
Abstract
The BAP1 gene has emerged as a major tumor suppressor mutated with various frequencies in numerous human malignancies, including uveal melanoma, malignant pleural mesothelioma, clear cell renal cell carcinoma, intrahepatic cholangiocarcinoma, hepatocellular carcinoma, and thymic epithelial tumors. BAP1 mutations are also observed at low frequency in other malignancies including breast, colorectal, pancreatic, and bladder cancers. BAP1 germline mutations are associated with high incidence of mesothelioma, uveal melanoma, and other cancers, defining the “BAP1 cancer syndrome.” Interestingly, germline BAP1 mutations constitute an important paradigm for gene–environment interactions, as loss of BAP1 predisposes to carcinogen-induced tumorigenesis. Inactivating mutations of BAP1 are also identified in sporadic cancers, denoting the importance of this gene for normal tissue homeostasis and tumor suppression, although some oncogenic properties have also been attributed to BAP1. BAP1 belongs to the deubiquitinase superfamily of enzymes, which are responsible for the maturation and turnover of ubiquitin as well as the reversal of substrate ubiquitination, thus regulating ubiquitin signaling. BAP1 is predominantly nuclear and interacts with several chromatin-associated factors, assembling multi-protein complexes with mutually exclusive partners. BAP1 exerts its function through highly regulated deubiquitination of its substrates. As such, BAP1 orchestrates chromatin-associated processes including gene expression, DNA replication, and DNA repair. BAP1 also exerts cytoplasmic functions, notably in regulating Ca2+ signaling at the endoplasmic reticulum. This DUB is also subjected to multiple post-translational modifications, notably phosphorylation and ubiquitination, indicating that several signaling pathways tightly regulate its function. Recent progress indicated that BAP1 plays essential roles in multiple cellular processes including cell proliferation and differentiation, cell metabolism, as well as cell survival and death. In this review, we summarize the biological and molecular functions of BAP1 and explain how the inactivation of this DUB might cause human cancers. We also highlight some of the unresolved questions and suggest potential new directions.

Similar content being viewed by others

Facts
-
BAP1 is the most frequently mutated deubiquitinase in human cancers and is a major tumor suppressor.
-
BAP1 germline mutations provide a model for gene–environment interactions in carcinogenesis.
-
BAP1 assembles multiple high molecular weight protein complexes, deubiquitinates histone H2A and coordinates gene expression and other chromatin-associated processes.
-
BAP1 orchestrates cell proliferation, cell differentiation, cell death, and cell metabolism.
-
BAP1 promotes DNA repair and genomic integrity.
-
BAP1 function is regulated by multiple post-translational modifications and interacting partners.
Open questions
-
How BAP1 is dynamically recruited to chromatin and gene regulatory regions?
-
Which signaling pathways orchestrate BAP1 transcriptional activities and how BAP1 function is coordinated to ensure proper gene transcription?
-
What is the full spectrum of BAP1 substrates?
-
How BAP1 regulates the balance between cell death and survival to ensure cellular homeostasis and tumor suppression?
-
How the BAP1 multi-protein complexes are assembled and structured in various cellular contexts and in response to stress?
-
Does BAP1 deficiency creates targetable vulnerabilities for cancer treatment?
Introduction
BAP1 is a widely expressed deubiquitinase (DUB) that belongs to the ubiquitin C-terminal hydrolase (UCH) domain-containing proteins [1] (Table 1). While BAP1 UCH domain and the C-terminal domain (CTD) are highly conserved throughout evolution, vertebrate BAP1 acquired a large insertion in the middle of the enzyme [2]. This insertion, termed non-organized regions (NORS), is predicted to be unstructured and contains binding motifs for several chromatin-associated proteins [3,4,5,6,7,8,9,10] (Fig. 1A). BAP1 interacts with numerous chromatin regulators, forming high molecular weight complexes, with the “core” complex being composed of ASXLs, HCF-1, and OGT [2,3,4,5,6, 9,10,11]. Other proteins including the transcription factors FOXK1/2 and YY1, the chromatin modifying enzymes HAT1 and KDM1B, and the ubiquitin ligase UBE2O are also associated with BAP1 with various stoichiometries [3, 5,6,7,8, 12] (Fig. 1A).

