
Abstract
Non-small cell lung cancer (NSCLC) has the highest mortality rate among all malignancies worldwide. The role of long noncoding RNAs (lncRNAs) in the progression of cancers is a contemporary research hotspot. Based on an integrative analysis of The Cancer Genome Atlas database, we identified lncRNA-RNA Component of Mitochondrial RNA Processing Endoribonuclease (RMRP) as one of the most highly upregulated lncRNAs that are associated with poor survival in NSCLC. Furthermore, N(6)-methyladenosine (m6A) was highly enriched within RMRP and enhanced its RNA stability. In vitro and in vivo experiments showed that RMRP promoted NSCLC cell proliferation, invasion, and migration. In terms of mechanism, RMRP recruited YBX1 to the TGFBR1 promotor region, leading to upregulation of the transcription of TGFBR1. The TGFBR1/SMAD2/SMAD3 pathway was also regulated by RMRP. In addition, RMRP promoted the cancer stem cells properties and epithelial mesenchymal transition, which promote the resistance to radiation therapy and cisplatin. Clinical data further confirmed a positive correlation between RMRP and TGFBR1. In short, our work reveals that m6A RNA methylation-mediated RMRP stability renders proliferation and progression of NSCLC through regulating TGFBR1/SMAD2/SMAD3 pathway.
Similar content being viewed by others

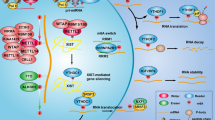

Introduction
Among malignant tumors, lung cancer has always been highly ranked in terms of incidence and mortality rate worldwide [1]. Lung cancer is histopathologically classified as non-small cell lung cancer (NSCLC) and small cell lung cancer. Approximately 85% of lung cancer patients are NSCLC. The advances in diagnostic and treatment modalities have helped improve the survival time of cancer patients; however, the 5-year survival rate of NSCLC is still 17.7% [2]. In addition, approximately 85% of all patients with NSCLC are diagnosed at an advanced stage [3]. Therefore, further research on the pathogenesis of NSCLC and identification of novel therapeutic targets and prognostic biomarkers is a key imperative to improve patient survival.
Long noncoding RNAs (lncRNAs) are RNA transcripts with lengths of more than 200 base pairs. Recently, lncRNAs and variations in epigenetic regulation were reported to be related to the incidence and progression of cancer [4]. In our previous studies, some lncRNAs were identified as potential prognostic biomarkers for cancer patients [5,6,7,8]. Furthermore, some lncRNAs also play an important role in the incidence and progression by influencing the expression of target genes. For example, lncRNA-CTS was shown to promote metastasis of cervical cancer by regulating the expression of ZEB2 [9]. LncRNA SATB2-AS1 was shown to inhibit metastasis by regulating SATB2 in colorectal cancer [10].
LncRNA-RNA Component of Mitochondrial RNA Processing Endoribonuclease (RMRP) was identified in the mitochondria of human tissues [11]. Subsequently, a study demonstrated the interaction between RMRP and telomerase reverse transcriptase [12]. Some recent studies have demonstrated upregulation of RMRP in cancerous tissues and its potential regulatory effect on tumorigenicity. RMRP promotes proliferation of bladder cancer cells by regulating miR-206 as a sponge [13]. RMRP was also shown to promote proliferation of NSCLC cells by regulating miR-1-3p or miR-206 as a sponge [14, 15]. However, further research is required to unravel the complex mechanism of RMRP.
Transforming Growth Factor Beta Receptor 1 (TGFBR1) is a receptor of Transforming Growth Factor Beta (TGFB) and modulates the cancer stem cell (CSC) properties and epithelial mesenchymal transition (EMT) [16]. The TGFB signaling played important role in tumorigenesis and progression. There are two types of cell-surface receptors in this pathway, namely, TGFBR1 and TGFBR2. After the binding of TGFB to its receptor, active TGFBR2 recruits and causes phosphorylation of TGFBR1, which can phosphorylate the transcription factors SMAD Family Member 2 (SMAD2) and SMAD Family Member 3 (SMAD3). Our previous work showed that TGFBR1 promotes EMT and cell cycle progression in NSCLC [17, 18]. However, there is a need to explore the upstream signaling pathways and the related mechanisms.
In the present study, based on bioinformatics analysis, firstly we found that RMRP may play an important role in NSCLC. Further research showed the m6A modification improved the stability of methylated RMRP transcripts by decreasing the RNA degradation rate. In addition, RMRP was found to promote proliferation, migration, and invasion. In terms of mechanism, RMRP promotes TGFBR1 transcription by recruiting YBX1 to TGFBR1 promoter. The TGFBR1/SMAD2/SMAD3 pathway was regulated by RMRP. Moreover, RMRP promoted the CSC properties and EMT which promoted the resistance to radiation therapy and cisplatin. Clinical data further confirmed the positive correlation between RMRP and TGFBR1. Hence, our work showed the clinical significance and regulatory mechanism of lncRNA RMRP in NSCLC, which may serve as a promising therapeutic target and as a biomarker to predict prognoses of NSCLC patients.
Results
Integrated analysis of NSCLC reveals that RMRP may be a biomarker
We performed an integrated analysis of the Cancer Genome Atlas (TCGA) lung adenocarcinoma (LUAD) database (535 NSCLC and 59 normal samples). RMRP was upregulated in the NSCLC tissues than in the normal tissues (Fig. 1A). High RMRP expression was associated with late T stages and worse survival rates (Fig. 1B and C). In addition, RMRP was upregulated in NSCLC tissues (n = 80) compared to normal lung tissues (n = 30) in GSE43458 [19], as shown in the Gene Expression Omnibus database (Fig. 1D) [20].

A. RMRP was significantly upregulated in NSCLC tissues (n = 535) as compared to normal lung tissues (n = 59) (Wilcoxon signed-rank test). B. RMRP was associated with tumor size in NSCLC patients (n = 335; Wilcoxon signed-rank test). C High RMRP expression was associated with worse survival rates by Kaplan–Meier method (n = 504). D RMRP was upregulated in NSCLC tissues (n = 80) compared to normal lung tissues (n = 30) in GSE43458, as shown in the Gene Expression Omnibus (GEO) database, which was analyzed by the online tool Lung Cancer Explorer (https://lce.biohpc.swmed.edu/lungcancer/). E Gene Ontology (GO) results showed that RMRP functions in protein heterodimerization activity, chromatin DNA binding, bitter taste receptor activity, Rab GTPase binding, histone binding, ubiquitin-like protein transferase activity, taste receptor activity, ubiquitin-protein transferase activity, Rab guanyl-nucleotide exchange factor activity, histone deacetylase binding, nucleosomal DNA binding, modification-dependent protein binding, transcription coactivator activity, nucleosome binding, and ubiquitin-like protein binding. F The KEGG results showed that RMRP was enriched in pathways of Alcoholism, Systemic lupus erythematosus, viral carcinogenesis, transcriptional misregulation in cancer, Necroptosis, Mitophagy—animal and Ubiquitin mediated proteolysis. G The GSEA research showed RMRP was enriched in the cell cycle, colorectal cancer, protein export, TGFB pathway, and ubiquitin-mediated proteolysis pathway. H The protein-coding potential of RMRP through the CPAT database. I The protein-coding potential of RMRP through the CPC database. LncRNA-MALAT1, LncRNA-HOTAIR, CCND1, and MYC were also explored as control.
Co-expression research is a way to explore the potential function of genes. We explored the mRNAs that are co-expressed with RMRP in the TCGA database (cor > 0.4, p < 0.01). The GO results showed that RMRP functions in protein heterodimerization activity, chromatin DNA binding, bitter taste receptor activity, etc. (Fig. 1E). The KEGG results showed that RMRP was enriched in pathways of alcoholism, systemic lupus erythematosus, viral carcinogenesis, transcriptional misregulation, etc. (Fig. 1F).
