
Abstract
Angiogenesis plays crucial roles in maintaining the complex operation of central nervous system (CNS) development. The architecture of communication between neurogenesis and angiogenesis is essential to maintain normal brain development and function. Hence, any disruption of neuron–vascular communications may lead to the pathophysiology of cerebrovascular diseases and blood–brain barrier (BBB) dysfunction. Here we demonstrate that neural differentiation and communication are required for vascular development. Regarding the cellular and molecular mechanism, our results show that PRDM16 activity determines the production of mature neurons and their specific positions in the neocortex. In the cortical plate (CP), aberrant neurons fail to secrete modular calcium-binding protein 1 (SMOC1), an important neuronal signal that participates in neurovascular communication to regulate CNS angiogenesis. Neuronal SMOC1 interacts with TGFBR1 by activating the transcription factors phospho-Smad2/3 to convey intercellular signals to endothelial cells (ECs) in the TGF-β-Smad signaling pathway. Together, our results highlight a crucial coordinated neurovascular development process orchestrated by PRDM16 and reveal the importance of intimate communication for building the neurovascular network during brain development.
Similar content being viewed by others


Introduction
The development of brain is a complex process supported by neurogenesis and angiogenesis. Moreover, in early brain development, the nervous system and vascular network display intriguing parallels in their functional organization [1]. During brain development, neurons are generated from neural progenitor cells (NPCs) and migrate along the basal radial glia (RG) to reach their final position in the cerebral cortex [2, 3]. The vascularization of the embryonic cortex begins from the perineural vascular plexus on the pial surface at embryonic day 9.5, followed by continued sprouting and remodeling until a hierarchical vascular network is formed [4, 5]. Blood vessels in the brain control the transport of nutrients, oxygen delivery, and energy metabolite supply from the blood into the brain [6, 7]. Importantly, the brain vessel system is distinct from that of other organs because the vessel walls are composed of specialized endothelial cells (ECs) that form a tightly sealed blood–brain barrier (BBB), which plays a key role in regulating the transport of substances between the blood and brain [8]. Furthermore, vascular dysfunction and BBB breakdown are frequently associated with complex neurodegenerative and disorders, including cerebrovascular disorder, Huntington’s disease, and Alzheimer’s disease (AD) [9,10,11]. The process of angiogenesis is associated with many signaling pathways. Among them, the multifunctional transforming growth factor β (TGF-β) signaling pathway plays many critical roles in vascular development [12,13,14]. TGF-β is a major extracellular signaling molecule in neuronal cells during CNS angiogenesis [15]. The effect of TGF-β signaling activation is mediated by the recruitment of endothelial TGF-β type I receptor (TGFBR1) to regulate endothelial proliferation and cerebrovascular integrity [16,17,18]. Similarly, neuronal cells can secrete cytokines to activate the endothelial TGF-β-Smad2/3 signaling pathway and thus regulate angiogenesis [19]. However, it remains unclear which specific genes affect vessel development by disturbing this process of neurogenesis.
During central nervous system (CNS) development and vascular remodeling, neural cells and the vasculature form the NVU, which plays an important role in maintaining brain homeostasis, and its dysfunction might result in a large number of neuropsychiatric diseases [20, 21]. Prior studies have indicated that neuronal progenitors, guide the ingression of blood vessels into the cerebral cortex, which further interact with growing vessels in regulating CNS angiogenesis [3, 22]. However, less is known about the bidirectional communication between the vascular and nervous systems that coordinates their functions. Although vascular cells play important roles in neurogenesis by forming a unique anatomical structure to regulate neural progenitor cell division and self-renewal [23, 24], how neural cell signals are transmitted to vascular components during angiogenesis remains elusive.
PRDM16 is a transcriptional regulator belonging to the larger PRDM family. The structure of the full-length PRDM16 includes an N-terminal PR domain and two clusters of zinc fingers [25, 26]. It was originally identified in human leukemias, in which chromosomal breakpoint mutations are oncogenic [27]. In addition, PRDM16 is also expressed in brown adipose tissue, where it acts as an important factor in regulating the cell-fate switch between skeletal myoblasts and brown adipose tissue [28,29,30]. Recent studies have focused on the role of PRDM16 in multiple stem cell lineages and the regulation of stem cell homeostasis [31, 32]. Moreover, PRDM16 is preferentially expressed in stem cells in the CNS and hematopoietic system [33]. However, how PRDM16 functions in multiple ways to orchestrate the neurovascular signaling that regulates the brain homeostasis remains largely unexplored. Here, we demonstrated the function of PRDM16 in regulating angiogenesis via niche neuron–EC communication during cortical development. We found that PRDM16 controlled neuronal production and determined the specific positions of these neurons in cortical plate (CP). Furthermore, we demonstrated that neuronal secrete modular calcium-binding protein 1 (SMOC1), through the TGF-β signaling pathway, played an important role to regulate angiogenesis in brain development.
Results
PRDM16 is expressed in neural progenitor cells and correlates with vessel development
During brain development, angiogenesis always occurs cooperatively with neuronal formation [34]. To evaluate blood vessel growth in the embryonic cortex, we systematically examined the patterns of angiogenesis using biotinylated isolectin B4 (IB4) to label neocortical blood vessels from E14 to P0. The results revealed that blood vessels sprouted from the pial perineural and soon invaded the CP (Fig. 1a–c). Previous studies have demonstrated that PRDM16 is preferentially expressed in the CNS [31]. We first examined where PRDM16 is expressed in the developing mouse neocortex. The expression pattern of PRDM16 was abundantly expressed in the ventricular zone (VZ) and subventricular zone (SVZ) (Fig. 1d). To further confirm the results, we also performed western blotting analysis of PRDM16 expression levels, and similar results were found (Supplementary Fig. 1a). Moreover, immunostaining assays revealed that PRDM16 was also expressed in dissociated neural progenitors labeled with Nestin, Pax6, and SOX2 (Fig. 1e and Supplementary Fig. 1b) [35], whereas PRDM16 was positively correlated with embryonic neurogenesis.

a Patterns of vascular development from E14.5 to P0 during mouse brain cortical development. IB4 was used to label growing vessels in the cortical plate (CP). Scale bars, 100 μm. Quantification of blood vessel length (b) and the number of branch points (c) in the CP. Increased numbers of vessels and branch points were observed during brain cortical development. The data are represented as the means ± SEM (n = 3 animals). d Immunofluorescence staining for PRDM16 in the developing cerebral cortex at E16.5, E18.5, and P0. Immunostaining showed that PRDM16 was abundantly expressed in the VZ/SVZ. e E13.5 neural progenitor cells were isolated from the mouse embryonic cortex. PRDM16 was co-labeled with Nestin and PAX6 in cultured cells, and Prdm16 was expressed in NPCs in vitro. Scale bar, 50 μm. f IB4 immunostaining images of E18.5 cortical sections electroporated with a control shRNA or Prdm16 shRNA plasmid at E13.5. The electroporation of Prdm16 shRNA results in abnormal GFP-positive cells distribution (left), which consequently caused vascular defects in the cortical plate (right). Scale bar, 100 μm. Quantification of the change in vessel length (g) and the number of branch points (h) in the cortical plate after electroporation. The data are represented as the means ± SEM (n = 3; one-way ANOVA; *P < 0.05).
