
Abstract
A deviated repertoire of the gut microbiome predicts resistance to cancer immunotherapy. Enterococcus hirae compensated cancer-associated dysbiosis in various tumor models. However, the mechanisms by which E. hirae restored the efficacy of cyclophosphamide administered with concomitant antibiotics remain ill defined. Here, we analyzed the multifaceted modes of action of this anticancer probiotic. Firstly, E. hirae elicited emigration of thymocytes and triggered systemic and intratumoral IFNγ-producing and CD137-expressing effector memory T cell responses. Secondly, E. hirae activated the autophagy machinery in enterocytes and mediated ATG4B-dependent anticancer effects, likely as a consequence of its ability to increase local delivery of polyamines. Thirdly, E. hirae shifted the host microbiome toward a Bifidobacteria-enriched ecosystem. In contrast to the live bacterium, its pasteurized cells or membrane vesicles were devoid of anticancer properties. These pleiotropic functions allow the design of optimal immunotherapies combining E. hirae with CD137 agonistic antibodies, spermidine, or Bifidobacterium animalis. We surmise that immunological, metabolic, epithelial, and microbial modes of action of the live E. hirae cooperate to circumvent primary resistance to therapy.
Similar content being viewed by others


Background
Over the past decade, it has become clear that the development and progression of cancer involved an incapacity of the immune system to eliminate malignant cells [1]. This paradigm shift has been accompanied by the demonstration that successful conventional and targeted anticancer therapies including immunogenic chemotherapy and immune checkpoint blockers rely on the reactivation or de-inhibition of latent anticancer immune responses [2, 3]. The primary purpose of the immune system resides in its capacity to keep internal organs in homeostasis, fighting off inflammation and infections arising from portals of entry. Located at the largest epithelial interface with the external environment, the gut microbiota has systemic effects throughout the meta-organism and influences whole-body functionalities [4]. The human gut microbiota contains approximately 3 × 1013 bacteria, most of which are commensals [5]. A “deviated” repertoire of the gut microbiome, that has been referred to as “intestinal dysbiosis” has been epidemiologically—and sometimes causally—associated with a variety of chronic inflammatory disorders [6]. From birth, the intestinal microbiota has an impact on the life-long programming of innate and acquired immune responses, as it fine-tunes the delicate balance between inflammation, immunity versus tolerance [7]. Some anticancer modalities and co-medications could affect the delicate equilibrium between the microbiota, the epithelium barrier and the immune system, for instance by promoting the gut exodus of bacteria or their products systemically, thereby influencing tumor immunosurveillance [8]. Numerous studies have now demonstrated that the gut microbiota modulates the efficacy of anticancer therapeutics [9,10,11,12].
In 2013, our team reported the first anticancer probiotic, Enterococcus hirae isolated from spleens of animals treated with the immunological adjuvant cyclophosphamide (CTX) [11]. The antitumor efficacy of metronomic dosing of this alkylating agent, already reported to be efficient in dampening tumor growth in eubiotic animals, was compromised in germ-free or specific pathogen-free (SPF) animals treated with broad spectrum antibiotics (ATBs) [11, 13]. By altering the integrity of the intestinal barrier, hence promoting the translocation of distinct Gram+ bacteria, CTX contributed to mount effector pathogenic CXCR3+CCR6+ (IL-17+IFNγ+) Th17 and a cytotoxic CD8+ T cell (CTL) response associated with tumor control [11, 13]. Colonization of jejunal and ileal intestines with distinct isolates of E. hirae (for instance, strain 13144, “EH13144”), but not with random bacteria, selectively restored the CTX-mediated antitumor effects in ATBs-treated tumor bearers [13].
The clinical development of microbes with anticancer properties will require to define appropriate pharmaco-dynamic and -kinetic parameters associated with their clinical benefit. Mechanistically, immunogenicity results from antigenicity and adjuvanticity, meaning that anticancer probiotics might provide both components to the immune system to prime T cell responses. In fact, we recently identified an epitope derived from a Siphoviridae phage which can lysogenize distinct strains of E. hirae in mice and Enterococcus faecalis in humans [14]. This antigen, TMP, shares a 78% molecular mimicry with an oncogenic driver, PSMB4, overexpressed in certain mouse tumors. TMP-specific CD8+ T cells elicited in the spleen after oral gavage with EH13144 during CTX therapy cross-reacted with tumor cells expressing PSMB4, contributing to keep in check tumor growth [14]. However, it remains unclear how the oral bacterium succeeds in breaking tolerance to this PSMB4 self-antigen.
Results
Pleiotropic immunological functions
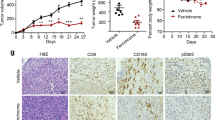
E. hirae is one of the first anticancer probiotics, as described in the setting of CTX-induced immunomodulatory effects [11, 13, 14]. Instead of using 14 days of ATBs to create the dysbiosis, we used a human fecal transfer into mice to probe the anticancer properties of EH13144. We selected stools from breast cancer (BC) patients who received alkylating agents in adjuvant chemotherapy. We then performed fecal microbial transfer (FMT) into mice previously treated with a 3-days broad-spectrum ATBs to allow human feces colonization (Fig. 1A). Stools from BC patients with dismal prognosis (triple negative BC or relapsing patients) were selected. The human ecosystem from BC patients could colonize the recipient mice with a relatively mild efficacy, yielding a mixed human/rodent microbiome at day 35 post-FMT (Fig. S1A). Fifteen days after FMT, mice were inoculated with MCA205 syngeneic tumor cells and optionally treated with weekly cycles of intraperitoneal (i.p.) CTX with a concomitant oral gavage with 109 CFU of EH13144 (Fig. 1A). At this dosing in ATBs-treated littermates, EH13144 colonized the host in a dominant manner for up to 1 week, as shown by serial culturomics of recipient stools after each cycle of CTX + oral gavage (Fig. S1B). Although the natural tumor growth was not impacted by FMT, CTX lost its capacity to reduce cancer growth in one donor FMT out of two, as exemplified in Fig. 1B, C. However, in both cases, EH13144 was effective in rescuing or ameliorating tumor growth reduction by CTX (Fig. 1D, E). Moreover, we did not observe weight loss, nor prolonged intestinal release of antimicrobial peptide lipocalin-2 or fecal IgA, nor systemic cytokine release, except CCL2, which increased as a surrogate marker of tumor progression in controls and EH13144-treated mice (Fig. S1C–H). Of note, we extended the bioactivity of EH13144 to other treatment modalities including anti-PD-1 antibody (Ab) against MCA205 [9, 14] (Fig. S2A, B) and against 4T1 where EH13144 mitigated the hyperprogression phenotype (Fig. S2C). In addition, EH13144 tended to ameliorate responses to anti-PD-1+CTLA-4 Abs in an orthotopic RENCA kidney cancer (Fig. S2D). Interestingly, random probiotics (such as Lactobacillus paracasei) failed to mediate any activity (Fig. S2C, D), demonstrating the specific antitumor potential of EH13144 across a broad array of transplantable tumors, genetic backgrounds, and therapeutics.

A Experimental setting of experiments including fecal microbial transfer (FMT). FMT from cancer patients was performed after 3 days of antibiotics (ATBs) in SPF mice. Two weeks later, the tumor cell line was inoculated and treatment started depending on the tumor type. B, C Individual tumor growth kinetics of MCA205 sarcoma treated or not with weekly administrations of cyclophosphamide (CTX) in animals reared in SPF conditions or in ATBs-treated mice subjected to FMT derived from two different breast cancer (BC) patients. Two/three experiments of 5–6 mice/groups were pooled for each FMT donor. D, E Mean ± SEM of tumor sizes at time of sacrifice (day 21) in SPF mice and animals transplanted with BC stools ± EH13144 during CTX-based therapy. Each dot represents one mouse. Statistics: *p < 0.05, **p < 0.01 (linear mixed-effects modeling or Mann–Whitney test).
We then performed a spatiotemporal analysis of the immunomodulatory potential of EH13144 in association with CTX across all lymphoid organs (including mesenteric lymph nodes (mLN), spleens, tumor draining lymph nodes (tdLN), and tumor beds) at days 3, 7, 10, and 14 post-CTX in FMT#1 mice. There were no major effects on mLN (Fig. S3A). Splenocytes were the first to react to the oral gavage at day 7, with a significant polarization of Th1 and IFNγ-producing CTL (Tc1) (Fig. 2A) and an increase of central memory CD8+ T cells (Fig. 2B). By day 7, EH13144 also induced the accumulation of Th1/Tc1 cells in tdLN, as shown by the upregulation of the chemokine receptors CXCR3 on CD4+ and CD8+ T (Fig. 2C) and, to some extent, of the intestinal CCR9 chemokine receptor in a subset of Th1/Tc1 cells peaking at day 7 (Fig. 2D, E). Intrigued by the CCR9 expression which was induced post-EH13144, we compared the abundance of the lymphocytic infiltrates of the ileal lamina propria in tumor-free versus tumor-bearing mice. In CTX + 13144 group, we did not observe intestinal lymphocytosis after inoculation of the sarcoma that could be ascribed to reduced CD8+ T cell infiltrates (as demonstrated using a CD8β cell-depleting Ab, Fig. S3B). Given the prompt activation of T cell responses by adding EH13144 to CTX, we analyzed thymopoïesis. Low-dose CTX induced a depletion of double-positive CD4+CD8+ thymocytes (Fig. S3C). EH13144 accelerated the CTX-induced accumulation of CD4 and CD8 single-positive (SP) cells and the maturation of CD8 SP thymocytes co-expressing TCRβ and CD3 by days 2–5 (Fig. 2F).