Then, Gene Set Enrichment Analysis (GSEA) of KEGG research was also conducted. RMRP was enriched in the cell cycle, colorectal cancer, protein export, TGFB pathway, and ubiquitin-mediated proteolysis pathways (Fig. 1G). These pathways were closely related to tumorigenesis and metastases, which suggested that RMRP may play a vital role in NSCLC. The protein-coding potential of RMRP was also explored which suggested that RMRP has no protein-coding potential (Fig. 1H and I).
m6A modification is enriched in RMRP and improves its transcript stability
Recent preliminary researches have reported that m6A modification is widespread and regulates the transcriptome to influence the splice of RNA, translation, export, localization, and stability. To explore the m6A modification of RMRP, we firstly used the online bioinformatics tool m6Avar (http://m6avar.renlab.org/) to predict the m6A sites located in RMRP, and identified two RMRP m6A sequence motifs (Supplementary Fig. S1).
Then, the methylated RNA immunoprecipitation (Me-RIP) assays in human lung epithelial cells (HBE) and two NSCLC cell lines (A549 and H1299) were performed. The m6A methylation level of RMRP in HBE cells was lower than that in the NSCLC cells (A549 and H1299) as revealed by MeRIP-qPCR assays (Fig. 2A). Furthermore, A549 and H1299 cell lines expressed higher levels of m6A than HBE (Supplementary Fig. S2). We then used small interfering RNA to target METTL3, a core component of the m6A methylase complex, and found the m6A levels in both the total RNA and RMRP RNA was decreased in A549 cells (Fig. 2B, C, D and Supplementary Fig. S3). The loss of RMRP RNA was then measured after the treatment of actinomycin D (5 μg/mL) to block new RNA synthesis in A549 cells. The results revealed that RMRP exhibited lower RNA stability after the downregulation of METTL3 (Fig. 2E).

A The m6A methylation level of RMRP in human normal lung epithelial cells (HBE) and NSCLC cells (H1299 and A549) were determined by MeRIP-qPCR assays. The input RNA fraction Ct value was used to account for RNA sample preparation differences; negative control groups (IgG) were used to adjust background fraction (n = 3). B Results of Western blot assay showing METTL3 protein expression in A549 cells transfected with two different short interference RNAs for METTL3 (si-METTL3-1 and si-METTL3-2) or negative controls (si-Control) (n = 3). C Results of qRT-PCR assays showing the mRNA levels of METTL3 and RMRP in the treated A549 cells (n = 3). β-actin was used as the reference for normalization. D The m6A level in the treated A549 cells (n = 3). E Reduction of RMRP RNA stability in METTL3-knockdown A549 cells as compared to control. Cells were treated with 5 μg/mL actinomycin D and RNA was isolated at 0 h, 2 h, and 4 h (n = 3). Data presented as mean ± SD from at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Furthermore, the expression of miR-122 or miR-206 was increased after the downregulation of METTL3, which could be rescued following RMRP overexpression in A549 cells. While METTL3 overexpression decreased the expression of miR-122 or miR-206 in H1299 cells (Supplementary Fig. S4). The above findings indicate the possibility that the m6A level of RMRP was upregulated in NSCLC cells compared to lung epithelial cells, and that m6A modification of RMRP improved its transcript stability, which may partially explain the significant upregulation of RMRP in NSCLC.
RMRP promotes NSCLC cell proliferation
To further explore the role of RMRP, we detected RMRP expression in NSCLC cells using quantitative RT-PCR analysis. RMRP expression in A549 cell lines was significantly higher than that in H1299 cells (Fig. 3A). Hence, we transfected A549 cells with two different shRNAs against RMRP (shRMRP-1 and shRMRP-2) or control scrambled shRNA (Scrambled) and transfected H1299 cells with an RMRP-overexpressing plasmid (RMRP) or matched Controls (Ctrl). The transfection efficiency was confirmed by quantitative RT-PCR analysis (Fig. 3B and C). Then CCK-8 and colony formation assays revealed significant inhibition of the growth and proliferation of NSCLC cells transfected with shRMRP compared with those transfected with scrambled, whereas the opposite effects were observed following transfection of RMRP-overexpressing plasmid (Fig. 3D–G, Supplementary Fig. S5, P < 0.05 for all).

A Results of quantitative RT-PCR (qRT-PCR) analysis showing RMRP expression in NSCLC cell lines (H1299, H460, H1915, A549) and human lung epithelial cell line (HBE). β-actin was used as the endogenous control (n = 3). B Results of qRT-PCR analysis showing RMRP expression in A549 cells transfected with two different shRNAs against RMRP (shRMRP-1 and shRMRP-2) or scrambled control (Scrambled). β-actin was used as the endogenous control (n = 3). C Results of qRT-PCR analysis showing RMRP expression in H1299 cells transfected with RMRP-overexpressing plasmid (RMRP) or matched Controls (Ctrl). β-actin was used as the endogenous control (n = 3). D CCK-8 assays of A549 cells transfected with shRMRP compared with those transfected with Scrambled (n = 3). E CCK-8 assays of H1299 cells transfected with RMRP or Ctrl (n = 3). F Representative images (Up) and quantification (Down) of colony formation assays in A549 cells transfected with shRMRP or Scrambled (n = 3). G Representative images (Up) and quantification (Down) of colony formation assays in H1299 cells transfected with RMRP or Ctrl (n = 3). Data presented as mean ± SD from at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
TGFBR1 is a critical target by which RMRP promotes proliferation, invasion, and migration
In our previous study, we found that TGFBR1 was associated with proliferation and metastasis in NSCLC, and the GSEA results showed that RMRP may participate in the TGFB pathway [17]. The TCGA database showed a positive correlation between the expressions of TGFBR1 and RMRP (Fig. 4A). Hence, we hypothesized that RMRP may target TGFBR1. In addition, the expression of TGFBR1 was altered after the knockdown or overexpression of RMRP in A549 or H1299 cells (Fig. 4B, C, and D), respectively. Furthermore, the expression of TGFBR1 was decreased, and BAX/Bcl-2 was increased after knockdown of RMRP in A549 cells, whereas the opposite effects were observed following TGFBR1 overexpression. In H1299 cells, the expression of TGFBR1 was increased, and that of BAX/Bcl-2 was decreased after RMRP overexpression, whereas the opposite effects were observed following TGFBR1 knockdown (Fig. 4D).

A The TCGA database of lung adenocarcinoma showed a positive correlation between the expressions of TGFBR1 and RMRP (n = 594). B Results of qRT-PCR analysis showing the expression of TGFBR1 mRNA in A549 cells after being transfected with shRMRP or TGFBR1-overexpressing plasmid (shRMRP + TGFBR1) as well as their respective negative controls. C Results of qRT-PCR analysis showing the expression of TGFBR1 mRNA in H1299 cells after being transfected with RMRP or short interference RNAs for TGFBR1 (RMRP + SiTGFBR1) as well as their respective negative controls. D Western blot analysis of the treated A549 and H1299 cells using the indicated antibodies (n = 3). E CCK-8 assays of the treated A549 cells (n = 3). F CCK-8 assays of the treated H1299 cells (n = 3). G Representative images (Left) and quantification (Right) of the wound-healing assays of the treated A549 cells (n = 3, Scale bar, 200 μm). H Representative images (Left) and quantification (Right) of colony formation assay of the treated A549 cells (n = 3). I Representative images (Left) and quantification (Right) of the wound-healing assays in the treated H1299 cells (n = 3). J Representative images (Left) and quantification (Right) of colony formation assay of the treated H1299 cells (n = 3). K Representative images (Left) and quantification (Right) of the transwell assays including invasion and migration of the treated A549 cells (n = 3). L The cell cycle assays of the treated A549 cells (n = 3). M The cell cycle assays of the treated H1299 cells (n = 3). N Representative images (Left) and quantification (Right) of the transwell assays including invasion and migration of the treated H1299 cells (n = 3, Scale bar, 200 μm). Data presented as mean ± SD from at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
In addition, we explored the involvement of TGFBR1 in the mechanism by which RMRP promotes NSCLC growth and proliferation. The results showed that the inhibition of growth and proliferation of NSCLC cells by shRMRP was rescued following TGFBR1 overexpression, whereas the opposite effects were found after TGFBR1 knockdown (Fig. 4E, F, H, and J).
