
Abstract
The basic function of β-arrestin 2 (Arrb2) is to negatively regulate the G-protein-coupled receptor signaling pathway through facilitating receptor desensitization and internalization. Arrb2 has also been reported to play various roles in cancer pathology including the proliferation, migration, invasion, metastasis, and apoptosis of solid tumors. However, the molecular mechanisms underlying the tumorigenic capacities of Arrb2 have not been elucidated. Here, we show a novel function of Arrb2: Arrb2 facilitates the degradation of HIF-1α, which is a master regulator of oxygen homeostasis. We also demonstrate that Arrb2 interacts with HIF-1α and stimulates ubiquitin-mediated 26S proteasomal degradation of HIF-1α by recruiting PHD2 and pVHL. Overexpression of Arrb2 in human glioblastoma cells suppresses HIF-1α signaling, tumor growth, and angiogenesis. Consistent with this antitumorigenic effect of Arrb2, low Arrb2 expression levels correlate with high HIF-1α expression and poor glioblastoma patient survival. These results collectively reveal a novel function of Arrb2 in the oxygen-sensing mechanism that directly regulates HIF-1α stability in human cancers and suggest Arrb2 as a new potential therapeutic target for glioblastoma.
Similar content being viewed by others


Introduction
Hypoxia is a physiologically relevant type of biological stress, and many solid tumors have hypoxic regions due to an imbalance between oxygen supply and oxygen demand. Hypoxia induces transcriptional and molecular alterations that promote processes related to tumor progression, such as cell proliferation, angiogenesis, metabolism, invasion, and metastasis. Under hypoxic conditions, most cellular responses are controlled by hypoxia-inducible factor-1 (HIF-1), which is a critical transcription factor that facilitates adaptation to low oxygen concentrations [1,2,3,4]. HIF-1 is a heterodimeric transcription factor that consists of oxygen-regulated α subunits and constitutively expressed nuclear translocator β subunits [2]. The interaction of HIF-1α and HIF-1β is required for the transcriptional function of HIF-1; HIF-1α serves as the major regulatory subunit responsible for hypoxic specific function. While HIF-1α protein is highly expressed in hypoxic cells, the protein is rapidly degraded by the ubiquitin-proteasomal degradation pathway in oxygenated cells. Oxygen-dependent degradation is triggered by prolyl hydroxylase domain protein 1, 2, and 3 (PHD-1, –2, and –3) [5]. Hydroxylation of HIF-1α at proline 402 and 564 of the oxygen-dependent degradation domain facilitates interaction with the von Hippel-Lindau tumor suppressor protein (pVHL), which recruits the Cullin2/Elongin-BC E3 ligase complex and consequently catalyzes ubiquitination and 26S proteasomal degradation [6, 7]. Because PHDs require oxygen for catalytic activity, HIF-1α is stabilized by inhibiting the enzyme activities of PHDs under hypoxic conditions, translocates to the nucleus, heterodimerizes with HIF-1β, and recruits co-activators such as histone acetyltransferase p300 and cAMP-response element-binding protein for transcriptional activation [8]. Transcription of several hundred of over 1500 known target genes increases in response to hypoxia [9].
Many of the proteins that interact with HIF-1α regulate HIF-1 activity by either promoting or inhibiting the interaction of HIF-1α with other proteins. Some HIF-1α interacting proteins facilitate oxygen-dependent degradation by stabilizing interactions with components of the hydroxylation complex or by stimulating ubiquitination of hydroxylated HIF-1α [10]. Other HIF-1α interacting proteins inhibit oxygen-dependent degradation by blocking the action of the pVHL-E3 ligase complex or by catalyzing deubiquitination [11]. Other HIF-1α interacting proteins facilitate oxygen-independent degradation by stimulating ubiquitination, SUMOylation, or lysosomal degradation [12, 13]. Another group of HIF-1α interacting proteins function as co-activators or co-repressors to regulate HIF-1α-mediated transactivation [14]. Although many HIF-1α interacting proteins have been identified, many cancer researchers still focus on finding a novel regulatory and binding protein of HIF-1α to generate a map of the HIF-1α regulatory pathway and identify a therapeutic target for cancer.
The nonvisual arrestins β-arrestin-1 and –2 (Arrb1 and Arrb2) are versatile adapter proteins that were initially reported to desensitize G-protein-coupled receptor (GPCR) signaling by regulating receptor internalization [15]. Upon GPCR activation, Arrb2 translocates to the cell membrane and binds to agonist-occupied receptors, and dissociation of these receptors from G proteins promotes internalization, thus causing desensitization [15]. Arrbs were initially known as negative regulators of GPCR-mediated signaling as mentioned above. However, new roles of Arrbs in receptor trafficking and signaling have recently been identified. They function as scaffolds in receptor endocytosis and adapters in signal transduction [16]. In addition, they bind to IκBα and MDM2 in the cytoplasm and can regulate the transcriptional activity of NFκB and p53 transcription factors [17, 18]. In particular, novel roles of Arrb2 have also been reported; these reports state that it regulates cellular processes, including cell proliferation, cancer cell migration, invasion and metastasis [19, 20]. Moreover, Arrb2 regulates cellular signal transduction, including the tyrosine kinase pathway, extracellular signal-regulated kinase 1/2 (ERK 1/2) signaling, gene transcription and cell death signaling [21,22,23]. Recently, the function of Arrb1 and Arrb2 in cancer progression was reported. Arrb1 promotes tumorigenesis of colorectal cancer by interacting with the cellular proto-oncogene c-SRC [24], increases secretion of vascular endothelial growth factor-A (VEGF-A) in breast cancer [25], and stimulates angiogenesis in lung cancer [26]. In the case of Arrb2, its expression is increased in breast cancer [27], and highly expressed Arrb2 mediates the progression of myeloid leukemia [28]. On the other hand, it has also been reported that Arrb2 inhibits tumor invasion and migration in hepatocellular carcinoma [29], and decreased levels of Arrb2 are related to poor prognosis in lung cancer [30]. Nonetheless, many aspects of the functional roles of Arrb2 remain poorly understood.
Gliomas are the most common primary malignant central nervous system tumors and are classified as grade II-IV according to their histology and molecular characteristics [31]. Grade IV glioblastomas (GBMs) are the most aggressive and show a median overall survival of <2 years [32]. Despite increasing morbidity and mortality rates for GBM, therapies for GBM, including surgery, radiotherapy, and chemotherapy, are still not optimal [33]. Therefore, the identification of novel regulators during GBM progression will provide a new therapeutic target for anticancer treatment in GBM patients. According to a recent report, the expression of Arrb1 is low in GBM [34]; however, there is no report regarding the expression and function of Arrb2 in GBM.
