of GSC [11,12,13]. The hypoxia-inducible factor (HIF) proteins are the major cellular regulators that response to hypoxia, and form a heterodimeric complex composed of an oxygen sensitive α subunit and constitutively expressed β subunit [14]. There are two HIF-α proteins, HIF-1α and HIF-2α, which are stabilized upon hypoxia treatment and dimerized with HIF-1β to regulate the transcription of multiple target genes through hypoxia-responsive elements (HREs) [15]. The 120 kDa pentaspan transmembrane protein CD133 (prominin-1), a protein with eight potential N-glycosylation sites on its two extracellular loops, has been shown to be associated with cancer stem cells in brain, lung, colon, liver, pancreas and prostate cancers [16, 17]. Importantly, GSCs marked by CD133, CD44, Sox2 or/and Oligo2, a subset of neoplastic cells with stem like properties, have been demonstrated to display potential tumor initiating capacity and be more resistant to traditional chemo- and radio-therapies [16, 18, 19]. In addition, accumulated evidence has shown that CD133 spatial distribution and protein stability are crucial determinants in tumor progression [20]. Therefore, elimination of GSC, by targeting CD133 or its associated posttranslational modifiers, may be a promising effective therapeutic treatment to inhibit tumor progression.
We have previously identified the glycosyltransferase 8 domain containing 1 (GLT8D1) as a schizophrenia risk gene by comprehensive integrative analyses [21]. In line with this finding, another study shows that ITIH3 polymorphism can confer susceptibility to psychiatric disorders through altering the expression of GLT8D1 [22]. In addition, as one of the candidates FOXM1-induced hypomethylated genes, GLT8D1 is upregulated in Head and Neck Squamous Cell Carcinomas (HNSCC) revealing its potential oncogenic role in these situations [23,24,25]. These findings prompted us to hypothesize that GLT8D1 plays pivotal roles in development and various human diseases, especially human central nervous system disorders. In this study, we found that the upregulation of GLT8D1 in cancerous cells and tissues induced by hypoxia or HIF-1α is associated with more aggressive disease in gliomas. Therefore, we decided to further characterize the functional role of GLT8D1 in gliomas.
Results
GLT8D1 is elevated in human gliomas
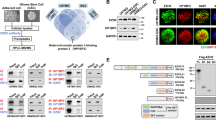
Previously, we found that GLT8D1 plays important roles in neural stem cells (NSCs) during the early developmental stage [26]. Spatio-temporal expression pattern analysis in the normal developing human brain showed that the mRNA expression of GLT8D1 is higher in post-conception weeks, which is significantly decreased after birth (Fig. S1a). In order to uncover the potential role of GLT8D1 during glioma progression, we performed immunohistochemistry (IHC) using a tissue microarray (Fig. 1a, S1b and Table S1). A strong association between increased GLT8D1 positive signals in higher-grade tumors was observed (Fig. 1b). Consistently, the analysis of the RNA dataset from the TCGA human glioma database [27] showed that the median survival time for patients with GLT8D1 high expression was 372 days, compared to 452 days for GLT8D1 low expression patients (Fig. 1c). To avoid bias caused by race differences, similar results were obtained by using both TCGA and Chinese Glioma Genome Atlas (CGGA) datasets (Fig. 1d, e). Importantly, we showed that GLT8D1 expression was higher in IDH1 WT gliomas compared with IDH1 mutant gliomas (Fig. 1f, g).

a Immunohistochemistry (IHC) was carried out to examine GLT8D1 proteins in the glioma tissue microarray. Scale bar: 50 μm. b The correlation between GLT8D1 protein level and malignant degree of gliomas. c GLT8D1 high expression is associated with adverse clinical outcome by Kaplan–Meier plot analysis. The mRNA levels of GLT8D1 were analyzed in different grades of gliomas based on the TCGA datasets (d) and the CGGA datasets (e). The mRNA levels of GLT8D1 were compared between IDH1 wild-type and IDH1 mutant glioma tissues in TCGA datasets (f) and CGGA datasets (g). h Relative GLT8D1 mRNA (top) and protein (bottom) expression in indicated cell lines, including glioma cancerous cell lines: U87, U251, A172 and glioma stem cell (GSC) lines: GBM2, GSC11, and GBM1, fetal normal human astrocytes (NHA) were used as control. i Both GLT8D1 mRNA and protein were increased under hypoxic conditions in indicated cells. N: normoxia; H: hypoxia. j Relative GLT8D1 mRNA expression was examined by qRT-PCR under indicated conditions in GSC11 cells. k GSC11 (left) or GBM1(right) cells expressing either control shRNA or HIF-1α shRNA were cultured under hypoxic or CoCl2 (100 μM) treatment conditions. Indicated gene expression was examined by immunoblot (IB) with indicated antibodies. l Scatter plot of expression of GLT8D1 and LDHA (left), GLUT1 (right) in the TCGA database. Pearson correlation coefficients are shown in the matrix. m Co-immunofluorescence staining of GLT8D1 (green), CD133 (red) and HIF-1α (purple) in human Glioma tissue. Scale bar: 100 μm. G8D = GLT8D1. Ctr shRNA = scrambled shRNA control, HIF-1α-sh#1=HIF-1α shRNA#1, HIF-1α-sh#2=HIF-1α shRNA#2. U251GSC = CD133+/CD44+ enriched U251 GSC cells. Means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001; t-test.
We next examined the mRNA and protein levels of GLT8D1 in glioma cell lines U87, U251, A172, one glioma stem cell line (GSC11) and two human primary cultured GSC cell lines GBM1 and GBM2 [28, 29], using fetal normal human astrocyte (NHA) as control. We discovered that both the mRNA and protein levels of GLT8D1 were significantly increased in cancerous cell lines, especially in patient-derived GSCs, which was confirmed in CD133+/CD44+ enriched-GSC cells compared to CD133−/CD44− tumor cells (Fig. S1c) [30] (Fig. 1h).
GLT8D1 high expression is induced under hypoxic conditions
To assess the potential regulation of GLT8D1 in GSCs by hypoxia or CoCl2, a well-known inducer of HIF-α expression [31], we silenced HIF-1α and HIF-2α using shRNAs, and found that downregulation of HIF-1α, but not HIF-2α, diminished GLT8D1 expression (Figs. 1i–k, S1d–e). In addition, we interrogated the expression of GLT8D1 and hypoxia-responsive genes [3, 32], and found that GLT8D1 expression positively correlated with expressions of LDHA, GLUT1, PDGF-β, VEGFA, and BNIP3 in the TCGA glioblastoma database (Figs. 1l, S1f). We also examined the expressions of GLT8D1, HIF-1α and 2 different putative stem cell markers, including Sox2 and CD133, in glioma tumor samples. GLT8D1 was found to be highly expressed in HIF-1α+ and carbonic anhydrase 9+ (CA9+) tumor cells expressing stem cell markers [33], although not all HIF-1α+ cells expressed GLT8D1 (Figs. 1m, S1g, Table S1). Furthermore, the co-expressions of GLT8D1, HIF-1α and CD133 proteins were detected in serial resected tumor tissues by IHC or GLT8D1+/CD133+ FACS analysis, respectively (Fig. S1h–j, Table S1).
We next determined whether HIF-1α promotes GLT8D1 expression at a transcriptional level. We inspected the upstream genomic sequence of the gene coding for GLT8D1. Three putative HREs (5’-RCGTG-3’; site 1, 2, 3) were identified within the promoter region of GLT8D1 (Fig. S1k). The subsequent chromatin immunoprecipitation (ChIP) assay corroborated the association of HIF-1α and the DNA fragments containing two of the three HREs within the GLT8D1 promoter (site 1 and 3, Fig. S1l). As expected, luciferase expression from the wild-type reporter, but not the mutant reporter, was markedly induced under hypoxic conditions (Fig. S1m). Consistently, the hypoxia-induced luciferase expression was dramatically reduced by HIF-1α knockdown (Fig. S1n).
GLT8D1 is crucial for GSC maintenance
To determine whether increased GLT8D1 expression has a direct impact on cancer cell proliferation, we inhibited GLT8D1 expression with 2 independent lenti-viral shRNAs in GSCs (Figs. 2a, S2a). As expected, stable knockdown of GLT8D1 consistently inhibited GSC cell growth using both growth curve and colony formation assays (Fig. 2b–d), and markedly decreased the ratio of BrdU-positive cells (Fig. 2e, f). Importantly, we found that GLT8D1 knockdown or overexpression, inhibited or promoted, CD133+/CD44+ enriched GSC, but not CD133−/CD44− tumor cell proliferation, respectively (Fig. S2b–f). Furthermore, we found that GLT8D1 knockdown increased the ratio of the G2/M cell population, with significantly accumulated CDK1 phosphorylation at Tyr15 (Cdk1Tyr15), and increased p21 proteins (Fig. 2g–h) [34]. In addition, we showed that inhibition of GLT8D1 increased cellular apoptosis (Fig. S2g–h).