TGFBR1 promotes the invasion, migration, and G1 cell cycle progression in NSCLC cells. Hence, we explored the effect of RMRP on invasion, migration, and G1 cell cycle progression. The wound-healing, transwell, and cell cycle assays suggested significant inhibition of the invasion, migration, and G1 cell cycle progression of NSCLC cells transfected with shRMRP compared with controls, whereas the opposite effects were observed in RMRP-overexpressing cells. Furthermore, inhibition of the invasion, migration, and G1 cell cycle progression of NSCLC cells by shRMRP was rescued following TGFBR1 overexpression, whereas opposite effects were observed following TGFBR1 knockdown (Fig. 4G, I, K, L, M, and N). In short, the above results revealed that TGFBR1 may be a critical target by which RMRP promotes proliferation, invasion, and migration.
RMRP is associated with the transcriptional factor YBX1
To further elucidate the mechanisms of RMRP, fractionated nuclear and cytoplasmic RNA, and fluorescence in situ hybridization (FISH) assays were conducted in NSCLC cells. The results showed that RMRP was mainly located in the cytosol (Fig. 5A and B). Several recent reports have found that many lncRNAs function by interacting with DNA or proteins. To explore the regulatory role of RMRP, we used catRAPID. The results showed that YBX1, which plays an important role in NSCLC, was a potential binding protein of RMRP (Fig. 5C). To further explore the interaction, the RIP assay showed that RMRP was significantly enriched by the anti-YBX1 antibody (Fig. 5D and E). RNA pull-down assays further confirmed the interaction between RMRP and YBX1 (Fig. 5F). In all, these above results suggested a close interaction between YBX1 and RMRP.

A qRT-PCR assays of the relative expression levels after the fractionation of nuclear and cytoplasmic RNA of H1299 and A549 cells. GAPDH was used as a cytoplasmic marker, and U1 was used as a nuclear marker (n = 3). B Representative RNA-FISH images showing the subcellular location of RMRP in H1299 and A549 cells. 18S and U6 were used as cytoplasmic and nuclear markers, respectively (Scale bar, 100 μm) (n = 3). C The analysis of RNA interaction profile showed the putative binding sequence of RMRP to YBX1 protein by catRAPID. D, E RNA immune precipitation (RIP) experiment was performed using the YBX1 and IgG antibodies to probe A549 and H1299 cells extracts, and the levels of the co-precipitated RNAs were determined using qRT-PCR (n = 3). F Total protein was extracted from A549 and H1299 cells and utilized in biotinylated RNA pull-down assay. Proteins that were associated with RMRP were explored by Western blot with anti-YBX1 antibody (n = 3). Data presented as mean ± SD from at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
RMRP promotes TGFBR1 transcription by recruiting YBX1 to TGFBR1 promoter
We explored the ATAC-seq data of TCGA-LUAD to identify the possible transcriptional factor of TGFBR1 (Supplementary Fig. S6). This analysis revealed a potential binding sequence for YBX1 in the promoter region of the TGFBR1 gene. The expression of YBX1 showed a positive correlation with TGFBR1 but not with RMRP in the TCGA-LUAD database by GEPIA (Fig. 6A–C). Hence, we presumed that YBX1 functions as a transcription factor that may be recruited to the TGFBR1 promoter by RMRP.

A The expression of YBX1 was not correlated with RMRP in TCGA-LUAD database by GEPIA (http://gepia.cancer-pku.cn/) using Pearson Correlation Coefficient. B The expression of YBX1 showed a positive correlation with TGFBR1 in TCGA-Lung adenocarcinoma (LUAD) database by GEPIA using Pearson Correlation Coefficient. C The expression of YBX1 showed a positive correlation with TGFBR1 in TCGA-Kidney Renal Clear Cell Carcinoma (KIRC) database by GEPIA using Pearson Correlation Coefficient. D Results of Western blot assays: knockdown of YBX1 inhibited the expression of TGFBR1, while overexpression of YBX1 increased the expression of TGFBR1 (n = 3). E, F YBX1-bound complex showed remarkable enrichment of the TGFBR1 promoter by ChIP and quantitative RT-PCR analysis in A549 and H1299 cells. IgG was used as a negative control. Enrichment was quantified relative to input controls (n = 3). G, H Luciferase activity assays were performed in A549 and H1299 cells transfected with wide type (TGFBR1-WT) or mutant-type (TGFBR1-MUT) TGFBR1 promoter-containing pGL3 reporter vector. These cells were further transfected with YBX1 (YBX1) or control (Ctrl). After 48 h, firefly luciferase activity was detected and normalized to Renilla luciferase activity (n = 3). I, J ChIP and quantitative RT-PCR analysis in the treated A549 and H1299 cells. IgG was used as a negative control. Enrichment was quantified relative to input controls (n = 3). K, L Luciferase activity assays in the treated A549 and H1299 cells were further transfected with TGFBR1-MUT or TGFBR1-WT. After 48 h, firefly luciferase activity was detected and normalized to Renilla luciferase activity, respectively (n = 3). Data presented as mean ± SD from three independent experiments. Student’s t test was used for statistical analysis: *P < 0.05; **P < 0.01; ***P < 0.001.
To further investigate the mechanism, siRNA (SiYBX1), pcDNA-YBX1 (YBX1), and their matched controls (SiNC or Ctrl) were transfected into the cells, respectively. Knockdown of YBX1 inhibited the expression of TGFBR1, while overexpression of YBX1 increased the expression of TGFBR1 (Fig. 6D, Supplementary Fig. S7). A ChIP assay and Luciferase reporter assay were also performed (Supplementary Fig. S8). The YBX1-bound complex showed remarkable enrichment of the TGFBR1 promoter (Fig. 6E and F). And YBX1 overexpression increased luciferase activity in cells expressing the wide-type TGFBR1 (TGFBR1-WT) but not in cells expressing the mutant TGFBR1 (TGFBR1-MUT) (Fig. 6G and H). Knockdown of RMRP decreased the enrichment of YBX1 on the TGFBR1 promoter, and this effect was reversed by co-transfection of YBX1 (Fig. 6I). In addition, the binding was increased by RMRP (Fig. 6J). Luciferase assay further revealed that the change in the luciferase activity was due to the knockdown or overexpression of RMRP (Fig. 6K and L).
In addition, the pull-down and RIP assays revealed that METTL3 interacted with RMRP (Supplementary Fig. S9A and B). Knockdown of METTL3 decreased the expression of TGFBR1, which was rescued following RMRP overexpression (Supplementary Fig. S9C). Overexpression of METTL3 augmented the enrichment of YBX1 on the TGFBR1 promoter, and this effect was reversed by treatment with siRMRP by ChIP assay (Supplementary Fig. S9D, E). Furthermore, after transfection with vector control or METTL3 for 24 h, interactions between RMRP and YBX1 in A549 or H1299 cells were investigated by RIP-qPCR using an antibody against YBX1. METTL3 overexpression increased the binding between RMRP and YBX1 in both A549 and H1299 cells (Supplementary Fig. S9F and G), which may account for m6A modification of RMRP improved its transcript stability.