Here, we report that Arrb2 binds to HIF-1α and decreases its protein stability. We demonstrate that Arrb2 interacts with HIF-1α and stimulates proteasome-mediated degradation by recruiting pVHL and PHDs. Consistently, high-level expression of Arrb2 correlates with reduced tumorigenesis in GBMs and a high survival probability in GBM patients. Collectively, this study reveals an important role of Arrb2 in the oxygen-sensing pathway regulated by HIF-1α stability in GBM and suggests that Arrb2 may be a potential therapeutic target for solid tumors, including GBM.
Materials and methods
Materials
Antibodies for human and mouse HIF-1α were purchased from BD Pharmingen (610958, San Diego, CA) and NOVUS Biologicals (NB100-479, Littleton, CO), respectively. Lamin A/C (4777), hydroxy-HIF-1α (3434S) and pVHL (68547S) antibodies were obtained from Cell Signaling Technology (Beverly, MA). Antibodies recognizing Flag (F3165) and α-tubulin (T5168) were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies for Arrb2 (SC-13140) and c-Myc (SC-789) were from Santa Cruz Biotechnology (Santa Cruz, CA). The PHD2 (NB100-137) and CD31 (550274) antibodies were obtained from Novus Biologicals and BD Pharmingen, respectively. Affinity anti-Flag gel and cycloheximide (CHX) were purchased from Sigma-Aldrich (St. Louis, MO). MG132 and G418 sulfate were purchased from Assay Designs Inc. (Ann Arbor, MI) and InvivoGen (San Diego, CA), respectively.
Cell culture and hypoxia treatment
Human glioblastoma cell lines (U87MG, T98G, and H4) were obtained from ATCC, and U251MG and immortalized primary human fetal astrocytes were kindly provided by Dr. Seok-Geun Lee (Kyung Hee University, Korea). These cells were maintained in high glucose Dulbecco’s modified Eagle medium (DMEM, WelGENE, Daegu, Korea). Human dermal microvascular endothelial cell line (HMEC-1) was purchased from Centers for Disease Control and Prevention (CDC) in USA and cells were maintained in low glucose MCDB131 medium (Gibco, Santa Clara, CA). Media were supplemented with 10% fetal bovine serum (FBS, HyClone Laboratories Inc., Logan, UT) and 1% penicillin and streptomycin (P/S, Mediatech Inc., Manassas, VA) in a humidified atmosphere of 95% air and 5% CO2 at 37 °C (normoxia). Cells passed for fewer than 6 months after receipt or resuscitation of validated cells were used in this study. To expose the cells to hypoxia, cells were transferred to a hypoxic workstation (INVIVO2, Ruskinn Technology, Bridgend, UK) supplemented with a triple gas mixture of 1% O2, 5% CO2, and 94% N2.
Construction of plasmids, transfections and stable cell lines
To construct the plasmid encoding human Arrb2, we amplified full-length human Arrb2 by PCR and subcloned it into pCMV-tag2B and pCMV-tag3B, which are Flag- and Myc-tagged vectors, respectively. Cells were transfected with each expression vector using polyethyleneimine (PEI, Sigma-Aldrich). Six hours after transfection, the media were replaced with fresh complete media. To make stable cell lines, transfected cells were selected with 1 mg/ml G418 for 1 week.
Preparation of cell extracts, cellular fractionation, and western blot analysis
Cells were lysed on ice using lysis buffer (10 mM Tris-pH 7.4, 150 mM NaCl, and 0.2% NP-40) containing a protease inhibitor cocktail (Sigma-Aldrich). To obtain the cytoplasmic fraction, cells were suspended in buffer C (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol (DTT), and 0.15% NP-40) containing a protease inhibitor cocktail and centrifuged at 1200 rpm for 30 s, and the supernatants were collected as a cytoplasmic fraction. The pellets were resuspended in buffer N (20 mM HEPES, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 0.5% NP-40) containing a protease inhibitor cocktail. The lysates were centrifuged at 12000 rpm for 15 min at 4 °C, and then the supernatant fraction was collected as the nuclear fraction. Western blot analysis was performed according to a standard protocol using a polyvinylidene fluoride membrane (Millipore Corporation, Billerica, MA).
Immunofluorescence staining
The cells were grown on coverslips under hypoxic conditions, fixed with 4% paraformaldehyde (Sigma-Aldrich) at room temperature for 15 min and then permeabilized with 0.3% Triton X-100 for 10 min. The cells were incubated with antibodies against Arrb2 and HIF-1α at 4 °C overnight and then incubated with appropriate Alexa Fluor-conjugated secondary antibodies at room temperature for 1 h. The nuclei were counterstained with Hoechst 33342 (Thermo Scientific) for 10 min. The coverslips were mounted with antifade solution, and the staining was determined using a LSM 700 confocal microscope (Carl Zeiss, Jena, Germany).
RT-PCR and qPCR
Total RNA was isolated using TRIzol reagent (Invitrogen Life Technologies; Carlsbad, CA) and was used to synthesize cDNA using MMLV-reverse transcriptase (Beams Biotechnology, Sungnam, Korea) and oligo-dT primers (Sigma-Aldrich). Synthesized cDNA was amplified, and the PCR product was then visualized on a 1% agarose gel. The sequences of each forward (F) and reverse (R) primer used for PCR were as follows: HIF-1α (F), CAGAAGATACAAGTAGCCTC and (R), CTGCTGGAATACTGTAACTG; Arrb2 (F), CTGTTCATCGCCACCTACCA and (R), TCGGCGTACTGTCTCACAGA; and 18S (F), CAGCCACCCGAGATTGAGCA and (R) TAGTAGCGACGGGCGGTGTG. For quantification, real-time PCR was performed using the TaKaRa SYBR Premix Ex Taq (Tli RNaseH Plus) kit (Takara, Japan). The primer sequences of each gene for qPCR were as follows: HIF-1α (F), GTGAACAGAATGGAATGGAGCA and (R), CTGGTCAGCTGTGGTAATCCA; and 18S (F), CAGCCACCCGAGATTGAGCA and (R) TAGTAGCGACGGGCGGTGTG.
Immunoprecipitation
Whole cell lysates were prepared using NETN lysis buffer (20 mM Tris-pH 8.0, 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40) containing protease inhibitor cocktail and phosphatase inhibitors (1 mM Na3VO4 and 10 mM NaF). The lysates were immunoprecipitated with anti-Flag conjugated to agarose beads (Sigma-Aldrich) or anti-HIF-1α antibody in the presence of protein A beads (Millipore Corporation) from 2 h at 4 °C with rotation. The immunoprecipitates were then washed 8 times with NETN buffer and analyzed by western blotting assays.