Flow cytometric determination of various subsets of lymphocytes in FMT#1-treated mice bearing MCA205 sarcoma and subject to CTX ± EH13144 treatment. A Flow cytometric analysis of Th1 (left panel) and Tc1 (right panel) cells in spleen 7 days post-CTX treatment. B Flow cytometric analysis of CD8+ T cells for CD44 and CD62L expression in spleen 7 days post-CTX treatment in mono-associated mice. TCM: central memory T cells co-expressing CD44 and CD62L. TE-TEM: effector- memory T cells expressing CD44 and negative for CD62L. C Flow cytometric analysis of Th1 (left panel) and Tc1 (right panel) cells in tumor draining lymph node (tdLN). D, E Flow cytometry analysis of double-positive CCR9+CXCR3+ expression in tumor draining lymph node-residing CD4+ (left panel) or CD8+ T cells (right panel) at day 7. F Flow cytometry of thymopoiesis showing CD4 simple positive (SP) thymocytes (left panel), CD8 SP cells (middle panel), and the mature population (TCRβ+ chain and CD3+) among CD8 SP (right panel) 2 or 5 days post-CTX treatment ± EH13144 (mono-associated mice). Each dot represents one mouse and mean ± SEM is depicted. Statistics: *p < 0.05, **p < 0.01, ***p < 0.001 (Mann–Whitney test).
Beyond its systemic immunostimulatory effects, EH13144 also ameliorated the CTX-mediated reprogramming of the tumor microenvironment (TME), including the accumulation and proliferation of CD8+ T cells within tumor-infiltrating lymphocytes (TILs) (Fig. 3A, B), likely as a consequence of its capacity to reduce the frequency of tumor-infiltrating Tregs, thereby increasing the CD8/Tregs ratio associated with tumor control (Fig. 3C). CCR9+CD4+ and CD8+ T cells, primarily observed in tdLN, also accumulated in the tumor beds (Fig. 3D). Importantly, EH13144 did not induce the expression of PD-1/LAG-3 exhaustion markers on CTX-primed T cells (Fig. 3E), but selectively upregulated the percentages and mean fluorescence intensity of the cell surface 4-1BB/CD137 on antigen-experienced (CD44+) TILs (Fig. 3F).

Flow cytometric determination of various subsets of tumor infiltrating lymphocytes in FMT#1-treated mice bearing MCA205 sarcoma and subject to CTX ± EH13144 treatment (A–D). A Flow cytometry analysis of CD8+ TILs (left panel) and Ki67+ expression in CD8+ TILs (right panel) 7 days post-CTX treatment. B Calculation of absolute numbers of CD8+ TILs normalized to tumor weight. C Flow cytometry analysis of TIL infiltrates, gating on Foxp3+CD25+ CD4+ T cells (Treg) (left panel) allowing to calculate the ratio between CD8+T/Treg (middle panel). Spearman correlation between CD8/Tregs ratio and tumor size at the time of killing (right panel). D Flow cytometry analysis of CCR9+ expression in CD8+ TILs 7 days post-CTX treatment. Flow cytometric determination of various subsets of tumor infiltrating lymphocytes in mono-associated mice bearing MCA205 sarcoma and subject to CTX ± EH13144 treatment (E, F). E Percentages of antigen-experienced CD8+ TILs expressing inhibitory receptors such as PD-1 and LAG-3, 7 days post-treatment. F Percentages of antigen-experienced CD8+ TILs expressing 4-1BB (left panel) and median of fluorescence intensity (MFI) of 4-1BB among 4-1BB+ cells (right panel). Each dot represents one mouse, and mean ± SEM is depicted. Statistics: *p < 0.05, **p < 0.01, ***p < 0.001 (Mann–Whitney test).
Altogether, live EH13144 ameliorated CTX immunological functions in primary and secondary lymphoid organs in the setting of a dysbiotic fecal microbial transplantation, contributing to reshaping the TME without inducing overt lymphocyte exhaustion nor systemic toxicity.
Role of NOD2/TLR2 in the adjuvanticity of EH13144
In the past, we reported the relevance of Th1 cytokines (IL-12p70 and IFNγ) for EH13144-induced T cell responses and antitumor effects [9, 13]. Furthermore, we previously showed that CTX promoted the translocation of E. hirae that mounted effector immune responses associated with tumor control. Of note, we demonstrated that the antitumor efficacy of CTX relied on E. hirae in a NOD2-dependent manner. Accordingly, we decided to decipher the role of pathogen recognition receptors ligands (including TLR2 and NOD2 ligands) in this scenario. The CTX + EH13144-induced splenic expansion of CD8+ Tc1 cell effectors was abolished in Nod2−/−xTlr2−/− gene-deficient mice compared with wild-type (WT) counterparts (Fig. 4A). We next compared the capacity of bacterial products to trigger the production of IL-12p70 production by dendritic cells (DC) derived from WT, Nod2−/−, Tlr2−/−, and Nod2−/−xTlr2−/− mice. Of note, the muropeptide profiles of EH17, an E. hirae strain devoid of immunogenicity [13, 14], and EH13144 were essentially identical, suggesting that the probiotic strain would be endowed with particular NOD2-modulatory effects (Fig. 4B, left). In contrast, we noted that filtered supernatants of EH13144 contained a more diverse proteome than EH17 (Fig. 4B, middle). Since lipoproteins function as TLR2 ligands, we hypothesized that membrane vesicles (MVs) excreted from the live enterococcus in the supernatants of EH13144 might act as vehicles for TLR2 and NOD2 ligands. We isolated MVs from supernatants by ultracentrifugation on a density gradient after a step of vacuum ultrafiltration. Two pellets sedimenting at different densities containing vesicles of various sizes were isolated (Fig. 4B, right). EH13144-derived MV exhibited a TLR2-dependent bioactivity on DC for IL-12p70 release (Fig. 4C). In contrast, heat-inactivated bacteria triggered DC in a NOD2-dependent manner (Fig. 4D). Nevertheless, intradermal vaccination using EH13144-derived MV directly injected into the skin was tolerogenic stimulating tumor outgrowth, regardless of dosage (Fig. 4E). Such MV-based vaccines decreased the pool of splenic CCR9+CXCR3+ Tc1 cells and of antigen-experienced PD-1+CD8+ T cell effectors in tdLN compared with naive, non-vaccinated animals (Fig. 4F).

A Flow cytometric analysis of Tc1 cells in spleen 7 days post-CTX ± EH13144 treatment in mono-associated WT versus Nod2−/−/Tlr2−/− mice. B Muropeptide profiles of EH17 and EH13144 (left panel). Schematic indicates the muropeptide composition of peaks of interest. Hexagons, GlcNAc (blue), MurNAc (pink); circles, l-Ala (blue), iso-Gln (green), l-Lys (yellow), d-Ala (purple), l-Asn or l-Asp (white). SDS-PAGE analysis of the supernatants of two strains of E. hirae (and media devoid of bacteria as negative control) (middle panel) with photograph of the sedimentation rings observed following discontinuous centrifugation gradient (right panel). Bands 1 and 2 were pooled and examined by transmission electron microscopy unveiling the presence of membrane vesicles (scale bar: 100 nm) (right panel). C, D ELISA measurements of IL12p70 concentrations in supernatants of bone marrow-derived dendritic cells (BMDCs) derived from WT versus Tlr2−/− or Nod2−/− or Nod2−/−/Tlr2−/−gene deficient mice stimulated with various concentrations, in μg/mL, of membrane vesicles (MV) (C) or boiled EH13144 or EH17 for 24 h (D). MOI: 1:20; 1:50; 1:100, n = 4/condition. E Experimental setting (top panel) and tumor growth kinetics of MCA205 after intra-dermal vaccination with EH13144 MV versus BMDCs incubated with heat-inactivated EH13144 (MOI 10 for 2 h) (bottom panel). F Flow cytometric determination of CCR9+CXCR3+ expression in CD8+ splenocytes (left panel) and PD-1 expression in CCR9+CXCR3+ in tumor draining lymph node-residing (tdLN) CD8+ (right panel). UT untreated, MV membrane vesicles. G Means ± SEM of tumor sizes at time of sacrifice in mice exhibiting “FMT-induced dysbiosis” and compensated with live (L) or pasteurized (P) EH13144 during CTX–based therapy. H Flow cytometric determination of CD4+ T cells producing IL-17 (left panel) and γδ+ T cells producing IL-17 or IFNγ (middle and right panels) in tdLN. Each dot represents one mouse, and mean ± SEM is depicted. Statistics: *p < 0.05, **p < 0.01 (linear mixed-effects modeling or Mann–Whitney test).