RMRP regulated the TGFBR1/SMAD2/ SMAD3 pathway in NSCLC
TGFBR1/SMAD2/SMAD3 pathway plays an important role in NSCLC. Hence, we performed SMAD-binding element (SBE) luciferase reporter gene activity assays and p-SMAD2/3 immunofluorescence assays to investigate the activation of the TGFB pathway by RMRP. The results revealed that TGFBR1 rescued TGFB pathway activation in NSCLC cells with RMRP knockdown (Supplementary Fig. S10). Western blot and immunofluorescence assays revealed that RMRP knockdown dramatically inhibited the nuclear translocation and expression of p-SMAD2/3 in NSCLC cells (Fig. 7A and B). To further explore whether RMRP can affect TGFB signaling in a different way other than regulation of TGFBR1 transcription, we treated NSCLC cells with TGFBR1 knockdown (siTGFBR1) or siRNA negative controls (siNC). Western blot results showed that alteration of RMRP had no effect on the expression levels of p-SMAD2 and p-SMAD3 in the siTGFBR1 group, which indicates that TGFBR1 was indispensable for the activation of SMAD2/SMAD3 signaling pathway by RMRP (Fig. 7C).

A A549/Scrambled, A549/shRMRP, H1299/Ctrl, and H1299/RMRP cells under the TGFB treatment were collected and subjected to Western blot analysis using the indicated antibodies. The TGFB treatment was stimulated with 5 ng/mL TGFB for 1 h (n = 3). B Representative immunofluorescence images of anti-p-SMAD2/3 (green) and DAPI (blue) staining of the treated NSCLC cells. The cells were also cultured under the TGFB treatment and were collected for immunofluorescence analysis (n = 3) (Scale bar, 5 μm). C A549/Scrambled, A549/shRMRP, H1299/Ctrl, and H1299/RMRP cells were treated with TGFBR1 knockdown (siTGFBR1) or siRNA negative controls (siNC). The samples under the TGFB treatment were collected and subjected to Western blot analysis using the indicated antibodies (n = 3). D Related cells were collected and analyzed by qRT-PCR (n = 3). β-actin was used as the reference for normalization. E The number of tumorspheres was counted, and the morphology was observed under a light microscope. Upper panel: Representative pictures of tumorspheres. Lower panel: Tumorspheres quantification results including the number and size of spheres. (Scale bar, 100 μm). F The treated A549 and H1299 cells were subjected to irradiation (6 Gy). Subsequently, immunofluorescence analysis was performed after 1 h (n = 3, Scale bar, 5 μm). G Results of CCK-8 assays showing cell viability after incubation with different concentrations of cisplatin for 24 h. *P < 0.05; **P < 0.01; ***P < 0.001. Data represent three independent experiments.
TGFB pathway is often associated with CSC properties and EMT. Hence, we explored whether RMRP promotes the CSC properties in NSCLC. We explored the expression of CSC-related genes (KLF4, SOX2, NANOG, CD133, and ALDH) by qRT-PCR, and found that the expressions of these genes were decreased after RMRP knockdown in A549 cells. In contrast, these genes were upregulated after overexpression of RMRP in H1299 cells (Fig. 7D). In addition, we further explored the EMT-related genes in these cells. The results revealed that RMRP knockdown increased the expressions of E-Cadherin, and decreased the expressions of N-Cadherin and Vimentin (Fig. 7A). Moreover, we also explored the sphere-forming capacity in these cells. The assays showed that RMRP knockdown decreased the sphere-forming capacity, while overexpression of RMRP increased this capacity (Fig. 7E).
Consistent with a pro-CSC effect, RMRP inhibition increased the sensitivity of A549 cells to cisplatinum and radiation therapy (Fig. 7F and G). These data suggest that RMRP facilitates NSCLC progression by potentiating CSC self-renewal and EMT.
RMRP in NSCLC tissues is positively correlated with TGFBR1 expression
We further explored the correlation between RMRP and TGFBR1 in a total of 81 NSCLC patients who had undergone surgery at our clinical center. The results revealed that RMRP and TNM stage were independent prognostic factors in univariate and multivariate analyses (Supplementary Fig. S11A, B). Kaplan–Meier analysis showed that patients with high RMRP expression had worse overall survival and disease-free survival (Supplementary Fig. S11C, D). Further research revealed a positive relation of RMRP with tumor size, lymphatic invasion, and worse TNM stage (Supplementary Fig. S11E–G). In addition, correlation analysis revealed a positive correlation (R = 0.445, n = 181) between RMRP and TGFBR1 expression (Supplementary Fig. S11H).
Knockdown of RMRP limited tumor growth in vivo
To further confirm that RMRP promotes tumor growth in vivo, A549-shRMRP cells and A549-Scrambled cells were injected into nude mice. After 30 days, the tumors in the shRMRP group were significantly smaller (Fig. 8A and B). In addition, the tumor volumes and weights were significantly lower in the shRMRP group (Fig. 8C, D). Furthermore, the expressions of Ki-67, MMP9, and TGFBR1 were higher in the A549-Scrambled group (Fig. 8E, Supplementary Fig. S12).

A A549 cells with stable expression of shRMRP (A549/shRMRP) or scrambled control (A549/Scrambled) were transplanted into the axilla of nude mice to construct tumor growth model. Representative images of nude mice and tumors. B A549/Scrambled and A549/shRMRP group samples were collected and analyzed by qRT-PCR (n = 3). C Tumor volumes in the A549/Scrambled and A549/shRMRP groups. D Tumor weight in the A549/Scrambled and A549/shRMRP groups. E Representative images of haematoxylin and eosin (H&E) staining, Ki-67, MMP9, and TGFBR1 expression by immunohistochemical analysis in xenograft tumors derived from A549/Scrambled and A549/shRMRP groups. F Flowchart of our experiments. Our study suggests the m6A mark improved the stability of methylated RMRP transcripts by decreasing the RNA degradation rate, which may partially account for the upregulation of RMRP in NSCLC. Furthermore, RMRP promotes TGFBR1 transcription by recruiting YBX1 to TGFBR1 promoter.
Discussion
The role of lncRNAs in cancer is a contemporary research hotspot [21]. For example, lncRNA MIR100HG was shown to mediate cetuximab resistance by Wnt/β-catenin signaling [22]. LncRNA AFAP1-AS1 was shown to regulate the proliferation of NSCLC cells by repressing p21 [23]. However, the role of lncRNAs in NSCLC is not well characterized. In the present study, bioinformatics analysis revealed significant upregulation of RMRP in NSCLC tissues compared to normal tissues; in addition, upregulated RMRP in NSCLC patients was associated with late T stage and worse overall survival.
RMRP, which is located on chr9, was known in cartilage hair hypoplasia. Recently, RMRP was reported to be an oncogene in the context of many cancers. In breast cancer, mutations in RMRP affect protein binding to their promoters and alter expression [24]. In gastric cancer, RMRP promotes carcinogenesis by sponging miR-206 [25]. In NSCLC, RMRP acts as an oncogene [15]. However, the detailed specific mechanisms have not been explored. In our current research, we found that RMRP promotes proliferation and invasion both in vitro and in vivo. In addition, RMRP was an independent prognostic factor of NSCLC patients. Hence, our work indicates that RMRP is a promoter of NSCLC progression.