Clonogenic assay
Five thousand stably transfected U87MG cells were plated in 60 mm dishes and then incubated for 2 weeks. After the medium was removed, cells were fixed with fixation solution (12.5% acetic acid in methanol) and then stained using crystal violet staining solution (Sigma-Aldrich). Colonies of U87MG cells were counted and areas occupied with colonies were measured by ImageJ software, and values are expressed as the mean ± S.D. of three independent experiments.
Tube formation and rat aortic ring sprouting assay
One hundred and fifty microliters of Matrigel (BD Pharmingen, San Diego, CA, USA) was polymerized on 24-well plates at 37 °C for 30 min. HMEC-1 cells (1 × 105) were seeded on polymerized Matrigel and incubated with conditioned medium collected from Arrb2-transfected U87MG cells under hypoxic conditions. After 9 h, morphological changes were observed, and the number of extremes and meshes in all areas were measured by Angiogenesis Analyzer in ImageJ software [35]. The results are expressed as the mean ± S.D. from three independent experiments. Aortas were excised from 6-week-old SD rats (Daehan Biolink, Chungbuk, Korea), and fibroadipose tissue was removed from the excised ring. Rings were sliced at a thickness of 1 mm and were then placed on polymerized Matrigel in each well of a 24-well plate and covered with an additional 50 μL of Matrigel. The rings were incubated in M199 medium containing conditioned medium-derived Arrb2-overexpressing U87MG cells for 4–7 days. At the end of the incubation period, the microvessel sprouts were measured using an inverted microscope, and the outgrowth area was measured using ImageJ software as described the previous report [36]. The results are expressed as the mean ± S.D. from three independent experiments.
Animal studies
All mouse procedures in this study were approved by the Institutional Animal Care and Use Committee protocol approved by the Committee for Care and Use of Laboratory Animals at Kyung Hee University (KHUASP(SE)-17-144, 24-11-2017). Six-week-old male BALB/c-nude mice were purchased from Daehan Biolink (Chungbuk, Korea) and maintained at 25 °C with a 12-h light/12-h dark cycle. Mice were monitored daily for their health by the animal staff. To generate a xenograft tumor mouse model, Arrb2-expressing lentivirus-infected U87MG cells (1 × 106 cells) were injected subcutaneously into the right flank of mice. Mice were assigned to groups with 5 mice for one independent experiment, and the experiments were repeated three times. Four weeks after the injection of U87MG cells, the mice were sacrificed, and tumors were isolated. Tumor volume (TV) was calculated by using the following formula: TV = length × (width)2 × 0.5.
Immunohistochemistry
The tumors were cut into 10 μm slices using a cryostat (Leica Microsystems; Wetzlar, Germany) and placed on gelatin-coated slides. The tumor tissues were fixed with 4% paraformaldehyde for 15 min, incubated with 0.3% H2O2 for 30 min, and then permeabilized with 0.3% Triton-X-100 for 20 min. The tumor tissues were incubated with antibodies against Arrb2 and HIF-1α at 4 °C overnight and then incubated with appropriate biotinylated secondary antibodies (Vector Laboratories) and incubated with peroxidase-conjugated ABC reagent (Vector Laboratories) for 1 h at room temperature. Immunoreactivity was detected with 3,3′-diaminobenzidine substrate (DAB, Sigma-Aldrich). A paraffin-embedded human glioma tissue microarray (Biomax Inc., Rockville, MD, USA) was deparaffinized and rehydrated before immunostaining as described above. One tissue section showing strong immunoreactivity in a pilot test was used as a positive control. Normal horse serum was substituted for the primary antibody as a negative control. The random fields in each section were calculated using ImageJ software and the relative expression level of each protein was quantified according to integrated optical density from three independent experiments. P values were calculated using the log-rank test.
The cancer genome atlas (TCGA) data analysis and recurrence-free survival analysis
For the survival analysis, we used gene expression data and survival information of GBM patients of TCGA. Recurrence-free survival data was obtained from cBioPortal (https://www.cbioportal.org) [37] and the gene expression data from UCSC XENA (https://xena.ucsc.edu) [38]. To divide the patients to ARRB2-high and -low groups, the optimal cutoff of ARRB2 expression for best log-rank statistics was obtained using maximally selected rank and statistics from R maxstat package [39]. Recurrence-free survival analysis between the two groups was performed by using the Kaplan–Meier method [40] and the log-rank test to compare survival rates between the ARRB2- high and -low groups. P value of the log-rank test was indicated with the Kaplan–Meier plot. Genomics data were analyzed using R2: Genomics analysis and visualization platform (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi).
Repetition and statistical analysis
All experiments were repeated more than three times. The results are expressed as the mean ± S.D. All statistical tests were carried out using SPSS software (version 25; IBM, Armonk, NY, USA). Differences between two groups were evaluated using an unpaired Student’s t test and were considered to be statistically significant when p < 0.05. For Kaplan–Meier survival analysis and immunohistochemical analyses, p values were calculated using the log-rank test.
Results
Arrb2 decreases HIF-1α expression in glioblastoma cells under hypoxic conditions
Similar to many solid tumors, HIF-1α mainly promotes tumor progression in GBM. Thus, we investigated the correlation between Arrb2 expression and HIF-1α protein levels in the human GBM cell lines U87MG, U251MG, H4, and T98G. The increase in HIF-1α protein levels in response to exposure to hypoxia were significantly decreased by overexpression of Arrb2 in all GBM cell lines we tested (Fig. 1A). Moreover, hypoxia-induced HIF-1α protein levels were highest in U87MG cells, where Arrb2 expression levels were the lowest (Fig. 1B). T98G and H4 cells with high expression levels of Arrb2 showed low levels of HIF-1α and were insensitive to hypoxic stress. Immortalized human astrocytes had low protein levels of both HIF-1α and Arrb2. From these results, we assumed that Arrb2 might be a negative regulator of HIF-1α protein expression in GBM, and we selected U87MG cells for further studies to verify Arrb2 as a novel negative regulator of HIF-1α. To examine the intracellular expression of Arrb2 and HIF-1α, a Flag-tagged Arrb2 expression vector was transfected into U87MG cells, and cells were exposed to hypoxia. Stabilized HIF-1α was specifically detected in the nucleus of hypoxia-exposed U87MG cells, and HIF-1α protein was diminished in Arrb2-overexpressing cells (Fig. 1C). The diminished nuclear expression of HIF-1α by Arrb2-overexpression was quantified by comparing the fluorescent intensities of HIF-1α in Flag-positive cells and Flag-negative cells (Fig. 1D). Next, we investigated whether Arrb2 reduces HIF-1A mRNA levels by Reverse transcription (RT)-polymerase chain reaction (PCR) and quantitative-PCR (qPCR). As shown in Fig. 1E, F, HIF-1A mRNA levels were not changed by Arrb2 overexpression, indicating that HIF-1α was regulated by Arrb2 at the protein level and not at the mRNA level.

