
Abstract
Renal tubular cell (RTC) death and inflammation contribute to the progression of obstructive nephropathy, but its underlying mechanisms have not been fully elucidated. Here, we showed that Gasdermin E (GSDME) expression level and GSDME-N domain generation determined the RTC fate response to TNFα under the condition of oxygen-glucose-serum deprivation. Deletion of Caspase-3 (Casp3) or Gsdme alleviated renal tubule damage and inflammation and finally prevented the development of hydronephrosis and kidney fibrosis after ureteral obstruction. Using bone marrow transplantation and cell type-specific Casp3 knockout mice, we demonstrated that Casp3/GSDME-mediated pyroptosis in renal parenchymal cells, but not in hematopoietic cells, played predominant roles in this process. We further showed that HMGB1 released from pyroptotic RTCs amplified inflammatory responses, which critically contributed to renal fibrogenesis. Specific deletion of Hmgb1 in RTCs alleviated caspase11 and IL-1β activation in macrophages. Collectively, our results uncovered that TNFα/Casp3/GSDME-mediated pyroptosis is responsible for the initiation of ureteral obstruction-induced renal tubule injury, which subsequentially contributes to the late-stage progression of hydronephrosis, inflammation, and fibrosis. This novel mechanism will provide valuable therapeutic insights for the treatment of obstructive nephropathy.
Similar content being viewed by others


Introduction
Ureteral obstruction is one of the most common problems in obstructive nephropathy. How to prevent the progression of obstructive nephropathy and help the kidney recover from the reversible ureteral obstruction is still a great challenge in clinical practice. If the obstruction can not be relieved in time, it will lead to hydronephrosis, eventually resulting in renal fibrosis and loss of renal function. Obstruction of the urinary tract may cause urinary flow disorder, leading to substantial damage to the tubular epithelial cells and the vascular network with rapid infiltration of inflammatory cells [1]. In the obstructed kidney, tubular cell death is also thought to induce the origin of tubular loss and tubulointerstitial atrophy and then promotes the development of hydronephrosis, interstitial inflammation, and fibrosis [2,3,4]. A large number of factors, including hypoxia, ischemia, inflammatory cytokines, reactive oxygen species, and mechanical stretch, can initiate tubular cell death [5,6,7,8]. However, during the progression of obstructive nephropathy, the causal relation between tubular cell death and fibrosis is still unclear.
Unilateral ureteral obstruction (UUO) in mice has been commonly utilized as an important model for exploring the pathophysiology of obstructive nephropathy and the mechanisms of renal fibrosis [9]. In this mouse model, an immediate and dramatic injury to tubular cells occurs during the early stage of acute unilateral obstruction [10]. Increasing evidence has shown that cell death plays a predominant role at the early stage of pathological progression of acute obstructive nephropathy. Mobilization and infiltration of macrophages and neutrophils and subsequent release of cytokines, including interleukin-1β (IL-1β) and IL-18 greatly contribute to the progression of fibrosis [10,11,12]. Danger-associated molecular patterns (DAMPs), such as high-mobility group box 1 (HMGB1) released from necrotic cells activate the innate immune system to produce inflammatory cytokines, which further amplify the inflammatory response or even trigger the cell death process [13, 14]. This loop has been considered as an important common relation of the cell death-inflammation cycle, which greatly contributes to the progression of renal fibrosis [15, 16]. Receptor-interacting protein kinase-3 (RIPK3), a key regulator of necroptosis, has been implicated in acute kidney injury and progression to CKD [16,17,18,19,20]. One study demonstrated that deletion of Ripk3 protected against UUO-induced kidney fibrosis in mice [8]. However, Ripk3 deficiency could not completely prevent tubular cell death and the development of renal fibrosis. Thus, we speculated that other cell death signaling pathways are involved in tubular cell damage and renal fibrosis progression after ureteral obstruction.
Pyroptosis, is now defined as gasdermin-mediated programmed necrosis [21]. The gasdermin family consists of Gasdermin A, B, C, D, E, and DFNB59. Except for DFNB59, all the gasdermin members possess an N-terminal pore-forming domain and a C-terminal repressor domain. The gasdermins execute proinflammatory cell death via the pore-forming activity after its cleavage by upstream proteases [22, 23]. The Gasdermin D (GSDMD), a substrate of caspase1/4/5/11, mediates the pyroptosis triggered by inflammasome activation [23,24,25]. GSDME, expressed in most normal tissue cells, could switch apoptosis to pyroptosis. Caspase-3 (Casp3) specifically cleaves GSDME in the linker, releasing a GSDME-N fragment to perforate membrane pores for triggering pyroptosis [26, 27].
Caspases and Gasdermins have been implicated in genetic association with diseases [22, 26,27,28,29,30]. Deletion Gsdme in mice reduced chemotherapy drugs-induced various tissue damages [26]. Casp3 deficiency mice were reported to prevent the development of IRI to CKD by reducing microvascular rarefaction and renal fibrosis [7]. In the current study, we established a persistent ureteral obstruction model and R-UUO model in Gsdme and Casp3 knockout mice and cell type-specific Casp3 or Hmgb1-deficient mice. We demonstrated Casp3/GSDME-mediated pyroptosis in renal tubular cells (RTCs), but not hematopoietic cells are responsible for the initiation of a ureteral obstruction-induced kidney. Pyroptotic RTCs lead to tubular loss and promote inflammation and fibrosis through HMGB1 release. Targeting renal tubular epithelial cell pyroptosis may be critical to interfere with the progression of ureteral obstruction-induced nephropathy.
Materials and methods
Mice
Gsdme−/− mice were provided by Prof. Feng Shao, National Institute of Biological Sciences, Beijing [26]. Ksp1.3/Cre (Ksp-cre) transgenic mice were obtained from Dr. Peter Igarashi, Division of Nephrology, UT Southwestern [31]. Hmgb1 c-KO, B6-Hmgb1 floxed (Hmgb1fl/fl, RBRC06240) were provided by Dr. Tadatsugu Taniguchi, University of Tokyo [32]. Casp3fl/fl mice were obtained from GemPharmatech Co., Ltd, and Casp3−/− mice were obtained by intercrossing the Casp3fl/fl mice with EIIa-Cre mice (Jackson Lab, 003724). Ksp-cre specifically delete floxed genes in a renal tubular system [31]. Vav-Cre mice are frequently used to specifically delete floxed genes in the hematopoietic system [33, 34]. Ksp-Cre × Casp3fl/fl mice were generated by intercrossing of Ksp-cre and Casp3fl/fl mice. Vav-cre × Casp3fl/fl mice were generated by intercrossing of Vav-icre (Jackson Lab, 008610) and Casp3fl/fl mice. Ksp-Cre × Hmgb1fl/fl mice were generated by intercrossing of Ksp-cre and Hmgb1fl/fl mice. Ksp-Cre×Casp3fl/fl and Vav-Cre × Casp3fl/fl mice were fully viable without any obvious phenotype. All mice were housed in a specific pathogen-free facility with 12-h light/dark cycles. All the mice were obtained on a C57BL/6 background. Genotypes were determined by tail-snip PCR amplification.