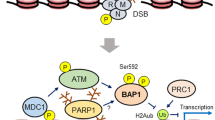
A BAP1 forms large chromatin-associated protein complexes with a “core complex” containing ASXLs and HCF-1/OGT. The HCF-1 binding motif (HBM) and phosphorylated threonine T493 are found in the unstructured loop of BAP1 (NORS non-organized regions). BAP1 interacts with ASXLs through its DEUBAD domain. Chromatin recruitment to promoters/enhancers is ensured by transcription factors and possibly epigenetic readers. BAP1 regulates transcription through interaction with other chromatin regulators such as MLL3 and NCOR1. B BAP1 regulates homologous recombination (HR). In response to ionizing radiations, BAP1 is phosphorylated on two SQ motif by ATM/ATR proteins. These modifications are important for targeting BAP1 at the site of DNA damage and for the recruitment of BRCA1 and RAD51 for DSB repair. C BAP1 promotes replication and recovery from replication stress by deubiquitinating and stabilizing INO80 at the replication fork and increasing RAD51 recruitment. BAP1 BRCA1-associated protein 1, NLS nuclear localization signal, ASXLs additional sex comb-like proteins, HBM HCF-1-binding motif, HCF-1 host cell factor 1, OGT O-linked N-acetylglucosamine transferase, UBE2O ubiquitin-conjugating enzyme 2O, HDACs histone deacetylases, NCOR1 nuclear receptor co-repressor 1, KMT2C lysine methyltransferase 2C, UTX KDM6A lysine demethylase 6A, KDM1B lysine demethylase 1B, HAT1 histone acetyltransferase 1, YY1 Ying Yang 1, NRF1 nuclear respiratory factor 1, KLF5 Kruppel-like factor 5, FOXK1/2 forkhead box K1/2, ELF1/2 E74-like ETS transcription factor 1/2, ATM ataxia-telangiectasia mutated, ATR ataxia telangiectasia and Rad3-related protein, DSBs double-strand breaks, BRCA1 breast cancer type 1 susceptibility protein, RAD51 INO80 INO80 complex ATPase subunit, Ub ubiquitin, KDM lysine demethylase, KMT lysine methyltransferase, KAT lysine acetyltransferase.
BAP1 regulates transcription, DNA repair, and replication through coordination of chromatin structure and function. While BAP1 deubiquitinates histone H2AK119, the mechanisms of its recruitment to specific chromatin locations and the coordination of its function by interacting partners remain incompletely understood. Moreover, while BAP1 functions predominantly in chromatin-associated processes, additional functions of BAP1 in the cytoplasm were also recently identified. BAP1 localizes at the endoplasmic reticulum (ER) regulating calcium signaling and cell death [13]. Thus, the mechanisms that govern BAP1 localization in the nucleus or the cytoplasm might play critical roles in regulating its function.
BAP1 is an important tumor suppressor as inactivating mutations causing loss of its function were identified in several human cancers [14,15,16,17,18,19,20,21,22,23,24]. BAP1 alterations in cancer generally follow the classical two-hit model of tumor suppression, with the highest incidence of BAP1 mutations in uveal melanoma (UM) and malignant pleural mesothelioma (MPM). The importance of BAP1 in tumor suppression is emphasized by the identification of the BAP1 cancer syndrome where families bear heterozygous BAP1 mutations that highly predispose affected individuals to develop malignant mesothelioma, UM, cutaneous melanoma, and melanocytic neoplasms termed “melanocytic BAP1-mutated atypical intradermal tumors” [15, 25,26,27,28,29,30,31] (see also a recent review [32]). Studies using mouse models with global or tissue-specific inactivation of the Bap1 gene have also established these tumor suppressor functions of this DUB and provided insights into the mechanisms of cancer development that result from BAP1 loss of function [33,34,35,36,37,38,39,40]. This review summarizes the current state of knowledge on BAP1 function and highlights potential mechanisms of tumor suppression by this DUB.