Importantly, in contrast to live bacteria, pasteurized EH13144 was not capable of boosting CTX-mediated immune responses and anticancer effects (Fig. 4G). Indeed, pasteurized EH13144 favored the differentiation of Th17 (Fig. 4H, left) and IL-17-producing γδT cells over that of IFNγ-producing γδT cells in tdLN (Fig. 4H, middle and right). It is noteworthy that IFNγ was previously shown to be pivotal for the EH13144-mediated antitumor efficacy [13].
Altogether, these data highlight the complexity of the bioactivity of various components of EH13144 in vitro and in vivo for cancer immuno-surveillance. Oral formulation of live EH13144 mediated Th1/Tc1 immune responses while dead bacteria induced Th17/γδT17 responses. Moreover, MV from EH13144 were not immunogenic.
EH13144 shifted the original microbiota ecosystem
We investigated the ability of EH13144 to modulate the composition of the gut microbiota resulting from the FMT of two independent BC patients into the intestines of MCA205-sarcoma-bearing mice. We performed quantitative metagenomics by shotgun metagenomic sequencing of 35 mouse stool samples harvested 35 days post-FMT in recipient groups treated or not EH13144 (Fig. 1). Accordingly, E. hirae was only detected in stools of recipient mice orally gavaged with EH13144, while it was absent from NaCl- or CTX-treated mice (Fig. 5A). Interestingly, E. hirae abundance was negatively correlated with the tumor size, emphasizing the impact of EH13144 on the anticancer effects of CTX (Fig. 5B). We next evaluated compositional differences between recipient and non-recipient of EH13144 using linear discriminant analysis effect size (LEfSe) [15] and pairwise comparison of relative taxonomic abundances. Several bacterial species were enriched or markedly depleted in recipients of EH13144 (Fig. 5C). While CTX alone mostly promoted an overrepresentation in Bacteroides sp., the addition of E. hirae specifically selected Bifidobacterium animalis, Lactobacillus sp. including L. reuteri and L. murinus as well as Candidatus arthromitus unclassified (Fig. 5C). Bacteroides nordii was underrepresented in E. hirae-treated mice when compared to CTX or NaCl-treated mice (Fig. 5C, D). Using culturomics, we cultivated L. reuteri only in stools of mice that were fed with E. hirae (Fig. 5E). When segregating animals into responders (R) versus non-responders (NR) (according to the tumor size, R being considered as displaying a tumor size below the mean tumor size of the whole group treated with CTX), we observed a trend toward an overrepresentation in E. hirae and B. animalis in R compared to NR mice (Fig. 5B, F). Altogether, E. hirae selected a specific gut microbiota that may play a role in its antitumor properties.

A Metagenomic analysis of E. hirae gene abundance in feces at day 14 post-cyclophosphamide (CTX). Gene abundance of 1750 E. hirae genes is shown (1 row for 1 gene, 1 column for 1 mice, n = 35 animals) (white: gene absent, light and dark blue: gene present). B Pearson correlations between E. hirae fecal abundance per mouse stool and tumor size at day 14 post-treatment on n = 35 animals contained in two pooled experiments. C, D Linear discriminant analysis of effect size (LEfSe) [15], and pairwise comparison of relative taxonomic abundances in three groups of 11 (NaCl) or 12 mice (CTX and CTX + EH13144) gathered from two independent experiments (n = 35 mice). Several bacterial taxa were enriched in recipients of E. hirae ordered by descending LDA score (C). Plots deriving from pairwise analysis at species level for the three groups (n = 35 mice, 11 untreated mice, 12 treated mice) (D). E Differential and common bacteria isolates identified by mass spectrometry following culturomics of mouse stools treated with CTX (with (red) or without (green) EH13144). F Relative abundance in percentages of Bifidobacterium animalis among responding (R) and non-responding (NR) mice in E. hirae-treated group (n = 12 mice). We considered responders (R), mice bearing tumors with surfaces less than the mean tumor surface in groups treated with CTX. Statistics: *p < 0.05, **p < 0.01 (Pearson correlation, Mann–Whitney test).
E. hirae modulated the local and systemic release of polyamines
We next monitored in a kinetic manner (day 3, day 7, day 14) the impact of oral gavages with E. hirae in mono-associated mice previously decontaminated by broad spectrum ATBs (versus continuous ATBs, Fig. S4A) on the local (ileal, colonic) and distal (liver, plasma) concentrations of various metabolites. ATBs mostly impacted colonic metabolites compared to SPF mice such as short-chain fatty acids (SCFA) and biliary acids (BA) (mostly secondary BA, such as T-hyodeoxycholic acid (T-HDCA) and T-deoxycholic acid (T-DCA)) were markedly downregulated by ATBs (Fig. S4B). The majority of secondary BA was also reduced in the plasma by ATBs (Fig. S4B). After withdrawing ATBs, the mono-association with E. hirae restored the primary BA β-muricholic acid found in all compartments by day 7 up to day 14 (Fig. S4C). E. hirae increased the plasma levels of the coenzyme nicotinamide (NAM) (Fig. S4D). Moreover, E. hirae specifically modulated the metabolism of polyamines (Fig. S4E–G). It induced the accumulation of ornithine and N8-acetylspermidine in the liver, that of putrescine, N-acetylputrescine, cadaverine in ilea and colons, culminating in plasma detection of spermine at day 14 (Figs. 6A–C, S4E, and S5A). E. hirae more rapidly induced the intestinal release of N1-acetylspermine and N1-acetylspermidine as well as spermidine than other commensals (such as Barnesiella intestinihominis or Catenibacterium. mitsuokella, Fig. S5B).

A–D Box plot representing normalized values of polyamines in the liver (A), ileum (B and C), and plasma (D) of E. hirae mono-associated mice compared to NaCl- (or SPF reared mice) and continuous antibiotics (ATBs)-treated mice at D7 and D14 (n = 12 mice in the E. hirae and SPF groups, n = 11 mice in ATB group) or D14 (plasma, n = 6 mice per group), experimental setting in Fig. S4A. E GFP-LC3 H4 cells were infected for 6 h with EH13144 or non-relevant E. hirae strain (depicted #10) (MOI 1:100), torin (1 µM) or media. Quantification of the GFP-LC3 puncta (mean of dot count/cells). F and G GFP-LC3 transgenic mice were subjected to 3-day ATBs treatment, followed by oral gavage with NaCl (UT group) or E. hirae (13144 group). On day 6, mice were intraperitoneally (i.p.) injected with leupeptin (40 mg/kg) and sacrificed 3 h later. Quantification of the GFP-LC3 puncta (dots/µm2) (F) and representative micrograph pictures are depicted (G). H WT mice were subjected to normal water (UT) or continuous ATBs (ATB) or followed by oral gavage with EH13144 (13144) ± i.p. CTX. Mice were sacrificed, ileal tissues were harvested to extract total proteins, and run western blotting to assess the relative expression of LC3B-II and LC3B-I. Data are expressed as mean ± SEM of two pooled independent experiments. I Relative efficacy of the combination of CTX + EH13144 against MCA205 tumor growth in WT versus atg4b−/−-deficient mice. The tumor sizes at sacrifice are depicted, one dot representing one mouse. A typical experiment comprising 6 mice/group is presented. Statistics: ***p < 0.001, **p < 0.01, *p < 0.05, +p < 0.1 (linear mixed-effects modeling, Mann–Whitney tests).
Given the capacity of spermidine to activate the autophagy machinery in many model systems across species and to synergize with chemotherapeutic agents [16, 17], we analyzed the capacity of E. hirae to induce autophagy in vitro and in vivo. We incubated H4 or Caco-2 cells engineered to express LC3 fused to GFP (LC3-GFP), an autophagosome marker, in the presence of EH13144. As shown in Figs. 6D and S5C, exposure to EH13144 induced the accumulation of LC3-GFP+ puncta in a manner comparable to the positive control whereas another strain of E. hirae (depicted #10) failed to do so. In addition, EH13144 induced LC3-GFP+ puncta in ileal enterocytes of GFP-LC3 transgenic mice (Fig. 6F, G). Next, we performed western blotting to evaluate the autophagy-associated lipidation of LC3 (which leads to an increase in the abundance of LC3-II, an electrophoretically more mobile form) (Fig. 6H). Indeed, exposure of ileal enterocytes to EH13144 in vivo induced increased LC3B-II/LC3B-I ratios, associated with autophagy (Fig. 6H). Since ATG4b is known to proteolytically activate LC3 to render possible its autophagy-relevant lipidation, and given the increased susceptibility of Atg4b-deficient mice to develop colitis [18], we compared the relative efficacy of the combination of CTX + EH13144 in WT versus Atg4b-deficient mice. ATG4 was required for tumor growth reduction by EH13144 (Fig. 6I). Similar findings were obtained using mice harboring a villin-driven Atg5 deficiency (not shown).
We conclude that colonization with E. hirae triggered the metabolism of polyamines in the gut, liver, and plasma, associated with the intestinal induction of autophagy. Moreover autophagy is mandatory for the tumor growth-reducing effect of E. hirae.