To study the functions of PRDM16 in embryonic cortex development, we generated two different Prdm16-specific shRNAs. Subsequently, we tested the efficiencies of the Prdm16-specific shRNAs in endogenous NPCs, and the knockdown efficiencies were examined by western blotting (Supplementary Fig. 1c, d). Next, we performed in utero electroporation (IUE) of Prdm16-specific shRNAs and control plasmids and found that knockdown of Prdm16 led to a significant change in the GFP-positive cells distribution in the whole cerebral cortex. There was a significant reduction in the number of GFP+ cells in the CP (Fig. 1f). Then, we performed immunostaining for IB4 to label blood vessels in the brain cortex. To our surprise, the results showed that both the vessel density and number of branch points in the vascular network were also disrupted (Fig. 1f–h). Signals from niche neurons have been implicated in the regulation of vascular proliferation and sprouting [36, 37]. It remains unclear whether and how PRDM16 exerts a distinct function in embryonic neurogenesis to influence angiogenesis.
Prdm16 deletion in neural progenitors results in angiogenesis defects during brain development
To further explore the function of PRDM16 in cortical development, we used Prdm16 conditional knockout (Prdm16cKO-Nes) mice to examine the role of PRDM16 (Fig. 2a). To confirm that the expression of PRDM16 was depleted in the Prdm16cKO-Nes brain cortex, we performed immunostaining of brain slices at E16.5 (Fig. 2b). Furthermore, western blot analysis and real-time PCR analysis of the brain cortex confirmed the decreased expression of PRDM16 in Prdm16cKO-Nes brains (Fig. 2c and Supplementary Fig. 2a, b). In addition, compared with Prdm16fl/fl brains, Prdm16cKO-Nes brains showed a reduction in size (Supplementary Fig. 2c). Cortical morphology was detected by H&E staining in coronal sections from P0 mice (Supplementary Fig. 2d). Although overall cortical lamination appeared normal, the size of the ventricles was increased and the thickness of the cortex was reduced in Prdm16cKO-Nes brains compared with Prdm16fl/fl littermate brains (Supplementary Fig. 2e, f).

a Schematic of the construction of Prdm16 conditional knockout mice. LoxP sites were inserted between the 9th exon in these mice. The Prdm16 gene was knocked out by nervous-tissue-specific Cre recombinase splicing. b Confocal images of PRDM16 immunostaining used to detect conditional knockout efficiency at E16.5. A reduction in the PRDM16 signal was observed in the cortical ventricular zone. Scale bar, 100 μm. c Western blot analysis of PRDM16 expression levels in Prdm16fl/fl and Prdm16cKO-Nes brain cortices at different stages. Evidently, PRDM16 expression was depleted in Prdm16cKO-Nes brains. d Representative confocal images of blood vessels in the brain cortex stained with IB4 at E18.5. The insets indicate higher-magnification images of the upper cortex in the delineated areas. Angiogenesis was reduced in the cortex in Prdm16cKO-Nes mice. Scale bar, 100 μm. Quantification of the vessel length (e) and the number of branch points (f) in the brain cortical plate. Reduction in vascularization in the Prdm16cKO-Nes cortical plate, as evidenced by changes in both the vessel length and the number of branch points, was observed in. The data are represented as the means ± SEM (n = 3 animals; unpaired Student’s t test; **P < 0.01). g IB4 labeling of blood vessels in the postnatal brain cortex. Prdm16 gene knockout during the development of the vasculature led to the persistent disruption of angiogenesis. Scale bar, 100 μm. Quantification of the vessel length (h) and the number branch points (i) showed reduced vascularization in the cortex of Prdm16cKO-Nes mice. The data are represented as the means ± SEM (n = 3 animals; unpaired Student’s t test; **P < 0.01). j Confocal images of collagen IV (Col IV) staining showed defective basement membrane maturation in Prdm16cKO-Nes mice compared with Prdm16fl/fl mice. Scale bar, 100 μm. k Quantification of the fluorescence intensity of Col IV staining showed reductions in Prdm16cKO-Nes mice. a.u., arbitrary units. The data are represented as the means ± SEM (n = 3 animals; unpaired Student’s t test; *P < 0.05). l PDGFRβ labeling of pericytes (green) showed similar coverage along blood vessels (red) in Prdm16fl/fl and Prdm16cKO-Nes brain cortices. The insets indicate higher-magnification images of the delineated areas. Scale bar, 100 μm. m Quantification of PDGFRβ-positive pericyte coverage along blood vessels. Pericyte coverage was quantified by analyzing the percent length of IB4. There were no significant differences between the Prdm16fl/fl and Prdm16cKO-Nes mice. The data are represented as the means ± SEM (n = 3 animals; unpaired Student’s t test; ns not significant).
Vessel growth is always accompanied by cerebral cortex development and regulated by niche neurons during vascularization [38, 39]. Therefore, we set out to address whether PRDM16 is involved in angiogenesis during brain development. We examined the vasculature of the cortex by labeling blood vessels with IB4 at E18.5. Clearly, the deletion of Prdm16 led to significant decreases in the vessel density compared with those of Prdm16fl/fl littermates (Fig. 2d). Subsequent quantification showed a dramatic reduction in the area of the CP covered by blood vessels (Fig. 2e, f). To verify the role of PRDM16 in vessel development, we analyzed the vascularization of the postnatal brain cortex. Immunostaining produced similar results in newborn Prdm16cKO-Nes mice (Fig. 2g–i). Moreover, another blood vessel markers, CD31 (PECAM-1) [40] and Flk1 (VEGFR2) [41], were used to label growing vessels in the cerebral cortex. Similar results were revealed in the Prdm16cKO-Nes developing neocortex (Supplementary Fig. 2g–l). To further investigate the long-term impact of PRDM16 on angiogenesis in cortical development, we examined the blood vessel networks in the brain cortex at P5 [42]. The immunostaining of blood vessels with IB4 showed a persistent disruption of angiogenesis (Supplementary Fig. 3a, b). We further evaluated the patterns of angiogenesis in the VZ/SVZ regions of Prdm16cKO-Nes brain cortices at different stages. Immunostaining with IB4 revealed similar angiogenesis defects in the VZ/SVZ regions when Prdm16 was absent (Supplementary Fig. 3d–i). To explore the potential influence of apoptosis, a TUNEL assay was performed to assess cell apoptosis. There was no significant difference between Prdm16fl/fl and Prdm16cKO-Nes cortices (Supplementary Fig. 3j, k).
The basement membrane plays fundamental roles in angiogenesis during cortical development. To evaluate potential vessel basement membrane defects, we performed immunostaining for collagen IV and the results showed that the basement membrane of Prdm16cKO-Nes vessels was discontinuous and patchy (Fig. 2j, k). In the CNS, neurovascular units couple with the BBB. We detected BBB integrity by the fluorescent tracer Alexa Fluor 555 cadaverine and found slightly elevated cadaverine extravasation from blood vessels in the Prdm16cKO-Nes brain cortex (Supplementary Fig. 3l–n). To further examine the recruitment of pericytes, we performed staining for PDGFRβ, which showed that the recruitment of pericytes to blood vessels was normal in the mutant brain (Fig. 2l, m). Together, these results indicate that the selective deletion of Prdm16 in neural progenitors leads to a significant and persistent disruption of vessel density and branch points.