Murine UUO and Reversible unilateral ureteral obstruction (R-UUO) models
UUO surgery was carried out as previously described [8, 33]. After male mice (10- to 12-week-old, 22–28 g body weight) were anesthetized, ureteral obstruction was performed by the ligation of the left ureter with 4–0 silk suture. Sham-operated mice had the same procedure but not ligated. R-UUO was conducted according to the previously published protocol [33]. A displaceable clip (Artikelnummer 00396-0 S&T Vascular Clamps, Fine Science Tools, Heidelberg, Germany) was used to ensure reversibility. This clip was placed on the left ureter on day 0, shifted in position on days 2 and 4 to avoid strictures, and finally removed on day 6. On day 12 after R-UUO, the contralateral kidney was removed to measure the function and access the restoration level of a kidney after relieving the obstruction. Blood and kidneys were collected and analyzed at the indicated time points. All the animal experiments were performed with the approval of the Laboratory Animal Management and Ethics Committee of Fujian Medical University, according to the Chinese Guidelines on the Care and Use of Laboratory Animals. After handling the mice, the investigators were blinded when assessing the outcome (histology, GFR measurement, qPCR, ELISA, and flow cytometry).
In the in vivo treatment study, some groups received intraperitoneal injection of HMGB1 neutralizing antibody (anti-HMGB1 mAb, 50 µg/mice) very other days [35]. Some groups were given mouse TNF-α neutralizing antibody (anti-TNF-α mAb, 20 µg/mice) every three days. Both anti-HMGB1 mab and anti-TNF-α mAb were given one day before ureteral obstruction performed.
Reagents and antibodies
Anti-GSDME antibody (ab215191, abcam), cleaved caspase-3 (Asp175) antibody (#9661, Cell Signaling), anti-mouse caspase-1 p20 (AG-20B-0042-C100, AdipoGen), cleaved caspase-8 (Asp387) antibody(#9429, Cell Signaling), anti-caspase-9 antibody (#9504, Cell Signaling), anti-caspase-11 antibody (17D9, Vovus Bio), anti-mouse IL-1β antibody (5129-100, BioVision), anti-HMGB1 (ab18256, abcam), anti-α-SMA (A5228, Sigma-Aldrich), anti-collagen I (ab34710, abcam), anti-tubulin (T9026, Sigma-Aldrich), anti-Lrp2/Megalin antibody [CD7D5] ab184676, and anti-GAPDH (3781, ProSci) antibodies were used for western blot. Mouse TNF-α neutralizing mAb (11969S) were obtained from Cell signaling Technology. Chicken anti-HMGB1 polyclonal antibody (Neutralizing antibody, 326052233) was obtained from SHINO-TEST Corporation. Mouse recombinant TNFα (mTNFα) was a kind gift from Dr. Jiahuai Han. Anti-F4/80 antibody [CI: A3-1] (ab6640, abcam), anti-Ly6G antibody [RB6-8C5] (ab25377, abcam) were used for immunohistochemical staining. Anti-Lrp2/Megalin (ab184676, abcam), anti-GSDME (13075-1-AP) antibody (Proteintech, China), and anti-HMGB1 (ab79823, abcam) were used for immunofluorescence staining. Mouse TNFα (MTA00B) and IL-1β (MLB00C) ELISA Kit provided by R&D Systems were used for detection of their secretion levels by ELISA. HMGB1 (ST51011) ELISA kit was obtained from TECAN. V500 (65-0867-14), Fixable Viability Dye eFluorTM 520, was purchased from eBioscienceTM. F4/80 (25- 4801-82, eBioscience™), and Ly-6G/Ly-6C (RB6-8C5, 11-5931-82, eBioscience™) antibodies were used for Flow cytometry. ApopTag fluorescein in situ apoptosis detection kit (S7110), was purchased from EMD Millipore.
Histologic analysis of kidney sections
Kidneys were embedded in paraffin or optimal cutting temperature compound (OCT, 4538, Leica). Paraffin sections (4 μm) were stained with PAS and Masson Trichrome for tubular injury and fibrosis assessment, according to our previous studies [16, 29]. Cell death was analyzed by terminal deoxynucleotidyl transferase-mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL). The kidney sections were stained with TUNEL according to the manufacturer’s protocol. Cryosections (4 μm) were used for immunofluorescence staining. After fixation with ice-cold acetone for 15 min, sections were washed with PBS and incubated with different primary antibodies: Anti-GSDME (1:250), anti-α-SMA (1:200), anti-collagen I (1:200), anti-HMGB1 (1:250), Anti-Megalin (1:200) for 1–2 h. As a detection antibody, Alexa Fluor® 488 and Alexa Fluor® 594 labeled secondary antibodies (abcam) were used. DNA was labeled with DAPI (Invitrogen). Immunohistochemical staining was performed by routine protocols. The number of F4/80-positive or Ly6G-positive cells was accessed with Image J software. All histologic sections were analyzed in a blinded manner.
Measurement of glomerular filtration rate (GFR)
To evaluate renal function, the GFR was assessed in the R-UUO-subjected kidney. GFR was measured in conscious mice by calculation the clearance of single-injection FITC-labeled sinistrin. One percent FITC-sinistrin (3.74 µl/g body weight) was intravenously injected into mice after R-UUO as the indicated time. Then, mice were placed in a restrainer after the injection, about 6 µl blood was drawn from the tail vein at 3, 7, 10, 15, 35, 55, and 75 min. Fluorescence intensity in each plasma sample was determined with a Thermo NanoDrop3300 spectrophotometer. Plasma concentrations of FITC-sinistrin were detected according to the fluorescence intensity obtained. As previously reported, a two-compartment model of two-phase exponential decay was used to calculated GFR [33, 36, 37].
Western blot analysis
For detecting the protein expression in primary cultured cells, the cells were harvested after treatment and then lysed with 1.2 × SDS buffer immediately. For measuring the protein expression in kidney tissues, approximate 50 mg tissues were homogenized in 300–500 μl RIPA lysis buffer containing protease inhibitors cocktail (Thermol Fisher). After protein concentrations were detected by Bradford protein assay, 5 × SDS was added. The lysates were subjected to electrophoresis and separated on 8–12% polyacrylamide gels and then transferred to polyvinylidene fluoride membranes (EMD Millipore). Membranes were blocked for 1 h in 5% BSA and incubated overnight at 4 °C with the primary antibodies. After washing, the membranes were then incubated for 1 h with horseradish peroxidase-labeled secondary antibodies. The blots were visualized with ECL.
Cell culture
Primary renal tubules were freshly isolated as described in our previous studies with minor modifications [16, 17]. The renal cortices were dissected and minced, and then digested with 0.75 mg/ml collagenase (Sigma-Aldrich) for 30 min at 37 °C. The suspension was filtered (pore size, 100 μm) and resuspended with HBSS. Enrichment of tubules was isolated by centrifugation on self-forming Percoll gradients. Finally, the freshly isolated primary tubule fragments were collected. If cultured, they were plated into collagen-coated dishes and cultured in DMEM-F12 as described. After ~5–6 days, the cultured renal primary tubular cells (RTCs) were used in this experiment.