a Individual GLT8D1 shRNA knockdown efficiency was verified by qRT-PCR (top) and IB (bottom) in indicated cells, scrambled shRNA was used as a control. Indicated cell lysates were probed with GLT8D1 and β-actin antibodies, representative images are shown. Green arrow: endogenous GLT8D1 proteins; Red arrow: exogenous GLT8D1 proteins. b Knockdown of GLT8D1 inhibited GSC11, GBM1, U251GSC and A172GSC cell growth. A172GSC = CD133+/CD44+ enriched A172 GSC cells; G8D sh#1 = GLT8D1 shRNA#1, G8D sh#2 = GLT8D1 shRNA#2, ove=overexpression. c, d GLT8D1 knockdown dramatically inhibits the colony formation ability of U251GSC and A172GSC in vitro. d Quantification data for c. Scale bar: 5 mm. e, f GLT8D1 knockdown inhibited the DNA synthesis in GSC11 cells by BrdU incorporation assay (e),(f) Quantification data for BrdU staining in GSC11 (top) and GBM1 (bottom) cells. DAPI: 4,6-diamidino-2-phenylindole (n ≥ 5). Scale bar: 100 μm. g, h GLT8D1 knockdown leads to accumulation of G2/M cells by FACS analysis in U251GSC (left) and A172GSC (right). Indicated cells were stained with PI for analyzing cell cycle distribution. h Depletion of GLT8D1 increased p21 and p-CDK1(Y15) expressions in U251GSC (left) and A172GSC (right) cells. Means ± SEM, *P < 0.05,**P < 0.01, ***P < 0.001; t-test.
We also examined the cell populations in GSC11 and GBM1 cells, and found that CD133+/CD44+ double positive cell populations were dramatically reduced upon GLT8D1 knockdown (Fig. 3a, b). Interestingly, we noticed that GLT8D1 knockdown reduced the protein but not the mRNA level of CD133 (Figs. 3c, S3a–b). However, we found that CD44, SOX2 and Oct4 were all downregulated in GSCs upon GLT8D1 knockdown (Figs. 3c, S3a–b). Consistently, the neurosphere-like structure forming ability in CD133+/CD44+ enriched GSC was significantly reduced, as validated by both sphere number and size statistical analysis (Fig. S3c–e). Considering patient-derived cell lines better reflect GSC key properties, we then used GSC11 and GBM1 to validate the potential role of GLT8D1 in GSC maintenance. Obviously, the in vitro extremely limiting dilution assay showed that GLT8D1 promoted GSC proliferation and the self-renewal ability (Figs. 3d–f, S3f–g).

a, b Indicated cells were stained with a PE-labeled anti-AC133 (130-113-186, Miltenyi Biotec) and FITC-labeled anti-CD44 (75122 S, CST) antibody followed by flow cytometry analysis (n = 3). b Quantification data for a. c Knockdown of GLT8D1 inhibits GSC marker gene expression in GSC11 cells verified by IB. Indicated antibodies were used. d–f Representative images of GSC11 and GBM1 tumor sphere expressing GLT8D1 shRNAs or control shRNA are shown (d). e, f Quantification data for d. Scale bar: 500 μm. g, h A subcutaneous limiting dilution tumor formation assay (1 × 102, 1 × 103, 1 × 104 and 1 × 105 cells/per mouse) was performed in indicated cells, tumor masses were harvested after 11 weeks. h Quantification data of tumor incidence for g. i Survival analysis of mice intracranially implanted with 5 × 105 indicated cells. X-axis represents days after cell injection. The survival difference was evaluated by log-rank analysis. n = 7. j Representative images of H&E staining for tumor formation of indicated xenografts for i. Scale bar: 2 mm. T = Tumor tissues, N = Normal tissues. Means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, t-test.
Next, we proceeded to look into the impact of GLT8D1 depletion on tumor initiation in vivo by injecting 1 × 102, 1 × 103, 1 × 104, and 1 × 105 GSC11 cells stably expressing control shRNA or GLT8D1 knockdown shRNAs into nude mice, followed by tumor initiation evaluation after 11 weeks. Our results showed that 100% of the mice developed tumors when injected with 1 × 104 control shRNA cells, whereas the tumor incidence was markedly reduced in GLT8D1 knockdown cells (Figs. 3g–h, S3h–i). To validate the above findings, we established an orthotopic xenograft mouse model by injecting 5 ×105 GSC11 cells intracerebrally, and found that GLT8D1 inhibition leads to a reduction of impact on normal brain structure, as well as prolonged survival rate (Fig. 3i, j).
Inhibition of CD133 trafficking into the endosomal-lysosomal pathway by GLT8D1 leads to the activation of Wnt/β-catenin signaling
To further explore the molecular event(s) regulated by GLT8D1 in GSCs, we decided to identify GLT8D1 interacting proteins by performing co-immunoprecipitated (co-IP) assay followed by mass spectrometry analysis in GSC11. Among the candidate GLT8D1 interacting proteins, CD133 caught our attention based on its essential role in sustaining cancer stem cell maintenance (Table S2) [16, 17]. As expected, we found that only endogenous CD133, but not Sox2, Oligo2, or CD44 physically associated with GLT8D1 (Figs. 4a, S4a), which was confirmed by co-IP experiments using exogenous proteins (Fig. 4b). The bulk of GLT8D1 proteins were detected to co-localize with both Golgi marker GM130 and CD133 determined by co-immunofluorescence staining (Fig. 4c, d). In line with previous studies that CD133 localizes in microvilli and cilia [35, 36], the high power immunofluorescence staining revealed that CD133 and GLT8D1 colocalized in microvilli, but not primary cilia due to incomplete ciliogenesis in human glioblastoma cells (Fig. S4b) [37, 38]. To map the region of CD133 protein binding to GLT8D1, we generated a series of CD133 mutants based on human prominin-1.s2 (NCBI Reference Sequence: NP_006008.1) [39, 40], and introduced them into HEK-293T cells (Fig. 4e). We found that a truncated mutant that contained the extracellular region of CD133 (CD1331-108) maintained the binding ability, whereas a truncated form lacking the part of first extracellular region of CD133 (CD133109-865) lost the binding affinity to GLT8D1 (Fig. 4f). Similarly, the CD133 binding region in GLT8D1 maps to a region from amino acid 221 to 371 (Fig. 4g). To validate the binding ability, we repeated the above experiment using flag and myristoylation motif double-tagged CD133 truncated mutants. Myristoylation motif has been recognized as a modification that anchors the eukaryotic cellular proteins on the cell membrane (Fig. S4c, d) [41]. In addition, we observed that human prominin-1.s1 (NCBI Reference Sequence: NP_001139319.1) [39], still bound to GLT8D1, suggesting that the missing 9 amino acids (CD13392-100) are not essential to mediate the interaction between GLT8D1 and CD133 (Fig. S4e).

a, b Co-immunoprecipitation assay confirms that both endogenous and exogenous GLT8D1 interact with CD133. Black arrow: heavy chain. Indicated constructs, cell lines and antibodies were shown. c, d Immunofluorescent staining for GLT8D1 (green), CD133 (red) and the Golgi apparatus marker GM130 (purple) in indicated cells. Enlarged pictures of indicated areas (white squares) were also indicated. Scale bar: 20 μm. d Quantification data for c. e Schematic representation of the C-terminal Flag-tagged full-length CD133 (CFL), N-terminal Myc-tagged full-length GLT8D1 (GFL), and various corresponding truncation mutants. CD133 (red), GLT8D1 (green), protein interaction domain (yellow). Grey box indicating transmembrane domain. f, g HEK-293T cells were transfected with the indicated constructs, followed by immunoprecipitation (IP) and immunoblot (IB) analysis with the indicated antibodies. h HEK-293T, expressing GLT8D1 knockdown or control shRNA, transfected with wild type CD133 or all sites CD133 N-glycan-deficient (CD133NallQ) mutant constructs, were treated with or without swainsonine (20 μM) or PNGase F (50 U μg−1), respectively. Red arrow: glycosylated CD133; green arrow: CD133 proteins have not undergone complex N-glycosylation; blue arrow: non-glycosylated CD133. i GSC11 cells, stably expressing GLT8D1 knockdown or control shRNAs, were treated with the lysosome inhibitor ammonium chloride (50 mM) or the proteasome inhibitor MG132 (20 μM), respectively, for 24 h, and the cell lysates were subjected to IB with indicated antibodies. The quantified ratios for CD133 expression normalized to reciprocal β-actin were indicated below. j GSC11 cells stably expressing GLT8D1 knockdown or control shRNAs were treated with 100 μg mL−1 cycloheximide (CHX), cell lysates were collected at the indicated time points and subjected to IB with the indicated antibodies. k–n Immunofluorescent staining for LAMP1 (green), Rab11 (green), CD133 (red) and transferrin receptor (red) in GSC11 cells. Nuclei were counterstained with DAPI (blue). Scale bar: 20 μm. TFR = transferrin receptor. l Quantification data for k. n Quantification data for m. The colocalization rates between indicated proteins were quantified using five randomly selected images.