A GBM cell lines were transfected with pCMV-Flag-Arrb2 and exposed to hypoxia for 8 h. Whole cell lysates were analyzed by immunoblotting for HIF-1α and Flag. B GBM cells and immortalized fetal astrocytes were exposed to hypoxia for the indicated times. Western blotting was performed using antibodies against HIF-1α and Arrb2. SE short exposure, LE long exposure. C U87MG cells were transfected with pCMV-Flag-Arrb2 and incubated in hypoxic conditions for 8 h. Immunofluorescence staining was performed using antibodies for HIF-1α (red) and Flag (green). The cellular nuclei were counterstained with Hoechst 33342 (blue). The scale bar is 20 μm. D The nuclear intensities of HIF-1α in Flag-Arrb2 overexpressing cells were quantified and plotted from four independent experiments. ***p < 0.001 versus Flag-negative cells. E, F Transient Arrb2-transfected U87MG cells were incubated in hypoxic conditions, and RT-PCR (E) and qPCR (F) were performed using specific primers as indicated. Relative HIF-1A expression from qPCR was divided and quantified as 18S rRNA expression from three independent experiments.
Arrb2 suppresses HIF-1α protein stability
Generally, protein levels can be regulated by the translation and stability of the protein. First, we examined whether Arrb2 decreases the synthesis of HIF-1α protein by treating cells with MG132, a specific inhibitor of the 26S proteasome. HIF-1α protein gradually accumulated with MG132 treatment over time (Fig. 2A), and the accumulation rate was not affected by Arrb2 overexpression (Fig. 2B). Under MG132 treatment and hypoxic conditions, the rate of HIF-1α protein accumulation was not changed by Arrb2 overexpression (Fig. 2C, D), suggesting that the decrease in HIF-1α by Arrb2 is not involved in HIF-1α translation. To identify the possibility that Arrb2 decreases HIF-1α protein stability, we examined the half-life of HIF-1α protein by treatment with CHX, an inhibitor of protein synthesis. HIF-1α protein was gradually decreased with increasing treatment time of CHX in normal cells and cells overexpressing Arrb2, and the level at the starting point was also decreased by Arrb2 overexpression (Fig. 2E). The rate of degradation of HIF-1α was significantly decreased by Arrb2 (Fig. 2F), supporting the notion that the reduction of HIF-1α level by Arrb2 is due to decreased HIF-1α stability. Moreover, the Arrb2-mediated reduction in HIF-1α protein was restored by treatment with MG132 (Fig. 2G). The reduction of HIF-1α by Arrb2 was mainly detected in the nucleus under hypoxic conditions, and the recovery of HIF-1α protein by MG132 was detected in the cytosol and nucleus (Fig. 2H). Because the main target sites of HIF-1α degradation are two proline residues in the ODD domain, the HIF-1α-DM expression vector, which is double mutated on P402 and P564, was transfected into U87MG cells. HIF-1α-DM protein was not decreased by Arrb2, but HIF-1α-WT protein was decreased by Arrb2 (Fig. 2I), suggesting that Arrb2 decreased HIF-1α protein levels by blocking the 26S proteasomal degradation pathway.

A, C Flag-Arrb2-transfected U87MG cells were treated with 10 μM MG132 for the indicated times under normoxic (A) and hypoxic conditions (C). Whole cell lysates were analyzed by immunoblotting for HIF-1α and Flag. E Flag-Arrb2-transfected U87MG cells were treated with 50 μM CHX under hypoxic conditions for the indicated times. B, D, F The relative expression of HIF-1α was quantified using ImageJ software and plotted from three independent experiments. *p < 0.05; **p < 0.01 versus Mock. Mock; empty-vector-transfected cells. G Flag-Arrb2-transfected U87MG cells were treated with 10 μM MG132 for 4 h and incubated in hypoxic conditions for the last 2 h. Cell lysates were harvested and subjected to western blot analysis for HIF-1α and Flag. H Nuclear and cytosolic lysates were prepared from Flag-Arrb2-transfected U87MG cells under normoxic or hypoxic conditions as indicated (left). Additionally, cells were treated with MG132 (10 μM) for 4 h (right). Immunoblotting for lamin A/C and α-tubulin was performed to determine the purity of nuclear and cytosolic extracts, respectively. I pcDNA-HIF-1α-WT, pcDNA-HIF-1α-DM, and pCMV-Myc-Arrb2 vectors were transiently transfected into U87MG cells as indicated, and western blotting was performed using anti-HIF-1α and anti-c-Myc antibodies.
pVHL-dependent HIF-1α degradation is triggered by Arrb2
To determine the interaction of Arrb2 with HIF-1α, an immunoprecipitation assay was performed based on exogenous expression of Flag-Arrb2 and GFP-HIF-1α which is a fusion protein between GFP and HIF-1α and produces the stabilized HIF-1α [41]. The results revealed that immunoprecipitated Arrb2 bound to HIF-1α in normoxic and hypoxic conditions (Fig. 3A). Moreover, endogenous HIF-1α was immunoprecipitated from MG132 treated cell extracts, endogenous Arrb2 was coimmunoprecipitated (Fig. 3B). In oxygenated cells, P402 and P564 of HIF-1α are hydroxylated by PHDs, and pVHL binds to hydroxylated HIF-1α and acts as the E3 ubiquitin ligase [6, 7]. From our results and previous reports, we hypothesized that Arrb2 gathers together with PHDs and pVHL to degrade HIF-1α protein. Therefore, we examined whether Arrb2 could stimulate the interaction of PHD2 or pVHL with HIF-1α while blocking the 26S proteasomal pathway with MG132. As shown in Fig. 3C, PHD2-HIF-1α and pVHL-HIF-1α complexes were increased by Arrb2 overexpression under normoxic and hypoxic conditions. In addition, the decrease in prolyl-hydroxylated HIF-1α in response to hypoxic exposure was significantly attenuated by Arrb2 overexpression (Fig. 3D), indicating that Arrb2 triggered the interaction of PHDs with HIF-1α. However, Arrb2 did not change the protein levels of PHD2 and pVHL (Fig. 3E). The overexpression of PHD2 and Arrb2 decreased HIF-1α protein levels, and the overexpression of both PHD2 and Arrb2 stimulated almost complete degradation of HIF-1α protein (Fig. 3F). Taken together, these results demonstrated that Arrb2 stimulates the interaction of HIF-1α with PHD2 and pVHL and finally promotes pVHL-dependent proteasomal degradation of HIF-1α in GBM.