Bone marrow-derived macrophages (BMDMs) were obtained from the bone marrow of the tibia and femur. For differentiating BM progenitors, BM was washed and resuspended, and then cultured in RPMI 1640 containing 30% L929-conditional medium. L929 was a gift from Prof. Jiahuai Han. After 6–7 days, BMDMs were divided and plated in a 12-well tissue plate for 24 h and then treated as indicated.
To stimulate RTC pyroptosis mimicing the in vivo condition after UUO, RTCs from mice were infected with vector or GSDME-expressing lentivirus and then treated with 100 ng/ml TNFα for 24 h under the condition of oxygen-glucose-serum deprivation (OGSD). To detect if HMGB1 release from the dying RTCs could trigger IL-1β secretion in BMDMs, the supernatants from the dying RTCs were collected and then used to induce caspase 11/IL-1β activation in BMDMs.
Lentivirus preparation and infection
For lentivirus production, pBOBI expression constructs and lentivirus-packing plasmids (PMDL/REV/ VSVG) were cotransfected to 293T cells by calcium phosphate precipitation. The virus-containing medium was harvested after transfection for 40–45 h. pBOBI-GSDME and pBOBI-GSDMED267A were provided by Prof. Jiahuai Han. RTCs were transfected with the virus-containing medium in presence of 10 μg/ml polybrene. The infectious medium was changed 16–18 h later, and the infected RTCs were further used for experiments after 36–48 h.
Flow cytometry
To obtain single-immune cell suspension, kidney tissues were dissected and minced, and then digested with a cocktail of 200 μg/ml DNase I (Roche, 11284932001) and 1.5 mg/ml collagenase D (Roche, 11088858001) in PBS at 37 °C for 30 min. The suspension was filtered (pore size, 40 μm) and resuspended with HBSS. Enrichment of immune cells was isolated by centrifugation on self-forming Percoll gradients. Single-cell suspensions were blocked for Fc-mediated reactions by using CD16/CD32 Monoclonal Antibody 93 (14-0161-85, eBioscienceTM). After washed, suspensions were stained with cell-surface antibodies for detecting the infiltration of F4/80 positive cells. V500 (eBioscienceTM Fixable Viability Dye eFluor™ 520, 65-0867-14) was added to the suspension to distinguish live or dead cells. All samples were run on a Navios (Beckman) flow cytometer and analyzed using FlowJo software.
For IL-1β secretion in macrophages, immune cells in CM were stimulated with PMA (50 ng/ml, LPL:250 ng/ml), Ionomycin (500 ng/ml, LPL:1ug/ml), and GolgiStop (1:1000 dilute) for 4–5 h. After stained with cell-surface antibodies, then cells were fixed and permeabilized.
Statistical analysis
Results were representative of at least three independently performed experiments. Statistical analysis was performed with Prism software (GraphPad Software, Inc.). Unpaired Student’s t tests or two-way analysis of variance (ANOVA) test with Bonferroni post-test was used to compare the means of two groups. Bars represent means ± SD. P < 0.05 was considered to indicate statistical significance.
Results
Both Casp3 and GSDME were activated in the obstructed kidney
To further investigate potential cell death pathways in this process, we focused on a recently described cell death pathway, Casp3-GSDME-mediated pyroptosis. All gasdermins except for DFNB59 share pyroptotic Gasdermin-N domain upon caspases cleavage [38,39,40]. We first examined the expression and activation of Casp3 and GSDME in the obstructed kidney of the UUO model. Immunofluorescence staining displayed evident GSDME expression in tubular epithelial cells of mice subjected to UUO (Fig. 1A), compared with those in mice subjected to sham surgery. UUO induced the activation of Casp3 on both day 3 and day 7, as evidenced by the presence of its cleaved form (Fig. 1B). The induction of cleaved GSDME on both day 3 and day 7 after UUO suggests that GSDME-mediated pyroptosis occurs in the obstructed kidneys. In line with this, kidneys with ureteral obstruction exhibited severe tubular necrosis, tubular dilation, and interstitial inflammation at day 3 and day 7 (Fig. 1C, D). This was further confirmed by electron microscope analysis (Fig. 1E), showing the necrotic morphology of tubular cells on day 3 and day 7. The enhanced severity of hydronephrosis was accompanied by increased tubular cell necrosis. After UUO, mitochondria in tubular cells became swollen, losing the integrity of cristae (Fig. 1F). The activation of Casp3 can occur through mitochondria-dependent and mitochondria-independent pathways, which are mediated by Casp9 and Casp8, separately [30, 39]. Both Casp8 and Casp9 were activated after UUO, as evidenced by the presence of their cleaved forms (Fig. 1G). The activation of Casp9 was dominant at the early stage (d1); while the activation of Casp8 was enhanced later (d3) (Fig. 1G). These results indicated that the cleavage of GSDME, possibly by Casp3 activated through both mitochondria-dependent and mitochondria-independent pathways, triggers pyroptosis in the obstructed kidneys after UUO.

A Immunostaining images of kidney sections from mice. Kidneys were isolated from mice on day 3 after sham or UUO surgery. Sections were stained using an anti-GSDME antibody (red) and DAPI (blue). n = 6 for Sham group; n = 10 for UUO group. Scale bar = 50 μm. B Freshly isolated tubules were collected for western blot to analyze the cleavage of GSDME and Caspase3. Antibody against GAPDH was used as a loading control. n = 4. C Representative PAS staining of kidney sections from sham-group mice and mice on day 3 or 7 after UUO. Scale bar (black) = 100 μm, scale bar (blue) = 50 μm. D Histologic renal injury scores are shown in panel (C). Scores were obtained by counting the percentage of tubules that displayed tubular necrosis and tubular dilation. ##P < 0.01 vs. sham group. Sham group, n = 6; UUO groups, n = 10. E Representative electron micrographs of a tubular necrotic cell. n = 5. Scale bar = 2.5 μm. F Representative electron micrographs of mitochondrial damage in proximal tubular cells. n = 5. Scale bar = 1 μm. G The activation of caspase 8 and caspase 9 were analyzed in freshly isolated tubules by the western blot at the indicated time. n = 4.