Roles of BAP1 in chromatin-associated processes
BAP1 is a transcriptional regulator
Both BAP1 and its Drosophila ortholog, calypso, deubiquitinate H2Aub [2], a transcriptional repressive histone mark catalyzed by the Polycomb group (PcG) complex PRC1 [41, 42]. Interestingly, calypso also represses transcription by forming a PcG repressive complex, termed the PR-DUB complex [2]. However, the mechanism by which calypso regulates gene expression remains incompletely understood as H2Aub was recently shown to be dispensable for PcG-mediated repression in Drosophila [43]. Mammalian BAP1 acts mainly as a transcriptional co-activator, at least partly, through H2Aub deubiquitination [6, 40, 44,45,46,47] (Fig. 1A). Consistent with this, BAP1 was recently found to interact with the H3K4 methyltransferase KMT2C, promoting its recruitment, along with the H3K27 lysine demethylase KDM6A, to gene enhancers, favoring transcriptional activation [44] (Fig. 1A). Indeed, BAP1 target genes are often associated with histone marks involved in transcriptional activation including H3K27ac, H3K4me1, and H3K4me3 as well as an open chromatin state [44, 45, 47].
However, the transcriptional activity of BAP1 is likely to be more complex due to its association with numerous chromatin-associated proteins. For instance, the highly abundant and major BAP1-interacting partner, HCF-1, regulates gene expression through association with several chromatin modifying enzymes with transcriptional activation or repression properties [4,5,6, 33, 48,49,50,51,52,53,54,55,56]. Thus, HCF-1 might play a key role in coordinating BAP1 function depending on the chromatin context of gene regulatory regions. In addition, BAP1 deubiquitinates HCF-1, and remains stably associated with this chromatin regulator, suggesting that BAP1/HCF-1 association nucleates the formation of multi-protein complexes that orchestrate transcriptional regulation [4,5,6, 33] (Fig. 1A). On the other hand, BAP1 interacts with and deubiquitinates the transcriptional co-repressor NCOR1, increasing its stability and chromatin recruitment, and this results in transcriptional repression of γ-globulin gene [57]. BAP1-mediated gene repression was also observed in other mammalian cell systems, although a causal link between H2Aub deubiquitination and transcriptional repression was not clearly demonstrated [8, 58, 59]. Finally, transcription factors that associate with the BAP1 complexes, e.g., FOXK1/2 and YY1, can also participate in directing the recruitment of BAP1 complexes to specific chromatin loci [6, 8, 12, 47].
Overall, BAP1 is mostly a transcriptional co-activator, but can also act as a transcriptional co-repressor. Consistent with its transcriptional regulatory function, gene expression profiling indicated that depletion of BAP1 or its inactivation induced up- or downregulation of hundreds of genes associated with cell cycle, DNA repair, cell survival, cell metabolism, and apoptosis [6, 16, 33, 45, 47, 59]. What are the direct BAP1 target genes and how BAP1 ensures these two opposite roles remains an outstanding question. In particular, it will be interesting to determine which co-factors favor its activator or repressive functions and which substrates are targeted by BAP1 during these transcriptional events.
BAP1 promotes DNA repair and maintenance of genomic integrity
The DNA damage response (DDR) involves the exquisite intervention of multiple signaling and repair factors, many of which are associated with chromatin function [60,61,62,63,64]. A role for BAP1 in DDR has been established in the context of DNA double-strand break (DSB) signaling and repair (Fig. 1B). BAP1 promotes the efficient assembly of the homologous recombination (HR) factors, BRCA1 and RAD51, at ionizing radiation (IR)-induced DNA repair foci [65, 66]. BAP1-deficient cells are sensitive to IR as well as PARP inhibitors, which are widely used to probe defects in HR and treat HR-defective cancers [16, 65, 66]. Consistently, BAP1-deficient cells are defective in HR-mediated immunoglobulin gene conversion and exhibit an increased frequency of chromosomal breaks [65]. Thus, the role of BAP1 in DNA repair provides a possible molecular basis for its tumor suppressor function. Mechanistically, BAP1 is directly recruited to chromatin near DSB sites and its recruitment is inversely correlated with the levels of H2Aub detected at sites of DNA damage [65, 66]. BAP1 is also phosphorylated by ataxia-telangiectasia mutated (ATM) kinase following DNA damage, and mutation of its phosphosites inhibits its recruitment to DSB sites [65, 66]. Moreover, both BAP1 catalytic activity and its phosphorylation state are critical for promoting the cellular recovery from DNA damage [65, 66]. BAP1 mode of action might involve its phosphorylation-induced recruitment to DNA damage sites to promote chromatin remodeling and subsequent assembly of the HR repair machinery [65, 66] (Fig. 1B). Nonetheless, other studies indicated that depletion of BAP1 results in deregulated expression of DDR genes [6, 22]. Thus, BAP1 might also indirectly contribute to DNA repair through the coordination of gene expression.