A U87MG cells were transfected with an expression vector containing GFP-tagged HIF-1α and/or Flag-tagged Arrb2 as indicated. Then, cells were treated with MG132 under normoxic or hypoxic conditions. Whole cell lysates were immunoprecipitated with anti-Flag agarose gel, and the immunoprecipitants were subjected to western blot analysis using a HIF-1α antibody. B MG132 treated cell lysates were immunoprecipitated with HIF-1α antibody or normal IgG as a negative control. C pCMV-Flag-Arrb2 vector was transfected into U87MG cells, and cells were treated with 10 μM MG132 for 4 h under normoxic or hypoxic conditions. Cell lysates were immunoprecipitated with HIF-1α antibody. Immunoprecipitated materials and total cell extracts were analyzed by western blotting with specific antibodies as indicated. D, E pCMV-Flag-Arrb2 was transiently transfected into U87MG cells and incubated with MG132 under normoxic or hypoxic conditions. Immunoblotting was performed with specific antibodies as indicated. F pCDNA-Flag-HIF-1α, pGFP-PHD2, and pCMV-Myc-Arrb2 vectors were transfected as indicated, and cell extracts were subjected to immunoblot analysis using specific antibodies as indicated.
Arrb2 inhibits the tumorigenic activities of GBM
Because HIF-1α is an essential factor for tumorigenesis in many solid tumors, including GBM, we tested whether Arrb2 affects the tumorigenic activity of U87MG cells. Before performing the clonogenic assay, stable Arrb2-overexpressing U87MG cells were selected with G418 for 1 week. Arrb2-overexpressing U87MG cells showed fewer colonies and smaller area occupied by colonies (Fig. 4A, B). Tumor angiogenesis is an essential event for tumor progression and is largely dependent on the function and expression of HIF-1α [42, 43]. To investigate whether Arrb2 modulates angiogenesis during tumor progression, ex vivo and in vitro angiogenesis assays were performed. Conditioned media were collected from stably Arrb2-transfected U87MG cells and treated with rat aortic rings. The number of microvessel sprouting from the aortic rings was increased by treatment with conditioned medium from hypoxia-exposed U87MG cells, and this induction was reduced by conditioned medium from hypoxia-exposed Arrb2-overexpressing U87MG cells (Fig. 4C, D). When a tube formation assay was performed using HMECs, the same results were obtained (Fig. 4E, F), suggesting that Arrb2 may reduce the tumorigenic activity of GBM and the secretion of angiogenic factors from GBM.

A U87MG cells were stably transfected with pCMV-Flag-Arrb2 and selected with G418 for 1 week. The transfected cells (1 × 104) were seeded on 60 mm dishes and incubated for 2 weeks. The scale bar is 5 mm. B The colony number was counted, and the area were measured using ImageJ software. Data are presented as the means ± S.D. of three independent experiments. *p < 0.05, ***p < 0.001 versus Mock. C Stable Arrb2-transfected cells were incubated in hypoxic conditions, and conditioned media were collected. Rat aorta rings were seeded on Matrigel and treated with each conditioned medium for 5 days. Representative images of microvessel sprouting are shown. The scale bar is 0.5 mm. D The number of sprouted microvessels was quantified from three independent assays using ImageJ software. **p < 0.01 versus normoxia; ###p < 0.001 versus Mock under hypoxic conditions. E HMEC-1s were seeded on Matrigel and treated with each conditioned medium as indicated. The scale bar is 0.25 mm. F The number of meshes or extremes was quantified from three independent experiments using ImageJ software. *p < 0.05 versus normoxia; #p < 0.05, ##p < 0.01 versus mock under hypoxic conditions. Mock, empty vector-transfected cells.
Arrb2 inhibits in vivo tumor growth
To further confirm the antitumorigenic effects of Arrb2, we generated a xenograft mouse tumor model. Lentiviruses for Arrb2 were collected and used to infect U87MG cells for animal experiments. Before making tumor-bearing mice, viral infection and expression of Arrb2 were confirmed in U87MG-Arrb2 cells. Infected cells exhibited a green fluorescent signal, and Arrb2 was effectively expressed in the infected cells (Fig. 5A). Tumor formation was induced by the injection of U87MG-Arrb2 cells into BALB/c-nude mice, and tumors were isolated from mice 4 weeks after injection. As shown in Fig. 5B, C, the tumors derived from Arrb2-expressing cells were smaller and paler than those derived from scramble virus-infected cells. Because tumor cell density also affects tumor malignancy as well as tumor size and volume, we examined the density of U87MG cells inside the tumor tissue stained by hematoxylin and eosin. We found that Arrb2-overexpressing tumors were less compact (Fig. 5D, E). Moreover, HIF-1α protein levels in tumor tissues were downregulated by overexpression of Arrb2 (Fig. 5F, G). To examine whether blood capillaries within the tumors were decreased by Arrb2 expression, the tissues were stained with CD31, a specific marker for endothelial cells. As shown in Fig. 5F, G, the capillaries inside the tumors were significantly decreased by Arrb2 overexpression. From these results, we confirmed the functional contribution of Arrb2 to HIF-1α expression and tumor growth in vivo.

A U87MG cells were infected with Arrb2-lentiviral particles for 72 h, and the infection efficacy was examined by GFP fluorescence under a confocal microscope. Arrb2 expression was examined by immunofluorescence staining (left) and western blotting (right). B BALB/c-nude mice were injected with Arrb2-expressing U87MG cells. Four weeks after injection, the tumors were isolated. The scale bar is 2.5 mm. C Tumor volume (left) and weight (right) were measured and plotted from three independent experiments. ***p < 0.001, compared to the control groups. D Tumor sections were stained with hematoxylin and eosin. The scale bar is 0.1 mm. E Tumor densities were quantified using stained images from three independent experiments and plotted. *p < 0.05, compared to the control groups. F Tumor sections were immunostained with specific antibodies for HIF-1α, CD31, and Arrb2. Lined-squares are the twofold magnified images for dotted-squares. The scale bar is 0.1 mm. G The immunostained signal was quantitatively analyzed using ImageJ software, and data are presented as the means ± S.D. from three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 compared to the control groups.
Arrb2 correlates with inhibition of HIF-1α and good prognosis on GBM
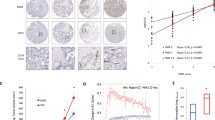
In an effort to identify the relationship between Arrb2 and HIF-1α expression in GBM, immunohistochemical analysis was performed using a GBM patient tissue array. We found that high Arrb2-expressing tumors contained decreased HIF-1α expression; in contrast, low Arrb2-expressing tumors showed increased HIF-1α protein expression (Fig. 6A). Analysis of immunohistochemistry for patients showed that Arrb2 expression was inversely proportional to HIF-1α expression in GBM patients (Fig. 6B). To confirm this negative correlation, we compared the mRNA expression of the ARRB2 and HIF-1α downstream genes VEGF and GLUT1 using R2: Genomic Analysis and Visualization Platform of GBM TCGA database. As shown in Fig. 6C, both relationships between ARRB2 and VEGF, ARRB2 and GLUT1 showed a significantly negative correlation, supporting that Arrb2 is a negative regulator of HIF-1α. When ARRB2 expression was sub divided in groups provided separately according to the cohort in TCGA database using R2: Genomic Analysis and Visualization Platform, the levels of ARRB2 mRNA in all glioma patients and GBM patients groups were relatively lower than the level in normal brain group (Fig. 6D). To further evaluate the potential correlation of ARRB2 expression with patient outcome, we performed Kaplan–Meier survival analysis using TCGA GBM datasets. The analysis showed that patients with high expression of ARRB2 have longer survival than those with low expression of ARRB2 (Fig. 6E). In addition, the level of ARRB2 expression was also negatively correlated with glioma grade from R2 analysis (Fig. 6F). These results strongly suggest that aberrantly elevated Arrb2 expression correlates with the downregulation of HIF-1α expression and function, consequently increasing the survival rate of GBM patients.