Gsdme deficiency ameliorated ureteral obstruction-induced hydronephrosis and undergoing fibrosis
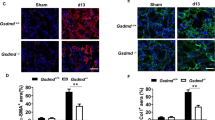
We examined whether Gsdme deficiency affected UUO-induced renal tubular epithelial cell death and pathological changes. The kidney from WT mice showed a higher percentage of TUNEL staining-positive cells (Fig. 2A, C), together with enhanced tubular necrosis and tubular dilation (Fig. 2B, D) on d3 and d7 after UUO; whereas such renal tubule damage was significantly ameliorated in the kidney from Gsdme−/− mice. The knockout efficiency of Gsdme was further confirmed by western blot (Fig. 2E). Exemplary photographs of obstructed kidneys in Fig. 2F displayed hydronephrosis. The kidney from WT mice showed a thinner renal cortex on d7 and became more severe on d13; while this was drastically prevented by Gsdme deficiency (Fig. 2F). Gsdme−/− mice developed significantly less UUO-induced renal fibrosis than WT mice, as showed in the protein levels of fibrotic markers, α-smooth muscle actin (α-SMA), and collagen I (Col I), which were significantly induced in the obstructed kidney (Fig. 2G). Overall fibrosis quantified by Masson Trichrome staining (Fig. 2H, I) and the deposition of α-SMA (Fig. 2J, K) and Col I (Fig. 2L, M) quantified by immunofluorescence were significantly reduced by Gsdme deletion. These data suggest that GSDME-mediated pyroptosis contributes to the progression of hydronephrosis, interstitial tubular injury, and fibrosis induced by ureteral obstruction.

A, C Representative images of TUNEL staining of kidney sections from mice. Kidneys were isolated from wild-type mice (Gsdme+/+) and Gsdme-deficient mice (Gsdme−/−) on days 0, 3, or 7 after UUO as indicated. Merged images of TUNEL signal (green) and DAPI (blue) were shown. The number of TUNEL-positive cells was counted in ten random fields in the obstructive kidney per mice. Scale bar = 50 μm. B, D Representative images of PAS-stained kidney and the scores of tubular damage. Scale bar = 100 μm. A–D, d 0 group, n = 6; UUO groups, n = 10. **P < 0.01 vs. Gsdme+/+ group. E The knockout efficiency of Gsdme was confirmed by western blot. n = 6. F Exemplary photographs were shown. n = 6. G Western blot analysis for α-smooth muscle actin (α-SMA) and type-I collagen (Col I) expression of kidney tissue lysates. Kidney tissue lysates were isolated on day 0, 3, 7, or 13 after UUO as indicated. GAPDH was used as a loading control. n = 4. H Representative images of Masson trichrome staining of kidney sections. Scale bar = 100 μm. I Quantification of fibrotic area evaluated by Masson trichrome staining shown in panel (H). J, L Representative images of immunofluorescence staining of kidney sections. Antibodies against α-SMA (I) and Col I (L) were used; DAPI was used for nuclear staining. Scale bar = 100 μm. K, M Quantification of αSMA (K) and Col I (M) expression by immunofluorescence. H–M, Sham group, n = 6; UUO groups, n = 10. **P < 0.01 vs. Gsdme+/+ group.
GSDME-mediated pyroptosis in bone marrow-derived cells did not contribute to the progression of obstructive nephropathy
To further determine whether GSDME-mediated pyroptosis in bone marrow (BM)-derived immune cells contributes to the progression of obstructive nephropathy, we performed bone marrow transplantation studies. Gsdme+/+ mice receiving Gsdme+/+ bone marrow (referred to Gsdme+/+ to Gsdme+/+ chimeric mice) developed a serious tubular injury in the kidney on day 3 after UUO, as determined by PAS staining (Fig. 3A, E). These mice also developed significant renal fibrosis on day 13 after UUO, as determined by Masson staining (Fig. 3B, F). Consistent with these data, a significant increase of α-SMA (Fig. 3C, G, I) and Col I (Fig. 3D, H, I) was detected. These parameters were significantly reduced in both Gsdme+/+ (WT) to Gsdme−/− (KO) and Gsdme−/− (KO) to Gsdme−/− (KO) chimeras (Fig. 3A–I). However, Gsdme−/− to Gsdme+/+ chimeric mice did not show significant renal protection on days 3 and 13 after UUO (Fig. 3A–I). These results indicate that GSDME-mediated pyroptosis in bone marrow-derived infiltrating immune cells is not responsible for the pathogenesis of obstructive nephropathy.

Chimeric mice were created, in which the BM was replaced with donor BM cells from WT or Gsdme-ko. A, E Representative images of PAS-stained obstructive kidney sections (Bar = 100 μM), and quantification of renal injury in BM chimeric mice at 3 days after UUO. n = 9. B, F Representative images of Masson trichrome-stained obstructive kidney sections (Bar = 100 μM), and assessment of renal fibrosis in BM chimeric mice at 13 days after UUO. n = 9. C, D Representative images of immunofluorescence staining with markers (α-SMA and Col-I) of Fibrosis. Scale Bar = 100 μM. G, H Quantification of α-SMA and Col I expression. n = 6. **P < 0.01 vs. WT to WT chimeric mice; ∆∆P < 0.01, vs. KO to WT chimeric mice. I Western blot analysis of protein expression of α-SMA and Col I. n = 4. WT wild type, KO Gsdme-ko.
Casp3/GSDME-mediated pyroptosis in tubules was responsible for the progression of obstructive nephropathy
We further investigated the role of Casp3 in renal tubular injury and fibrogenesis in mice subjected to UUO. As expected, Casp3 deficiency mice displayed a lower percentage of TUNEL staining-positive RTCs than WT mice on day 3 after UUO (Fig. 4A, B). Consistently, deletion of Casp3 significantly prevented tubular injury (Fig. 4C, D) and cleavage of GSDME (Fig. 4G). There was also a significant reduction of renal fibrosis in Casp3−/− mice than that in Casp3+/+ mice on day 13 after UUO (Fig. 4E, F).

A–F Kidneys were collected as the indicated time after UUO in Casp3+/+ and Casp3−/− mice. A–B Representative images of TUNEL staining and quantitative analysis of TUNEL-positive cells are shown in the obstructive kidney. Scale bar = 50 μM. C–D Representative images of PAS-stained kidney sections and the scores of tubular damage. E–F Representative images of Masson’s trichrome-stained obstructive kidney sections and quantification of renal fibrosis. A–F n = 6 for Sham group; n = 10 for UUO group. Scale bar = 100 μM. **P < 0.01 vs. Casp3+/+ group. G, L Freshly isolated tubules were collected for western blot to analyze the expression and activation of GSDME and Casp3. n = 6. H, J Mice with specific Casp3 deletion in renal tubular cells (Ksp-cre × Casp3fl/fl) or hematopoietic cells (Vav-Cre × Casp3fl/fl) were subjected to sham operation or UUO as the indicated time. Representative PAS-stained images of kidney sections and quantification of renal tubular injury were shown. I, K Representative images of Masson’s trichrome-stained obstructive kidney sections and quantitative analysis of renal fibrosis. H–K, n = 6 for Sham group; n = 10 for UUO group. Scale bar = 100 μM. **P < 0.01 vs. Casp3fl/fl UUO-group.