Our previous research described TGFBR1 as a crucial inducer of the progression of NSCLC [17, 18]. The TGFBR1 inhibitor SD208 effectively reduced bone metastases of prostate cancer [26]. MiR-140-5p was shown to suppress tumor growth and metastases of hepatocellular carcinoma by targeting TGFBR1 [27]. The above research revealed that TGFBR1 is a key factor in the progression of NSCLC. Recent studies have found a regulatory effect of lncRNAs on the TGFBR1 expression. Linc00462/miR-665 was found to regulate TGFBR1/2 to promote invasiveness of pancreatic cancer [28]. Here, we found that RMRP promotes the expression of TGFBR1. In terms of mechanisms, TGFBR1 is transcriptionally regulated by RMRP by recruiting the transcriptional factor YBX1 to the TGFBR1 promotor. Furthermore, RMRP and TGFBR1 showed a significantly positive correlation.
RMRP is a component of the nuclear RNase MRP complex, which participates in the processing of ribosomal RNA to generate the short mature 5.8S rRNA and cleaves B-cyclin (CLB2) mRNA in yeast, lowering B-cyclin levels during mitosis [29, 30]. In our study, we found that RMRP was mainly located in the cytosol. Our data are in accordance with several studies that have shown RMRP acts as competing endogenous RNAs (ceRNAs) or natural microRNA sponges, which are mainly located in the cytoplasm [14, 15, 31]. In general, the location of RMRP could be regulated by many signal pathways. For example, a previous study demonstrated export of Human antigen R (HuR) and GRSF1 bound RMRP from the nucleus to the cytosol [32]. Moreover, there are many other factors except HuR and GRSF1 that influence the location of RMRP which merit further work.
Furthermore, pull-down and RIP assays further proved the interaction between YBX1 and RMRP. YBX1 is a member of the RNA-binding proteins. In sarcoma, YBX1 promotes the translation of HIF1A, which promotes metastases [33]. In gastric cancer, lncRNA HOXC-AS3 mediates tumorigenesis by binding to YBX1 [34]. The exact roles of YBX1 in NSCLC are not well characterized. Our study demonstrated that RMRP recruits YBX1 to the TGFBR1 promoter and, consequently, upregulates TGFBR1 transcription.
TGFB/SMAD2/SMAD3 pathway is often associated with CSCs and EMT, which promotes resistance to radiation therapy and chemotherapy. Hence, we explored the association between RMRP and CSC properties. The SBE luciferase reporter gene activity assays and p-SMAD2/3 immunofluorescence assays showed that RMRP regulated the TGFBR1/SMAD2/SMAD3 pathway. In addition, RMRP promoted the sphere-forming capacity, EMT, and resistance to radiation therapy and chemotherapy.
In addition, many types of research showed RMRP was associated with T cell. In rheumatoid arthritis patients, RMRP is significantly high expression in T cells [35]. In response to infection, the DDX5-RMRP complex is recruited to RORγt-occupied DNA to drive the expression of genes that promote host defense and autoimmunity in Th17 cells [36]. And RMRP mutations have a drastic effect on lymphocyte maturation and function, and that they interfere with thymic generation of T lymphocytes [37]. In addition, TGFB pathway plays an important role in Vasculature and Cancer-associated fibroblasts [38]. As well known, T cell, Vasculature, and Cancer-associated fibroblasts are important components of tumor microenvironment [39, 40]. And recently work showed some lncRNAs play key role in tumor microenvironment such as lncRNA CamK-A or lncRNA H19 [41,42,43]. Hence, there is a possibility that RMRP may play an important role in changing the microenvironment of NSCLC which merits further work.
The m6A methylation in mRNA controls gene expression by readers, writer-complex components, and erasers. Recently, writer methyltransferases have been observed to participate in tumor progression by mediating the m6A modification of RNA. For example, METTL3 was shown to contribute to the progression and development of acute myeloid leukemia by increasing m6A [44]. METTL14 suppresses the metastases of hepatocellular carcinoma by modulating m6A [45]. Sun et al found that the m6A modification led to upregulation of lncRNA FAM225A by improving its stability [46]. In our study, we found that modification of m6A led to upregulation of RMRP, which was similar to the results of Sun’s study.
Overall, the current study suggests that the m6A modification promoted the lncRNA-RMRP/TGFBR1/SMAD2/SMAD3 pathway. The underlying mechanism involves transcriptional regulation of TGFBR1 by RMRP by recruiting the transcriptional factor YBX1 to the TGFBR1 promotor. Furthermore, RMRP increased the sphere-forming capacity and EMT, which are related to the resistance to radiation therapy and chemotherapy. In addition, our research suggests that RMRP is a biological marker with potential prognostic and therapeutic relevance in the context of NSCLC.
Materials and methods
Biological information analysis
Level 3 expression profiles of genes were downloaded from the TCGA LUAD Data Portal (https://tcga-data.nci.nih.gov/tcga/; 535 NSCLC and 59 normal samples) and normalized [47]. The R software (version 4.0) was used for data processing. Patients with NSCLC for whom complete clinical information was not available were excluded from our current work. In total, 335 NSCLC patients were enrolled. Overall survival analysis was performed using the Kaplan–Meier method. The clusterProfiler package was used to explore the GO and KEGG of RMRP. GSEA was used to explore significantly different genes according to the expression of RMRP. The permutations were set at 1000. The value of P and normalized enrichment score (NES) were used to evaluate the enrichment KEGG pathways.
The related cells
The normal lung epithelial cell line HBE and the human lung cancer cell lines H1299, H460, H1915, and A549 were used. The above cell lines were obtained from the Institute of Cell Biology of the Chinese Academy of Sciences (Shanghai, China). The cell lines were authenticated regularly by short tandem repeat DNA profiling and did not have any mycoplasma contamination. The culture environment was RPMI-1640 (Gibco, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS; 0500, ScienCell, USA) and 1% penicillin–streptomycin (Beyotime, Shanghai, China). The incubation parameters were 37 °C and 5% CO2.
Transfection
Stable knockdown of RMRP (ShRMRP) or control scrambled shRNA (Scrambled) was accomplished by lentiviral constructs in the presence of polybrene (107689, Sigma) containing two different RMRP shRNAs or no targeting shRNA (Genechem, Shanghai, China). The transduced cells were cultured in puromycin (Calbiochem, USA) for 2 weeks and validated by qRT-PCR.
Short interference RNAs for human METTL3 (siMETTL3), TGFBR1 (siTGFBR1), YBX1 (siYBX1), and matched negative controls (siNC) were provided by Genechem (Shanghai, China). The transfections were performed with INTERFERin® (Polyplus-transfection® SA) according to the manufacturer’s instructions. The target sequences used for the shRNAs or siRNAs are listed in Tables S1 and S2. This method is identical to that described in our previous work [47].
Overexpression of RMRP, YBX1, TGFBR1, and matched Controls (Ctrl) were cloned into the pcDNA3.1 vector (Genechem, China). The transfections were performed with jetPRIME® (Polyplus-transfection® SA) according to the manufacturer’s instructions.
Quantitative real time-PCR (qRT-PCR)
The E.Z.N.A.® Total RNA Kit I (Omega Bio-Tek, USA) was used to obtain RNA from cultured cells lysates. The RNA samples were then reverse transcribed into cDNA using a cDNA Synthesis Kit (Roche, USA). Polymerase chain reaction (PCR) was performed using the ABI 7500 FAST Real-Time PCR system. The expression levels of mRNAs or lncRNAs were determined by the cycle threshold (Ct) method (2-ΔΔCT).