A Immunohistochemical detection of HIF-1α and Arrb2 was performed using a paraffin-embedded human glioma patient tissue array (GL806e, US Biomax). Lined-squares are the 2-fold magnified images for dotted-squares. The scale bar is 0.1 mm. B The positive areas of HIF-1α and Arrb2 for 56 patients were analyzed using ImageJ software and plotted. P values were calculated using the log-rank test. C Gene correlation between VEGF (left) or GLUT1 (right) and ARRB2 was plotted using R2: Genomics Analysis and Visualization Platform (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi). Datasets were exported from Tumor Glioma-Sun-153_MAS5.0-u133p2. D ARRB2 expression was represented in glioma and GBM groups using R2: Genomics Analysis and Visualization Platform (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi). Datasets were exported from Normal Brainregions-Berchtold-172_MAS5.0-u133p2 (NB), Tumor Glioma-French-284_MAS5.0-u133p2 (G1), Tumor Glioma-Gleize-30_MAS5.0-u133p2 (G2), Tumor Glioma-Paugh-37_MAS5.0-u133p2 (G3), Tumor Glioma-Kawaguchi-50_MAS5.0-u133p2 (G4), Tumor Glioblastoma-Pfister-46_MAS5.0-u133p2 (GBM1), Tumor Glioblastoma-Loeffler-70_MAS5.0-u133p2 (GBM2), and Tumor Glioblastoma-Hegi-84_MAS5.0-u133p2 (GBM3). n number of patients. E ARRB2 mRNA expression data set was obtained from TCGA for GBM patients. Recurrence-free survival analysis between the two groups was performed by using the Kaplan-Meir method and the log-rank test to compare survival rates between the ARRB2 high and low groups. F ARRB2 expression was represented with the grade of glioma using R2: Genomics Analysis and Visualization Platform (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi). Datasets were exported from Tumor Glioma-Sun-153_MAS5.0-u133p2. Patients were divided into three groups: low-grade (g2, n = 7), grade 3 (g3, n = 19), and grade 4 (g4, n = 77).
Discussion
The present study demonstrated for the first time that Arrb2 binds to HIF-1α, thereby stimulating the degradation of HIF-1α by recruiting pVHL and PHDs in GBMs. Consequently, Arrb2 inhibits tumorigenesis and angiogenesis during GBM progression. Our data explain why the level of Arrb2 negatively correlates with HIF-1α and HIF-1α targets in clinical GBM samples and correlates with TV, weight, and density in a xenograft GBM tumor model. Thus, we propose that Arrb2 obstructs GBM growth and progression through destabilization of HIF-1α and downregulation of angiogenesis, ultimately providing potential strategies and biomarkers for optimal development of novel antitumorigenic therapies for GBM and potentially for other solid tumors.
Hypoxia is a common condition in solid tumors and is frequent in GBMs due to its rapid growth [44]. Consistent with the notion that hypoxia is a major challenge for the growth, malignancy, and metastasis of solid tumors, HIF-1 signaling is involved in almost all steps of tumor progression [45]. In addition, HIF-1α protein levels and the levels of a large number of target genes are upregulated in clinical specimens [46]. HIF-1α has also been associated with increased mortality in most human cancers through multiple aspects of the protumorigenic process, including not only angiogenesis, tumor growth, and metastasis but also chemoresistance and radioresistance [4, 47]. Therefore, many oncologists and basic researchers have tried to find novel HIF-1α partners and establish regulatory mechanisms to overcome the hypoxia hurdle. Originally, to identify candidate proteins that interact with HIF-1α and function in the oxygen-dependent degradation of HIF-1α, we performed yeast two-hybrid screening using the ODD domain of HIF-1α as bait in a mouse embryonic library [48, 49]. Although Arrb2 was one of the HIF-1α binding candidates, we dropped the study on Arrb2 because it was known as an adapter protein of GPCRs.
Arrbs are ubiquitously expressed and have been identified as essential multifunctional adaptor molecules for the desensitization, sequestration, recycling, and downregulation of most GPCRs [50]. Subsequently, functions of Arrb2 independent of GPCRs have revealed additional roles in cell signal regulation, such as cell growth, proliferation, migration, invasion, metastasis, angiogenesis, and cell death signaling in a variety of cancer cells. For example, Arrb2 mediates LPA-induced migration and invasion through the Ral GTPase-dependent pathway in breast cancer cells [27] and increases tumor growth and progression via Wnt signaling in myeloid leukemia and colorectal cancer [51]. In a recent study, the cell cycle and tumor growth in renal cell carcinoma were regulated by Arrb2 [20]. On the other hand, other reports demonstrated a suppressive effect of Arrb2 in different types of cancer. Arrb2 inhibits cell migration, invasion, and tumorigenesis via downregulation of the AKT signaling pathway in hepatocellular carcinoma [29], and its downregulation was associated with poorer prognosis in lung cancer patients [30]. Additionally, apoptosis through the NFκB pathway is promoted by Arrb2 in colorectal cancer [52]. The data in the present study strongly support that Arrb2 negatively regulates tumorigenic activities, including proliferation, migration, and angiogenesis, in GBM. We assume that these contradictory functions of Arrb2 may be cell context- and cancer type-dependent, and the function of Arrb2 in other types of cancer should be elucidated.
In the hypoxic tumor microenvironment, HIF-1 induces the transcription of target genes involved in crucial aspects of cancer biology, including cell survival, glucose metabolism, angiogenesis, migration, and invasion [53, 54]. Moreover, HIF-1α-mediated tumor angiogenesis has been shown to play an essential role in tumor growth [54]. Because HIF-1 plays multiple roles in tumor progression, inhibition of HIF-1 activity in preclinical studies has marked effects on tumor growth and angiogenesis. Notably, GBM is a highly vascularized brain cancer and mainly promotes angiogenesis by upregulating angiogenic factors such as VEGF [55]. Therefore, angiogenesis inhibition has become an important therapeutic strategy for GBM therapy. Previous studies have demonstrated that Arrb1 elevates angiogenesis by increasing of HIF-1α stability in breast cancer and that its expression is decreased in GBM patients [25, 34]. Here, we first found that Arrb2 significantly decreases HIF-1α protein stability and reduces angiogenesis in GBM cell lines. In addition, our data support the notion that Arrb2 significantly suppresses GBM growth and that Arrb2 negatively correlates with HIF-1α expression and prognosis in GBM patients.