Since GLT8D1 encodes a potential member of the glycosyltransferase family, whose enzymatic activity or candidate substrate remains elusive [42], we decided to examine whether or not the posttranslational modification of CD133 including N-glycosylation, proteasomal and/or endosomal-lysosomal dependent modification is regulated by GLT8D1 [20, 43]. Consistent with the former finding that swainsonine or PNGase F treatment led to a decrease in the molecular mass of CD133 [44], we found that, GLT8D1 inhibition resulted in the same reduced molecular mass of CD133 as that in the swainsonine treatment group, whereas the CD133 all sites N-glycan-deficient (CD133NallQ) mutant collapsed to a molecular mass of ~100 kDa, similar to the PNGase F treatment group, indicating that GLT8D1 mediates the α-mannosidase II and complex N-glycan biosynthesis of CD133 (Fig. 4h). Furthermore, with the exception of the CD133NallQ mutant, wild-type or single-site N-glycan deficient mutant CD133 proteins in GLT8D1 knockdown groups were unanimously reduced compared to the control group (Fig. S4f), indicating that the N-glycan modification mediated by GLT8D1 is important for CD133 stability.
As we did not observe obvious change in CD133 mRNA level upon GLT8D1 depletion, we decided to examine whether the post transcriptional modification of CD133 protein was regulated by GLT8D1 (Fig. S3a). In GLT8D1 knockdown cells, CD133 proteins were less stable and persisted in the presence of ammonium chloride but not MG132, compared to control cells, suggesting that CD133 degradation through the endosomal-lysosomal pathway, but not ubiquitin-proteasome system dependent signaling pathway [45], is mediated by GLT8D1 (Figs. 4i–j, S4g–h). In line with previous finding, inhibition of GLT8D1 led to an increased CD133 colocalization with CD63 [46], and lysosomal associated membrane protein 1 (LAMP1, a lysosomal-dependent degradation marker) [47], whereas decreased CD133 colocalization with Rab11 (marker for recycling vesicles) [48], respectively (Figs. 4k–l, S4i). However, inhibition of GLT8D1 did not impair the expression and trafficking of transferrin receptor (TRF), a canonical membrane protein and model cargo for endosomal-lysosomal pathway (Fig. 4m, n) [49]. While the expressions of EEA1, LAMP1, CD63 and Rab11, were not affected after GLT8D1 knockdown (Fig. S4j). As documented before, CD133-containing exosomes play important roles in cell differentiation [50] and tumor progression [51], we revealed that the extracellular vesicles (EVs) containing CD133 proteins were also markedly decreased after GLT8D1 knockdown (Fig. S4k, l) [50, 52]. However, GLT8D1 knockdown did not affect the phosphorylation at Y828 site of CD133, which is important for GSC maintenance via activating PI3K/Akt signaling (Fig S4m) [53]. In addition, we found that previously documented enzymatic mutant forms of GLT8D1 [54] were still able to interact with CD133 leading to partially reversed CD133 protein degradation compared to wild-type GLT8D1 (Fig. S4n–o).
Consistent with former findings [43, 55], we confirmed that CD133 physically interacts with and stabilizes β-catenin proteins (Fig. S5a, b). Furthermore, we found that Wnt3a ligand and LiCl activated signaling, together with the expressions of Wnt downstream targets c-Myc, Axin2 and cyclin D1 were decreased in GSCs (Figs. 5a–c, S5c–e). We also tracked β-catenin localization by immunofluorescence and immunoblot in GLT8D1 depleted cells and found a robust decrease in cytoplasmic and nuclear localized β-catenin (Figs. 5d, S5f). In addition, both the decreased cell proliferation and tumor sphere formation ability caused by GLT8D1 knockdown were restored by CD133 overexpression (Figs. 5e–g, S5g–i). These results strongly suggest that GLT8D1 promotes Wnt/β-catenin signaling in GSC for tumor progression.

a Wnt signaling response luciferase reporter TOPFlash was examined in GSC11 cells after Wnt3a or LiCl treatment. b The β-catenin, LaminB and GAPDH proteins were examined by IB with indicated antibodies in GSC11 cells. c The mRNA expressions of indicated genes in GSC11 cells were examined by qRT-PCR after PBS or NH4Cl treatment. Mock=PBS treatment. d Immunofluorescent staining of β-catenin proteins in GSC11 cells expressing GLT8D1 knockdown or control shRNAs. White arrows: β-catenin nuclear-accumulated signals. Scale bar: 50 μm. CD133 forced overexpression in GLT8D1 knockdown cells (e) promotes cell proliferation (f), and tumor sphere formation (g) in GSC11. h Cellular apoptosis was determined using Annexin V staining followed by FACS analysis in GSC11 cells treated by DMSO or TMZ. i Cell lysates were examined by IB with indicated antibodies. tPARP = total PARP, cPARP = cleaved PARP. CC3 = cleaved caspase 3. j The scheme for xenograft tumors generated from GSC11 cells responding to TMZ, TMZ: 60 mg kg−1, once a week for 5 weeks. k–m GLT8D1 knockdown tumors were more sensitive to TMZ treatment compared to control shRNA tumors. Indicated xenograft tumor masses were harvested, representative xenograft tumors are pictured (k), weights (l) and tumor volumes (m) were shown. Means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, t-test.
GLT8D1 knockdown promotes TMZ-induced cellular apoptosis and inhibits xenograft tumor formation in vivo
Previous findings suggested that TMZ preferentially depletes GSC or tumor initiating cell (TIC) [56]. We postulated that GLT8D1 knockdown might sensitize cancerous cells responding to TMZ treatment. In deed, the percentages of TMZ-induced apoptotic cells were higher in GLT8D1 knockdown cells compared to control shRNA expressing cells (Figs. 5h–i, S5j–k). To verify this efficacy in vivo, we performed a xenograft tumor formation assay in nude mice. When the xenograft tumors reached about 50 mm3 in size, nude mice were treated weekly by intraperitoneal injection of TMZ (60 mg kg−1). Mice were monitored each day and tumor growth measured every 3 days. After 5 weeks, this regimen resulted in an obvious reduction in gross tumor extent and in a statistically significant reduction in both tumor weights and volumes (Fig. 5j–m). Furthermore, the proliferative marker Ki67 decreased whereas apoptotic marker cleaved caspase 3 (CC3) increased in GLT8D1 knockdown xenografts compared to the scrambled shRNA control group (Fig. S5l, m).
A rationally designed peptide “FECD133” potently inhibits tumor growth
Based on our findings that (1) a truncated mutant CD1331-108 is required and sufficient for CD133 interaction with GLT8D1, and (2) GLT8D1 regulates CD133 protein stability through the endosomal-lysosomal pathway (Figs. 4, S4), we designed an inhibitor peptide termed “FECD133 (the first extracellular region of CD133)” for a therapeutic purpose. As shown by GST pull-down and co-IP experiments, the FECD133 physically interacted with endogenous GLT8D1, and reduced the exogenous interaction between GLT8D1 and CD133 (Fig. 6a, b). We also detected that FECD133 preferentially inhibited the survival of CD133+/CD44+ enriched GSCs, but not CD133−/CD44− tumor cells (Figs. 6c, S6a). As predicted, the FECD133 inhibited CD133 and β-catenin protein stability, cell viability, sphere and colony formation abilities of GSC11, GBM1 and CD133+/CD44+ enriched U251 and A172 cells (Figs. 6d, S6b–f). Interestingly, we observed that TMZ inhibited almost equally the growth and colony formation of all the cells tested, whereas the FECD133 preferentially inhibited GSC but not Human Neural Progenitor Cells (NHNP) and NHA using the same higher dosage (Fig. 6d). We also found that the FECD133 treatment in GSC11 cells blocked the activity of Wnt/β-catenin signaling pathway (Fig. 6e, f), and the FECD133 alone or in combination with TMZ promoted cellular apoptosis, but inhibited cell survival and tumor sphere formation in vitro (Figs. 6g–h, S6g–i).