Relative quantification method was used to analyze the qRT-PCR data. The primer sequences were synthesized by Shanghai Generay Biotech Co. Ltd. (Generay, Shanghai, China) and are listed in Table S3. The details are described in our previous study [17]. β-actin was used as the reference for normalization of expressions of mRNAs or lncRNAs. The relative fold changes were analyzed using 2−ΔΔCT method. All samples were analyzed in triplicate.
Western blot analysis
The related proteins were lysed in ice-cold RIPA buffer with 0.1 M PMSF and measured using the BCA kit. Lysates of cells were separated by SDS polyacrylamide gels. The separated proteins were then transferred to a polyvinylidene fluoride membrane (Millipore) by 300 mA for 60–90 min. The membranes were then blocked by 5% skimmed milk and probed overnight with matched antibodies against the target proteins at 4 °C. Subsequently, the membranes were incubated with relevant antibodies. Finally, we used the FluorChem HD2 (Protein Sample, USA) to explore the proteins. The primary detection antibodies that were used in the work are listed in Table S4.
Subcellular fractionation
Firstly, the cells required for the experiments were prepared. Then, the nuclear and cytoplasmic RNA were separated using Nuclear and Cytoplasmic Extraction Reagents (Norgen Belmont, CA) by the manufacturer’s instructions. Then, quantitative real time-PCR was explored to determine the RNA. U1 and GAPDH were used as the internal references for the nucleus and cytoplasm, respectively.
M6A RNA methylation quantification
The m6A levels were determined using the EpiQuik m6A RNA Methylation Quantification Kit (Colorimetric; America) according to the manufacturer’s instructions. Firstly, 200 ng of RNA samples were added to assay wells. Then, the diluted capture antibody, the diluted detection antibody, and the diluted enhancer solution were added to each well. M6A levels were determined according to the absorbance of each sample at 450 nm, and a standard curve was then used to calculate the relative m6A content.
Transwell assay
Transwell assay was performed to evaluate the invasion and migration of cells (Costar 3422, Corning, United States). A polycarbonate membrane (8 μm pore size) with or without Matrigel (BD Biosciences, San Jose, CA, USA) separated the upper and lower compartments. Then, 5 × 104 cells/well suspended in a serum-free medium were seeded into the upper chamber. Mitomycin C (0.5 mg/mL) was also added (M5353, Sigma, United States).
In the bottom chamber, 750 μL of medium containing 20% serum was added. The plate was incubated at 37 °C for the indicated amount of time. Then, the membrane was rinsed using a PBS-soaked cotton swab and the lower side of the membrane was immobilized with 1 mL of 4% paraformaldehyde in each well for 30 min at room temperature. The fixative was removed, the cells were washed once with PBS, and 1 mL of 0.5% crystal violet solution (PAB180004, Bio-Swamp) was added to each well. After 90 min of staining, the cells were washed three times with PBS. Five randomly selected visual fields on each insert were photographed, and cells were counted manually.
Fluorescence in situ hybridization
We conducted the FISH assay using the FISH Kit (RiboBio, China) according to the manufacturer’s instruction. Firstly, we cultured cells in 24-well plates for 24 h. Then the treated cells were hybridized with 20 μM Cy3-labeled U6, 18S and RMRP. Finally, the images were observed with a confocal laser scanning microscope (FV1200, Olympus, Japan).
Chromatin immunoprecipitation (ChIP)
We performed the ChIP assays using EZ-ChIPTM ChIP Kit (Millipore, USA) according to the manufacturer’s instruction. A549 cells, H1299 cells, A549 Scrambled cells, A549 shRMRP-1 cells, H1299 cDNA, and H1299 RMRP cells were used in this assay.
We first crosslinked the cells by 1% formaldehyde for 10 min. Related cells were added into lysis buffer for 5 min. Then, cell lysates were subjected to pulsed ultrasonication to break nuclear membrane and shear DNA into fragments (300–500 bp fragments). The final lysates were incubated overnight with YBX1 (Santa Cruz, USA) or IgG (Santa Cruz, USA) antibody at 4 °C. Cross-linked DNA released from the protein-DNA complex was purified using Universal DNA Purification Kit (Tiangen, China) and the eluted DNA was further detected by qRT-CR. Input and IgG were used simultaneously to confirm that the detected signals were derived from the specific bonding of chromatin and YBX1. All ChIP assays were repeated independently three times. The results were reported as % input ± standard error of the mean (S.E.M.). The primers used for PCR are listed in Supplementary Table S5.
RNA immunoprecipitation (RIP)
RIP assays were performed using Magna RIPTM RNA-Binding Protein Immunoprecipitation Kits (Millipore, USA) according to the manufacturer’s instructions. Briefly, the related cells were harvested to perform RIP experiments using m6A antibody (2 μg/sample; Synaptic Systems, Goettingen, German), YBX1 antibody (Abcam, ab76149), and IgG (Santa, USA) antibody. The co-precipitated RNAs were then explored by magnetic beads.
Finally, qRT-PCR assay was used to explore the total RNA. The input fraction Ct value was used to normalize the RNA sample preparation differences in each group, and the negative controls (IgG) Ct was used to adjust for background fraction.
RNA pull-down assay
The T7 RiboMAXTM Express Large Scale RNA Production System (Catalogue Number P1320, Promega, USA) was used to produce abundant RMRP in vitro according to the manufacturer’s instructions. The 3′ ends of RMRP RNAs were then labeled using Pierce™ RNA 3′ End Desthiobiotinylation Kit (Thermo Fisher Scientific, USA). Then the compounds were further incubated with the protein of the treated cells by Pierce™ Magnetic RNA-Protein Pull-Down Kit (Thermo Fisher, USA). Finally, the precipitated proteins of NSCLC cells were explored by Western blot assays. We used the antisense RNA as the negative control.
Dual-Luciferase reporter
Dual-Luciferase reporter assay was performed as described in our previous report [17]. The promoter region of TGFBR1 containing 200 bp putative binding area (wild type, named TGFBR1-WT) or mutant area (mutant type, named TGFBR1-MUT) was constructed into pGL3-basic vectors by GeneCreate (Wuhan, China). These were then transfected in related cells. Finally, the dual luciferase reporter assay was performed using the Dual-Luciferase Reporter Assay system (Promega, Madison, USA) according to the manufacturer’s instructions. The firefly luciferaseactivity was normalized to Renilla activity. The sequences of related genes are listed in Supplementary Table S6. As for SMAD-binding element (SBE) luciferase reporter gene activity, the related cancer cells were transfected with a luciferase reporter plasmid carrying three copies of a SBE, pGL4.48-luc2P/SBE/Hygro (Promega, Madison, USA). The relative luciferase activity was explored by the Dual-Luciferase Reporter Assay System (Promega) following the manufacturer’s instructions. The sequences of the relevant genes are listed in Supplementary Table S6.
CCK8 and colony formation assays
CCK-8 (Dojindo, Kyushu, Japan) and colony formation assays were used to assess cell proliferation. For CCK-8 assays, the related cells were seeded in 96-well plates (1 × 103 cells per well) in triplicate and cultured at the indicated concentrations. At the end of each time-point (1 day, 2 days, 3 days, and 4 days), CCK-8 was added for 1 h at 37 °C. The absorbance was detected at 450 nm.
For cisplatin sensitivity, suspensions of NSCLC cell lines (1 × 104 cells per well) were plated in 96-well plates. The cells were treated with different concentrations of cisplatin for 24 h. After treatment, the media were removed, and RPMI1640 (90 μL) and CCK-8 (10 μL) were added. The plates were incubated for 3 h in the incubator. The absorbance at 450 nm was measured using an automated reader. The inhibition rate was calculated using the following formula:
For the colony formation assays, 600 cells were inoculated into six-well plates for ~2 weeks. The cells were subsequently fixed with 4% paraformaldehyde and stained with 0.5% crystal violet. The photographs of the wells were obtained and the number of colonies was counted by three different individuals [17].