From modes of action to improved therapeutic strategies of E. hirae
Our findings indicate that EH13144 (combined with CTX) induces the accumulation of CD137-expressing CD8+ TIL (Fig. 3), favors the fecal enrichment in B. animalis (Fig. 5), and triggers systemic release of polyamines and NAM (Fig. 6). To implement these pleiotropic modes of actions in the optimization of EH13144-mediated anticancer effects, we designed combinatorial treatments. Firstly, to test the functional relevance of 4-1BB+ T cells elicited by EH13144 7 days post-oral gavage, we treated mice only once with the comibnation of CTX + EH13144 and waited 7 days to start the supplementation with i.p. administrations of agonistic CD137 mAb. Secondly, we treated mice with CTX + EH13144 concomitantly with spermidine (i.p.) or NAM in drinking water. Thirdly, in order to test the added value of B. animalis onto CTX + EH13144, we gavaged mice with both bacteria (following the classical schedule depicted in Fig. 1A). Successful additive bioactivity was achieved when CTX + EH1344 was combined with an agonistic anti-CD137 Ab (Fig. 7A), or with oral supplementation of spermidine or NAM (Fig. 7B) or with B. animalis (Fig. 7C).

MCA205 tumor growth kinetics were monitored in animals receiving intraperitoneal (i.p.) CTX and oral gavages with E. hirae 13144 alone (only one gavage in A) (A–C) or followed by i.p. administration of anti-4-1BB/CD137 agonistic Abs (or isotype control Abs) (A) or concomitant with spermidine (i.p., 50 mg/kg) or nicotinamide (NAM) in drinking water (0.5%) (B) or with Bifidobacterium animalis (C). One representative experiment out of two yielding similar results, or pooled two independent experiments yielding the same conclusions, is depicted for each combinational regimen, comprising at least 6 mice/group. Statistics: *p < 0.05, **p < 0.01, ***p < 0.001 (linear mixed-effects modeling).
These results indicate that E. hirae has a pleiotropic action on the immune system, whole-body metabolism, and the gut microbiota that can be exploited to further enhance its favorable immuno-oncological profile (Fig. S6).
Discussion
A number of functional modalities targeting the gut microbiome are being harnessed to restore the homeostasis of the gut and the meta-organism during cancer progression and treatment, to boost therapeutic effects or alleviate side effects [19]. Our group focused on the small intestine resident E. hirae (phylum Firmicutes, class Bacilli, order Lactobacillales, family Enterococcaceae), which is endowed with the capacity to translocate into secondary lymphoid organs and to stimulate the anticancer immune response [11, 13]. This initial discovery has paved the way to cultivation of highly homologous strains of EH13144 from human stools in lung and kidney cancer patients [9]. The clinical relevance of this anticancer microbe has been confirmed in advanced lung and ovarian cancer patients treated with CTX [13] or anti-PD-1 Ab [9], for whom circulating memory Th1/Tc1 immune responses against E. hirae (and not nine other commensals) were associated with prolonged progression-free survival. Other investigators reported correlative studies between E. hirae-specific CD8+ T cell responses in blood and tumors and prognosis in hepatocarcinoma (HCC) [20]. Disease-free survival time of HCC patients after surgery was associated with the frequency of E. hirae-reactive CD8+ T cell. Of note, less than 20% of normal volunteers harbor cultivatable E. hirae colonies (unpublished observation), while shotgun metagenomics of feces hardly detect these enterococci, probably because they are residing in the upper tract of the bowels at low frequency and are subdominant in the stool.
In ATBs-induced dysbiosis, we found that oral gavage with 109 CFU of EH13144 maintained stable colonization and culturomics-based detection of EH13144 for up to 7 days (Fig. S1B). This was sufficient to trigger a tumor antigen-specific, cytotoxic IFNγ+ CD8+ T cells response and a decrease in intratumor regulatory T cells associated with tumor control [13]. We have now extended these immunological properties to mice rendered dysbiotic by FMT derived from BC patients. These immune adjuvant effects appear to depend on several membrane components (such as NOD2 ligands, TLR2 ligands). While NOD2 ligands may be the drivers of IL-12 release using killed bacteria, TLR2 ligands appear to account for the bioactivity of MV. Of note, NOD2 expression by epithelial cells is known to mitigate CTX-induced enterococci translocation, thereby reducing the immunomodulatory effects of oral gavage with E. hirae [13]. TLR2 lipoproteins are known to dampen Treg-mediated immunosuppression, for instance during infection with Candida albicans [21], and may directly engage TLR1/2 receptors in viral or tumor antigen-specific CD8+ T cells, enhancing their cytolytic function [22]. Moreover, the quality of cognate T cell responses elicited by “priming” with oral E. hirae was quite different from that reported with other Gram+ bacteria such as Listeria monocytogenes expressing a neoantigen [23], where effector memory CD8+ TIL not only upregulated Granzyme B, IFNγ, KLRG1 but also LAG-3, CTLA4, and PD-1, suggestive of “terminally differentiated exhausted T cells”. Instead, E. hirae-mediated T cell activation was associated with TNFRSF9 (CD137) expression. Co-stimulation of TIL through TNFRSF9 is a major signaling pathway associated with TCR cognate interactions, preventing premature apoptosis, and favoring long-term recirculation in adoptively transferred CAR or TCR engineered T cells [24,25,26,27].
Interestingly, the capacity of E. hirae to elicit a productive anticancer immune response depended on its effective colonization and survival in vivo, and its delivery as live bacteria. Pasteurized E. hirae induced immunosuppressive or inefficient γδT17 immune responses, as previously reported [28,29,30]. The novel concept of “vita-PAMPs” unveiled by Blander’s group relies on the notion that live attenuated vaccines elicit stronger protective immunity. This effect requires the sensing of bacterial mRNA and metabolites by innate immune effectors, thus allowing for proper ignition of the inflammasome cascade culminating in type 1 IFN responses [31]. Since this process involves an Endoplasmic Reticulum stress response engaging autophagy in phagocytes, it remains possible that E. hirae not only triggers autophagy in intestinal epithelial cells but also in host antigen-presenting cells, as suggested by the reduction of their anticancer effects in Atg4b-deficient hosts.
Metabolic remodeling can also contribute to systemic and intratumor immune functions. While the bacterium-derived butyrate has been reported to enhance the expression of CD8+ T cell-associated effector molecules [32], the polyamine spermidine could activate Th1-dependent anticancer effects by inducing autophagy in tumor cells [17]. Here, oral gavage with E. hirae increase the abundance of polyamines that in turn might stimulate anticancer immune responses. NAM was also increased in the plasma of E. hirae-treated mice and boosted the antitumor effects of CTX combined with E. hirae. A phase III study showed that NAM reduced the incidence of non-melanoma skin cancers, but the underlying mechanism has not been elucidated [33].
Another interesting mode of action of E. hirae resided in its ability to promote the fecal abundance of B. animalis. B. animalis subsp lactis was reported to mediate multiple actions on body mass and gut functions [34, 35].
Regardless of these considerations, investigating the local and systemic “abscopal” effects of probiotics will allow to better integrate these new modes of action into the oncological armamentarium and perhaps to develop new cancer treatments exploiting specific bacterial species or their products.
Methods
Mice
Experiments were performed in accordance with Government and institutional guidelines and regulations (French and European laws and regulations). The institutional board of animal ethics and “Ministère de la Recherche” approved all mouse experimental settings (permission numbers: 2014-074-501, 2016-109-7450, 2018-013-13366, and 2019-076-23184). Female C57BL/6 were purchased and used between 7 and 12 weeks of age from Envigo (France) and bred in SPF conditions (unless otherwise specified) in the animal facility of Gustave Roussy Cancer Campus. Mice reaching the endpoint established by the ethical committee before the experiment time were sacrificed and so removed from the analysis.
ATB treatments
Intestine decontamination was achieved with an ATB solution containing ampicillin (1 mg/mL), streptomycin (5 mg/mL), colistin (1 mg/mL), and vancomycin (0.25 mg/mL) (Sigma-Aldrich, Saint-Louis, Missouri, USA) added to the sterile drinking water of mice. ATB activity was confirmed by cultivating fecal pellets resuspended in Brain Heart Infusion (BHI) + 15% glycerol at 0.1 g/mL on COS (Columbia Agar with 5% sheep blood, COS ref: 43049, BioMérieux, France) plates for 48 h at 37 °C in aerobic and anaerobic conditions (GENbox Jar and GENbag, BioMérieux, France).
FMT experiments
FMT was performed by thawing fecal material. Two hundred microliters of the suspension was then transferred by oral gavage into ATB pre-treated recipient. In addition, another 100 μL was applied on the fur of each animal. Two weeks after FMT, tumor cells were injected and mice were treated with CTX or NaCl after at least 1 week when reaching a 25-mm2 surface, as mentioned below.
Cell culture and reagents
MCA205 WT fibrosarcoma cell line (syngeneic from C57BL/6J mice) was cultured at 37 °C under 5% CO2 in RPMI 1640 (GIBCO Invitrogen), containing 10% FCS, 2 mM l-glutamine, 100 UI/mL penicillin/streptomycin, 1 mM sodium pyruvate, and MEM non-essential amino acids (henceforth referred to as complete RPMI-1640), all reagents being purchased from Gibco-Invitrogen, Carlsbad, CA, USA.