Neuronal SMOC1 mediates angiogenesis during central nervous system (CNS) development
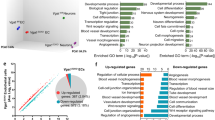
Previous studies have found that vascular development depends on some signals from niche neurons [36], and we hypothesize that PRDM16 regulates angiogenesis through some exogenous factors. To identify candidate signals, we performed RNA-seq to analyze the genome-wide changes when Prdm16 was deleted. Gene Ontology analysis of the downregulated genes in biological processes showed enrichment of terms related to neurogenesis, blood vessel development, and secretion by cells (Fig. 3a, b). The heat map and volcano plots indicated that transcripts of genes were differentially expressed in Prdm16fl/fl and Prdm16cKO-Nes cortices (P < 0.05, fold change > 1.5; Fig. 3c, d). We performed further analysis of the RNA-seq results and found a dramatic decrease in the expression of SMOC1 which belongs to the SPARC family of proteins, and the whole genome showed that SMOC1 potentially linked to altered vascular structure [43, 44]. We excavated 15 differentially expressed genes rated by significance which had been depicted as a heat map (Fig. 3e). The SMOC1 gene was a promising candidate for regulating critical events and modulating signaling events during brain development [45]. Besides, SMOC1 has been linked to angiogenesis in the murine retina by inhibiting ALK5 signaling [46], whereas little is known about its potential role in the CNS angiogenesis.

a, b Gene Ontology (GO) analysis of biological processes related to the downregulated or upregulated genes in the Prdm16-depleted forebrain at E16.5. The log10 (P value) is indicated by the bar plots. c Heat map showing global genes were sorted by fold change (fold change > 2.0). d Volcano plots showing differentially expressed genes between Prdm16fl/fl and Prdm16cKO-Nes cortices. Upregulated genes are indicated by red dots and downregulated genes are indicated by green dots. The Smoc1 gene indicated by the red dots highlights the significant reduction in downregulated genes. e Heat maps were further analyzed and classified to show the top 15 differentially expressed genes in the mutants. The Smoc1 gene was one of the significantly differentially expressed genes. f Western blotting confirmed the dramatic reduction in SMOC1 expression levels in Prdm16fl/fl and Prdm16cKO-Nes cortices at E18.5 and P0. g Quantification of SMOC1 expression levels in Prdm16fl/fl and Prdm16cKO-Nes cortices. The data are represented as the means ± SEM (n = 3; unpaired Student’s t test; *P < 0.05). h SMOC1 secretion from the cultured neurons was measured by an ELISA kit. As a result, SMOC1 secretion from Prdm16cKO-Nes neurons was decreased when compared with SMOC1 secretion from Prdm16fl/fl neurons. The data are represented as the means ± SEM (n = 4; unpaired Student’s t test; *P < 0.05). i The SMOC1 concentration in cultured neurons was measured by an ELISA kit on different days. E13.5 NPCs were isolated from the mouse brain cortex and cultured in differentiation medium for 1, 3, 5, or 7 days to differentiate NPCs into neurons. The data are represented as the means ± SEM (n = 3).
To confirm that Smoc1 was a downregulated gene, we validated the expression level of SMOC1 in Prdm16fl/fl and Prdm16cKO-Nes cortical samples at different stages (E18.5 and P0). Western blotting and real-time PCR analysis showed that SMOC1 was obviously reduced in knockout samples (Fig. 3f, g and Supplementary Fig. 4a, b). We also performed immunostaining of brain slices with SMOC1 at E18.5, and the results showed a consistent reduction of SMOC1 (Supplementary Fig. 4c, d). Because SMOC1 is a secretory protein, we measured SMOC1 secreted by cultured neurons from Prdm16fl/fl and Prdm16cKO-Nes mice through ELISA kit. As expected, there was also a reduction in SMOC1 secretion upon Prdm16 knockout (Fig. 3h). To further confirm the source of secreted SMOC1, neurons were isolated from embryonic brains and we collected supernatants from neurons cultured to measure secreted SMOC1 by ELISA. The results showed that the expression of SMOC1 was increased upon neuronal differentiation in cell culture (Fig. 3i). Subsequently, we asked whether the deletion of Prdm16 in vitro reduces the expression level of secreted SMOC1. The results revealed that the secreted SMOC1 level was decreased in vitro cultured cells (Supplementary Fig. 4e). Furthermore, to tested whether the reduced level of SMOC1 was due to individual neurons in CP. Neurons and NPCs were isolated from Prdm16fl/fl and Prdm16cKO-Nes mice embryos. And then, we performed western blotting analysis for the detection of SMOC1 in neurons and NPCs. The results showed that the reduced level of SMOC1 in the position of CP came from specific-type neurons (Supplementary Fig. 4f, g).
PRDM16 regulates vessel development through disturbances in neurogenesis
In the CNS, the absence of PRDM16 caused a decrease in the vessel density and number of branch points during cortical development (Fig. 2d–i). Previous results have shown that PRDM16 organizes stage-specific gene expression programs in RG and regulates the production of neuronal subtypes in the cortex [45]. However, we focused on nervous system defects that result in vessel development defects. To verify the function of PRDM16 in neurogenesis, we electroporated GFP plasmids into the embryonic Prdm16fl/fl and Prdm16cKO-Nes mice and the GFP+ cells showed an aberrant distribution in the Prdm16cKO-Nes cortex (Supplementary Fig. 5a, b). To examine whether the upper layers were impaired, we analyzed the cortex by labeling neurons with CUX1 at E18.5. There was a dramatic decrease in the thickness of CUX1+ neurons in the CP and the presence of abnormal upper layer neurons caused disruptions of vessel networks (Fig. 4a, b). To confirm the persistent disruption, we performed immunostaining for CUX1 and IB4 in the cortices of newborn mice. Similar results were found in postnatal mice (Supplementary Fig. 5c, d). Furthermore, immunostaining of deep layer neurons with CTIP2 showed a slight reduction in the Prdm16cKO-Nes cortex (Fig. 4c, d). To examine whether Prdm16 deletion led to a widespread decrease in neurons, immunofluorescence staining for Tuj1 and MAP2 was performed, and the results showed that neurons in CP layers were decreased (Fig. 4e, f and Supplementary Fig. 5e, f).