Oxygen-dependent hydroxylation of HIF-1α by PHDs and recruitment of pVHL have been well-identified as central oxygen-sensing HIF-1α degradation pathways. In this work, we demonstrated that Arrb2 is involved with HIF-1α degradation pathway-associated factors, such as PHD2 and pVHL. A key question is how Arrb2 works in the HIF-1α degradation pathway. In a previous study, Arrb2 delocalized nuclear proteins, such as MDM2 and JNK3. Nucleocytoplasmic shuttling of Arrb2 for cytoplasmic relocalization of nuclear protein occurs through protein/protein interactions [22, 56]. We propose that Arrb2 forms a multicomplex with HIF-1α, PHDs, and pVHL, thereby stimulating the ubiquitin-mediated 26S proteasomal pathway for HIF-1α. We found that the interactions of pVHL with HIF-1α and PHD2 with HIF-1α were increased and the hydroxylation of HIF-1α was increased during Arrb2 overexpression. PHD2 can hydroxylase prolines of ODD whenever it binds to ODD, and pVHL specifically binds to hydroxylated ODD. However, under hypoxic condition, the interaction of pVHL and PHD2 with HIF-1α is minimal because of low oxygen. It means that O2-dependent hydroxylation of HIF1-α by PHDs is reduced, not completely blocked. According to the several reports. the activity of PHDs in hypoxia is reduced in a graded manner, but significant PHD activity is still observed at 1% oxygen because of their high affinity for oxygen, and HIF-1α still become hydroxylated under nearly completely anoxic conditions [57,58,59,60,61]. Therefore, we could assume that Arrb2 stimulates the PHD2 and HIF-1α interaction under both normoxic and hypoxic conditions. Moreover, it was reported that Arrb2 regulates internalization of β2-adrenoceptor (β2-AR) through direct binding to PHD2. Arrb2 is also hydroxylated at proline residues by PHD2 and is recruited to β2-AR [62]. From our data and recent reports, we speculate that Arrb2 directly interacts with PHD2, recruits HIF-1α and affects the docking of HIF-1α and the pVHL complex. Perhaps other factors are presumed to directly or indirectly affect the determination of the substrate to be hydroxylated by PHD2.
To better understand the potential clinical implications of Arrbs for the prognosis of human cancer, we visited publicly available datasets for GBM. The ARRB2 level in GBM specimens was significantly lower than that in normal brain tissue (Fig. 6D). We also observed that the cancer tissues expressing high Arrb2 levels expressed lower levels of HIF-1α protein, and the tissues expressing low Arrb2 levels expressed higher levels of HIF-1α protein (Fig. 6A, B). In summary, our findings indicate that Arrb2 enhances posttranslational hydroxylation of the proline residues of HIF-1α by PHD2 and enhances the interaction with pVHL. Ultimately, HIF-1α degradation is increased in the tumor microenvironment. Additionally, our study revealed that Arrb2 has an antitumorigenic function in GBM not only through the reduction of HIF-1α but also through the inhibition of angiogenesis. Therefore, our study identified an important novel function of Arrb2 as a downregulator of HIF-1α, which supports the idea that Arrb2 could be a target for antitumor therapy of GBM.
References
Semenza GL. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. 2010;2:336–61.
Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–4.
Chan DA, Sutphin PD, Yen SE, Giaccia AJ. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2005;25:6415–26.
Melillo G. Targeting hypoxia cell signaling for cancer therapy. Cancer Metastasis Rev. 2007;26:341–52.
Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–40.
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5.
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8.
Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA. 1996;93:12969–73.
Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56.
Semenza GL. A compendium of proteins that interact with HIF-1alpha. Exp Cell Res. 2017;356:128–35.
Li Z, Wang D, Messing EM, Wu G. VHL protein-interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1alpha. EMBO Rep. 2005;6:373–8.
Hubbi ME, Hu H, Kshitiz, Ahmed I, Levchenko A, Semenza GL. Chaperone-mediated autophagy targets hypoxia-inducible factor-1alpha (HIF-1alpha) for lysosomal degradation. J Biol Chem. 2013;288:10703–14.
Kang X, Li J, Zou Y, Yi J, Zhang H, Cao M, et al. PIASy stimulates HIF1alpha SUMOylation and negatively regulates HIF1alpha activity in response to hypoxia. Oncogene. 2010;29:5568–78.
Liu W, Shen SM, Zhao XY, Chen GQ. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int J Biochem Mol Biol. 2012;3:165–78.
Barak LS, Ferguson SS, Zhang J, Caron MG. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J Biol Chem. 1997;272:27497–500.
Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–7.
Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. beta-Arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc Natl Acad Sci USA. 2004;101:8603–7.
Wang P, Gao H, Ni Y, Wang B, Wu Y, Ji L, et al. Beta-arrestin 2 functions as a G-protein-coupled receptor-activated regulator of oncoprotein Mdm2. J Biol Chem. 2003;278:6363–70.
Rosano L, Cianfrocca R, Masi S, Spinella F, Di Castro V, Biroccio A, et al. Beta-arrestin links endothelin A receptor to beta-catenin signaling to induce ovarian cancer cell invasion and metastasis. Proc Natl Acad Sci USA. 2009;106:2806–11.
Masannat J, Purayil HT, Zhang Y, Russin M, Mahmud I, Kim W, et al. betaArrestin2 mediates renal cell carcinoma tumor growth. Sci Rep. 2018;8:4879.
Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem. 2002;277:9429–36.
Ma L, Pei G. Beta-arrestin signaling and regulation of transcription. J Cell Sci. 2007;120:213–8.
Zhang Z, Hao J, Zhao Z, Ben P, Fang F, Shi L, et al. beta-Arrestins facilitate ubiquitin-dependent degradation of apoptosis signal-regulating kinase 1 (ASK1) and attenuate H2O2-induced apoptosis. Cell Signal. 2009;21:1195–206.
Buchanan FG, Gorden DL, Matta P, Shi Q, Matrisian LM, DuBois RN. Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proc Natl Acad Sci USA. 2006;103:1492–7.
Shenoy SK, Han S, Zhao YL, Hara MR, Oliver T, Cao Y, et al. beta-arrestin1 mediates metastatic growth of breast cancer cells by facilitating HIF-1-dependent VEGF expression. Oncogene. 2012;31:282–92.