a GLT8D1 physically interacts with the FECD133 as assessed by GST pull-down assay. GST and GST-FECD133 were expressed and purified in E.coli BL21 (DE3), and then visualized by Coomassie blue staining. b Co-IP assay examining the interaction between GLT8D1 and CD133 in the presence of different doses of the FECD133 (40 nM, 200 nM). GST (200 nM) proteins were used as control. GSC11 cells were lysed, and then different doses of the FECD133 were added into the indicated cell lysates. After incubation for 30 min, cell lysates were immunoprecipitated with GLT8D1 antibodies and then subjected to IB with the indicated antibodies. c Evaluation of the efficacies of the FECD133 and TMZ in regulating cell viability using SRB assay in indicated cells. The IC50 in each cell line was indicated. d GSC11 cells were treated with different doses of the FECD133 (30 nM, 60 nM), and the indicated cell lysates were analyzed by IB with the indicated antibodies. e The indicated doses of the FECD133 that inhibit the expression of c-Myc and Axin2 examined by qRT-PCR. f β-catenin proteins were reduced upon different doses of the FECD133 (0, 30, 60 nM) treatment. g The FECD133 alone or with TMZ induces GSC11 and GBM1 cellular apoptosis. GST (40 nM), TMZ (200 μM), the FECD133 (40 nM), or TMZ plus FECD133 for 48 h. h GSC11 cells were treated under the indicated conditions, and the cell lysates subjected to IB with the indicated antibodies. GST (40 nM), TMZ (200 μM), FECD133 (40 nM), or TMZ plus FECD133 for 48 h. i–k Xenograft tumors after subcutaneously injecting the indicated cell lines and treated by the indicated conditions, mice received tail vein injections of GST (5 mg kg−1), TMZ (60 mg kg−1), the FECD133 (5 or 50 mg kg−1) alone, or the FECD133 (5 mg kg−1) with TMZ (60 mg kg−1), once every week for five 5 weeks. Xenograft tumors were harvested and photographed (i), tumor weights (j) and volumes (k) were plotted. l Schematic representation of the intracranial mouse model. GSC11 cells (5 × 105 cells per mouse) were intracranially injected into NOD-SCID mice. Mice were in situ injected with GST (100 μg kg−1), the FECD133 (100 μg kg−1), TMZ (1 mg kg−1) or the FECD133 with TMZ. m The survival of mice was evaluated (n = 5, Kaplan–Meier model with a log-rank test). P values were analyzed by comparing TMZ or the FECD133 alone versus the combination of TMZ with the FECD133. The X-axis represents days after cell injection. n Representative images of H&E staining for tumor formation following indicated treatments. Scale bar: 2 mm. o PDXs tumors treated by GST (5 mg kg−1) or FECD133 (5 mg kg−1) after tail vein injection were harvested and photographed, tumor weights and volumes were plotted. Mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, t-test.
Next, we carried out a preclinical study using previous xenograft models. The subcutaneous xenograft model in nude mice showed that a dosage of 5 mg kg−1 FECD133 was equally effective as 60 mg kg−1 of TMZ in terms of reducing tumor mass and volume, whereas a higher dosage of 50 mg kg−1 FECD133 alone or 5 mg kg−1 FECD133 synergizing with TMZ inhibits tumor growth in vivo more efficiently (Figs. 6i–k, S6j, k). In the orthotopic xenograft model, on day 15, pairs of mice were randomized to receive orthotopic injections of GST (100 μg kg−1), TMZ (1 mg kg−1) and/or FECD133 (100 μg kg−1) every other day (Fig. 6l). FECD133 alone or with TMZ repressed tumor growth in vivo, validated by prolonged survival rate and reduced tumor area in the brain sections (Fig. 6m, n). Furthermore, the FECD133 peptide (5 mg kg−1) treatment resulted in obvious reduction in tumor growth using primary patient-derived xenografts (PDXs) mouse model (Figs. 6o, S6l and Table S1).
Drug screening identifies lercanidipine as an inhibitor of GSC
To improve the clinical therapy of gliomas, we decided to test 200 compounds from FDA-Approved Drug Library Mini for their potential activities inhibiting GLT8D1 expression (Table S3). The screening was performed using immunoblot to determine GLT8D1 protein expression after treatment with 20 μM concentrations of a single application in GSC11. Every compound that reduced GLT8D1 protein expression to less than 70% of control level was considered as a positive candidate. 21 compounds fulfilled this criterion (Fig. S7a), and 16 of these have been documented to inhibit glioma cell proliferation (Table S3). The IC50 values of the rest 5 compounds were evaluated in GSC11 and GBM1 comparing with NHA and NHNP, and compound lercanidipine was identified as the only one specifically inhibiting the viability of GSC (Fig. 7a, S7b). Importantly, the survival of CD133+/CD44+ enriched GSCs, but not CD133−/CD44− tumor cells were preferentially inhibited by lercanidipine (Fig. S7c). Furthermore, we verified that CD133 and β-catenin protein stabilities were significantly suppressed by lercanidipine treatment (Fig. 7b–e). Lercanidipine is a lipophilic third-generation DHP-CCB (dihydropyridine-calcium channel blocker), which has been broadly used for treating patients with different degrees of hypertension [57]. In line with previous finding [58], we revealed that the expression levels of HIF-1α, GLUT1 and LDHA, but not HIF-2α, proteins were decreased upon lercanidipine treatment (Fig. S7d, e). We also found that lercanidipine treatment, in combination with TMZ, decreased sphere numbers and cell survival (Figs. 7f–h, S7f–h). In addition, lercanidipine (5 mg kg−1) treatment dramatically suppressed tumor growth in both PDXs and orthotopic glioma xenografts mouse models by tail vein injection (Fig. 7i–k, Table S1). Immunohistochemical staining of representative tumor sections from lercanidipine treated groups showed less GLT8D1, CD133, Ki67, while more cleaved caspase 3 (CC3) expressions (Fig. S7i–k).

a Lercanidipine specifically inhibits GSC cell viability, compared to NHNP and NHA cells. b, c Membrane tethered form of CD133 were reduced by lercanidipine (7 μM) treatment. c Quantification data for b. d IB detecting CD133, β-catenin and GLT8D1 expressions after lercanidipine (7 μM) treatment. e β-catenin proteins were examined in cytoplasm (C) and nuclear fractions (N) of indicated cells following 7 μM lercanidipine treatment by IB. f, g Tumor spheres derived from indicated GSC cell lines were significantly inhibited following lercanidipine treatment. g Quantification data for f. Scale bar: 500 μm. h Lercanidipine alone or synergizes with TMZ to induce cellular apoptosis. Lercanidipine (7 μM), TMZ (200 μM). i Patient-derived xenograft tumors treated with 5 mg kg−1 lercanidipine via tail vein injection twice every week for four weeks. Indicated xenograft tumors were harvested, tumor weights and volumes were plotted. j GSC11 cells were implanted intracranially into NOD-SCID mice. Mice were treated with vehicle (PBS) or 5 mg kg−1 lercanidipine by tail vein injection daily for 7 days, and the relative survival curves were plotted (n = 5, Kaplan–Meier model with log-rank test). k Representative images of H&E staining for tumor formation of indicated treatments. Scale bar: 2 mm. l Proposed mechanism of GLT8D1 in GSC during glioma progression. Under normoxia condition, low expression of GLT8D1 results in less efficient glycosylation modification in CD133, leading to CD133 degradation through endosomal-lysosomal pathway. On the contrary, GLT8D1 transcripts and proteins are increased under hypoxic conditions in a HIF-1α dependent manner. Therefore, abundant GLT8D1 proteins physically interact with CD133 to promote high glycosylation and stabilization of CD133, which activates Wnt signaling in GSC leading to glioma progression. However, the FECD133 peptide and lercanidipine can inhibit GSC maintenance and tumor growth by blocking GLT8D1-CD133 complex formation or GTL8D1 expression, respectively. Means ± SEM, *P < 0.05,**P < 0.01, ***P < 0.001, t-test.
Discussion
Gliomas are debilitating tumors associated with a poor prognosis, short median survival and limited response to differential therapies [59, 60]. Hypoxia occurs in the majority of solid tumors including gliomas, and HIF-1α with its downstream factors are frequently upregulated and closely associated with poor clinical prognosis [61]. Recent findings by our group and others showed that GLT8D1 is frequently disregulated or mutated in human schizophrenia or Amyotrophic lateral sclerosis (ALS) [21, 54], suggesting that GLT8D1 plays important role in central nervous system.
The existence of the glioma initiating cell or stem cell (GIC or GSC) is an important evidence for chemo-, radio- or drug-therapy resistance, and CD133 is one of the “stemness” biomarkers for gliomas and cancer stem cells [62]. CD133 has been identified to be involved in the formation or maintenance of protrusions, such as microvilli and primary cilia [35, 63,64,65]. The direct interaction between CD133 and important factors leading the deregulation of related signaling pathways have been documented, including cholesterol [66, 67], GM(1) and GM(3) gangliosides [68], actin-related protein 2/3 (Arp2/3) complex [36], ADP-ribosylation factor-like protein 13B (Arl13b) and histone deacetylase 6 (HDAC6) [55, 69], and p85 subunit of phosphoinositide 3-kinase (PI3K) complex [53]. For examples, the critical role of CD133 in retina development, intestinal inflammation and tumorigenesis, photoreceptor degeneration, have been thoroughly characterized in different model organisms [70,71,72]. In this study, we revealed that GLT8D1 colocalizes with CD133 in microvilli, indicating that GLT8D1 might orchestra cell membrane structure.