Tumor challenge and treatment
About 0.8 × 106 MCA205 tumor cells were inoculated subcutaneously in the right flank of syngeneic C57BL/6 mice. When tumors reached a size of 20–40 mm2 (day 0), mice were randomized based on their tumor sizes to homogenize the groups. Mice were injected weekly i.p. with 100 mg/kg (mice weight were taken at that time) of CTX (ENDOXAN, Baxter, Deerfield, Illinois, USA, provided by Gustave Roussy Cancer Center) or NaCl. Tumor size was routinely monitored by means of a calliper (Ref: 530-122, Mitutoyo). For some experiments, the measurement of tumor size was performed blinding by the investigator. In some experiments, spermidine (Sigma Aldrich) was injected i.p. (at 50 mg/kg) on the same day as each oral gavage.
Bacterial preparations and gut colonization with dedicated bacterial species
Colonization of ATBs-pre-treated or naïve C57BL/6 mice was performed by oral gavage with 100 µL of suspension containing 1 × 109 bacteria. Efficient colonization was confirmed by culture of feces 48 h post-gavage. Fecal pellet contents were harvested and resuspended in BHI + 15% glycerol at 0.1 g/mL. Serial dilutions of feces were plated onto sheep’s blood agar plates (COS, BioMérieux) and incubated for 48 h at 37 °C with 5% CO2 in aerobic or anaerobic conditions. After 48 h, single colonies were isolated and Gram staining was performed. The identification of specific bacteria was accomplished through the combination of morphological tests and analysis by means of a matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometer (Andromas, Beckman Coulter, Brea, California, USA, or Microflex, Bruker Daltonics, Leipzig, Germany). E. hirae 13144 isolate used in the experiments was originally isolated from secondary lymphoid organs of SPF mice treated with CTX. E. hirae were grown on COS plates for 24 h at 37 °C in aerobic conditions. The other species were grown on COS plate in accordance with their requirement in oxygen. E. hirae were inactivated by pasteurization (30 min at 70 °C) [36] or boiling (20 min at 100 °C) or heat (2 h at 65 °C). Bacterial preparations were then immediately frozen and stored at −80 °C.
Culture and propagation of bone marrow-derived dendritic cells
Bone marrow-derived dendritic cells (BMDCs) were generated using bone marrow from the femurs and tibias of 8–12-week-old female C57Bl/6 WT mice. Bones were collected in sterile PBS and washed in alcohol and Iscove’s medium (IMDM, Sigma-Aldrich) baths. Then, the extremities of bones were cut and flushed using a 26G needle. After red blood cell lysis, cells were cultured in IMDM supplemented with 10% of FCS, 2 mM l-glutamine, 100 UI/mL penicillin/streptomycin, and 50 μM 2-mercaptoethanol (Sigma-Aldrich) (referred herein as complete IMDM medium) at 0.5 × 106 cells/mL and treated with 40 ng/mL of GM-CSF (supernatant of GM-CSF transfected-cells J558) and 10 ng/mL of IL-4 (PeproTech, Hamburg, Germany). Cells were split every 3–4 days.
Vaccinations
BMDCs were activated with poly I:C (10 µg/mL) overnight before infection with heat-inactivated E. hirae 13144 at a multiplicity of infection (MOI) 1:10. After 6 h of incubation, BMDCs were washed three times with PBS before subcutaneous injection in the left flank of mice. Mice were vaccinated twice at 10 days intervals with EH13144-pulsed-BMDCs or MVs (5 μg/mice), then they were subcutaneously injected with tumor MCA205 (0.8 × 106 cells/mice) in right flank, 30 days after second vaccination with BMDCs.
Cytokine release post-BMDCs stimulation with MVs or boiled bacteria
At day 8, BMDCs derived from WT, or Tlr2−/− or Nod2−/− or Tlr2−/−Nod2−/− dKO mice were exposed with the isolated MVs) from either EH13144 or EH17 at different concentrations (0.8 μg/mL, 2 μg/mL, or 4 μg/mL) or with boiled bacteria (EH13144 or EH17) at different MOIs (1:20, 1:50, or 1:100) for 24 h at 37 °C in complete medium. Then, culture supernatants from BMDCs were monitored for IL12p70 secretion as indicated by ELISA (BD Biosciences, San Jose, CA, USA).
Flow cytometry analyses
Tumors, mLN, tdLN, and spleens were harvested at different time points after the first injection of CTX into mice bearing MCA-205 tumors. Excised tumors were cut into small pieces and digested in RPMI medium containing Liberase TM at 25 μg/mL (Roche) and DNase1 at 150 UI/mL (Roche) for 30 min at 37 °C, and then crushed and filtered twice using 100- and 70-μm cell strainers (Becton & Dickinson). Lymph nodes and spleen were crushed in RPMI medium and subsequently filtered through a 100-μm cell strainer. About 0.5–2 × 106 splenocytes (after red blood cell lysis through ACK regimen), tdLN, mLN, and tumor cells were preincubated with purified anti-mouse CD16/CD32 (clone 93; eBioscience) for 15 min at 4 °C, before membrane staining. For intracellular staining, the Foxp3 staining kit (eBioscience) was used. Dead cells were excluded using the Live/Dead Fixable Yellow dead cell stain kit (Life Technologies TM, Carlsbad, California, USA). Anti-mouse antibodies for CD3ε (145-2C11, ref: 557596), CXCR3 (CXCR3-173, ref: 17-1831-82), CCR9 (CW-1.2, ref: 128706), CD4 (GK1.5, ref: 17-0041-82 or 100428), CD8α (53-6.7, ref: 100722, 553036), CD8β (H35-17.2, ref: :550798), IL-17 (eBio17B7, ref: 17-7177-81), IFNγ (XMG1.2, ref: 45-7311-82), CD45.2 (104, ref: 45-0454-82), CD25 (PC61.5.3, ref: 102022), Ki67 (FITC mouse anti-human KI67 set, ref: 556026), Foxp3 (FJK-16s, ref: 17-5773-82), CD44 (IM7, ref: 103032), CD137 (17B5, ref: 17-1371-82), LAG3 (c9b7w, ref: 17-2231-82), PD-1 (J43, ref: 11-9985-82), TCRβ (H57-597, ref: 11-5961-82), and TCRδ (eBioGL3, 12-5711-82) were obtained from BD, BioLegend, eBioscience and R&D. For cytokine staining, cells were stimulated for 4 h at 37 °C with 50 ng/mL of phorbol 12-myristate 13-acetate (PMA; Calbiochem), 1 μg/mL of ionomycin (Sigma-Aldrich), and BD Golgi STOPTM (4 µL/6 mL), BD Biosciences. Stained samples were acquired on a Canto II or on a Cytoflex (Beckman Coulter) cytometer and analyses were performed with FlowJo software (Tree Star, Ashland, OR, USA).
Peptidoglycan purification and muropeptide analysis
Highly purified peptidoglycan was prepared based on a previously published protocol (Wheeler et al. [37]). Peptidoglycan (500 μg) was digested overnight with 100 U/mg of mutanolysin (from Streptomyces globisporus ATCC 21553, Sigma Aldrich) in 12.5 mM sodium phosphate buffer pH 5.6 (37 °C with continuous agitation). The reaction was stopped by boiling for 3 min in a waterbath and the supernatant which contains the soluble muropeptides collected, following centrifugation for 5 min at 16,000 × g. Muropeptides were separated by reversed-phase high-pressure liquid chromatography (HPLC) using a Shimadzu LC-20 system with a Hypersil GOLD aQ column (250 × 4.6 mm, 5 μm particle size, Thermo Fisher Scientific, Waltham, Massachusetts, USA), flow rate 0.5 mL/min at 52 °C. Mobile phases were prepared using HPLC-super gradient-grade reagents (HiPerSolv CHROMANORM, VWR). The mobile phase gradient was H2O + 0.05% trifluoroacetic acid with 0–25% acetonitrile over 135 min. Elution of muropeptides was detected by UV absorption measured at 206 nm.
Electron microscopy
For transmission electron microscopy, the MV suspension (in 10 mM HEPES-NaCl buffer) was spotted onto carbon film-coated 300 mesh copper grids (Agar Scientific, England) pretreated by glow-discharge, and subjected to negative staining with uranyl acetate. Once dry, the grids were imaged using a FEI Tecnai T12 microscope.
SDS-PAGE analysis
The Laemmli Tris-glycine method [38] was used for SDS-PAGE analysis. A 12% polyacrylamide gel was used with a mini PROTEAN Tetra System (Bio-Rad) and a Bio-Rad PowerPac Basic power supply. Proteins were visualized using Imperial protein stain (Thermo Fisher Scientific).
Culturomic analysis
To identify the bacterial species present in the fecal samples of mice treated by CTX with or without E. hirae, feces were crushed and diluted in 1 mL of PBS. Tenfold serial dilutions of the bacterial suspension were inoculated on 5% sheep blood-enriched Columbia Agar (BioMerieux) at 37 °C in aerobic and anaerobic conditions. The anaerobic atmosphere was created using the GENbag Anaer system (BioMerieux). Moreover, 100 μL were added to blood culture bottles (aerobic and anaerobic conditions) at 37 °C. The liquid cultures were then seeded on 5% sheep blood-enriched Columbia agar. After a 24-h and 48-h incubation for aerobic and anaerobic conditions, respectively, colonies were subcultured on 5% sheep blood-enriched Columbia Agar for purification and identified using a MALDI-TOF mass spectrometer (Microflex spectrometer, Bruker Daltonics, Leipzig, Germany).