To further determine whether Casp3/GSDME-mediated pyroptosis in tubular cells or hematopoietic cells contribute to renal fibrosis in obstructive nephropathy, we generated RTCs-specific and hematopoietic cells-specific Casp3-deficient mice by crossing Casp3 floxed mice (Casp3fl/fl mice) with Ksp-Cre- and Vav-Cre-expressing mice on a C57BL/6 background, separately [31, 33, 34]. As expected, RTCs-specific Casp3-deficient mice (Ksp-Cre×Casp3fl/fl) inhibited GSDME activation (Fig. 4L) and developed significantly less tubular injury at the early stage (Fig. 4H, J) and reduced renal fibrosis at a late stage after UUO than wild type mice (Ksp-Cre × Casp3wt/wt) (Fig. 4I, K). However, hematopoietic cells-specific Casp3-deficient mice (Vav-Cre × Casp3fl/fl mice) did not show significant changes in renal tubular injury and interstitial fibrosis when compared with wild-type mice (Vav-Cre × Casp3wt/wt) on day 3 and 13 after UUO (Fig. 4H–K). Taken together, these data provide evidence that Casp3/GSDME-mediated pyroptosis in RTCs, but not infiltrating immune cells, predominantly contributes to the progression of kidney injury and fibrosis induced by ureteral obstruction.
Deletion of Gsdme/Casp3 improved the recovery of renal function after ureteral obstruction relieved
To further investigate the effects of Casp3/GSDME-mediated pyroptosis in RTCs on the initiation of early renal injury and the inflammation which contributes to the development of renal fibrosis, we applied the R-UUO model in mice to study their recovery from renal tubular injury and fibrosis after the removal of the obstruction. As shown in Fig. 5A, R-UUO was achieved with a clip that was placed on day 0 and removed on day 6 after UUO, as the previous study showed [33]. On day 12, the contralateral kidney was removed in all mice. The extent of renal tubular damage and fibrosis progression in Gsdme−/− and Casp3−/− mice, as determined by PAS (Fig. 5B) and Masson’s trichrome (Fig. 5C) staining, was lighter than those in WT mice subjected to R-UUO (Fig. 5D, E). On day 13 after R-UUO, WT mice exhibited a significantly reduced GFR when compared with Gsdme−/− and Casp3−/− mice; on day 34 after R-UUO, the renal function of Gsdme−/− and Casp3−/− mice was significantly better than that of WT mice (Fig. 5F). Similar results on tubular damage and fibrosis (Fig. 5B, C, G, H) and renal function (Fig. 5I) were obtained in RTCs-specific Casp3-deficient mice subjected to R-UUO. These data showed that blockage of Casp3/GSDME-mediated tubular cell pyroptosis could improve the recovery of renal function after ureteral obstruction relieved.

A Scheme of the experimental design for the model of R-UUO. The right ureteral obstruction was performed on day 0, obstruction was relieved on day 6, and the left kidney was removed on day 12. B–C Representative images of PAS and Masson’s trichrome-stained kidney sections subjected to R-UUO as depicted in WT, Gsdme-ko, Casp3-ko, Casp3fl/fl, Ksp-cre × Casp3wt/wt, and Ksp-cre × Casp3fl/fl mice. Scale bar = 100 μM. D, E, G, H Assessment of the renal tubular injury and renal fibrosis scores. n = 9. **P < 0.01, ##P < 0.01. F GFR (one kidney) per 100 g body was measured in WT, Gsdme-ko, and Casp3-ko mice. Basel GFR was obtained by the mice with the left kidney removed, but not undergoing with R-UUO procedure. Mice for measuring Basel GFR, n = 6. Mice for R-UUO model, n = 9. **P < 0.01 vs. Gsdme-ko group; ##P < 0.01 vs. Casp3-ko group. I GFR (one kidney) per 100 g body was measured in Casp3fl/fl, Ksp-cre × Casp3wt/wt, and Ksp-cre × Casp3fl/fl mice. GFR was obtained as described in (F). Mice for measuring Basel GFR, n = 6. Mice for R-UUO model, n = 9. **P < 0.01 vs. Casp3fl/fl group; ##P < 0.01 vs. Ksp-cre × Casp3wt/wt.
Tubular cell pyroptosis promoted HMGB1 secretion and macrophages and neutrophils recruitment post-UUO
Inflammation plays an important role in the pathophysiology of obstructive nephropathy and influences the severity and prognosis of the obstructive kidney [11, 12, 41, 42]. The number of macrophages and neutrophils, as indicated by F4/80+ cells (Fig. 6A, Fig. S1A) and Ly6G+ cells (Figs. 6B and S1B), mildly increased on day 5 and persistently increased up to day 10 post-UUO in WT mice; while these were significantly reduced by deletion of Gsdme or Casp3. RTC-specific deletion of Casp3 also similarly reduced the infiltration of macrophages and neutrophils (Fig. 6C, D and S1C, D), suggesting that persistent inflammation in the kidney after subjected to UUO could result from pyroptotic tubular cells which trigger the migration and recruitment of inflammatory cells. Therefore, we further monitored the production of pro-inflammatory cytokines and HMGB1 in the kidney. Compared with WT mice, both Gsdme- or Casp3-deficient mice produced much less TNFα and IL-1β (Fig. 6E, F) after UUO. RTC-specific deletion of Casp3 resulted in a similar reduction of TNFα and IL-1β (Fig. 6G, H). Very interestingly, the level of HMGB1 increased at an early stage but decreased at a late stage after UUO. Gsdme or Casp3 deficiency decreased HMGB1 level from the early time point after UUO; while there were no obvious differences between WT mice and Gsdme−/− or Casp3−/− mice at the late stage (on day 10) after UUO (Fig. 6I). Similar results were obtained in RTCs-specific Casp3-deficient mice (Fig. 6J). The HMGB1 mRNA and protein levels in tubules were confirmed by qPCR and western blot (Fig. S1E, F, Fig. 6K, L). These reveal that blockage of Casp3/GSDME-mediated tubular cell pyroptosis prevents HMGB1 secretion and the inflammatory cells infiltration after ureteral obstruction.

A, C Representative images of kidney tissues stained with the monocytes-macrophage marker F4/80+ and (B, D) leukocyte marker Ly6G by immunohistochemistry at 5 and 10 days following UUO. Scale bar = 100 μM. A–D n = 10. E–J TNFα, IL-1β, and HMGB1 production were detected by ELISA. n = 6. **P < 0.01; ##P < 0.01. K, L Freshly renal tubules were isolated and HMGB1 expression was analyzed by western blot. n = 4.
HMGB1 released by pyroptotic tubular cells promoted the progression of obstructive nephropathy
HMGB1, a nuclear protein, functions as a DAMP, once it is released from damaged or necrotic cells [14]. Extracellular HMGB1 induces the production and release of proinflammatory cytokines and chemokines from macrophages and monocytes [32, 43]. HMGB1 is highly expressed in RTCs on day 3 after UUO, but not in RTC-specific Hmgb1-deficient mice (Ksp-Cre × Hmgb1fl/fl) (Fig. 7A, B). To further determine the roles of HMGB1 during the development of renal fibrosis, we subjected RTC-specific Hmgb1-deficient mice to UUO and R-UUO models. Deletion of Hmgb1 in renal tubules did not show an obvious effect on cell death (Fig. 7C, I) and renal damage on day 4 after UUO (Fig. 7D, J), but greatly alleviated the renal interstitial fibrosis on day 27 after R-UUO (Fig. 7E, K). Renal tubules-specific Hmgb1-deficient mice also showed a dramatic decrease of macrophages infiltration (Fig. 7F, G, L) and IL-1β release (Fig. 7H). These results indicate that HMGB1 released from pyroptotic tubular cell death triggers the inflammatory response, which drives the progression of fibrosis after UUO.