Cell cycle analysis
The Cell Cycle Staining Kit (Heygen, China) was used. We first washed the cells with cold PBS fixed with ethanol overnight (−20 °C). On the next day, the cells were centrifugated for 10 min. We further used PI/RNase to stain the cells. Finally, a flow cytometer (Becton-Dickinson) was used to evaluate the cell cycle in each sample following the manufacturer’s instructions. The details were described as previously described [17].
Patients
181 NSCLC samples were collected between May 2013 and August 2015 at the department of thoracic surgery in Harbin Medical University Cancer Hospital. The patients enrolled in the study all signed informed consent forms for medical record review and tissue sample donation according to their preference. The current work was also approved by the Institutional Research Ethics Committee of the Harbin Medical University Cancer Hospital.
Xenograft analysis
The animal experiments were approved by the Institutional Animal Care and Use Committee of the Harbin Medical University Cancer Hospital. The assays conformed to all the regulatory standards. All animals (n = 10) were bred under pathogen-free conditions at the Animal Center of Harbin Medical University. BALB/c athymic nude mice (male, age: 5 weeks) were purchased from Beijing Charles River (https://www.vitalriver.com/). We injected 4 × 106 cells suspended in 0.1 mL of PBS into ~5-week-old BALB/c mice. When the tumors became palpable, the tumor volume (V) was assessed using the following equation:
Furthermore, each mouse was weighed. The tumors were harvested for immunohistochemistry and qRT-PCR assays. Tissue sections were immersed in minocycline-ethylenediaminetetraacetic acid. Subsequently, the sections were heated at 100 °C for 15 min and incubated in methanol for 15 min. The slides were incubated with anti-Ki67 (abcam, ab92742, 1:1000), anti-MMP9 (abcam, ab228402, 1:1000), and anti-TGFBR1 (abcam, ab235178, 1:500) primary antibody. Relative staining intensity was judged according to the percentage of immunoreactive cells. The details were as previously described [17].
Statistical analysis
Statistical analyses were performed using SPSS 17.0 and GraphPad Prism 8. All data are presented as mean ± standard deviation. All experiments (except the xenografts) were performed at least three times. Between-group differences with respect to continuous variables were assessed using Student’s t test. P values < 0.05 were considered indicative of statistical significance. The Kaplan–Meier method was used to determine overall survival and disease-free survival. The correlation between RMRP and TGFBR1 was estimated using Pearson’s correlation analysis. More materials and methods were supplied in the part of supplement.
Data availability
The data sets in this study are available from the corresponding author upon reasonable request.
Change history
10 October 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41418-022-01075-z
References
DeSantis CE, Miller KD, Goding Sauer A, Jemal A, Siegel RL Cancer statistics for African Americans, 2019 CA Cancer J Clin. 2019;69:211–33.
Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376:2109–21.
Zhou M, Wang H, Zeng X, Yin P, Zhu J, Chen W, et al. Mortality, morbidity, and risk factors in China and its provinces, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2019;394:1145–58.
Beermann J, Piccoli MT, Viereck J, Thum T. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev. 2016;96:1297–325.
Yin H, Wang X, Zhang X, Wang Y, Zeng Y, Xiong Y, et al. Integrated analysis of long noncoding RNA associated-competing endogenous RNA as prognostic biomarkers in clear cell renal carcinoma. Cancer Sci. 2018;109:3336–49.
Wang Y, Liu Y, Guan Y, Li H, Liu Y, Zhang M, et al. Integrated analysis of immune-related genes in endometrial carcinoma. Cancer Cell Int. 2020;20:477.
Wang X, Yin H, Zhang L, Zheng D, Yang Y, Zhang J, et al. The construction and analysis of the aberrant lncRNA-miRNA-mRNA network in non-small cell lung cancer. J Thorac Dis. 2019;11:1772–8.
Xu Q, Yin H, Ao H, Leng X, Liu M, Liu Y, et al. An 11-lncRNA expression could be potential prognostic biomarkers in head and neck squamous cell carcinoma. J Cell Biochem. 2019;120:18094–103.
Feng S, Liu W, Bai X, Pan W, Jia Z, Zhang S, et al. LncRNA-CTS promotes metastasis and epithelial-to-mesenchymal transition through regulating miR-505/ZEB2 axis in cervical cancer. Cancer Lett. 2019;465:105–17.
Xu M, Xu X, Pan B, Chen X, Lin K, Zeng K, et al. LncRNA SATB2-AS1 inhibits tumor metastasis and affects the tumor immune cell microenvironment in colorectal cancer by regulating SATB2. Mol Cancer. 2019;18:135.
Ridanpaa M, van Eenennaam H, Pelin K, Chadwick R, Johnson C, Yuan B, et al. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell. 2001;104:195–203.
Maida Y, Yasukawa M, Furuuchi M, Lassmann T, Possemato R, Okamoto N, et al. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature. 2009;461:230–5.
Cao HL, Liu ZJ, Huang PL, Yue YL, Xi JN. lncRNA-RMRP promotes proliferation, migration and invasion of bladder cancer via miR-206. Eur Rev Med Pharm Sci. 2019;23:1012–21.
Wang Y, Luo X, Liu Y, Han G, Sun D. Long noncoding RNA RMRP promotes proliferation and invasion via targeting miR-1-3p in non-small-cell lung cancer. J Cell Biochem. 2019;120:15170–81.
Meng Q, Ren M, Li Y, Song X. LncRNA-RMRP acts as an oncogene in lung cancer. PLoS One. 2016;11:e0164845.
Moore-Smith L, Pasche B. TGFBR1 signaling and breast cancer. J Mammary Gland Biol Neoplasia. 2011;16:89–95.
Wang X, Chen X, Meng Q, Jing H, Lu H, Yang Y, et al. MiR-181b regulates cisplatin chemosensitivity and metastasis by targeting TGFbetaR1/Smad signaling pathway in NSCLC. Sci Rep. 2015;5:17618.
Zhao Y, Qiao W, Wang X, Yin H, Cui J, Cui Y, et al. 14-3-3zeta/TGFbetaR1 promotes tumor metastasis in lung squamous cell carcinoma. Oncotarget. 2016;7:82972–84.
Kabbout M, Garcia MM, Fujimoto J, Liu DD, Woods D, Chow CW, et al. ETS2 mediated tumor-suppressive function and MET oncogene inhibition in human non-small cell lung cancer. Clin Cancer Res. 2013;19:3383–95.
Cai L, Lin S, Girard L, Zhou Y, Yang L, Ci B, et al. LCE: an open web portal to explore gene expression and clinical associations in lung cancer. Oncogene. 2019;38:2551–64.
Mendell JT. Targeting a long noncoding RNA in breast cancer. N Engl J Med. 2016;374:2287–9.
Lu Y, Zhao X, Liu Q, Li C, Graves-Deal R, Cao Z, et al. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/beta-catenin signaling. Nat Med. 2017;23:1331–41.
Yin D, Lu X, Su J, He X, De W, Yang J, et al. Long noncoding RNA AFAP1-AS1 predicts a poor prognosis and regulates non-small cell lung cancer cell proliferation by epigenetically repressing p21 expression. Mol Cancer. 2018;17:92.
Rheinbay E, Parasuraman P, Grimsby J, Tiao G, Engreitz JM, Kim J, et al. Recurrent and functional regulatory mutations in breast cancer. Nature. 2017;547:55–60.