Metagenomic analysis
Total fecal DNA was extracted as described [39] and sequenced using ion-proton technology (Thermo Fisher) resulting in 23.2 ± 2.5 million (mean ± SD) single-end reads with a mean length of 150 bases. Reads were cleaned using [31] AlienTrimmer in order (i) to remove resilient sequencing adapters and (ii) to trim low-quality nucleotides at the 3ʹ side using a quality and length cut-off of 20 and 45 bp, respectively. Cleaned reads were subsequently filtered from human, mice, and other possible food contaminant DNA (using Human genome RCh37-p10, Mus musculus, Bos taurus, and Arabidopsis thaliana and an identity score threshold of 97%). The gene abundance profiling was performed using the 9.9 million gene integrated reference catalog of the human gut microbiome [40], the 2.6 million gene mouse gut catalog [41], and against E. hirae ATCC9790 genes. Filtered high-quality reads were mapped with an identity threshold of 95% to the different gene catalogs using Bowtie 2 [42] included in METEOR software [43]. The gene abundance profiling table was generated by means of a two-step procedure using METEOR. First, the unique mapped reads (reads mapped to a unique gene in the catalog) were attributed to their corresponding genes. Second, the shared reads (reads that mapped with the same alignment score to multiple genes in the catalog) were attributed according to the ratio of their unique mapping counts. The gene abundance table was processed for normalization and further analysis using the MetaOMineR (momr R) package [44]. To decrease technical bias due to different sequencing depth and avoid any artefacts of sample size on low abundance genes, normalization was performed according to the FPKM strategy (normalization by the gene size and the number of high-quality reads reported in frequency) to give the gene abundance profile table. E. hirae abundance was estimated as the mean abundance of 1750 genes.
Metabolomics
Standard and reactives
Acetonitrile (Sigma Aldrich)/isopropanol (Sigma Aldrich)/methanol (Sigma Aldrich)/chloroform (Sigma Aldrich)/acetic acid (Sigma Aldrich)/formic acid (Sigma Aldrich)/dibutylamine acetate concentrate (DBAA) (Sigma Aldrich)/methoxyamine hydrochloride (Sigma Aldrich)/MSTFA—N-Methyl-N-(trimethylsilyl) trifluoroacetamide (Sigma Aldrich)/pyridine (Sigma Aldrich)/3 nitrophenylhydrazine (3-NPH) (Sigma Aldrich)/N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) (Sigma Aldrich)/sulfosalicylic acid (SSA) (Sigma Aldrich).
Sample preparation, tissue (liver)
Tissues were weighted and 30 mg was solubilized in 1.5 mL of polypropylene microtubes containing ceramic beads and 1 mL of cold lysate buffer with ISTD (MeOH/water/chloroform, 9/1/1, −20 °C), then homogenized three times for 20 s at 5500 r.p.m. using a Precellys 24 tissue homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France), followed by a centrifugation (10 min at 15,000×g, 4 °C). The upper phase of the supernatant was split in three parts: 250 µL were used for Gas Chromatography coupled by Mass Spectrometry (GC/MS), while 50 µL and 250 µL, respectively, were used for ultra-high-pressure liquid chromatography coupled by mass spectrometry (UHPLC/MS) experimentations. For GC-MS, 150 µL were transferred from the microtube to glass tube, evaporated and 50 µL of methoxyamine (20 mg/mL in pyridine) were added on the dried extracts. The solution was then stored at room temperature in dark for 16 h. The following day, 80 µL of MSTFA were added and incubated for final derivatization at 40 °C for 30 min. Samples were then transferred into vials and directly injected into GC-MS. For the detection of SCFA, 50 µL of UHPLC-MS aliquots were transferred from the injection vial. Then 25 µL of 3-NPH (200 mM) and 25 µL of EDC (150 mM) were added. The mixture was incubated at 40 °C for 1 h, and following 100 µL of water were added before injection into UHPLC-MS. Additional 250 µL of UHPLC-MS aliquots were evaporated in microtubes at 40 °C in a pneumatically assisted concentrator (Techne DB3, Staffordshire, UK). UHPLC-MS dried extracts were solubilized with 450 µL of MilliQ water and aliquoted into microtubes for each LC method. Aliquots were transferred in LC vials and injected into an UHPLC-MS or kept at −80 °C until injection.
Sample preparation, polyamines tissue (liver)
About 30 mg of tissues were first weighted and solubilized in 1.5 mL of polypropylene microtubes containing ceramic beads and 1 mL of cold lysate buffer with ISTD (5% SSA in MeOH/water, 9/1, −20 °C), then homogenized three times for 20 s at 5500 r.p.m. using a Precellys 24 tissue homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France), followed by a centrifugation (10 min at 15,000 × g, 4 °C). Then, the upper phase (600 µL) of the supernatant was evaporated in microtubes at 40 °C in a pneumatically assisted concentrator (Techne DB3, Staffordshire, UK). UHPLC-MS dried extracts were solubilized with 300 µL of 10% SSA in MilliQ water, centrifugated (10 min at 15,000 × g, 4 °C), and 50 µL were transferred in polypropylene vial injection. Aliquots were transferred in polypropylene vials and injected into an UHPLC-MS or kept at −80 °C until injection.
Sample preparation, fecal content (ileum/colon content)
Material for each condition was first weighted and 30 mg per sample were solubilized in 1.5 mL of polypropylene microtubes containing 1 mL of cold lysate buffer with ISTD (MeOH/water/chloroform, 9/1/1, −20 °C). After being vortexed for 10 min, the samples were centrifugated (10 min at 15,000 × g, 4 °C). The upper phase of the supernatant was split in three parts: 250 µL were used for GC-MS experiment in microtube, while 50 µL and 250 µL, respectively, were used for UHPLC-MS experimentations. Concerning the GC-MS aliquots, 150 µL were transferred from the microtube to a glass tube and evaporated; 50 µL of methoxyamine (20 mg/mL in pyridine) were added on dried extracts, then stored at room temperature in dark, during 16 h. The following day, 80 µL of MSTFA were added and final derivatization was achieved by an incubation at 40 °C for 30 min. Samples were then transferred in vials and directly injected into GC-MS. UHPLC-MS aliquots (50 µL for SCFA method) were transferred from the injection vial; 25 µL of 3-NPH (200 mM) and 25 µL of EDC (150 mM) were added. The whole was heated at 40 °C during 1 h; 100 µL of water was added and injected into UHPLC-MS. Concerning UHPLC-MS aliquots, the collected supernatant was evaporated in microtubes at 40 °C in a pneumatically assisted concentrator (Techne DB3, Staffordshire, UK). UHPLC-MS dried extracts were solubilized with 450 µL of MilliQ water and aliquoted in three microtubes (100 µL) for each LC method and one microtube as backup. Aliquots for analysis were transferred in LC vials and injected into an UHPLC-MS or kept at −80 °C until injection.
Sample preparation, polyamines fecal content (ileum/colon content)
About 30 mg of tissues were first weighted and solubilized into 1.5 mL polypropylene microtubes containing ceramic beads and 1 mL of cold lysate buffer with ISTD (5% SSA in MeOH/Water, 9/1, -20 °C), vortexed followed by a centrifugation (10 min at 15000 g, 4 °C). The upper phase (600 µL) of supernatant was evaporated in microtubes at 40 °C in a pneumatically-assisted concentrator (Techne DB3, Staffordshire, UK). UHPLC-MS dried extracts were solubilized with 300 µL of 10% SSA in MilliQ water, centrifugated (10 min at 15000 g, 4 °C) and 50 µL were transferred into polypropylene vial injection. Aliquots were transferred into polypropylene vials and were injected into an UHPLC-MS or kept at −80 °C until injection.
Sample preparation, plasma (lithium heparin)
A volume of 50 µL of plasma was mixed with 500 µL a cold solvent mixture containing ISTD (MeOH/water/chloroform, 9/1/1, −20 °C), in a 1.5-mL microtube, vortexed and centrifugated (10 min at 15,000 × g, 4 °C). Then the upper phase of the supernatant was split in three parts: first 200 µL were used for GC-MS experimentation in glass tubes, the rest was used for UHPLC-MS experimentations. GC-MS aliquots (150 µL) were transferred from the microtube to a glass tube and evaporated; 50 µL of methoxyamine (20 mg/mL in pyridine) were added on the dried extracts, then stored at room temperature in the dark, for 16 h. The following day, 80 µL of MSTFA was added and final derivatization was achieved by incubation at 40 °C for 30 min. Samples were then transferred into vials and directly injected into GC-MS. UHPLC-MS aliquots (50 µL for SCFA method) were transferred from the injection vial and 25 µL of 200 mM 3-NPH and 25 µL of EDC (150 mM) were added. The mixture was heated at 40 °C for 1 h before the addition of 100 µL of water and injection into an UHPLC-MS. For UHPLC-MS, the collected supernatant was evaporated in microtubes at 40 °C in a pneumatically assisted concentrator (Techne DB3, Staffordshire, UK) and dried extracts were solubilized with 450 µL of MilliQ water and then aliquoted into microtubes (100 µL) for each LC method. Aliquots for analysis were transferred into LC vials and injected into UHPLC-MS or kept at −80 °C until injection.