A Immunofluorescence staining for HMGB1 (red), Megalin (green), and DAPI (blue) in the kidney subjected to UUO for 3 days. The tubules were labeled with megalin. n = 6. Scale bar = 50 μm. B Western blot analyzed HMGB1 levels. n = 6. C, I Representative images of TUNEL staining and quantitative analysis of TUNEL-positive cells are shown in the obstructive kidney. Scale bar = 50 μM. n = 6. D, J Representative images of PAS-stained obstructive kidney sections and the scores of tubular damage. E, K Representative images of Masson’s trichrome-stained obstructive renal tissues and quantification of renal fibrosis. F, L Representative images immunohistochemistry staining with F4/80+ of kidney tissues and quantification of the number of F4/80-positive cells per hpf. D–F, n = 8, Scale bar = 100 μM. **P < 0.01 vs. Hmgb1fl/fl group; ##P < 0.01 vs. Ksp-cre × Hmgb1wt/wt group. G, H The number of macrophages (F4/80+) and IL-1β secretion in the kidney were detected by flow cytometry. n = 6.
TNFα played an important role in triggering pyroptosis after UUO
TNFα-induced apoptosis could be switched to pyroptosis when the level of GSDME protein is upregulated [26]. TNFα neutralizing antibody treatment inhibited RTC death (Fig. S2A–D) and Casp3/GSDME activation at an early stage (Fig. S2E, F), suggesting TNFα played a very important role in triggering pyroptosis after UUO. Interestingly, TNFα neutralizing antibody reduced kidney injury on day 6 after subjected to ureteral obstruction; while HMGB1 neutralizing antibody treatment did not show a significant effect, and combination use of anti-TNFα and anti-HMGB1 antibodies did not show additive protection (Fig. S3A, C), which is in line with the results obtained in renal tubules-specific Hmgb1-deficient mice. However, in the R-UUO model, both the anti-TNFα antibody and anti-HMGB1 antibody reduced kidney fibrosis on day 27 after the obstruction relieved, even though the anti-HMGB1 antibody showed milder protection than the anti-TNFα antibody (Fig. S3B, D). Combination use of anti-TNFα and anti-HMGB1 antibodies revealed the best inhibition of renal fibrosis progression. These data provided evidence for a major role of the TNFα/Casp3/GSDME/HMGB1 axis in the development of obstructive nephropathy.
GSDME promoted RTC death in vitro under the UUO-mimicking condition
As shown above (Fig. 1A, B), the expression of GSDME is upregulated during the pathogenesis of obstructive nephropathy. Previous studies have shown that ischemia, hypoxia, and TNFα contributed to the pathophysiology changes in the kidney after UUO in vivo [9, 10]. Very interestingly, the combination of OGSD with TNFα, but not TNFα alone, induced pyroptosis in GSDME-expressing RTCs in vitro. Thus, to mimic UUO in vitro, the combination of OGSD with TNFα treatment was applied. We used OGSD and 100 ng/ml TNFα to induce pyroptosis in GSDME-overexpressing RTCs. GSDME-overexpressing RTCs (Fig. 8A) responded effectively to TNFα treatment under the condition of OGSD. The GSDME expression level determined the susceptibility of RTCs to OGSD and TNFα-induced cell death (Fig. 8A–C). GSDME mostly located in the cytoplasm in control cells, but is translocated to the plasma membrane in cells after OGSD and TNFα treatment (Fig. 8D). The morphology of necrotic cell death of GSDME-overexpressing RTCs was confirmed by electron microscopy (Fig. 8E). Without the addition of TNFα, OGSD alone did not significantly induce RTC death, suggesting that TNFα is essential in triggering RTC pyroptosis. OGSD and TNFα treatment induced the activation of Casp3 in RTCs (Fig. 8F). Wild type GSDME, but not the GSDME D270A mutant (deficient in cleavage by Casp3), was cleaved in response to OGSD and TNFα treatment (Fig. 8F). Consistently, expression of GSDME wild type, but not D270A mutant, dramatically induced pyroptosis in RTCs in responses to OGSD and TNFα treatment (Fig. 8G). Furthermore, deletion of Casp3 completely blocked cell death of RTCs and cleavage of GSDME after OGSD and TNFα treatment (Fig. 8H, I). Thus, GSDME cleavages by Casp3 are essential for TNFα-induced pyroptosis in RTCs in vitro. These results further consolidate the important role of TNFα/Casp3/GSDME-mediated pyroptosis during the pathogenesis of obstructive nephropathy.

A–E RTCs from WT mice were infected with lentivirus encoding vector or GSDME for 44 h and then treated with or without 100 ng/ml TNFα for 24 h under the condition of oxygen-glucose-serum deprivation (OGSD). The control groups were treated with OGSD but without TNFα. Western blot analysis of GSDME expression in (A). PI (red) and Hoechst (blue) staining were used to assessed cell death cells in (B) (Scale bar = 100 μm). The percentage of PI-positive cells was counted as (C). n = 6. **P < 0.01 vs. Vector group; ##P < 0.01 vs. Low dose virus encoding GSDMD group. Cells were immunostained for GSDME and representative images obtained by confocal microscope were shown as (D) (Scale bar = 25 μm). Representative electron micrographs of live and necrotic RTCs are shown in (E) (Scale bar = 3 μm). F–G RTCs from WT mice were infected with lentivirus encoding nothing (vector) or GSDMEwt, or GSDMED267A for 44 h and then treated as (A). Western blot analyzed the cleavage of GSDME as (F). PI-positive cells were counted in (G). n = 6. **P < 0.01, ##P < 0.01. H–I RTCs from Casp3+/+ and Casp3−/− mice were infected with lentivirus encoding GSDME for 44 h and then treated as (A). Western blot analyzed the cleavage of caspase-3 and GSDME (H) and cell death was evaluated by counting PI-positive cells (I). n = 6. **P < 0.01, ##P < 0.01. J RTCs were isolated from Hmgb1fl/fl or Hmgb1Ksp−/− (Ksp-cre × Hmgb1fl/fl) mice, and then treated as (A). ELISA analyzed the level of HMGB1 in the extracellular supernatants. n = 6. **P < 0.01. K BMDMs from WT mice were pre-incubated with LPS (500 ng/ml) for 4 h and then were treated with culture supernatants for 24 h, which were collected from wild type or Hmgb1-deficient RTCs after treated as (A). Western blot analyzed the expression of caspase 11 and IL-1β. n = 4. L Schematic model: Caspase3/GSDME-mediated pyroptosis in the development of obstructive nephropathy.