Shao Y, Ye M, Li Q, Sun W, Ye G, Zhang X, et al. LncRNA-RMRP promotes carcinogenesis by acting as a miR-206 sponge and is used as a novel biomarker for gastric cancer. Oncotarget. 2016;7:37812–24.
Fournier PG, Juarez P, Jiang G, Clines GA, Niewolna M, Kim HS, et al. The TGF-beta signaling regulator PMEPA1 suppresses prostate cancer metastases to bone. Cancer Cell. 2015;27:809–21.
Tang YH, He GL, Huang SZ, Zhong KB, Liao H, Cai L, et al. The long noncoding RNA AK002107 negatively modulates miR-140-5p and targets TGFBR1 to induce epithelial-mesenchymal transition in hepatocellular carcinoma. Mol Oncol. 2019;13:1296–310.
Zhou B, Guo W, Sun C, Zhang B, Zheng F. Linc00462 promotes pancreatic cancer invasiveness through the miR-665/TGFBR1-TGFBR2/SMAD2/3 pathway. Cell Death Dis. 2018;9:706.
Schmitt ME, Clayton DA. Nuclear RNase MRP is required for correct processing of pre-5.8S rRNA in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:7935–41.
Gill T, Cai T, Aulds J, Wierzbicki S, Schmitt ME. RNase MRP cleaves the CLB2 mRNA to promote cell cycle progression: novel method of mRNA degradation. Mol Cell Biol. 2004;24:945–53.
Yang M, Ke H, Zhou W. LncRNA RMRP promotes cell proliferation and invasion through miR-613/NFAT5 axis in non-small cell lung cancer. OncoTargets Ther. 2020;13:8941–50.
Noh JH, Kim KM, Abdelmohsen K, Yoon JH, Panda AC, Munk R, et al. HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes Dev. 2016;30:1224–39.
El-Naggar AM, Veinotte CJ, Cheng H, Grunewald TG, Negri GL, Somasekharan SP, et al. Translational activation of HIF1alpha by YB-1 promotes sarcoma metastasis. Cancer Cell. 2015;27:682–97.
Zhang E, He X, Zhang C, Su J, Lu X, Si X, et al. A novel long noncoding RNA HOXC-AS3 mediates tumorigenesis of gastric cancer by binding to YBX1. Genome Biol 2018;19:154.
Moharamoghli M, Hassan-Zadeh V, Dolatshahi E, Alizadeh Z, Farazmand A. The expression of GAS5, THRIL, and RMRP lncRNAs is increased in T cells of patients with rheumatoid arthritis. Clin Rheumatol. 2019;38:3073–80.
Atianand MK, Fitzgerald KA. IMMUNOLOGY. An RNA twist to T(H)17 cells. Science. 2016;351:1032.
de la Fuente MA, Recher M, Rider NL, Strauss KA, Morton DH, Adair M, et al. Reduced thymic output, cell cycle abnormalities, and increased apoptosis of T lymphocytes in patients with cartilage-hair hypoplasia. J Allergy Clin Immunol. 2011;128:139–46.
Huang X, Zhang G, Liang T. Cancer environmental immunotherapy: starving tumor cell to death by targeting TGFB on immune cell. J Immunother Cancer. 2021;9:e002823.
Li J, Lee Y, Li Y, Jiang Y, Lu H, Zang W, et al. Co-inhibitory molecule B7 superfamily member 1 expressed by tumor-infiltrating myeloid cells induces dysfunction of anti-tumor CD8(+) T Cells. Immunity 2018;48:773–86 e5.
Amelio I, Mancini M, Petrova V, Cairns RA, Vikhreva P, Nicolai S, et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc Natl Acad Sci USA. 2018;115:E10869–E78.
Sang LJ, Ju HQ, Liu GP, Tian T, Ma GL, Lu YX, et al. LncRNA CamK-A regulates Ca(2+)-signaling-mediated tumor microenvironment remodeling. Mol Cell. 2018;72:71–83 e7.
Ghafouri-Fard S, Esmaeili M, Taheri M. H19 lncRNA: roles in tumorigenesis. Biomed Pharmacother. 2020;123:109774.
Wan P, Su W, Zhang Y, Li Z, Deng C, Li J, et al. LncRNA H19 initiates microglial pyroptosis and neuronal death in retinal ischemia/reperfusion injury. Cell Death Differ. 2020;27:176–91.
Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. 2017;552:126–31.
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65:529–43.
Zheng ZQ, Li ZX, Zhou GQ, Lin L, Zhang LL, Lv JW, et al. Long noncoding RNA FAM225A promotes nasopharyngeal carcinoma tumorigenesis and metastasis by acting as ceRNA to sponge miR-590-3p/miR-1275 and upregulate ITGB3. Cancer Res. 2019;79:4612–26.
Yin H, Wang X, Zhang X, Zeng Y, Xu Q, Wang W, et al. UBE2T promotes radiation resistance in non-small cell lung cancer via inducing epithelial-mesenchymal transition and the ubiquitination-mediated FOXO1 degradation. Cancer Lett. 2020;494:121–31.
Acknowledgements
We thank TCGA database. This study was supported by the National Natural Science Foundation of China (81903120 to HY; 81902317 to XW), Young Innovative Talents Training Program of Higher Education Institutions in Heilongjiang Province (UNPYSCT-2020161 to HY), Haiyan Foundation of Harbin Medical University Cancer Hospital (No. JJQN2019-01 to XW; JJZD2021-13 to HY), Postdoctoral Foundation of Heilongjiang Province (LBH-Z18224 to XW; LBH-Z20076 to HY), the Natural science funding of Heilongjiang (LH2019H041 to XW; YQ2021H024 to HY), China Postdoctoral Science Foundation (2018M641861to XW; 2020M681117 and 2021T140170 to HY), Young Talents Foundation of Harbin Medical University Cancer Hospital (No. BJQN2019-06 to XW; No. BJQN2020-01 to HY).
Author information
Authors and Affiliations
Contributions
HY supervised the project. HY and XYW designed the overall study, interpreted the experiments, and wrote the paper. LC, SQP, YRW, ZGL, HYZ and YL performed in vitro experiments and analyzed data. XQT, HJZ and HYZ performed in vivo experiments and analyzed data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
All procedures followed were in accordance with the ethical standards approved by Institutional Ethics Committee on human experimentation and with the Helsinki Declaration. All institutional and national guidelines for the care and use of laboratory animals were followed.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Due to a figure error.
Edited by R De Maria
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yin, H., Chen, L., Piao, S. et al. M6A RNA methylation-mediated RMRP stability renders proliferation and progression of non-small cell lung cancer through regulating TGFBR1/SMAD2/SMAD3 pathway. Cell Death Differ 30, 605–617 (2023). https://doi.org/10.1038/s41418-021-00888-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-021-00888-8
This article is cited by
-
The m6A modification mediated-lncRNA POU6F2-AS1 reprograms fatty acid metabolism and facilitates the growth of colorectal cancer via upregulation of FASN
Molecular Cancer (2024)
-
KIT mutations and expression: current knowledge and new insights for overcoming IM resistance in GIST
Cell Communication and Signaling (2024)
-
Crosstalk between m6A modification and autophagy in cancer
Cell & Bioscience (2024)
-
Single-cell transcriptomic sequencing data reveal aberrant DNA methylation in SMAD3 promoter region in tumor-associated fibroblasts affecting molecular mechanism of radiosensitivity in non-small cell lung cancer
Journal of Translational Medicine (2024)
-
Emerging role of RNA modification and long noncoding RNA interaction in cancer
Cancer Gene Therapy (2024)