GTL8D1 has been predicted to be a single-pass type II membrane protein (UniProt dataset), with its N-terminus in the cytoplasmic side of the cell membrane. In this study, we demonstrate that GLT8D1 promotes CD133 glycosylation, and the glycosyltransferase activity deficient GLT8D1 mutant can partially reverse GLT8D1 depletion-induced CD133 degradation, indicating that both the enzymatic and membrane anchoring roles of GLT8D1 are critical to inhibit CD133 degradation through the endosomal-lysosomal pathway. Although hypoxic conditions have been shown to enhance CD133 expression and glycosylation previously [73, 74], our mechanistic study for the first time identifies that hypoxia-induced GLT8D1 is required for CD133 stabilization and the stemness maintenance in GSCs (Fig. 7l).
A number of combination therapies with TMZ have been developed to increase the therapeutic efficacy [75]. Our data identified the clinical relevance of GLT8D1 as a CD133 activator or agonist, and showed that the FECD133 peptide sufficiently blocks GLT8D1 binding with its enzymatic substrate CD133, while lercanidipine decreases the overall GLT8D1 expression indirectly via inhibiting HIF-1α expression, leading to dramatic antitumor activities in vitro and in vivo. Importantly, the ability of lercanidipine crossing blood-brain barrier may facilitate its clinical application in the future [76, 77]. From the clinical point of view, both GLT8D1 and CD133 expression levels should be restrained within an appropriate range. Our results show that the Wnt/β-catenin signaling pathway is also downregulated upon GLT8D1 depletion or addition of FECD133 in GSC (Fig. 7l). Based on the enzymatic activity of GLT8D1 and its biological functions in gliomas, it is worth defining other candidate substrates glycosylated and regulated by GLT8D1 in order to facilitate the development of optimal combined therapeutic strategies. Taken together, our study supports GLT8D1 as a potential novel glioma therapeutic target, and its rapid clinical translation will be important for patients in the future.
Methods
Analysis of tissue microarrays, IHC staining and bioinformatics
Glioma tissue microarrays, fully annotated with clinical and pathological information, were obtained from Shanghai Outdo Biotech Co., Ltd. (HBraG090PG01). Glioma cases were assessed from Sep 2001 to Mar 2016, the median age of patients was 48 ± 27 years, ranging from 21 to 75 years old. All specimens were graded by the pathologic and clinic stage, spanning grade I (n = 1), grade I-II (n = 2), grade II (n = 9), grade II-III (n = 4), grade III (n = 22), grade III-IV (n = 7) and grade IV (n = 42). The description of the patient represented in each tissue microarray and of the scoring system used to evaluate GLT8D1 expression is provided. The Immunohistochemical staining was performed following the former standard protocol [78]. Simply, tissue specimens were deparaffinized in xylene, rehydrated through graded ethanol solutions, antigen retrieval and immersed in a 0.3% hydrogen peroxide solution. The slides were washed by phosphate-buffered saline (PBS), and nonspecific binding was then blocked by incubation with 10% goat serum for 20 min at room temperature. The indicated primary and secondary antibodies were used. To comprehensively study the expression pattern and prognostic implications of GLT8D1 in gliomas, whole transcriptome sequencing and corresponding clinical data (IDH mutation, WHO grades and survival information) were downloaded from TCGA (https://cancergenome.nih.gov) and CGGA (http://www.cgga.org.cn).
Constructs, cell culture, immuno-precipitation and immunoblotting
Independent GLT8D1, HIF-1α, HIF-2α, and CD133 targeting shRNAs were subcloned into the pLKO.1 vector, and all the constructs were sequence verified. The detailed sequences of shRNAs used in this study were designed as previously study [79] and shown in supplementary Table S4. Human GLT8D1 cDNA was amplified by PCR and introduced into pCDH-MCSV-E2F-eGFP and verified by sequencing. The information for truncated forms of GLT8D1 and CD133 was also provided in supplementary Table S4. pLv-CD133-FLAG and related mutant plasmids were kind gifts from Dr. Jianhai Jiang at FuDan university, China. GLT8D1 1KB promoter were synthesized and cloned into pGL3 basic vector. For the lenti-viral expression system, briefly, the indicated lenti-viruses were packaged and collected from HEK-293T (ATCC) cells following the manufacturer’s protocol, and viral infected cells were then selected by puromycin. NHNP cell line was purchased from Lonza and cultured in ReNcell NSC maintenance medium (Merck, SCM005), supplemented with 20 ng mL−1 EGF (Gbico, PHG0311L), 20 ng mL−1 bFGF (Gbico, PHG0266). The NHA cell line was a kind gift from Dr. Xiaozhong Peng at Institute of Medical Biology, Chinese Academy of Medical Sciences [80]. NHA cells were cultured using commercial astrocyte medium (catalog 1801, ScienCell) supplemented with 2% FBS (ScienCell), 1% AGS (ScienCell), and 1% P/S solution (ScienCell). Human primary glioma stem cell lines (GBM1 and GBM2 were gifts from Drs. Xiuwu Bian and Yu Shi at Institute of Pathology and Southwest Cancer Centre, Southwest Hospital, The Third Military Medical University, Chongqing 400038, China) [18, 29]. U251, U87, A172 and HEK-293T cells were all cultured in DMEM medium (Hyclone) supplemented with 10% fetal bovine serum (FBS, Gibco, 10270–106) and 1% penicillin/streptomycin. The GSC11 cells were cultured in serum-free DMEM/F12, supplemented with B27 (1:50, Invitrogen, 2175161), 20 ng mL−1 EGF (Gbico, PHG0311L), 20 ng mL−1 bFGF (Gbico, PHG0266), 4 μg mL−1 heparin (Sigma, H3149-500KU-9) and 1% penicillin/streptomycin. The GBM1 and GBM2 were cultured in neurobasal A medium (Gibco, 2085380) supplemented with B27, GlutaMAX (1:100, Gibco 35-50-061), Sodium pyruvate (1:100, Gibco R25-0000-CI), MEM NEAA (1:100, Gibco, 11140−050), 20 ng mL−1 bFGF and 1% penicillin/streptomycin. Except for the hypoxia treatment, all cells were cultured at 37 ˚C in a 5% CO2 humidified environment. Cells were lysed and the supernatants were collected, indicated samples were separated by electrophoresis and transferred to a polyvinylidene fluoride membrane (Millipore, IPVH00010).
For immuno-precipitation assay, indicated cells were collected and lysed in lP buffer (0.5% NP 40; 20 mM Hepes; 150 mM NaCl; 2 mM EDTA; 1 mM NaF) supplemented with protease inhibitors (Roche, 11836170,001), incubated on ice for 45 min, and cleared by centrifugation at 14,000 g at 4˚C for 20 min. Protein lysate was subjected to immunoprecipitation with the primary antibody, overnight at 4˚C. The precipitates were extensively washed with ice cold PBS, boiled with SDS loading buffer and subjected to immunoblot. The detailed antibody information used in this study was shown in supplementary Table S4.
Chromatin immunoprecipitation (ChIP), luciferase assay and qRT-PCR
ChIP assay was performed using the Kit (Abcam, ab500) according to the manufacturer’s protocol. Indicated cells were cultured under hypoxic conditions for 24 hr before harvest. 5 μg of pre-immune mouse IgG and anti HIF-1α antibody was used for ChIP reaction. Primers used for ChIP assay were designed [79] and the sequences were provided in supplementary Table S4. To investigate whether GLT8D1 is transcriptionally regulated by HIF-1α, indicated cells expressing either control shRNA or two independent HIF-1α shRNAs were transfected with the pGL3-basic construct containing wild type or mutant GLT8D1 promoters plus a Renilla luciferase plasmid. 24 h later, cells were cultured under normoxic or hypoxic conditions for another 24 h. Luciferase activities were measured with a Dual Luciferase Kit (Promega, E1960). For qRT-PCR assay, indicated total RNAs were extracted from cells using RNAiso Plus (Takara, 108-95-2), following which they were reverse transcribed using an PrimeScript RT reagent Kit (Takara Bio, RR047A). cDNA was subjected to RT-qPCR analysis using FastStart Universal SYBR Green Master Mix (Roche, 04194194001). All reactions were performed in triplicate using an Applied Biosystems 7500 machine. β-actin was used to normalize expression levels. The detailed primer sequences used in this study were shown in supplementary Table S4.
Cell proliferation assay
For the cell proliferation assay, a total of 2 × 104 indicated cells were plated into 12-well plates in triplicate, the exact cell numbers for each day were determined by an automatic cell analyzer countstar (Shanghai Ruiyu Biotech Co., IC 1000).
BrdU incorporation assay
For BrdU incorporation assay, a total of 3 × 104 indicated cells were plated. 20 min before fixation, indicated cells were pre-treated with 10 μM BrdU (Abcam, ab142567, 1:100) at 37 ˚C, followed by fixation with 4% PFA and then incubation with BrdU primary antibody overnight at 4 ˚C (CST, 5292 s, 1:1000). The slides were then further stained by secondary antibody (Abclonal, 61303, 1:500). DAPI was used to stain the cell nuclei. More than five fields/samples randomly selected were imaged by a Nikon Ti fluorescence microscope and quantified.