BAP1 is also involved in the progression of the DNA replication fork [67, 68]. BAP1 deubiquitinates and stabilizes the chromatin-remodeling factor INO80 at the replication fork [67] (Fig. 1C). INO80 is part of a multi-protein chromatin-remodeling complex that regulates several DNA-associated processes and plays a key role in the maintenance of genomic stability [69, 70]. BAP1 interaction with INO80 facilitates the anchorage of the complex onto chromatin through interaction with H2Aub, promoting the progression of the DNA replication fork [67]. On the other hand, BAP1 promotes the assembly of RAD51 foci to facilitate DNA repair and replication fork restart following replication stress [68]. However, the exact mechanisms by which BAP1/INO80 regulate DNA replication under normal or stress conditions remain largely unknown.
In summary, BAP1 regulates DNA repair and replication, likely through deubiquitination of H2Aub and possibly other factors. Consistent with these findings, BAP1-deficient cells are characterized by numerous chromosomal aberrations and aneuploidy [65], which emphasize the importance of BAP1 for the maintenance of genomic integrity and tumor suppression.
Physiological roles of BAP1
Dual functions of BAP1 in regulating cell proliferation
Initial studies indicated that reestablishing BAP1 expression in BAP1-null cancer cell lines leads to defects in cell-cycle progression and notably an accumulation of the cells in S phase [71]. The authors postulated that BAP1 might promote G1 to S cell-cycle transition and, as a consequence, this might result in DNA damage, eventually leading to growth arrest and cell death [71]. Conversely, depletion of BAP1 in cells normally expressing this DUB also reduces cell proliferation and delays G1 to S transition, suggesting that BAP1 exerts a tight control on cell-cycle progression [5, 9, 16, 22, 67, 72]. Mechanistically, BAP1 promotes cell-cycle progression by directly acting at the replication fork, as outlined above [67], although it is still unclear whether this activity is an universal function of BAP1 in diverse cell systems. On the other hand, cumulating evidence also indicates that BAP1 acts through its transcriptional activity to regulate the expression of genes that coordinate cell cycle. Indeed, BAP1 regulates the expression of E2F target genes, which play critical roles in orchestrating G1 to S cell-cycle progression [6, 22, 72, 73] (Fig. 2A). BAP1 interacts with HCF-1 and YY1, both of which regulate E2F target genes and promote G1 to S progression [4,5,6, 52, 74,75,76]. BAP1 seems to promote cell proliferation by forming a complex with both HCF-1 and YY1, protecting HCF-1 from degradation and promoting the expression of E2F target genes [4,5,6] (Fig. 2A). However, the link between BAP1 and E2F pathway might be more convoluted than expected. For instance, normal mesothelial cells from Bap1+/− mice display a significant downregulation in the mRNA and protein levels of the retinoblastoma tumor suppressor RB, indicating a negative role of BAP1 in regulating the expression of E2F target genes [77]. Moreover, mesothelioma tumor cells derived from asbestos-treated Bap1+/− acquire a biallelic inactivation of Bap1 with a more pronounced effect on RB downregulation [77]. In addition, BAP1 deubiquitinates and stabilizes KLF5 transcription factor, which is known to promote cell proliferation [78]. In this context, BAP1/HCF-1/KLF5 complex promotes cell-cycle progression, at least in part, through inhibiting the expression of the cell-cycle inhibitor p27Kip1 [78] (Fig. 2A). Overall, BAP1 might coordinate the RB/E2F pathway by exerting both positive and negative effects, involving direct action on E2F target genes as well as indirect effects on the regulation of cell-cycle inhibitors, perhaps depending on the cell type and/or stage of carcinogenic transformation.