Sample preparation, polyamines plasma (lithium heparin)
A volume of 50 µL of plasma was mixed with 500 µL of a cold solvent mixture with ISTD (5% SSA in MeOH/water, 9/1, −20 °C) into 1.5 mL microtubes, vortexed and centrifugated (10 min at 15,000 × g, 4 °C). Then upper phase (300 µL) of supernatant was evaporated in microtubes at 40 °C in a pneumatically assisted concentrator (Techne DB3, Staffordshire, UK). UHPLC-MS dried extracts were solubilized with 300 µL of 10% SSA in MilliQ water, centrifugated (10 min at 15,000 g, 4 °C), and 50 µL were transferred into polypropylene vial for injection into an UHPLC-MS or kept at −80 °C until injection.
Quality control policy
A daily qualification of the instrumentation was set up with automatic tune and calibration processes. These qualifications were completed with double injections of standards mixes, at the beginning and end of the run, as for a blank extracted sample to control background impurities. Mixtures were adapted for each chromatographic method. After extraction, fractions of each biological sample were pooled to create a quality control (QC) sample, used to passivize the column before analysis with biological matrix and re-injected regularly during batch analysis to monitor and correct analytical bias (m/z, retention time, and sensitivity drifts) during post acquisition data normalization.
Widely targeted analysis of intracellular metabolites gas chromatography (GC) coupled to a triple quadrupole (QQQ) mass spectrometer
GC-MS/MS method was performed on a 7890A gas chromatograph (Agilent Technologies, Santa Clara, CA, US) coupled to a triple quadrupole 7000C (Agilent Technologies) equipped with a high-sensitivity electronic impact source (EI) operating in positive mode. Front inlet temperature was 250 °C, injection was performed in splitless mode. Transfer line and ion-source temperature were 250 °C and 230 °C, respectively. Septum purge flow was fixed at 3 mL/min, purge flow to split vent operated at 80 mL/min during 1 min and gas saver mode was set to 15 mL/min after 5 min.
Helium gas flowed through column (J&W Scientific, HP-5MS, 30 m × 0.25 mm, i.d. 0.25 mm, d.f., Agilent Technologies Inc.) at 1 mL/min. Column temperature was held at 60 °C for 1 min, then raised to 210 °C (10 °C/min), followed by a step to 230 °C (5 °C/min) and reached 325 °C (15 °C/min), and was held at this temperature for 5 min. Collision gas was nitrogen. Scan mode used was MRM for biological samples. Peak detection and integration of analytes were performed using Agilent Mass Hunter quantitative software (B.07.01).
Targeted analysis of bile acids by ion pairing UHPLC coupled to a Triple Quadrupole (QQQ) mass spectrometer
Targeted analysis was performed on a RRLC 1260 system (Agilent Technologies) coupled to a Triple Quadrupole 6410 (Agilent Technologies) equipped with an electrospray source operating in positive mode. Gas temperature was set to 325 °C with a gas flow of 12 L/min. Capillary voltage was set to 4.5 kV. Then 10 μL of the sample were injected on a Column Poroshell 120 EC-C8 (100 mm × 2.1 mm, particle size 2.7 µm) from Agilent technologies, protected by a guard column XDB-C18 (5 mm × 2.1 mm, particle size 1.8 μm) and heated at 40 °C by a Peltier oven. Gradient mobile phase consisted of water with 0.2% of formic acid (A) and acetonitrile/isopropanol (1/1; v/v) (B) freshly prepared. The flow rate was set to 0.3 mL/min, and gradients as follows: initial condition was 70% phase A and 30% phase B, maintained for 1.5 min. Molecules were then eluted using a gradient from 30 to 60% phase B over 9 min. The column was washed using 98% mobile phase B for 2 min and equilibrated using 30% mobile phase B for 2 min. After each injection, the needle was washed twice with isopropanol and thrice with water. The autosampler was kept at 4 °C. At the end of batch analysis, the column was rinsed with 0.3 mL/min of MilliQ water (phase A) and acetonitrile (phase B) as follows: 10% phase B for 20 min, to 90% phase B in 20 min, and maintained during 20 min before shutdown. Collision gas was nitrogen. Scan mode used was the MRM for biological samples. Peak detection and integration of the analytes were performed using the Agilent Mass Hunter quantitative software (B.07.01).
Targeted analysis of polyamines by ion pairing UHPLC coupled to a Triple Quadrupole (QQQ) mass spectrometer
Targeted analysis was performed on a RRLC 1260 system (Agilent Technologies) coupled to a Triple Quadrupole 6410 (Agilent Technologies) equipped with an electrospray source operating in positive mode. The gas temperature was set to 350 °C with a gas flow of 12 L/min. The capillary voltage was set to 3.5 kV; 10 μL of sample was injected on a Column Kinetex C18 (150 mm × 2.1 mm, particle size 2.6 µm) from Phenomenex, protected by a guard column C18 (5 mm × 2.1 mm) and heated at 40 °C by a Peltier oven. Heating the column above the room temperature allowed rigorous control of the column temperature. The gradient mobile phase consisted of water with 0.1% of heptafluorobutyric acid (HFBA, Sigma-Aldrich) (A) and acetonitrile with 0.1% of HFBA (B) freshly prepared. The flow rate was set to 0.2 mL/min, and gradients as follows: initial condition was 95% phase A and 5% phase B. Molecules were then eluted using a gradient from 5 to 40% phase B over 10 min. The column was washed using 90% mobile phase B for 2.5 min and equilibrated using 5% mobile phase B for 4 min. The autosampler was kept at 4 °C. The collision gas was nitrogen. The scan mode used was the MRM for biological samples. Peak detection and integration of the 14 analytes were performed using the Agilent Mass Hunter quantitative software (B.07.01).
Targeted analysis of SCFA by ion pairing UHPLC coupled to a Triple Quadrupole (QQQ) mass spectrometer
Targeted analysis was performed on a RRLC 1260 system (Agilent Technologies) coupled to a Triple Quadrupole 6410 (Agilent Technologies) equipped with an electrospray source operating in positive mode. Gas temperature was set to 350 °C with a gas flow of 12 L/min. Capillary voltage was set to 4.0 kV; 10 μL of the sample was injected on a Column Zorbax Eclipse XBD C18 (100 mm × 2.1 mm, particle size 1.8 µm) from Agilent technologies, protected by a guard column XDB-C18 (5 mm × 2.1 mm, particle size 1.8 μm) and heated at 50 °C by a Peltier oven. Gradient mobile phase consisted of water with 0.01% of formic acid (A) and acetonitrile with 0.01% of formic acid (B). Flow rate was set to 0.4 mL/min, and gradients as follows: initial condition was 80% phase A and 20% phase B, maintained for 6 min. Molecules were then eluted using a gradient from 20 to 45% phase B over 7 min. Column was washed using 95% mobile phase B for 5 min and equilibrated using 20% mobile phase B for 4 min. The autosampler was kept at 4 °C. At the end of batch analysis, the column was rinsed with 0.3 mL/min of MilliQ water (phase A) and acetonitrile (phase B) as follows: 10% phase B during 20 min, to 90% phase B in 20 min, and maintained during 20 min before shutdown. Peak detection and integration of analytes were performed using Agilent Mass Hunter quantitative software (B.07.01).
Quantification of autophagy in Ileum
GFP- LC3 transgenic mice or WT C57BL/6 mice were subjected to 3-day ATB treatment, followed by oral gavage with NaCl or E. hirae on day 4 and day 5. On day 6, the mice were i.p. injected with leupeptin (40 mg/kg). Three hours later, the mice were sacrificed. For the quantification of GFP-LC3 puncta, the ileum tissues from GFP-LC3 transgenic mice were collected, fixed, and dehydrated. Ileum swiss-roll sections were done and counterstained with DAPI for the nucleus. Images displayed in the figures were acquired as whole slide images (WSI) with a digital slide scanner Olympus VS120. Image analysis of GFP-LC3 puncta (dots/µm2) was performed using QuPath [45]. Regions of Interest (ROI) were defined for villi, crypts, and lamina propria in each WSI by hand. Cells were detected based on the DAPI intensity. Next, GFP-LC3 puncta were detected by using subcellular detection command on QuPath.
For the western blot analysis of LC3bII, total proteins of ileum tissues (for each sample, 1 cm of the ileum tissue from the end close to cecum was used) were extracted and subjected to western blot analysis of the expression level of LC3bII (Cell Signaling Technology, #2775, 1:1000). GAPDH was used as an internal loading control. Quantification of the intensity of the bands was done by Fiji [46].
Statistics
Data analyses were performed either with the statistical environment R (http://www.R-project.org/) or Prism 6 (GraphPad, San Diego, CA, USA). Tumor growth differences were calculated using a dedicated software (https://kroemerlab.shinyapps.io/TumGrowth/). Briefly, tumor growth was subjected to a linear mixed-effect modeling applied to log pre-processed tumor surfaces. p-Values were calculated by testing jointly whether both tumor growth slopes and intercepts (on a log scale) were different between treatment groups of interest. All reported tests are two-tailed and were considered significant at p < 0.05.