Colony formation assay
For the colony formation assay, a total of 3000 indicated cells were seeded on soft agar in 6-well plates, and colonies were counted 3 weeks after seeding. The resulting colonies were then washed twice with phosphate-buffered saline (PBS) and fixed with 4% formaldehyde for 30 min at room temperature and stained for 12 h with 0.01% crystal violet. The exact colony number of colonies was then quantified.
FACS assay
For cell cycle analysis, indicated cells were harvested and then fixed with 70% ethanol at 4 ˚C overnight. The fixed cells were washed by cold PBS, stained with PI for 30 min at 37 °C in dark and then examined by flow cytometer. For the cellular apoptosis assay, cell suspensions were stained with Annexin V (BD Pharmingen) and propidium iodide (PI). A total of 30,000 events were recorded for each experiment and analyzed by FlowJo software. For CD133+/CD44+ cell population selection, U251 and A172 cells were cultured in serum-free DMEM/F12, supplemented with B27, 20 ng mL−1 EGF, 20 ng mL−1 bFGF, 10 ng mL−1 heparin and 1% penicillin/streptomycin for 2 weeks. Indicated cells were then collected and incubated with phycoerythrin (PE) conjugated anti-AC133 (130-113-186, Miltenyi Biotec) and FITC (fluorescein isothiocyanate) conjugated anti-CD44 antibody (75122 S, CST) for 30 min at 37˚C in dark, followed by washing twice in PBS supplemented with 1% FBS. CD133+/CD44+ GSCs and CD133−/CD44− tumor cells were separated by FACS Aria III (BD Immunocytometry Systems, San Jose, CA, USA) sorting. In addition, fresh GBM specimen were washed and dissociated into single cells, and the isolated tumor cells were collected and subjected for incubation with PE-AC133 (130-113-186, Miltenyi Biotec) and GLT8D1 (ab236974, Abcam) antibodies, followed by secondary antibody staining and sorting with FACS Aria III.
Xenograft tumor formation
Male nude or NOD/SCID mice at 5 weeks of age were purchased from GemPharmatech. Co. Ltd (Nanjin, China). The mice were randomly divided into indicated groups and injected with differential cell lines subcutaneously or intracerebrally. Subcutaneously injected tumor sizes were measured with Vernier calipers (Suzhou, China) every 3 days for 4–6 weeks. At the end of the experiment, all mice were sacrificed and tumors were collected, weighed, and tumor volumes were calculated by the formula length × (width)2/2. For the Temozolomide (TMZ), sensitivity assay; when subcutaneous xenograft tumors reached around 50 mm3 in volume, TMZ (60 mg kg−1: MedChem Express. HY-17364/CS-0943) was intraperitoneally injected once per week for 6 weeks. All mice were sacrificed at the end of the experiment and tumors were harvested, pictured and weighed. Orthotopic glioma xenografts were established through intracranial implantation of GSC cells. Briefly, 5 × 105 GSC11 cells (in 3 μL of PBS) per mouse were transplanted into the left frontal lobes of each mouse through intracranial injection (1 mm anterior, 2 mm lateral to bregma and at a depth of 3 mm) using a Hamilton microsyringe (5 μL). Mice were maintained until the development of neurologic signs that significantly inhibited their quality of life (e.g., ataxia, lethargy, seizures, inability to feed). The FECD133 or/and TMZ were added in situ. Time of survival was recorded and the mouse brains were collected, fixed in 4% formaldehyde and embedded in paraffin after the mice were sacrificed. Brain sections were examined by H&E staining. For patient-derived xenografts formation (PDXs), tumor specimens were transplanted into nude mice. 8 weeks later, the xenografts termed as P1 (PDX passage 1) were cut out as isometric tumor specimens, and then transplanted into nude mice and treated with lercanidipine (5 mg kg−1) by tail vein injection. All clinicopathological information is provided in Table S1.
Mass spectrometry analysis
For sample preparation, sequencing-grade modified trypsin (Promega) was added to each sample (enzyme to protein ratio 1:25, w/w) and incubated at 37 °C for 18 h. 20 μg of the digested peptide mixture was desalted via StageTipC18 (3 M Empore, USA). All experiments were performed on a Q-Exactive mass spectrometer with an ancillary EASY-nLC 1000 HPLC system (Thermo Fisher Scientific). The tryptic digested peptides were loaded on a 75 μm × 200 mm fused silica column packed in-house with 3 μm ReproSil-Pur C18 beads (Dr. Maisch GmbH, Ammerbuch, Germany) and separated with a 90-min gradient at a flow rate of 300 nL/min. Solvent A contained 100% H2O and 0.1% formic acid; Solvent B contained 100% acetonitrile and 0.1% formic acid. The gradient was 2–4% B, 1 min; 4–30% B, 75 min; 30–45% B, 7 min; 45–90% B, 1 min, 90% B, 6 min. The mass spectrometry instrument parameters were: MS1 full scan resolution, 70000 at m/z 200; automatic gain control target, 3 × 106; maximum injection time, 120 ms. MS2 scan resolution 17,500 at m/z 200; automatic gain control target, 1 × 105; maximum injection time, 120 ms; isolation window, 2.0 m/z; dynamic exclusion, 30 s. The precursor ions were fragmented by higher energy collisional dissociation (HCD) with a normalized collision energy of 27%. The MS data was analyzed via software MaxQuant (http://maxquant.org/, version 1.6.5.0). Carbamidomethyl (C) was set as a fixed modification, while oxidation (M) and protein N-term acetylation as variable modifications. The GLT8D1 interacting proteins were shown in supplementary Table S2.
Purification and characterization of exosome
For exosome preparation, supernatants were collected from 48–72 h cell cultures, and indicated exosomes were purified through a standard centrifugation protocol. In brief, the supernatants were centrifuged at 2000 g for 20 min to remove cell debris. Supernatants were then centrifuged at 100,000 g for 2 h at 4 °C (Hitachi, Tokyo, Japan) . The exosome containing pellets were washed and re-suspended in PBS after centrifugation at 100,000 g for 2 h. The exosome morphology was identified by Transmission Electron Microscopy (Leica, German) using negative staining, the size distribution and concentration were determined by Flow NanoAnalyzer NanoFCM instrument (Xiamen, China).
Immunofluorescence staining
A tyramide signal amplification method was used in immunofluorescence staining for Paraffin-embedded glioma cancerous tissue samples and cells. Briefly, tissues or cells were incubated with 3% hydrogen peroxide solution for 20 min to quench the endogenous horseradish peroxidase (HRP) activity. Tissues or cells were then blocked with 10% normal goat serum for another 25 min and incubated with the indicated primary antibody overnight. Samples were washed 3 times with PBS plus 0.2% Tween-20, and incubated with the indicated HRP conjugated secondary antibody (1 μg mL−1) for 1 h at room temperature, following which they were washed 3 times with PBS plus 0.2% Tween-20 and incubated with tyramide working solution (Alexa Fluor tyramide, 1% 4-Iodophenol, 0.03% H2O2) for 10 min at room temperature to label the target proteins. Samples were further stained with other indicated proteins repeatedly using the above procedure.
Tumor sphere formation and in vitro extremely limiting dilution assays
Cells 3 × 104/well were plated in ultralow-attachment 6-well plates (Corning; 3471) and grown in serum-free DMEM/F12, supplemented with B27, 20 ng mL−1 EGF and 20 ng mL−1 bFGF, and 4 μg mL−1 heparin. The spheres were cultured for 14 days, and then pictured and counted. For the in vitro extremely limiting dilution assay, indicated cells at a density of 10, 20, 50, 100 cells well−1, with 3 replicates, were cultured in the above medium. 12 days after culture, the numbers of tumor spheres in each well were pictured and counted.
Design and production of the FECD133
The basic rationale for the FECD133 design lies in that the CD133N1~108 domain is sufficient for GLT8D1 binding, and therefore inhibiting CD133 degradation through the endosomal-lysosomal pathway. DNA fragment containing CD133N1~108 (or FECD133) was cloned into the bacterial expression plasmid pGEX-4T-1 with BamH I and EcoR I restriction sites, and GST fusion proteins were expressed in E. coli BL21 (DE3) by induction with 0. 3 mM isopropyl β-d-1-thiogalactopyranoside at 16˚C for 12 h. The E. coli were collected after centrifugation and re-suspended in PBS, subjected to ultrasonic decomposition followed by ultracentrifugation (15,000 g) for 1 h. The supernatant was collected for purifying GST fusion proteins using glutathione beads (GE, 17-0756-01) according to the manufacturer’s protocol. In brief, the purified proteins bound on the resin were eluted with elution buffer (50 mM Tris pH8.8, 1 mM EDTA, 5% glycerol, 0.01% Triton ×100, 50 mM NaCl, 5 mM DTT, 20 mM Glutamine) and dialysed in PBS supplemented with 10% glycerol. The purified GST or GST-FECD133 proteins were then stored for indicated assays.