A An important step during cell-cycle progression is the phosphorylation of RB by Cyclin D/CDK4/6 complex. Phosphorylated RB dissociates from E2F proteins to induce transcription of genes important for cell-cycle progression. BAP1 interaction with HCF-1 and YY1 might regulate the G1 to S cell-cycle progression through modulating the expression of E2F target genes. ASXLs proteins might also regulate E2F target genes since these factors are involved in G1 to S progression. BAP1 was also shown to regulate RB expression. BAP1/HCF-1 interaction with KLF5 inhibits the expression of p27Kip1, which increases cell proliferation. Overall, BAP1 acts during the G1/S transition to orchestrate cell proliferation by acting on both activators and inhibitors of the cell cycle. B Bap1 inactivation in mice is embryonic lethal. Depletion of BAP1 in adult mice causes important changes in the hematopoietic and immune systems including myeloid cell transformation. BAP1 is also critical for the development and homeostasis of other tissues and organs, including the liver and the pancreas. In the hematopoietic system, BAP1 was shown to stabilize the Polycomb group protein L3MBTL2 at Ezh2 promoter causing increased deposition of H4K20me, which inhibits Ezh2 gene transcription. BAP1 inactivation in thymocytes is associated with decreased expression of E2F and c-Myc proteins. E2F retinoblastoma-associated protein 1, RB retinoblastoma protein, CycD cyclin D, CDK4/6 cyclin-dependent kinase, CycE cyclin E, p27Kip1 cyclin-dependent kinase inhibitor 1B, EZH2 enhancer of Zeste 2 is a Polycomb repressive complex 2 subunit, L3MBTL2 lethal(3)malignant brain tumor-like protein 2, HSCs hematopoietic stem cells, MDS myelodysplastic syndrome, CMML chronic myelomonocytic leukemia.
BAP1 interaction with ASXLs might also be critical for its ability to regulate cell proliferation [9, 10] (Fig. 2A). Defects in BAP1 interaction with ASXL delay G1 to S progression [9, 10]. Moreover, overexpression of BAP1 in normal fibroblasts, and remarkably its catalytic dead form, induces cell-cycle arrest and senescence, effects that require BAP1 interaction with ASXLs [9]. Whether BAP1/ASXLs act in conjunction with HCF-1/YY1 in regulating E2F target gene expression remains to be determined.
Finally, there are also evidence for BAP1 negatively regulating cell proliferation through (i) downregulation of ERK1/2 and JNK signaling pathways [79], (ii) deubiquitination-mediated stabilization of the tumor suppressor LATS, a component of the Hippo signaling pathway [80], and (iii) deubiquitination and stabilization of the tumor suppressor PTEN, a negative regulator of the kinase AKT [81].
In summary, BAP1 appears to have multiple roles in the coordination of cell proliferation. This can be possibly explained by cell-context and/or cell-type-specific functions of BAP1 and its interacting partners. BAP1 might therefore exert some of its tumor suppressor functions by altering the mechanisms of cell-cycle control in a cell-type-dependent manner.
Roles of BAP1 in development and cell differentiation
The mechanisms underlying stem/progenitor cell renewal, lineage commitment, and differentiation are complex and involve intricate networks of transcriptional regulations and feedback loops that govern cell fate decisions [82,83,84,85,86]. Increasing evidence suggests that BAP1 is a critical regulator of stem/progenitor self-renewal and cell fate in multiple tissues and at multiple stages of development. Bap1 gene deletion in mice induces embryonic lethality between days 8.5 (E8.5) and 9.5 (E9.5), indicating that BAP1 is required for mammalian embryogenesis and tissue specification [33]. To bypass this embryonic lethality and further investigate the roles of BAP1 in development, conditional ablation of Bap1 was conducted in adult mice or in specific organs [33, 34, 37, 46, 72, 87]. For instance, systemic or hematopoietic-restricted Bap1 deletion in adult mice recapitulates the development of a hematological phenotype with similar features of the human myelodysplastic syndrome (MDS) [33]. BAP1 inactivation impairs normal hematopoietic stem cell (HSC) differentiation toward the myeloid lineage and this is accompanied by an increased proliferation of BAP1-deficient myeloid progenitors, resulting in extramedullary hematopoiesis and splenomegaly [33, 88]. Mechanistically, BAP1 stabilizes the atypical PcG protein L3MBTL2, resulting in higher levels of monomethylated histone H4K20 at the Ezh2 gene locus, and subsequent downregulation of EZH2 and H3K27 trimethylation [88] (Fig. 2B). In addition, EZH2 inhibition prevents the myeloid transformation phenotype caused by the loss