a Representative image of the laminar distribution of CUX1 staining and IB4-stained blood vessels in Prdm16fl/fl and Prdm16cKO-Nes cortices at E18.5. At this stage of brain development, the cortical plate in Prdm16cKO-Nes brains is thinner than that in Prdm16fl/fl brains (white dotted line). Scale bar, 100 μm. b Quantification of the changes in CUX1+ neurons in Prdm16fl/fl and Prdm16cKO-Nes cortical plates. The data are represented as the means ± SEM (n = 4 animals; unpaired Student’s t test; **P < 0.01). c Representative confocal images of CTIP2 expression in the deep layers of Prdm16fl/fl and Prdm16cKO-Nes cortices at E18.5. Scale bar, 100 μm. d Quantification of the decrease in the number of CTIP2+ cells in the deep layers of the Prdm16cKO-Nes cortex when compared with that in the deep layers of the Prdm16fl/fl cortex. The data are represented as the means ± SEM (n = 4 animals; unpaired Student’s t test; *P < 0.05). e Tuj1 staining of Prdm16fl/fl and Prdm16cKO-Nes cortices at E18.5. Scale bar, 100 μm. f Quantification of the fluorescence intensity of Tuj1 staining showed a reduction in Prdm16cKO-Nes mice. The data are represented as the means ± SEM (n = 3 animals; unpaired Student’s t test; *P < 0.05). g Immunostaining for the mitotic marker pH3 in Prdm16fl/fl and Prdm16cKO-Nes cortices at E18.5. Scale bar, 100 μm. h Quantification of the number of pH3+ mitotic cells in the ventricular zone and subventricular zone (VZ/SVZ). The data are represented as the means ± SEM (n = 4 animals; unpaired Student’s t test; *P < 0.05). i High-magnification confocal images of GFP+ cells in the upper layer showed abnormal branched processes in the Prdm16cKO-Nes cortex. Scale bar, 100 μm. j Quantification of the total dendritic length of upper layer neurons in Prdm16fl/fl and Prdm16cKO-Nes cortices. The data are represented as the means ± SEM (n = 12 images; unpaired Student’s t test; *P < 0.05). k Representative images of neuronal morphology in vitro. GFP plasmids were electroporated into the lateral ventricles of Prdm16fl/fl and Prdm16cKO-Nes brains at E13.5. After 24 h, GFP+ cells were isolated from the cortex and cultured in differentiation medium for 4 days. Scale bar, 50 μm. l Quantification of the number of primary dendrites showed a reduction when Prdm16 was deleted. The data are represented as the means ± SEM (n = 10 images; unpaired Student’s t test; **P < 0.01). m Quantification showed that the total dendritic length of neurons was decreased when Prdm16 was deleted. The data are represented as the means ± SEM (n = 10 images; unpaired Student’s t test; **P < 0.01).
Because PRDM16 is mainly expressed in the VZ/SVZ regions and regulates both the organization of upper layer neurons, we want to know the role of Prdm16 in NPCs. We used phospho-Histone3 (pH3) to label mitotically active cells, and the results indicated that dividing cells were increased when Prdm16 was absent (Fig. 4g, h). Similar results were also found in postnatal Prdm16cKO-Nes mice (Supplementary Fig. 5g, h). The effect of premature NPCs on the regulation of neuronal production was explored by a BrdU birthdating experiment [47]. The immunostaining showed that later-born neurons exhibited aberrant positioning when Prdm16 was absent (Supplementary Fig. 6a, b). In a long-term experiment, immunostaining for CUX1 confirmed that defects of upper layer neurons in Prdm16cKO-Nes mice (Supplementary Fig. 6c, d). To test whether the absence of PRDM16 affects cortical neuronal morphology, we performed IUE for long-term tracing of GFP+ neurons. We found that the upper layer neurons exhibited abnormal dendritic morphology after their migration (Fig. 4i, j). These neurons with abnormal morphology failed to exert normal functions in the NVU, which plays an important role in maintaining brain homeostasis [48]. To further assess neuronal cell morphology, primary progenitor cells isolated from GFP plasmid-electroporated Prdm16fl/fl and Prdm16cKO-Nes embryonic brains. Quantification revealed that both the number of primary dendrites and the total dendritic length were reduced in Prdm16-deleted neurons (Fig. 4k–m). Together, the data demonstrates that Prdm16 deletion impairs both the number and organization of upper layer neurons and damages cortical neuronal morphology.
Prdm16 deletion impairs vessel growth, and exogenous SMOC1 affects EC proliferation
Vessel growth relies on coordinated collective proliferation of endothelial stalk cells, and new vessels promote the stability of vascular connections [49]. We then assessed whether the proliferation of stalk cells is affected by the loss of PRDM16 during brain development. To test this, we injected BrdU into pregnant Prdm16fl/fl and Prdm16cKO-Nes mice and analyzed EC proliferation at E18.5. Immunostaining for IB4 and BrdU showed that BrdU+ECs were reduced in the Prdm16cKO-Nes CP (Fig. 5a, b). Similar results were obtained by co-immunostaining with anti-IB4 and anti-BrdU antibodies in P0 (Supplementary Fig. 7a, b). Additional experiments were performed to investigate whether exogenous SMOC1 acts on EC proliferation. E13.5 embryonic brains were dissected and digested into single-cell suspensions, and ECs were sorted by fluorescence-activated cell sorting (FACS) for further analysis (Fig. 5c). There was a high enrichment of the desired cell types in the sorted cell suspensions (Fig. 5d). We cultured sorted ECs stimulated with SMOC1 and the BrdU was used to examine the proliferation of ECs. These results demonstrated that the direct application of exogenous SMOC1 promoted a specific increase in the proliferation of brain ECs (Fig. 5e, f). Furthermore, we used two culture systems, a direct co-culture system and an indirect culture system, to study whether neurons secrete SMOC1 to regulate EC proliferation. Neurons from Prdm16fl/fl and Prdm16cKO-Nes cortices were co-cultured with ECs. Immunofluorescence showed that there was decreased proliferation of Prdm16cKO-Nes ECs in neurovascular co-culture systems (Supplementary Fig. 7c, d). In the indirect co-culture system, we treated ECs with neuron-conditioned medium, and the results also showed that Prdm16cKO-Nes EC proliferation was decreased (Supplementary Fig. 7e, f). These findings indicate that neurons regulate EC proliferation via secreted factors but not in a direct cell–cell contact-dependent manner.

a Representative confocal images of cortical ECs proliferation at E18.5. A reduced number of BrdU-labeled ECs was observed in the Prdm16cKO-Nes cortical plate. Scale bar, 100 μm. b Quantification of the decrease in the number of BrdU+ ECs when compared with that in the Prdm16fl/fl mouse cortex. The data are represented as the means ± SEM (n = 3 animals; unpaired Student’s t test; *P < 0.05). c Schematic overview of the strategy used to sort ECs in cell suspensions with a FACSCalibur cytometer. d Representative confocal images of cell suspensions costained with CD31 and DAPI presorting and postsorting. e Immunofluorescence staining with IB4/BrdU in ECs showed increased endothelial cell proliferation upon the addition of SMOC1 (0, 10, 20 ng/ml) to the culture medium. Scale bar, 50 μm. f Quantification of the increased proliferation of BrdU-positive ECs upon SMOC1 (0, 10, 20 ng/ml) application. The data are represented as the means ± SEM (n = 3; unpaired Student’s t test; *P < 0.05; **P < 0.01).
SMOC1 participates in angiogenesis through the TGF-β signaling pathway and rescues Prdm16 depletion-induced defects
What are the molecular signaling mechanisms associated with the loss of Prdm16 in neural progenitors that contribute to impaired angiogenesis in the embryonic cortex? To explore this question, we performed KEGG pathway analysis on the RNA-seq dataset and found potential responses involving neuroactive ligand–receptor interactions in Prdm16cKO-Nes cortical tissues (Fig. 6a). In addition, the key pathway involving TGF-β-induced gene expression is the canonical TGF-β-Smad pathway. We examined whether the TGF-β-Smad pathway is impaired in the Prdm16cKO-Nes cortex, and the data showed that the phosphorylation level of Smad2/3 was reduced in the mutant cortex (Fig. 6b, c). To better explore the interaction between SMOC1 and the TGF-β-Smad pathway, sorted ECs were cultured in vitro and then treated with exogenous SMOC1. We measured the levels of pSmad2/3 in ECs and found that exogenous SMOC1 upregulated the expression of pSmad2/3 in the TGF-β-Smad pathway (Fig. 6d, e). Immunostaining showed that exogenous SMOC1 and the cytokine TGF-β promoted ECs proliferation (Supplementary Fig. 8a, b). Together, these findings suggest that exogenous SMOC1-mediated activation of the TGF-β-Smad pathway is important for CNS angiogenesis.