Statistical assessment of microbial biomarkers using LEfSe and pair-wise analysis
The LEfSe method was used to compare abundances of all bacterial, viral, archeal, and eukaryotic species according to the three groups studied (NaCl, CTX, CTX ± EH13144). Species with differential abundance between study groups were used as input for the linear discriminant analysis (LDA) to calculate an effect size (LDA score >2). Benjamini–Hochberg False Detection Rate (FDR) at 10% was applied on normalized and standardized relative abundances before LEfSe analysis. LEfSe analysis was performed with an in-house Python v2.7 script based on the original ones available at the following link: https://bitbucket.org/nsegata/lefse. For pair-wise analysis of relative abundances, two-tailed Mann–Whitney U test was employed to assess significance (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
Culturomic
For each bacterial taxa, a mean frequency was calculated in two groups of mice, treated by CTX with or without E. hirae. A relative frequency difference was calculated for each species in order to determine which species were enriched in the two groups. Statistical significance of the relative frequency difference was determined using uncorrected chi-square test, comparing the proportion of each taxa in the two groups.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70.
Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28:690–714.
Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol. 2011;33:369–83.
Belkaid Y, Naik S. Compartmentalized and systemic control of tissue immunity by commensals. Nat Immunol. 2013;14:646–53.
Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e1002533.
de Vos WM, de Vos EAJ. Role of the intestinal microbiome in health and disease: from correlation to causation. Nutr Rev. 2012;70 Suppl 1:S45–56.
Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14:667–85.
Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE. et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 2018;555:623–8.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7.
Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–84.
Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D. et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971–6.
Derosa L, Routy B, Fidelle M, Iebba V, Alla L, Pasolli E, et al. Gut bacteria composition drives primary resistance to cancer immunotherapy in renal cell carcinoma patients. Eur Urol. 2020;78:195–206.
Daillère R, Vétizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V. et al. Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity. 2016;45:931–43.
Fluckiger A, Daillère R, Sassi M, Sixt BS, Liu P, Loos F. et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science. 2020;369:936–42.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60.
Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 2009;11:1305–14.
Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE. et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30:147–60.
Cabrera S, Fernández AF, Mariño G, Aguirre A, Suárez MF, Español Y, et al. ATG4B/autophagin-1 regulates intestinal homeostasis and protects mice from experimental colitis. Autophagy. 2013;9:1188–200.
Zitvogel L, Ma Y, Raoult D, Kroemer G, Gajewski TF. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science. 2018;359:1366–70.
Rong Y, Dong Z, Hong Z, Jin Y, Zhang W, Zhang B. et al. Reactivity toward Bifidobacterium longum and Enterococcus hirae demonstrate robust CD8+ T cell response and better prognosis in HBV-related hepatocellular carcinoma. Exp Cell Res. 2017;358:352–9.
Sutmuller RPM, den Brok MHMGM, Kramer M, Bennink EJ, LWJ Toonen, Kullberg B-J. et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–94.
Asprodites N, Zheng L, Geng D, Velasco-Gonzalez C, Sanchez-Perez L, Davila E. Engagement of Toll-like receptor-2 on cytotoxic T-lymphocytes occurs in vivo and augments antitumor activity. FASEB J. 2008;22:3628–37.
Deng W, Lira V, Hudson TE, Lemmens EE, Hanson WG, Flores R. et al. Recombinant Listeria promotes tumor rejection by CD8+ T cell-dependent remodeling of the tumor microenvironment. Proc Natl Acad Sci USA. 2018;115:8179–84.
Turcotte S, Gros A, Hogan K, Tran E, Hinrichs CS, Wunderlich JR. et al. Phenotype and function of T cells infiltrating visceral metastases from gastrointestinal cancers and melanoma: implications for adoptive cell transfer therapy. J Immunol. 2013;191:2217–25.
Hernandez-Chacon JA, Li Y, Wu RC, Bernatchez C, Wang Y, Weber JS, et al. Costimulation through the CD137/4-1BB pathway protects human melanoma tumor-infiltrating lymphocytes from activation-induced cell death and enhances antitumor effector function. J Immunother. 2011;34:236–50.
June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64–73.
Chacon JA, Wu RC, Sukhumalchandra P, Molldrem JJ, Sarnaik A, Pilon-Thomas S, et al. Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS ONE. 2013;8:e60031.
Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–41.
Punt S, Langenhoff JM, Putter H, Fleuren GJ, Gorter A, Jordanova ES. The correlations between IL-17 vs. Th17 cells and cancer patient survival: a systematic review. Oncoimmunology. 2015;4:e984547
Silva-Santos B, Serre K, Norell H. γδ T cells in cancer. Nat Rev Immunol. 2015;15:683–91.
Blander JM, Barbet G. Exploiting vita-PAMPs in vaccines. Curr Opin Pharmacol. 2018;41:128–36.
Luu M, Weigand K, Wedi F, Breidenbend C, Leister H, Pautz S, et al. Regulation of the effector function of CD8+ T cells by gut microbiota-derived metabolite butyrate. Sci Rep. 2018;8:14430.
Chen AC, Martin AJ, Choy B, Fernández-Peñas P, Dalziell RA, McKenzie CA. et al. A phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N Engl J Med. 2015;373:1618–26.
Takahashi S, Anzawa D, Takami K, Ishizuka A, Mawatari T, Kamikado K, et al. Effect of Bifidobacterium animalis ssp. lactis GCL2505 on visceral fat accumulation in healthy Japanese adults: a randomized controlled trial. Biosci Microbiota Food Health. 2016;35:163–71.
Szajewska H, Guandalini S, Morelli L, Van Goudoever JB, Walker A. Effect of Bifidobacterium animalis subsp lactis supplementation in preterm infants: a systematic review of randomized controlled trials. J Pediatr Gastroenterol Nutr. 2010;51:203–9.
Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L. et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med. 2017;23:107–13.
Wheeler R, Veyrier F, Werts C, Boneca IG. ‘Peptidoglycan and Nod Receptor’ In Taniguchi N, Endo T, Hart GW, Seeberger PH, Wong CH, editors. Glycoscience: Biology and Medicine. Springer; 2014. pp. 737–47.
Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5.
Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD. et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65:4799–807.
Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32:834–41.
Xiao L, Feng Q, Liang S, Sonne SB, Xia Z, Qiu X. et al. A catalog of the mouse gut metagenome. Nat Biotechnol. 2015;33:1103–8.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E. et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500:585–8.
Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G. et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–6.
Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7:16878.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T. et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82.
Acknowledgements
We thank the animal facility team and V. Rouffiac from the imaging platform of Gustave Roussy.
Funding
A-GG was supported by Fondation pour la Recherche Médicale (FRM). LZ and GK were supported by the Ligue contre le Cancer (équipes labellisées); Agence National de la Recherche (ANR)—Projets blancs; CANTO study (ANR-10-COHO-0004); ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), FRM; a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085 and GDW20181100051), Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology (ANR-18-IDEX-0001); the RHU Torino Lumière (ANR-16-RHUS-0008); the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM).
Author information
Authors and Affiliations
Contributions
LZ, RD, and AGG conceived the study, analyzed the data, provided the intellectual guidance, and wrote the paper with the help of GK. AGG, RD, AF, RW, BQ, FL, KI, LM, MTA, EP, LD, SD, FAp, NB, MM-N, DD, GC, SY, OK, ML, and AJ performed the experiments and analyzed the data. LD and FAn provided patients’ feces. ELC, NP, NS, and VL analyzed the bioinformatics data. DR and IGB provided the mice, bacteria, and reagents. IGB and GK also provided intellectual guidance.
Corresponding authors
Ethics declarations
Conflict of interest
RD, LZ, and GK are cofounders of EverImmune, a biotech company focused on the use of commensal bacteria for cancer treatment. FL and RD were employees of EverImmune.
Ethical approval and consent to participate
The institutional board of animal ethics and Ministère de la Recherche approved all mouse experimental settings (permission numbers: 2014-074-501, 2016-109-7450, 2018-013-13366, and 2019-076-23184). Stools from breast cancer patients were collected from CANTO study (NCT01993498). The study was approved by the Comité de Protection des Personnes IDF7 (Bicêtre, project: 11-039). All patients gave their consent for the use of their fecal material and the related data, to be published.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Piacentini
Rights and permissions
About this article

Cite this article
Goubet, AG., Wheeler, R., Fluckiger, A. et al. Multifaceted modes of action of the anticancer probiotic Enterococcus hirae. Cell Death Differ 28, 2276–2295 (2021). https://doi.org/10.1038/s41418-021-00753-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-021-00753-8
This article is cited by
-
Targeting the gut and tumor microbiota in cancer
Nature Medicine (2022)
-
The role of the tumor microbe microenvironment in the tumor immune microenvironment: bystander, activator, or inhibitor?
Journal of Experimental & Clinical Cancer Research (2021)
-
The involvement of oncobiosis and bacterial metabolite signaling in metastasis formation in breast cancer
Cancer and Metastasis Reviews (2021)