of BAP1 [88]. Thus, loss of BAP1 in the hematopoietic compartment promotes EZH2 expression and repression of polycomb target genes, notably the Hoxa gene cluster, which might promotes the expansion and transformation of myeloid progenitors [88]. Interestingly, loss of BAP1 in adult mice is also coupled with severe thymic atrophy and impaired maintenance of the lymphoid lineage [72]. A strong depletion of immature thymocytes cell populations, including early thymic progenitors, double-negative cells, immature single-positive cells, and double-positive cells, was observed. Specific deletion of BAP1 in double-positive cells indicated that BAP1 is also required for mature T cells homeostasis as well as their response to antigen stimulation. T-cell differentiation in vitro, using bone marrow-derived HSCs and progenitors, showed that inactivation of BAP1 blocks thymocytes development at the double-negative 3 (DN3) stage and prevents their maturation. The failure of DN3 thymocytes to undergo subsequent maturation steps is correlated with increased levels of H2Aub and a significant downregulation of E2F and MYC target genes [72] (Fig. 2). Notably, inactivation of BAP1 does not appear to enhance H3K27 trimethylation by EZH2 in thymocytes, suggesting that BAP1 governs different mechanisms of regulation in myeloid and lymphoid lineages [72]. On the other hand, B-cell development is also abrogated in BAP1-deficient cells, although additional studies are required to further define the function of BAP1 in B-cell lineage commitment [72]. Finally, inactivation of BAP1 in pancreatic progenitor cells results in a progressive tissue damage leading to chronic pancreatitis characterized by loss of acinar architecture and identity, tissue damage, obstruction of the ducts and immune cell infiltration, all of which are indicative of an extensive inflammation of the pancreas [40]. The molecular basis of these changes might be associated with diminished DNA repair in the absence of BAP1 [40]. Clearly, BAP1 is required for the development and homeostasis of diverse mammalian tissues and various cell types might respond differently to BAP1 inactivation during oncogenic transformation.
BAP1 is also crucial for Xenopus laevis embryogenesis. BAP1 depletion in this model organism induces severe gastrulation defects and developmental abnormalities associated with ectoderm, mesoderm, and neural crest lineages [89]. Partial depletion of BAP1 results in less penetrant embryonic lethality and animals that completed development exhibit several malformations and organ abnormalities. At the molecular level, BAP1 promotes the expression of key developmental genes regulating the switch from pluripotency to differentiation by preventing the deacetylation of histone H3K27 at gene regulatory regions [89]. Consistent with these results, inhibition of histone deacetylase (HDAC) activity rescues the phenotypes associated with BAP1 deficiency by restoring normal expression of genes regulating embryonic lineages [89].
Overall, BAP1 is tightly associated with multiple processes related to stem/progenitor cell maintenance, development, and lineage commitment. However, additional investigations, using mouse models, are needed to further define the exact mechanisms by which BAP1 coordinates these processes and how their deregulation following inactivation of this DUB promotes cancer development.
BAP1 regulates different modes of cell death
BAP1 has recently emerged as a key factor orchestrating, through multiple mechanisms, the balance between cell survival and death. Systemic inactivation of BAP1 in adult mice is associated with liver damage and pancreatic atrophy with the presence of cleaved caspase 3, a hallmark of apoptosis [46]. Interestingly, BAP1 deficiency in mouse embryonic stem cells, primary keratinocytes, or E1A oncogene-immortalized mouse embryonic fibroblasts induces apoptosis through transcriptional downregulation of the anti-apoptotic genes Bcl2 and Mcl1, both of which are direct target genes of BAP1 [46]. In contrast, melanocytes and mesothelial cells are resistant to apoptosis induced by BAP1 inactivation, likely due to an inability of BAP1 to modulate the expression of anti-apoptotic genes in these cells [46]. Thus, diverse cell types respond differently to BAP1 inactivation and cell types with higher cell death thresholds might be more prone to malignant transformation, perhaps explaining the tumor spectrum of BAP1 mutation-associated human cancers (Fig. 3). Nonetheless, additional studies are needed to further demonstrate the importance of cell-type susceptibility to apoptosis in BAP1 mutation-associated carcinogenesis.