a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was used to evaluate enriched pathways. The neuroactive ligand–receptor interaction pathway was dramatically enriched (red rectangle). b Western blot analysis of the expression levels of pSmad2/3, Smad4, and Smad2/3 in Prdm16fl/fl and Prdm16cKO-Nes cortices at E18.5. c Quantification of pSmad2/3, Smad4, and Smad2/3 expression levels in Prdm16fl/fl and Prdm16cKO-Nes cortices. The data are represented as the means ± SEM (n = 3; unpaired Student’s t test; *P < 0.05; ns not significant). d Western blot analysis of ECs with or without SMOC1 (10 ng/ml) stimulation. The expression levels of phosphorylated Smad2/3 were increased upon SMOC1 stimulation. e Quantification of pSmad2/3, Smad4, and Smad2/3 expression levels in SMOC1 stimulated ECs. The data are represented as the means ± SEM (n = 3; unpaired Student’s t test; *P < 0.05; ns not significant). f Western blotting of forward co-immunoprecipitation of SMOC1 and TGFBR1. g The disruption of angiogenesis caused by Prdm16 knockdown was partly rescued by PRDM16. A Prdm16 shRNA plasmid was coelectroporated with a PRDM16-expressing plasmid at E13.5, and then the brains were isolated at E18.5. Scale bar, 100 μm. Quantification of the vessel length (h) and the number of branch points (i) in the cortical plate after coelectroporation. The data are represented as the means ± SEM (n = 3; one-way ANOVA; *P < 0.05). j Model showing how PRDM16 regulates angiogenesis through neuronal SMOC1 in the developing cortex. PRDM16 regulates SMOC1 secretion from neurons, and then neuronal SMOC1 activates the vascular TGF-β signaling pathway through interactions with the TGFBR1 receptor during brain development.
How does SMOC1 interact with the TGF-β-Smad pathway to regulate angiogenesis? Next, we tested whether SMOC1 interacts with TGFBR1 to activate downstream cascade signaling. We performed forward and reverse co-immunoprecipitation assays, and the data showed that there was a direct interaction between SMOC1 and TGFBR1 (Fig. 6f and Supplementary Fig. 8e). To further confirm that the effects of SMOC1 are mediated by TGFBR1, we examined the proliferation of ECs in the presence of SMOC1 or SMOC1 plus the TGFBR1 inhibitor LY-364947. Despite the presence of SMOC1, the proliferation of endothelial was not increased after the addition of the TGFBR1 inhibitor LY-364947 (Supplementary Fig. 8c, d). To determine whether forced PRDM16 overexpression can partially rescue the Prdm16 knockdown phenotypes, we performed IUE. Both the distribution of GFP-positive cells and the disruption of the vessel were partially rescued by PRDM16 overexpression (Fig. 6g–i). We next detected whether exogenous SMOC1 can rescue the developmental deficiency caused by Prdm16 deletion. ECs and neurons were co-cultured and treated with exogenous SMOC1. Immunofluorescence staining showed that the proliferation of ECs in mutant cultures was partly rescued by exogenous SMOC1 stimulation (Supplementary Fig. 8f, g). Collectively, these results support a neuron-to-vascular model, in which PRDM16 regulates CNS angiogenesis through the interaction of neuronal SMOC1 with TGFBR1 to activate the TGF-β-Smad signaling pathway during brain development (Fig. 6j).
Discussion
The brain is a complex and highly organized organ containing multiple systems that communicate with each other to ensure brain development. Among them, the nervous and vascular systems display coordination and parallelism in their development. In addition, the precise communication between the two systems is crucial for proper function and formation [38, 50,51,52]. Dysregulation of this communication might result in a series of vascular dysfunctions, including cerebrovascular disorder, BBB breakdown, and AD [9,10,11].
PRDM16 plays an important role in regulating the cell-fate switch and stem cell homeostasis [30, 31]. However, the functions of PRDM16 in mutually dependent neurovascular systems remain largely unexplored. Previous studies have shown that pericytes, neurons, and astrocytes are crucial for the development and maintenance of angiogenesis during brain development [36, 53, 54]. In the present study, we demonstrated that PRDM16 regulated angiogenesis by disrupting the process of neuron formation. We found that PRDM16 was abundantly expressed in the VZ/SVZ and exhibited a tendency to decrease during brain development. However, a recent study showed that PRDM16 leads to defects of progenitor specification by modulating oxidative stress [55]. Other functions of PRDM16 in maintaining brain homeostasis remain largely unknown. Our findings demonstrate that the deletion of Prdm16 in NPCs leads to many neuronal defects that are the cause of aberrant angiogenesis. In the VZ/SVZ regions of Prdm16cKO-Nes brain cortices, we also found angiogenesis defects. Similar results showing that late-born upper layer neurons failed to reach their final positions after their migration and exhibited abnormal dendritic morphology when PRDM16 activity was lacking [45]. However, there were some incompatibilities in the labeling of mitotic cells with pH3 at different stages. The timing of cortical development is very important in the embryonic brain. The transition of RG cells from a neurogenic to a gliogenic state may lead to incompatibilities about mitotic cells that labeled by pH3 in later embryogenesis. However, even more importantly, our data show that the absence of cortical upper layer neurons can cause disruptions in angiogenesis. These results indicate that PRDM16 is required for angiogenesis in a manner dependent on specific signals from niche neurons. Consistently, previous studies have shown that neuron–vascular communication regulates brain vessel development in the neocortex [36, 56]. To assess the molecular signaling mechanisms by which PRDM16 regulates vascular development, we found many downstream genes were involved in neuroactive ligand–receptor interactions. Among them, SMOC1 was prominently reduced in the mutant cortex. A previous study also reported that secretory factors contribute to vascular structure reconstruction [44]. To test the source of SMOC1, neurons, and NPCs were isolated and purified from Prdm16fl/fl and Prdm16cKO-Nes mice embryos. The results showed that the reduced level of SMOC1 in the position of CP came from specific-type neurons. Although there was also a decrease of SMOC1 in NPCs, the lowered expression level of SMOC1 in CP was due to specific-type neurons. Moreover, our data show that exogenous SMOC1 specifically promotes the proliferation of brain ECs. The source of secreted SMOC1 in CP was confirmed, with the results showing that CP SMOC1 was secreted by specific-type neurons. The data suggest that abnormal cortical upper layer neurons cause a decrease in the production of secreted SMOC1 to induce vascularization during brain development.