We further determined the role of HMGB1 in the cross-talk between RTCs and macrophages during the development of obstructive nephropathy. The release of HMGB1 from RTCs significantly increased after treatment with OGFD and TNFα (Fig. 8J). Bone marrow-derived macrophages (BMDMs) were stimulated with culture supernatants collected from wild type and Hmgb1-deficient (Hmgb1Ksp−/−) RTCs after treatment with OGFD and TNFα. As expected, the supernatants from wild type, but not Hmgb1-deficient RTCs efficiently induce the activation of caspase11 and the cleavage and release of IL-1β in BMDMs (Fig. 8K). This suggests that HMGB1 released by pyroptotic RTCs could amplify inflammatory responses by inducing the activation of the inflammasome, the release of cytokines, and pyroptosis in macrophages.
Discussion
Tubular cell death rapidly increases in the kidney when subjected to ureteral obstruction. Several tubular pathological changes, including dilatation, atrophy and loss, inflammatory cell infiltration, and interstitial fibrosis, occur in prolonged ureteral obstruction [1, 10, 44]. These observations suggested that tubular cell death might be pathogenetically related to tubular loss and renal fibrosis. GSDME is expressed in various normal tissues, including kidneys [26]. In the current study, we showed that activation of Casp3 and cleavage of GSDME were markedly induced in the renal tubules in mice subjected to ureteral obstruction. Moreover, deficiency of either Gsdme or Casp3 inhibited hydronephrosis and fibrosis in response to UUO, accompanied by reduced renal tubular pyroptosis and inflammatory cell recruitment in kidneys in response to UUO. Our findings revealed the important link between tubular cell pyroptosis and obstructive nephropathy.
We also showed that Casp3/GSDME-mediated bone marrow-derived hematopoietic cell death did not contribute to renal injury and fibrosis after UUO through bone marrow transplantation experiments and application of tissue-specific Casp3 knockout mice. Renal tubules-specific, but not hematopoietic cell-specific, deletion of Casp3 protected mice from renal tubule damage and renal fibrosis after UUO. Thus, Casp3/GSDME-mediated tubular cell pyroptosis plays a predominant role in the development of obstructive nephropathy. In the model of R-UUO with subsequent reversible obstruction, mice deficient in either Casp3 or Gsdme displayed better renal protection and higher-level GFR, confirming that the severity of the early injury determines the later outcome of renal function and fibrosis.
Recent studies demonstrated the expression level of GSDME determines the type of cell death in Casp3-activated cells. GSDME-high expressed cells undergo pyroptosis upon “apoptotic stimulation” like TNFα treatment [26]. We showed that TNFα neutralizing antibody inhibited RTC death and Casp3/GSDME activation at an early stage after ureteral obstruction, and prevented renal tubular injury and fibrosis. We overexpressed GSDMD in RTCs in vitro to mimic the high GSDME expression condition in the kidney after UUO. Casp3 deficiency inhibited TNFα-induced cell death in high GSDME-expressing RTCs. These results confirmed that active Casp3 cleaves GSDME to liberate the GSDME-N domain, which directly triggers pyroptosis in RTCs after ureteral obstruction.
Non-microbial (sterile) inflammation is an important event during the development of AKI to CKD [11, 42]. DAMPs, including HMGB1 production, contributed to the activation of inflammasome in macrophages [13, 43]. Interestingly, besides the extracellular release, the mRNA and protein levels of HMGB1 in renal tubules following UUO induction increased in vivo. However, in vitro, the changes of HMGB1 mRNA levels in GSDME-mediated pyroptotic RTCs were not observed (data not shown). In vivo, UUO leads to more complex outcomes in the kidney, including RTC cell death, tubular loss, and hydronephrosis, which lead to a decrease in renal blood flow, ischemia, and hypoxia [2, 10]. One possible mechanism could be that ischemia and hypoxia-induced by hydronephrosis following ureteral obstruction triggered the transcription of HMGB1, which has been previously reported [45,46,47]. Deletion of Gsdme or Casp3 might primarily ameliorate hydronephrosis through prevention of RTC death and tubular loss, subsequently alleviated ischemia and hypoxia indirectly, which contributed to downregulating HMGB1 mRNA and protein levels. We also gained important insights into the functional relevance between tubular pyroptosis and renal fibrosis by directly and specifically interfering with the production of HMGB1 from tubular cells. Specific deletion of Hmgb1 in renal tubules reduced macrophages infiltration and IL-1β release, which markedly contributed to renal fibrogenesis. Consistently, the specific inhibition of HMGB1 production from RTCs displayed less efficiency to trigger an inflammatory response in macrophages. Hmgb1 in renal tubules deficiency or HMGB1 neutralizing antibody prevented interstitial fibrosis progression after relieving the ureteral obstruction. These findings suggest HMGB1 released from the TNFα/Casp3/GSDME-mediated dying tubular cells drives the recruitment and activation of immune cells which promote the progression of obstructive nephropathy.
In summary, our study delineated the important mechanisms underlying ureteral obstruction-induced nephropathy. After ureteral obstruction, TNFα/Casp3/GSDME signaling mediates pyroptosis in renal tubules, triggering the release of HMGB1 and inflammasome activation, finally resulting in tubular loss and progressing into hydronephrosis and renal fibrosis (Fig. 8L). Therapeutic strategies targeting Casp3/GSDME-mediated pyroptosis may prove useful for progressive obstructive nephropathy.
References
Stevens S. Obstructive kidney disease. Nurs Clin North Am. 2018;53:569–78.
Capelouto CC, Saltzman B. The pathophysiology of ureteral obstruction. J Endourol. 1993;7:93–103.
Park HC, Yasuda K, Ratliff B, Stoessel A, Sharkovska Y, Yamamoto I, et al. Postobstructive regeneration of kidney is derailed when surge in renal stem cells during course of unilateral ureteral obstruction is halted. Am J Physiol Ren Physiol. 2010;298:F357–364.
Cochrane AL, Kett MM, Samuel CS, Campanale NV, Anderson WP, Hume DA, et al. Renal structural and functional repair in a mouse model of reversal of ureteral obstruction. J Am Soc Nephrol. 2005;16:3623–30.
Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol. 2010;21:1732–44.
Xiao X, Du C, Yan Z, Shi Y, Duan H, Ren Y. Inhibition of necroptosis attenuates kidney inflammation and interstitial fibrosis induced by unilateral ureteral obstruction. Am J Nephrol. 2017;46:131–8.
Yang B, Lan S, Dieudé M, Sabo-Vatasescu J-P, Karakeussian-Rimbaud A, Turgeon J, et al. Caspase-3 is a pivotal regulator of microvascular rarefaction and renal fibrosis after ischemia-reperfusion injury. J Am Soc Nephrol. 2018;29:1900–16.
Imamura M, Moon J-S, Chung K-P, Nakahira K, Muthukumar T, Shingarev R, et al. RIPK3 promotes kidney fibrosis via AKT-dependent ATP citrate lyase. JCI Insight. 2018;3:e94979.
Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009;75:1145–52.
Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Ren Physiol. 2002;283:F861–875.
Mack M. Inflammation and fibrosis. Matrix Biol. 2018;68–69:106–21.
Marchal P-O, Kavvadas P, Abed A, Kazazian C, Authier F, Koseki H, et al. Reduced NOV/CCN3 Expression limits inflammation and interstitial renal fibrosis after obstructive nephropathy in mice. PLoS ONE. 2015;10:e0137876.
Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21:1229–39.
Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 2019;26:99–114.
Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015;11:264–76.
Chen H, Fang Y, Wu J, Chen H, Zou Z, Zhang X, et al. RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis. 2018;9:878.
Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, et al. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol. 2015;26:2647–58.
Tonnus W, Belavgeni A, Xu Y, Linkermann A. Don’t trick me twice! Kidney Int. 2019;95:736–8.
Xu Y, Han J. The necrosome in acute kidney injury. Semin Nephrol. 2016;36:199–207.
Mulay SR, Honarpisheh MM, Foresto-Neto O, Shi C, Desai J, Zhao ZB, et al. Mitochondria permeability transition versus necroptosis in oxalate-induced AKI. J Am Soc Nephrol. 2019;30:1857–69.
Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016;535:111–6.
Orning P, Lien E, Fitzgerald KA. Gasdermins and their role in immunity and inflammation. J Exp Med. 2019;216:2453–65.
Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20:143–57.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, et al. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell. 2020;180:941–955.e20.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103.
Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415–20.
Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial endotoxin activates the coagulation cascade through gasdermin D-dependent phosphatidylserine exposure. Immunity. 2019;51:983–996.e6.
Chen H, Li Y, Wu J, Li G, Tao X, Lai K, et al. RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. 2020;27:2568–85.
Julien O, Wells JA. Caspases and their substrates. Cell Death Differ. 2017;24:1380–9.
Shao X, Somlo S, Igarashi P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol. 2002;13:1837–46.
Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462:99–103.
Buchtler S, Grill A, Hofmarksrichter S, Stöckert P, Schiechl-Brachner G, Rodriguez Gomez M, et al. Cellular origin and functional relevance of collagen I production in the kidney. J Am Soc Nephrol. 2018;29:1859–73.
de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, et al. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol. 2003;33:314–25.
Cai J, Yuan H, Wang Q, Yang H, Al-Abed Y, Hua Z, et al. HMGB1-driven inflammation and intimal hyperplasia after arterial injury involves cell-specific actions mediated by TLR4. Arterioscler Thromb Vasc Biol. 2015;35:2579–93.
Pill J, Kraenzlin B, Jander J, Sattelkau T, Sadick M, Kloetzer H-M, et al. Fluorescein-labeled sinistrin as marker of glomerular filtration rate. Eur J Med Chem. 2005;40:1056–61.
Chen L, Kim SM, Oppermann M, Faulhaber-Walter R, Huang Y, Mizel D, et al. Regulation of renin in mice with Cre recombinase-mediated deletion of G protein Gsalpha in juxtaglomerular cells. Am J Physiol Ren Physiol. 2007;292:F27–37.
He W, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–98.
Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10:1689.
de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019;26:146–61.
Krajewski W, Wojciechowska J, Dembowski J, Zdrojowy R, Szydełko T. Hydronephrosis in the course of ureteropelvic junction obstruction: an underestimated problem? Current opinions on the pathogenesis, diagnosis and treatment. Adv Clin Exp Med. 2017;26:857–64.
Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Investig. 2014;124:2299–306.
Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, et al. The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity. 2018;49:740–753.e7.
Hesketh EE, Vernon MA, Ding P, Clay S, Borthwick G, Conway B, et al. A murine model of irreversible and reversible unilateral ureteric obstruction. J Vis Exp. 2014;20:52559.
Tian S, Li C, Ran R, Chen S-Y. Surfactant protein A deficiency exacerbates renal interstitial fibrosis following obstructive injury in mice. Biochim Biophys Acta Mol Basis Dis. 2017;1863:509–17.
Zhang C, Dong H, Chen F, Wang Y, Ma J, Wang G. The HMGB1-RAGE/TLR-TNF-α signaling pathway may contribute to kidney injury induced by hypoxia. Exp Ther Med. 2019;17:17–26.
Wyczanska M, Lange-Sperandio B. DAMPs in unilateral ureteral obstruction. Front Immunol. 2020;11:581300.
Acknowledgements
We are grateful to Pro. Feng Shao (Beijing National Institute of Biological Sciences) and Jiahuai Han (Xiamen University) for research information and experimental materials.
Funding statement
This work was supported by grants from National Natural Science Foundation of China (No. 82070720, No. 81870472 and No. 81700596), Joint Funds for the Innovation and Natural Science Foundation of Science and Technology of Fujian province (No. 2019Y9019 and No. 2020J02020), Fujian Province finance project (2020B009) and Startup Fund for Scientific Research of Fujian Medical University (No. 2019QH2037).
Author information
Authors and Affiliations
Contributions
YX, TWM, and HYL conceived and designed the research. YL, YY, ZXH, HC, KL, HBM, and ZC performed animal experiments. YL, YY, RL, ZW, ZC, and HBM performed all in vitro experiments. RL, HC, ZZ, and YX performed histologic analysis and flow cytometry. YL, KL, HC, ZZ, HYL, and YX analyzed the data. TWM and YX supervised the research. YL, YY, and YX wrote the original draft. TWM and YX performed writing-review and editing. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
The manuscript reporting studies did not involve human participants, human data or human tissue. The animal experiments were accomplished in compliance with ethical standards. All the animal experiments were performed with the approval of the Laboratory Animal Management and Ethics Committee of Fujian Medical University, according to the Chinese Guidelines on the Care and Use of Laboratory Animals.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Y. Shi
Supplementary information
Rights and permissions
About this article

Cite this article
Li, Y., Yuan, Y., Huang, Zx. et al. GSDME-mediated pyroptosis promotes inflammation and fibrosis in obstructive nephropathy. Cell Death Differ 28, 2333–2350 (2021). https://doi.org/10.1038/s41418-021-00755-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-021-00755-6
This article is cited by
-
(Pro)renin receptor mediates tubular epithelial cell pyroptosis in diabetic kidney disease via DPP4-JNK pathway
Journal of Translational Medicine (2024)
-
The pyroptosis mediated biomarker pattern: an emerging diagnostic approach for Parkinson’s disease
Cellular & Molecular Biology Letters (2024)
-
Gasdermin E promotes translocation of p65 and c-jun into nucleus in keratinocytes for progression of psoriatic skin inflammation
Cell Death & Disease (2024)
-
CHOP-mediated Gasdermin E expression promotes pyroptosis, inflammation, and mitochondrial damage in renal ischemia-reperfusion injury
Cell Death & Disease (2024)
-
A pyroptosis-related gene expression signature predicts immune microenvironment and prognosis in head and neck squamous cell carcinoma
European Archives of Oto-Rhino-Laryngology (2024)