The tumor suppressor BAP1 stabilizes IP3R3 at the ER membrane through its DUB activity, which promotes the physiological release of Ca2+ into the cytoplasm and the mitochondria. Excessive Ca2+ release from ER results in high mitochondrial concentrations of Ca2+, which induces the release of cytochrome C, thus leading to apoptosis. BAP1 activity promotes the expression of pro-survival genes. BAP1 interaction with 14-3-3 releases BAX from 14-3-3 and promotes apoptosis. Conversely, loss of BAP1 induces apoptosis, in a cell-type-dependent manner, by inducing the transcriptional repression of pro-survival genes. On the other hand, BAP1 prevents apoptosis following glucose deprivation and ER stress by repressing the expression of proapoptotic factors. Finally, BAP1 promotes ferroptosis, a non-apoptotic form of cell death, by suppressing the expression of the SLC7A11 glutamate/cystine antiporter. Reduced expression of SLC7A11 leads to reduced levels of cystine uptake, which in turn leads to low levels of reduced GSH, therefore resulting in increased lipid peroxidation, which triggers ferroptosis. ATF3 activating transcription factor 3, CHOP C/EBP homologous protein, ER endoplasmic reticulum, GPX4 glutathione peroxidase 4, GSH reduced glutathione, GSL glutamate cysteine ligase, H2O2 hydrogen peroxide, iP3R3 inositol-1,4,5-triphosphate receptor, SLC7A11 solute carrier family 7 member 11, BAX BCL2-associated X, MCL1 myeloid cell leukemia 1.
BAP1 also negatively regulates the expression of the ER-associated stress-response genes including ATF3 and CHOP. ATF3 and CHOP are induced by glucose deprivation and mediate the unfolded protein response (UPR), a transcriptional program that results in ER stress-induced apoptosis by provoking ATP depletion and ROS production. BAP1-deficient cells become sensitive to metabolic-stress-induced UPR activation and cell death [58] (Fig. 3). It will be interesting to further identify how BAP1 is dynamically recruited to ATF3 and CHOP promoters to repress their expression and how BAP1 transcriptional activity responds to cell-signaling events. On the other hand, BAP1 can also act directly at the ER, whereby it controls Ca2+ signaling and apoptosis [13]. BAP1 deubiquitinates and stabilizes type 3 inositol-1,4,5-triphosphate receptor, promoting Ca2+ release from the ER [13]. It is established that the ER is the major store of intracellular Ca2+ and coordinated release of this second messenger regulates several processes in the mitochondria and cytosol [90,91,92]. Excessive release of Ca2+ by the ER is absorbed by mitochondria through multiple Ca2+ channels, leading to mitochondrial Ca2+ overload, which induces cytochrome C release and cell death [90, 91, 93, 94]. Inhibition of BAP1 reduces cellular sensitivity to Ca2+-dependent apoptosis, possibly contributing to the survival of cells that experienced stress conditions [13]. Therefore, cytoplasmic BAP1 mediates apoptosis and this activity might account for BAP1 tumor suppressor function (Fig. 3). It is unclear at the present time what the relative contribution of BAP1 is in direct (IP3R-Ca2+ signaling) versus indirect (UPR signaling) effects in ER-associated cell death. It will be interesting to determine how BAP1 function is regulated at the ER and whether the DUB activity of BAP1 requires additional co-factors. Moreover, BAP1 might also promote apoptosis through modulating the interaction between the proapoptotic factor Bax and 14-3-3 protein [95]. BAP1 appears to prevent 14-3-3 from binding Bax, releasing the latter for inducing cytochrome c-dependent apoptosis.