The brain NVU is a complicated microenvironment with many signals that transduce into adjacent cells through a number of signaling pathways. As previously reported, the TGF-β signaling pathway is essential for CNS angiogenesis development through the TGF-β receptors [57]. However, TGF-β receptors in different cells may have various functions, demonstrating that the response to signaling molecules relies on signaling mechanisms that produce signals within individual ECs [58]. Therefore, we examined whether some neural signals in these signaling pathways are impaired and thus affect the developing vasculature. The findings demonstrate that aberrant neurons failed to secrete SMOC1 in close proximity to peripheral ECs. We reason that secreted SMOC1 may participate in the molecular mechanism that regulates vessel development. Consistent with this reasoning, our results show that the stimulation of endothelial proliferation by SMOC1 was blocked when TGFBR1 was inhibited in EC cultures. Furthermore, the SMOC1–TGFBR1 interaction via the transcription factors phospho-Smad2/3 activated downstream cascade signaling in the TGF-β signaling pathway. Angiogenesis is an important component for maintaining the complex operation of the brain. Neural signals are essential for regulating and maintaining a functional vascular network during brain cortical development. Some neural signals have been found to regulate CNS angiogenesis, while it remains unknown how neural signals control vessel development and what molecular determinants trigger vascularization in neurovascular communication. Our present study elucidated that PRDM16 regulates CNS angiogenesis by communicating with adjacent ECs through neuronal signaling. Our data provide new insights into the regulatory link between neurons and blood vessels, and the molecular mechanisms may hold a promising perspective for the treatment of cerebrovascular defects in neurological research and the clinic.
Materials and methods
Animals
The Prdm16 floxed (Prdm16fl/fl) mice (JAX stock # 024992: B6.129-Prdm16tm1.1Brsp/J) and Nestin-Cre mice (JAX stock # 003771: B6.Cg-Tg(Nes-cre)1Kln/J) were obtained from the Jackson Laboratory. To generate Prdm16cKO-Nes mice, homozygous Prdm16fl/fl mice were crossed with the Nestin-Cre mice line. Genotyping for Prdm16cKO-Nes was using the following primers: 5′-TAT GGA GCT AGG CAG GGA CA-3′ and 5′-TCC ATA CAT CAG GGA GCA GA-3′. The Institute of Cancer Research (ICR) pregnant mice which used for IUE were obtained from Vital River Laboratories. All studied animals were housed on a 12 h light/dark cycle at proper growth temperature with the standard rodent chow. All animal experiments were approved and performed according to the guidelines and protocols, which approved by the Experiment Animal Center of Institute of Zoology, Chinese Academy of Sciences.
In utero electroporation
The detailed protocols of IUE have been described in previously study [59]. Briefly, pregnant ICR female mice or Prdm16cKO-Nes mice were deeply anesthetized. Firstly, recombinant plasmid DNA mixed with Venus-GFP plasmid were microinjected into fetal brain lateral ventricles with a paddle electrode. Every embryonic brain was electroporated with five 50 ms pulses at 40 V with 950 ms interval by using an electroporator (Manual BTX, ECM830). The electroporation brains were fixed in 4% paraformaldehyde (PFA) and then dehydrated in 30% (w/v) sucrose at 4 °C. Brains were cryosectioned at 40 μm or 15 μm by using a cryostat microtome (Leica CM1950).
BrdU Labeling
For BrdU labeling, the E13.5 pregnant mice were injected with BrdU (Sigma-Aldrich) at a dose of 50 mg/kg. And then, the mice brains were harvested at E18.5 or newborns (P0). For cell lines BrdU labeling, cells were treated with 10 μg/ml BrdU for 2 h. Finally, the brain slices or cells were stained with anti-BrdU primary antibodies for further analysis.
Cell cultures
The human embryonic kidney cells (HEK293FT) were cultured in high-glucose DMEM medium (Gibco), supplemented with 10% FBS, 1% nonessential amino acids (NEAA, Invitrogen), 0.5% GlutaMAX (Invitrogen), and 1% penicillin/streptomycin (Life Technologies). NPCs were isolated from E12.5 pregnant mice embryos and seeded into plate which coated with laminin (10 g/ml, Sigma) and poly-D-lysine (10 g/ml, Sigma) for 12 h at 37 °C. For NPCs proliferation, we need the proliferation medium which consists of 50% Neurobasal-A medium (Invitrogen) and 50% DMEM/F12 medium (Invitrogen), supplemented with 0.5% GlutaMAX (Invitrogen), 2% B27 supplement (Invitrogen), 1% penicillin/streptomycin (Invitrogen), 1% nonessential amino acids (Invitrogen), epidermal growth factor (EGF, 10 ng/ml, Invitrogen), and basic fibroblast growth factor (bFGF, 10 ng/ml, Invitrogen). For NPCs differentiation, we need the differentiation medium which includes low-glucose DMEM medium (Gibco), supplemented with 1% penicillin/streptomycin (Invitrogen), 2% B27 supplement, and 1% fetal bovine serum (Invitrogen). For SMOC1 (Abnova) stimulation experiments, ECs were pretreated for 12 h with serum-free medium and treated with SMOC1 (10 ng/ml, Abnova) for 30 min as indicated to analyze the levels of pSmad2/3.
Isolation and culture of primary mouse brain endothelial cells
The isolation and culture of primary mouse brain ECs were performed as previously described [60]. E13.5 embryonic brains were dissected on a 10 cm dish and the telencephalon was removed. Removing the pial membranes is necessary and important for the isolation of primary mouse brain ECs. Subsequently, ventral and dorsal of telencephalon were minced into 1–2 mm fragments with a scalpel. The cleaned tissues were digested with 1 mg/mL collagenase/dispase supplemented with 30 U/mL DNase I for 30 min at 37 °C. After washing three times with DMEM, the cells were filtered through a sterile 70 μm nylon mesh. Filtered cells were collected and resuspended in 2 ml red blood cell lysing buffer (Sigma-Aldrich) for 3 min at room temperature. Washing cells three times in DMEM, the cells was resuspended in 1 ml of 10% FCS-DMEM and labeled by anti-CD31-FITC (PECAM-1) (BD Biosciences). The labeled cells were sorted by FACSCalibur cytometer (Becton Dickinson), according to the manufacturer’s instructions. Finally, primary ECs were cultured with EGM-2 media (EGM-2 BulletKit; Lonza) in 24-well plate which coated with rat tail collagen type I (Sigma-Aldrich).
Immunostaining
Embryonic brains were dissected on ice and fixed in 4% (w/v) PFA. And then, the brains were cryoprotected in 30% (w/v) sucrose for 48 h at 4°C. The brains were embedded in Tissue-Tek OCT and sectioned on a freezing microtome (Leica, CM1950, Germany). 40 μm-thick brain cryosections were used to label blood vessels and 15 μm-thick brain sections were used to label nerve cells. Briefly, brain sections were washed with 1M PBS and fixed with 4%PFA for 30 min. After washing three times for 10 min with PBST (1%Triton X-100 in 1M PBS), the brain cryosections were incubated with 5% bovine serum albumin blocking solution. After three 1%PBST washes, brain sections were incubated with primary antibodies at 4 °C for overnight. The second day, sections were incubated with secondary antibodies which conjugate fluorochrome. Finally, the slices were incubated with DAPI for 5 min and mounted.