GST pull-down assay
GST or GST-FECD133 proteins bound to glutathione resin were incubated with GSC11 cell total extracts at 4 °C, overnight. The resin bound proteins were then washed three times with ice-cold PBS. Specific GST and GST-FECD133 interacting proteins were then eluted with elution buffer (50 mM Tris pH8.8, 1 mM EDTA, 5% glycerol, 0.01% Triton × 100, 50 mM NaCl, 5 mM DTT, 20 mM Glutamine), respectively. The eluted solutions were then loaded to SDS-PAGE and detected by immunoblot.
Statistical analyses
Data are represented as the mean ± SEM, and error bars indicate SEM. P values were calculated by either unpaired or paired two-tailed Student’s t test, *P < 0.05, **P < 0.01, and ***P < 0.001. All analyses were performed using GraphPad Prism software (GraphPad Software, Inc.). All experiments were performed at least three independent times.
Data availability
The data are available to academic researchers from corresponding author upon reasonable request.
References
Stupp R, Tonn JC, Brada M, Pentheroudakis G, Group EGW. High-grade malignant glioma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol: Off J Eur Soc Med Oncol. 2010;21:v190–193.
Bocangel DB, Finkelstein S, Schold SC, Bhakat KK, Mitra S, Kokkinakis DM. Multifaceted resistance of gliomas to temozolomide. Clin Cancer Res: Off J Am Assoc Cancer Res. 2002;8:2725–34.
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164:550–63.
Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. 2015;372:2499–508.
Fine HA. Bevacizumab in glioblastoma-still much to learn. N Engl J Med. 2014;370:764–5.
Owonikoko TK, Arbiser J, Zelnak A, Shu HK, Shim H, Robin AM, et al. Current approaches to the treatment of metastatic brain tumours. Nat Rev Clin Oncol. 2014;11:203–22.
Varki A, Kannagi R, Toole B, Stanley P Glycosylation Changes in Cancer. In: rd, Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, et al. (eds). Essentials of Glycobiology: Cold Spring Harbor (NY), 2015, pp 597-609.
Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer. 2005;5:526–42.
Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16:1373–82.
Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra34.
Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer cell. 2009;15:501–13.
Seidel S, Garvalov BK, Wirta V, von Stechow L, Schanzer A, Meletis K, et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain: a J Neurol. 2010;133:983–95.
Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene. 2009;28:3949–59.
Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol cell Biol. 2004;5:343–54.
Maxwell PH, Pugh CW, Ratcliffe PJ. Activation of the HIF pathway in cancer. Curr Opin Genet Dev. 2001;11:293–9.
Ferrandina G, Petrillo M, Bonanno G, Scambia G. Targeting CD133 antigen in cancer. Expert Opin Ther targets. 2009;13:823–37.
Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–12.
Shi Y, Ping YF, Zhou W, He ZC, Chen C, Bian BS, et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat Commun. 2017;8:15080.
Liu YP, Zheng CC, Huang YN, He ML, Xu WW, Li B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. Med Comm. 2021;2:315–40.
Yang F, Xing Y, Li Y, Chen X, Jiang J, Ai Z, et al. Monoubiquitination of cancer stem cell marker CD133 at lysine 848 regulates its secretion and promotes cell migration. Mol Cell Biol. . 2018,38:e00024-18.
Yang CP, Li X, Wu Y, Shen Q, Zeng Y, Xiong Q, et al. Comprehensive integrative analyses identify GLT8D1 and CSNK2B as schizophrenia risk genes. Nat Commun. 2018;9:838.
Sasayama D, Hori H, Yamamoto N, Nakamura S, Teraishi T, Tatsumi M, et al. ITIH3 polymorphism may confer susceptibility to psychiatric disorders by altering the expression levels of GLT8D1. J Psychiatr Res. 2014;50:79–83.
Teh MT, Gemenetzidis E, Patel D, Tariq R, Nadir A, Bahta AW, et al. FOXM1 induces a global methylation signature that mimics the cancer epigenome in head and neck squamous cell carcinoma. PloS one. 2012;7:e34329.
Hwang S, Mahadevan S, Qadir F, Hutchison IL, Costea DE, Neppelberg E, et al. Identification of FOXM1-induced epigenetic markers for head and neck squamous cell carcinomas. Cancer. 2013;119:4249–58.
Chen R, Jiang X, Sun D, Han G, Wang F, Ye M, et al. Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J proteome Res. 2009;8:651–61.
Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. Spatio-temporal transcriptome of the human brain. Nature 2011;478:483–9.
Goswami CP, Nakshatri H. PROGgene: gene expression based survival analysis web application for multiple cancers. J Clin Bioinforma. 2013;3:22.
Bhat KPL, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer cell. 2013;24:331–46.
Zhou K, Yao YL, He ZC, Chen C, Zhang XN, Yang KD, et al. VDAC2 interacts with PFKP to regulate glucose metabolism and phenotypic reprogramming of glioma stem cells. Cell death dis. 2018;9:988.
Chen ZX, Wang HW, Wang S, Fan LG, Feng S, Cai XM, et al. USP9X deubiquitinates ALDH1A3 and maintains mesenchymal identity in glioblastoma stem cells. J Clin Investig. 2019;129:2043–55.
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–22.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401.
Man J, Yu X, Huang H, Zhou W, Xiang C, Huang H, et al. Hypoxic Induction of Vasorin Regulates Notch1 turnover to maintain glioma Stem-like Cells. cell stem cell. 2018;22:104–18 e106.
Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14:1878–91.
Joszai J, Thamm K, Karbanova J, Janich P, Fargeas CA, Huttner WB, et al. Prominins control ciliary length throughout the animal kingdom: New lessons from human prominin-1 and zebrafish prominin-3. J Biol Chem. 2020;295:6007–22.
Thamm K, Simaite D, Karbanova J, Bermudez V, Reichert D, Morgenstern A, et al. Prominin-1 (CD133) modulates the architecture and dynamics of microvilli. Traffic. 2019;20:39–60.
Gradilone SA, Pisarello MJL, LaRusso NF. Primary Cilia in Tumor Biology: the Primary Cilium as a therapeutic target in cholangiocarcinoma. Curr drug targets. 2017;18:958–63.
Yang Y, Roine N, Mäkelä TP. CCRK depletion inhibits glioblastoma cell proliferation in a cilium-dependent manner. EMBO Rep. 2013;14:741–7.
Fargeas CA, Huttner WB, Corbeil D. Nomenclature of prominin-1 (CD133) splice variants - an update. Tissue antigens. 2007;69:602–6.
Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M, et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010;70:719–29.
Meinnel T, Dian C, Giglione C. Myristoylation, an ancient protein modification mirroring eukaryogenesis and evolution. Trends Biochem Sci. 2020;45:619–32.
Liu Y, Ren S, Xie L, Cui C, Xing Y, Liu C, et al. Mutation of N-linked glycosylation at Asn548 in CD133 decreases its ability to promote hepatoma cell growth. Oncotarget. 2015;6:20650–60.
Mak AB, Nixon AM, Kittanakom S, Stewart JM, Chen GI, Curak J, et al. Regulation of CD133 by HDAC6 promotes beta-catenin signaling to suppress cancer cell differentiation. Cell Rep. 2012;2:951–63.
Mak AB, Blakely KM, Williams RA, Penttila PA, Shukalyuk AI, Osman KT, et al. CD133 protein N-glycosylation processing contributes to cell surface recognition of the primitive cell marker AC133 epitope. J Biol Chem. 2011;286:41046–56.
Karbanova J, Laco J, Marzesco AM, Janich P, Vobornikova M, Mokry J, et al. Human PROMININ-1 (CD133) Is Detected in Both Neoplastic and Non-Neoplastic Salivary Gland Diseases and Released into Saliva in a Ubiquitinated Form. PloS one. 2014;9;e98927.
Fonseca AV, Bauer N, Corbeil D. The stem cell marker CD133 meets the endosomal compartment-new insights into the cell division of hematopoietic stem cells. Blood Cells Mol Dis. 2008;41:194–5.
Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–35.
Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol cell Biol. 2004;5:121–32.
Hsu VW, Bai M, Li J. Getting active: protein sorting in endocytic recycling. Nat Rev Mol Cell Bio. 2012;13:1–6.
Marzesco AM, Janich P, Wilsch-Brauninger M, Dubreuil V, Langenfeld K, Corbeil D, et al. Release of extracellular membrane particles carrying the stem cell marker prominin-1 (CD133) from neural progenitors and other epithelial cells. J Cell Sci. 2005;118:2849–58.
Chao OS, Chang TC, Di Bella MA, Alessandro R, Anzanello F, Rappa G, et al. The HDAC6 Inhibitor tubacin Induces release of CD133(+) extracellular vesicles from cancer cells. J Cell Biochem. 2017;118:4414–24.