Qiu C, Zheng C, Zhu L, Qu X, Shen H, Du J. beta-arrestin1 over-expression is associated with an unfavorable prognosis in lung adenocarcinomas and correlated with vascular endothelial growth factor. Int J Clin Exp Pathol. 2015;8:3785–93.
Li TT, Alemayehu M, Aziziyeh AI, Pape C, Pampillo M, Postovit LM, et al. Beta-arrestin/Ral signaling regulates lysophosphatidic acid-mediated migration and invasion of human breast tumor cells. Mol Cancer Res. 2009;7:1064–77.
Fereshteh M, Ito T, Kovacs JJ, Zhao C, Kwon HY, Tornini V, et al. beta-Arrestin2 mediates the initiation and progression of myeloid leukemia. Proc Natl Acad Sci USA. 2012;109:12532–7.
Sun WY, Hu SS, Wu JJ, Huang Q, Ma Y, Wang QT, et al. Down-regulation of beta-arrestin2 promotes tumour invasion and indicates poor prognosis of hepatocellular carcinoma. Sci Rep. 2016;6:35609.
Wu Z, Tong W, Tan Z, Wang S, Lin P. The clinical significance of beta-arrestin 2 expression in the serum of non-small cell lung cancer patients. Zhongguo Fei Ai Za Zhi. 2011;14:497–501.
Wen PY, Huse JT. 2016 World Health Organization classification of central nervous system tumors. Continuum. 2017;23:1531–47.
Noiphithak R, Veerasarn K. Clinical predictors for survival and treatment outcome of high-grade glioma in Prasat Neurological Institute. Asian J Neurosurg. 2017;12:28–33.
Alphandery E. Glioblastoma treatments: an account of recent industrial developments. Front Pharmacol. 2018;9:879.
Mandell JW, Glass G, Gianchandani EP, Locke CN, Amos S, Bourne TD, et al. Dephosphorylation of beta-arrestin 1 in glioblastomas. J Neuropathol Exp Neurol. 2009;68:535–41.
Carpentier G. Angiogenesis Analyzer for ImageJ. 2012. http://image.bio.methods.free.fr/ImageJ/?Angiogenesis-Analyzer-for-ImageJ.
Kapoor A, Chen CG, Iozzo RV. A simplified aortic ring assay: a useful ex vivo method to assess biochemical and functional parameters of angiogenesis. Matrix Biol. 2020;6–7:100025.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Goldman M, Craft B, Swatloski T, Cline M, Morozova O, Diekhans M, et al. The UCSC Cancer Genomics Browser: update 2015. Nucleic Acids Res. 2015;43:D812–7.
Hothorn T, Zeileis A. Generalized maximally selected statistics. Biometrics. 2008;64:1263–9.
Goel MK, Khanna P, Kishore J. Understanding survival analysis: Kaplan-Meier estimate. Int J Ayurveda Res. 2010;1:274–8.
Kallio PJ, Okamoto K, O’Brien S, Carrero P, Makino Y, Tanaka H, et al. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998;17:6573–86.
Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59:5830–5.
Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–62.
Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005;7:134–53.
Carroll VA, Ashcroft M. Targeting the molecular basis for tumour hypoxia. Expert Rev Mol Med. 2005;7:1–16.
Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol. 2014;49:1–15.
Wouters A, Pauwels B, Lardon F, Vermorken JB. Review: implications of in vitro research on the effect of radiotherapy and chemotherapy under hypoxic conditions. Oncologist. 2007;12:690–712.
Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, et al. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2002;111:709–20.
Bae MK, Ahn MY, Jeong JW, Bae MH, Lee YM, Bae SK, et al. Jab1 interacts directly with HIF-1alpha and regulates its stability. J Biol Chem. 2002;277:9–12.
Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–65.
Bonnans C, Flaceliere M, Grillet F, Dantec C, Desvignes JP, Pannequin J, et al. Essential requirement for beta-arrestin2 in mouse intestinal tumors with elevated Wnt signaling. Proc Natl Acad Sci USA. 2012;109:3047–52.
Ren W, Wang T, He X, Zhang Q, Zhou J, Liu F, et al. betaarrestin2 promotes 5FUinduced apoptosis via the NFkappaB pathway in colorectal cancer. Oncol Rep. 2018;39:2711–20.
Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;9:10–7.
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32.
Zhang M, Ye G, Li J, Wang Y. Recent advance in molecular angiogenesis in glioblastoma: the challenge and hope for anti-angiogenic therapy. Brain Tumor Pathol. 2015;32:229–36.
Song X, Raman D, Gurevich EV, Vishnivetskiy SA, Gurevich VV. Visual and both non-visual arrestins in their “inactive” conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J Biol Chem. 2006;281:21491–9.
Zhao L, Liu Z, Yang F, Zhang Y, Xue Y, Miao H, et al. Intrabody against prolyl hydroxylase 2 promotes angiogenesis by stabilizing hypoxia-inducible factor-1alpha. Sci Rep. 2019;9:11861.
Tian YM, Yeoh KK, Lee MK, Eriksson T, Kessler BM, Kramer HB, et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J Biol Chem. 2011;286:13041–51.
Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–80.
Kwon HS, Choi YK, Kim JW, Park YK, Yang EG, Ahn DR. Inhibition of a prolyl hydroxylase domain (PHD) by substrate analog peptides. Bioorg Med Chem Lett. 2011;21:4325–8.
Bruick RK. Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor. Genes Dev. 2003;17:2614–23.
Yan B, Huo Z, Liu Y, Lin X, Li J, Peng L, et al. Prolyl hydroxylase 2: a novel regulator of beta2—adrenoceptor internalization. J Cell Mol Med. 2011;15:2712–22.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MIST) (NRF-2021R1A2C1003297, J-WJ) and by the Ministry of Education (NRF-2019R1I1A1A01062164, W-YB).
Author information
Authors and Affiliations
Contributions
W-YB performed experimental procedures, paper writing, and data analysis. J-SC performed experimental procedures, and data analysis. SN analyzed TCGA data. J-WJ supervised the entire project, designed the experiment, analyzed the data, revised the paper, and approved the final version of the paper for publication.
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing interests.
Ethical approval
All mouse procedures in this study were approved by the Institutional Animal Care and Use Committee protocol approved by the Committee for Care and Use of Laboratory Animals at Kyung Hee University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by R. De Maria
Rights and permissions
About this article

Cite this article
Bae, WY., Choi, JS., Nam, S. et al. β-arrestin 2 stimulates degradation of HIF-1α and modulates tumor progression of glioblastoma. Cell Death Differ 28, 3092–3104 (2021). https://doi.org/10.1038/s41418-021-00802-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-021-00802-2
This article is cited by
-
Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis
Journal of Experimental & Clinical Cancer Research (2023)
-
Alleviating hypoxia to improve cancer immunotherapy
Oncogene (2023)