Western blotting and co-immunoprecipitation
Brain cortices or cultured cells were lysed with RIPA buffer (Solarbio, China). The concentration of protein was measured Pierce BCA Protein Assay Reagent according to the manufacturer’s instructions. Next, the protein samples in SDS-PAGE loading buffer were boiled for 10 min. Finally, the lysates were loaded into 12% SDS-PAGE gels. The protein bands were transferred to polyvinylidene fluoride or nitrocellulose (NC) membranes. Membranes were blocked in 5% nonfat milk in PBST (PBS containing 0.05% Tween-20) and incubated with primary antibodies at 4 °C for overnight. Next day, the bands were visualized by IRDye 680CW or 800CW (LI-COR Biosciences) donkey secondary antibodies. The gray density of bands was measured by the Odyssey (LI-COR Biosciences) software.
For co-immunoprecipitation, protein samples were lysed with co-IP lysis buffer (Beyotime Biotechnology) and supernatants were collected. Supernatants were incubated with anti-HA-tag or anti-Flag-tag magnetic beads (MBL) at 4 °C for overnight. Samples were washed with precold washing buffer for three times. The magnetic beads–proteins complex was suspended with SDS protein loading buffer, and protein samples from IP or total lysates were boiled for 10 min. The immunoprecipitates were separated by SDS-PAGE and transferred to NC membranes for western blot analysis.
Antibodies
The following antibodies were used for immunostaining: goat anti-PRDM16 (R&D Systems), biotinylated IB4 (Vector Laboratories; B-1205; 1:600), mouse anti-Nestin (Millipore; MAB353; 1: 1000), rabbit anti-Tuj1 (Millipore; MAB1637; 1:2000), rabbit anti-Pax6 (Abcam; ab5790; 1:1000), rabbit anti-β-actin (Proteintech; 20536-1-AP; 1:5000), mouse anti-Sox2 (R&D Systems; MAB2018; 1:1000), rabbit anti-PDGFRβ (Abcam; ab32570; 1:1000), goat anti-Flk1(R&D Systems; AF644; 1:1000), rat anti-CD31 (BD Biosciences; 553370; 1:400), rabbit anti-collagen IV (Abcam; ab6586; 1:1000), rabbit anti-Smad4 (Cell Signaling Technology; 46535; 1:1000), rabbit anti- Smad2/3 (Cell Signaling Technology; 3102; 1:500), rabbit anti-Phospho-Smad2 (Ser465/467)/Smad3 (Ser423/425) (Cell Signaling Technology; 8828; 1:500), rabbit anti-SMOC1 (ABGENT; AP13673B; 1:1000), rabbit anti-CUX1 (Santa Cruz; sc-13024; 1:1000), mouse anti-MAP2 (Millipore; MAB3418; 1:400), rabbit anti-phospho-Histone H3 (pH3) (Cell Signaling Technology; 1:500), rat anti-BrdU (Abcam; ab6326; 1:1000), mouse anti-Flag (Sigma; F1804; 1:2000), and rabbit anti-HA (Cell Signaling Technology; 1:1000).
ELISA
NPCs were isolated from Prdm16fl/fl and Prdm16cKO-Nes brain cortices and cultured in neuronal differentiation medium for 4 days at 37 °C. Supernatants from neuronal cell cultures were collected for ELISA. SMOC1 concentrations were quantitatively determined by a commercially available ELISA kit according to the manufacturer’s instructions. The absorbance was measured by an automatic microplate fluorescence reader at 450 nm. The SMOC1 concentration was calculated by interpolating values into a standard curve which generated from the commercially ELISA kit.
Quantitative real-time PCR
The total RNA of brain cortices or cultured cells was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Total RNA was reverse-transcribed into cDNA by the FastQuant RT Kit (with DNase; Tiangen), following the manufacturer’s instructions. Quantitative real-time PCR assays were performed using a SYBR qPCR master mix (Tiangen) in an ABI7500 real-time PCR system (Applied Biosystems). β-Actin expression served as an endogenous control for normalization of real-time PCR reactions. All real-time PCR reactions were performed at least in three independent biological repeats. The primers (forward sequence and reverse sequence) used for real-time PCR were listed in Supplementary Table S1.
RNA-sequencing and data analysis
Total RNA was extracted from E16.5 telencephalon of Prdm16fl/fl and Prdm16cKO-Nes mice using TRIzol reagent (Invitrogen) following the manufacturer’s instructions. RNA quality was determined by Agilent 2100 Bioanalyzer. High-throughput sequencing was performed by Beijing Annoroad Corporation through using the Illumina HiSeq 2500 platform. Data analysis was performed according to Cuffdiff and R as previously described. For heat maps, expression levels were normalized by the SCAN method. All sequencing-derived data reported in this paper has been deposited in NCBI’s GEO (Gene Expression Omnibus) under accession number GSE130802.
Imaging and statistical analysis
All the confocal images were obtained through Zeiss LSM780 and analyzed with Photoshop CS6 (Adobe). For analysis of the vessel length and number of branch points, brain sections were used 40 μm and images were acquired with a 10× objective. Images were analyzed blind with Imaris 9.0 software. For each image, a cortical region immediately surrounding blood vessels was drawn the length of vessel and branch points were measured with the module of Filament in Imaris 9.0 software. The area of cortex was measured with the module of Surface in Imaris 9.0 software. All data are presented as means ± SEM. Statistical analysis were performed with the two-tailed unpaired Student’s t test. For multiple comparisons, the data were analyzed with one-way ANOVAs. In the figures, differences were considered significant with a P value and star signs were used to represent different ranges of P values. *P < 0.05; **P < 0.01; ***P < 0.001. All statistical analyses were performed using GraphPad Prism 6.0.
References
Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature. 2005;436:193–200.
Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–44.
Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–20.
Cullen M, Elzarrad MK, Seaman S, Zudaire E, Stevens J, Yang MY, et al. GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proc Natl Acad Sci USA. 2011;108:5759–64.
Hill J, Cave J. Targeting the vasculature to improve neural progenitor transplant survival. Transl Neurosci. 2015;6:162–7.
Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–43.
Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–66.
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell. 2015;163:1064–78.
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302.
Drouin-Ouellet J, Sawiak SJ, Cisbani G, Lagace M, Kuan WL, Saint-Pierre M, et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: potential implications for its pathophysiology. Ann Neurol. 2015;78:160–77.
Sweeney MD, Sagare AP, Zlokovic BV. Cerebrospinal fluid biomarkers of neurovascular dysfunction in mild dementia and Alzheimer’s disease. J Cereb Blood Flow Metab. 2015;35:1055–68.
Jakobsson L, van Meeteren LA. Transforming growth factor beta family members in regulation of vascular function: in the light of vascular conditional knockouts. Exp Cell Res. 2013;319:1264–70.
ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8:857–69.
Gaengel K, Genove G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–8.
Garcia CM, Darland DC, Massingham LJ, D’Amore PA. Endothelial cell-astrocyte interactions and TGF beta are required for induction of blood-neural barrier properties. Brain Res Dev Brain Res. 2004;152:25–38.
Goumans MJ, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009;19:116–27.
Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20:556–67.
Li F, Lan Y, Wang Y, Wang J, Yang G, Meng F, et al. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev Cell. 2011;20:291–302.
Moya IM, Umans L, Maas E, Pereira PN, Beets K, Francis A, et al. Stalk cell phenotype depends on integration of Notch and Smad1/5 signaling cascades. Dev Cell. 2012;22:501–14.
Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci. 2016;19:771–83.
Ma S, Huang Z. Neural regulation of CNS angiogenesis during development. Front Biol. 2015;10:61–73.
Haigh JJ, Morelli PI, Gerhardt H, Haigh K, Tsien J, Damert A, et al. Cortical and retinal defects caused by dosage-dependent reductions in VEGF-A paracrine signaling. Dev Biol. 2003;262:225–41.
Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, et al. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–40.
Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–94.
Nishikata I, Sasaki H, Iga M, Tateno Y, Imayoshi S, Asou N, et al. A novel EVI1 gene family, MEL1, lacking a PR domain (MEL1S) is expressed mainly in t(1;3)(p36;q21)-positive AML and blocks G-CSF-induced myeloid differentiation. Blood. 2003;102:3323–32.
Fog CK, Galli GG, Lund AH. PRDM proteins: important players in differentiation and disease. Bioessays. 2012;34:50–60.
Mochizuki N, Shimizu S, Nagasawa T, Tanaka H, Taniwaki M, Yokota J, et al. A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t(1;3)(p36;q21)-positive leukemia cells. Blood. 2000;96:3209–14.
Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Investig. 2011;121:96–105.
Kajimura S, Seale P, Spiegelman BM. Transcriptional control of brown fat development. Cell Metab. 2010;11:257–62.
Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–7.
Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol. 2010;12:999–1006.
Aguilo F, Avagyan S, Labar A, Sevilla A, Lee DF, Kumar P, et al. Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood. 2011;117:5057–66.
Shimada IS, Acar M, Burgess RJ, Zhao Z, Morrison SJ. Prdm16 is required for the maintenance of neural stem cells in the postnatal forebrain and their differentiation into ependymal cells. Genes Dev. 2017;31:1134–46.
Ma S, Kwon HJ, Johng H, Zang K, Huang Z. Radial glial neural progenitors regulate nascent brain vascular network stabilization via inhibition of Wnt signaling. PLoS Biol. 2013;11:e1001469.
Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, et al. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–51.
Xu B, Zhang Y, Du XF, Li J, Zi HX, Bu JW, et al. Neurons secrete miR-132-containing exosomes to regulate brain vascular integrity. Cell Res. 2017;27:882–97.
Ogunshola OO, Stewart WB, Mihalcik V, Solli T, Madri JA, Ment LR. Neuronal VEGF expression correlates with angiogenesis in postnatal developing rat brain. Brain Res Dev Brain Res. 2000;119:139–53.
Eichmann A, Thomas JL. Molecular parallels between neural and vascular development. Cold Spring Harb Perspect Med. 2013;3:a006551.
Segura I, De Smet F, Hohensinner PJ, de Almodovar CR, Carmeliet P. The neurovascular link in health and disease: an update. Trends Mol Med. 2009;15:439–51.
Williams KC, Zhao RW, Ueno K, Hickey WF. PECAM-1 (CD31) expression in the central nervous system and its role in experimental allergic encephalomyelitis in the rat. J Neurosci Res. 1996;45:747–57.
Yamashita J, Itoh H, Hirashima M, Ogawa M, Nishikawa S, Yurugi T, et al. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature. 2000;408:92–6.
Harb R, Whiteus C, Freitas C, Grutzendler J. In vivo imaging of cerebral microvascular plasticity from birth to death. J Cereb Blood Flow Metab. 2013;33:146–56.
Gersdorff N, Muller M, Schall A, Miosge N. Secreted modular calcium-binding protein-1 localization during mouse embryogenesis. Histochem Cell Biol. 2006;126:705–12.
Sherva R, Miller MB, Lynch AI, Devereux RB, Rao DC, Oberman A, et al. A whole genome scan for pulse pressure/stroke volume ratio in African Americans: the HyperGEN study. Am J Hypertens. 2007;20:398–402.
Baizabal JM, Mistry M, Garcia MT, Gomez N, Olukoya O, Tran D, et al. The epigenetic state of PRDM16-regulated enhancers in radial glia controls cortical neuron position. Neuron. 2018;99:239–41.
Awwad K, Hu J, Shi L, Mangels N, Abdel Malik R, Zippel N, et al. Role of secreted modular calcium-binding protein 1 (SMOC1) in transforming growth factor beta signalling and angiogenesis. Cardiovasc Res. 2015;106:284–94.
Duque A, Rakic P. Different effects of bromodeoxyuridine and [3H]thymidine incorporation into DNA on cell proliferation, position, and fate. J Neurosci. 2011;31:15205–17.
Wu KW, Mo JL, Kou ZW, Liu Q, Lv LL, Lei Y, et al. Neurovascular interaction promotes the morphological and functional maturation of cortical neurons. Front Cell Neurosci. 2017;11:290.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307.
Adams RH, Eichmann A. Axon guidance molecules in vascular patterning. Cold Spring Harb Perspect Biol. 2010;2:a001875.
Quaegebeur A, Lange C, Carmeliet P. The neurovascular link in health and disease: molecular mechanisms and therapeutic implications. Neuron. 2011;71:406–24.
Walchli T, Wacker A, Frei K, Regli L, Schwab ME, Hoerstrup SP, et al. Wiring the vascular network with neural cues: a CNS perspective. Neuron. 2015;87:271–96.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–6.
Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani J, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. Mult Scler J. 2012;18:125–6.
Inoue M, Iwai R, Tabata H, Konno D, Komabayashi-Suzuki M, Watanabe C, et al. Prdm16 is crucial for progression of the multipolar phase during neural differentiation of the developing neocortex. Development. 2017;144:385–99.
Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood–brain barrier. Nat Med. 2013;19:1584–96.
Ma S, Santhosh D, Kumar TP, Huang Z. A brain-region-specific neural pathway regulating germinal matrix angiogenesis. Dev Cell. 2017;41:366–81.e4.
Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. 2013;61:1939–58.
Saito T. In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc. 2006;1:1552–8.
Vasudevan A, Long JE, Crandall JE, Rubenstein JL, Bhide PG. Compartment-specific transcription factors orchestrate angiogenesis gradients in the embryonic brain. Nat Neurosci. 2008;11:429–39.
Acknowledgements
We thank H.L. offered reagents about the experiments. This work was supported by grants from the National Key R&D Program of China (2019YFA0110300), the National Science Fund for Distinguished Young Scholars (81825006), CAS Strategic Priority Research Program (XDA16010301), the National Natural Science Foundation of China (31730033 and 31621004), and K.C. Wong Education Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by S. Fulda
Rights and permissions
About this article

Cite this article
Su, L., Lei, X., Ma, H. et al. PRDM16 orchestrates angiogenesis via neural differentiation in the developing brain. Cell Death Differ 27, 2313–2329 (2020). https://doi.org/10.1038/s41418-020-0504-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-0504-5
This article is cited by
-
Prognostic DNA mutation and mRNA expression analysis of perineural invasion in oral squamous cell carcinoma
Scientific Reports (2024)
-
A Single-Cell Transcriptomic Analysis of the Mouse Hippocampus After Voluntary Exercise
Molecular Neurobiology (2024)
-
A review of migraine genetics: gathering genomic and transcriptomic factors
Human Genetics (2022)