Bauer N, Wilsch-Brauninger M, Karbanova J, Fonseca AV, Strauss D, Freund D, et al. Haematopoietic stem cell differentiation promotes the release of prominin-1/CD133-containing membrane vesicles-a role of the endocytic-exocytic pathway. Embo Mol Med. 2011;3:398–409.
Wei YY, Jiang YZ, Zou F, Liu YC, Wang SS, Xu N, et al. Activation of PI3K/Akt pathway by CD133-p85 interaction promotes tumorigenic capacity of glioma stem cells. P Natl Acad Sci USA. 2013;110:6829–34.
Cooper-Knock J, Moll T, Ramesh T, Castelli L, Beer A, Robins H, et al. Mutations in the glycosyltransferase domain of GLT8D1 are associated with familial amyotrophic lateral sclerosis. Cell Rep. 2019;26:2298–306 e2295.
Mak AB, Nixon AML, Kittanakom S, Stewart JM, Chen GI, Curak J, et al. Regulation of CD133 by HDAC6 promotes beta-Catenin signaling to suppress cancer cell differentiation. Cell Rep. 2012;2:951–63.
Beier D, Rohrl S, Pillai DR, Schwarz S, Kunz-Schughart LA, Leukel P, et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008;68:5706–15.
Grassi G, Robles NR, Seravalle G, Fici F. Lercanidipine in the management of hypertension: an update. J Pharmacol Pharmacotherapeutics. 2017;8:155–65.
Robador PA, Jose GS, Rodriguez C, Guadall A, Moreno MU, Beaumont J, et al. HIF-1-mediated up-regulation of cardiotrophin-1 is involved in the survival response of cardiomyocytes to hypoxia. Cardiovasc Res. 2011;92:247–55.
Cancer Genome Atlas Research N. Comprehe`nsive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77.
Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207–14.
Grosse-Gehling P, Fargeas CA, Dittfeld C, Garbe Y, Alison MR, Corbeil D, et al. CD133 as a biomarker for putative cancer stem cells in solid tumours: limitations, problems and challenges. J Pathol. 2013;229:355–78.
Dubreuil V, Marzesco AM, Corbeil D, Huttner WB, Wilsch-Brauninger M. Midbody and primary cilium of neural progenitors release extracellular membrane particles enriched in the stem cell marker prominin-1. J Cell Biol. 2007;176:483–95.
Freund D, Bauer N, Boxberger S, Feldmann S, Streller U, Ehninger G, et al. Polarization of human hematopoietic progenitors during contact with multipotent mesenchymal stromal cells: Effects on proliferation and clonogenicity. Stem Cells Dev. 2006;15:815–29.
Corbeil D, Roper K, Fargeas CA, Joester A, Huttner WB. Prominin: a story of cholesterol, plasma membrane protrusions and human pathology. Traffic. 2001;2:82–91.
Roper K, Corbeil D, Huttner WB. Retention of prominin in microvilli reveals distinct cholesterol-based lipid micro-domains in the apical plasma membrane. Nat Cell Biol. 2000;2:582–92.
Karbanova J, Lorico A, Bornhauser M, Corbeil D, Fargeas CA. Prominin-1/CD133: lipid raft association, detergent resistance, and immunodetection. Stem Cell Transl Med. 2018;7:155–60.
Janich P, Corbeil D. GM(1) and GM(3) gangliosides highlight distinct lipid microdomains within the apical domain of epithelial cells. Febs Lett. 2007;581:1783–7.
Singer D, Thamm K, Zhuang H, Karbanova J, Gao Y, Walker JV, et al. Prominin-1 controls stem cell activation by orchestrating ciliary dynamics. Embo J. 2019;38:e99845.
Gurudev N, Florek M, Corbeil D, Knust E. Prominent role of prominin in the retina. Prominin-1 (Cd133): N Insights Stem Cancer Stem Cell Biol. 2013;777:55–71.
Zacchigna S, Oh H, Wilsch-Brauninger M, Missol-Kolka E, Jaszai J, Jansen S, et al. Loss of the cholesterol-binding protein prominin-1/cd133 causes disk dysmorphogenesis and photoreceptor degeneration. J Neurosci. 2009;29:2297–308.
Fargeas CA, Buttner E, Corbeil D. Commentary: “Prom1 function in development, intestinal inflammation, and intestinal tumorigenesis”. Front Oncol. 2015;5:91.
Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol. 2010;177:1491–502.
Lehnus KS, Donovan LK, Huang X, Zhao N, Warr TJ, Pilkington GJ, et al. CD133 glycosylation is enhanced by hypoxia in cultured glioma stem cells. Int J Oncol. 2013;42:1011–7.
Gaspar N, Marshall L, Perryman L, Bax DA, Little SE, Viana-Pereira M, et al. MGMT-independent temozolomide resistance in pediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer Res. 2010;70:9243–52.
Seravalle G, Brambilla G, Pizzalla DP, Casati A, Riva M, Cuspidi C, et al. Differential effects of enalapril-felodipine versus enalapril-lercanidipine combination drug treatment on sympathetic nerve traffic and metabolic profile in obesity-related hypertension. J Am Soc Hypertens. 2016;10:244–51.
McClellan KJ, Jarvis B. Lercanidipine - a review of its use in hypertension. Drugs. 2000;60:1123–40.
Shi Y, Fan S, Wu M, Zuo Z, Li X, Jiang L, et al. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun. 2019;10:4892.
Yang F, Zhang HF, Mei YD, Wu M. Reciprocal regulation of HIF-1 alpha and LincRNA-p21 Modulates the Warburg Effect. Mol cell. 2014;53:88–100.
Hu Y, Zhang M, Tian N, Li D, Wu F, Hu P, et al. The antibiotic clofoctol suppresses glioma stem cell proliferation by activating KLF13. J Clin Investig. 2019;129:3072–85.
Acknowledgements
We thank Drs. Xiuwu Bian and Yu Shi at Institute of Pathology and Southwest Cancer Centre, The Third Military Medical University, China, for providing us the human primary glioma stem cell lines: GBM1 and GBM2. We thank Dr. Hu Zhou and Jin Gao at Shanghai Institute of Materia Medica, Chinese Academy of Sciences for the Mass spectrometry analysis. We thank Dr. Nigel W. Fraser (Dept of Microbiology, Pereleman School of Medicine, university of Pennsylvania, USA), Dr. Jumin Zhou (Kunming Institute of Zoology, CAS) and Dr. Dangsheng Li (Deputy editor-in-chief of Cell Research) for their instructive comments on the manuscript writing. This study was supported by National Key Research and Development Program of China (2021YFF1000602), National Nature Science Foundation of China (U2102206, U1902216, 82173110, 82160512), Yunnan Applied Basic Research Projects (2019FJ009, 202001AS070037, 2019FB106, 2019FB111 and 2019HB076). C.P.Y was also supported by Youth Innovation Promotion Association, CAS; Yunnan Ten Thousand Talents Plan Young & Elite Talents Project. Y.B.C was supported by grant from the Strategic Priority Research Program of the Chinese Academy of Sciences XDPB17, and YJKYYQ20190048; Science & Technology Department of Sichuan Province Research Program (2020YFSY0009).
Author information
Authors and Affiliations
Contributions
YBC supervised and wrote the manuscript. KL designed and performed the biochemical functional analysis for GLT8D1 in vitro and xenograft tumor models in vivo. LPJ performed the tumor sphere, qRT-PCR, immunoblot, and PDXs analysis, YLS, BYL, YMH, QSS, XLJ, ZN, JP, and CPY performed the bioinformatics analysis, and provided clinical tumor samples.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Mouse care and treatment was approved by the Animal Care and Use Committee at the Kunming Institute of Zoology, Chinese Academy of Sciences. Human resected tissues were obtained from Kunming medical university, China, with informed consent.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M Piacentini
Supplementary information
Rights and permissions
About this article

Cite this article
Liu, K., Jiang, L., Shi, Y. et al. Hypoxia-induced GLT8D1 promotes glioma stem cell maintenance by inhibiting CD133 degradation through N-linked glycosylation. Cell Death Differ 29, 1834–1849 (2022). https://doi.org/10.1038/s41418-022-00969-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-022-00969-2
This article is cited by
-
Emerging roles of prominin-1 (CD133) in the dynamics of plasma membrane architecture and cell signaling pathways in health and disease
Cellular & Molecular Biology Letters (2024)
-
Functional Roles of CD133: More than Stemness Associated Factor Regulated by the Microenvironment
Stem Cell Reviews and Reports (2024)
-
Glycosyltransferase GLT8D1 and GLT8D2 serve as potential prognostic biomarkers correlated with Tumor Immunity in Gastric Cancer
BMC Medical Genomics (2023)
-
Alleviating hypoxia to improve cancer immunotherapy
Oncogene (2023)
-
USP7-mediated JUND suppresses RCAN2 transcription and elevates NFATC1 to enhance stem cell property in colorectal cancer
Cell Biology and Toxicology (2023)