Finally, BAP1 also enhances ferroptosis, a newly identified non-apoptotic form of cell death associated with metabolic stress [59]. Ferroptosis could be caused by cystine depletion and subsequent lipid peroxidation in an iron-dependent manner [96,97,98,99]. Cystine is normally imported into the cytosol through the cystine/glutamate antiporter system Xc− composed of two subunits, SLC7A11 and SLC3A2. Imported cystine is converted to cysteine, which is then used for the synthesis of the anti-oxidant glutathione (GSH). To prevent excessive membrane lipid peroxidation and ferroptosis, the glutathione peroxidase 4 converts the lipid hydroperoxides to lipid alcohol in a GSH-dependent manner. SLC7A11 gene transcription is repressed by BAP1 in a DUB activity-dependent manner [59]. This inhibition results in reduced cystine uptake, enhanced lipid peroxidation and ferroptosis (Fig. 3). While the underlying mechanisms by which BAP1 DUB activity represses SLC7A11 gene expression remain to be further defined, these data revealed, nonetheless, that BAP1 might act as a tumor suppressor by promoting ferroptosis [59].
In summary, apoptosis and ferroptosis are two different mechanisms of cell death that can be coordinated by BAP1. Furthermore, while BAP1 can modulate cell death through different programs, it is interesting to note that BAP1 can also promote cell survival following DNA damage [65, 66]. Thus, further studies are required to establish how these processes work, either independently or in concert, to ensure tumor suppression by BAP1.
Roles of BAP1 in cell metabolism
Alterations of cell metabolism contribute to the development of multiple diseases such as inflammation, diabetes, and cancer [100,101,102,103]. BAP1 and several of its interacting partners regulate cell metabolism, although the roles of BAP1 in metabolic regulations might be more pleiotropic than anticipated. BAP1 depletion results in deregulated expression of numerous metabolism-associated genes including those involved in mitochondrial function [6]. Indeed, a BAP1/HCF-1 complex is recruited by YY1 transcription factor to regulate the expression of Cox7c, a gene encoding a critical factor of the mitochondrial respiratory chain [6] (Fig. 4). Consistent with a role of BAP1 in mitochondrial function, it was found that primary fibroblasts derived from patients with heterozygous germline BAP1 mutations shift their source of ATP production from oxidative respiration to aerobic glycolysis, a phenomenon known as the ‘’Warburg effect,” which constitutes a hallmark of cancer cells [104]. BAP1 can also promote gluconeogenesis by protecting PGC-1α, a key regulator of gluconeogenesis, from ubiquitin-mediated degradation [105]. Mechanistically, the OGT/HCF-1 complex binds to and O-GlcNAcylates PGC-1α to facilitate BAP1 recruitment and subsequent stabilization of PGC-1α [105]. Interestingly, PGC-1α O-GlcNAcylation and its protein levels as well as gluconeogenesis are all concomitantly regulated by glucose availability indicating the importance of BAP1-mediated PGC-1α stabilization in nutrient sensing [105]. Moreover, hepatic knockdown of HCF-1/OGT in diabetic mice promotes PGC-1α proteasomal degradation and significantly reduces the expression of PGC-1α gluconeogenic target genes, therefore improving glucose homeostasis [105] (Fig. 4). In accordance with these observations, inducible liver-specific BAP1 deletion in adult mice was found to promote perinatal lethality accompanied by severe metabolic alterations before the onset of liver damage [87]. BAP1 deficiency in the liver results in reduced gluconeogenesis, hypoglycemia, and depleted hepatic lipid and glycogen contents, indicating an important role of this DUB in regulating both lipid and glucose homeostasis [87]. Surprisingly, BAP1 deletion also significantly reduces PGC-1α mRNA levels suggesting that the function of BAP1 as a regulator of cell metabolism is more complex than anticipated [87]. On the other hand, ASXL2, which is a major BAP1-interacting factor, also regulates glucose and lipid homeostasis [106]. Whether ASXL2-BAP1 interaction is critical for metabolic regulation remains to be determined. Last, BAP1 was also shown to deubiquitinate and stabilize the tumor suppressor LKB1, a serine/threonine kinase that phosphorylates and activates AMPK kinase, a metabolic sensor that regulates glucose and lipid metabolism [107].

The nuclear BAP1-HCF-1-YY1 complex regulates the expression of mitochondrial genes, thus promoting oxidative phosphorylation. ER-localized BAP1 stabilizes IP3R3 and promotes Ca2+ signaling, which might also regulate mitochondrial function. The BAP1-HCF-1-OGT complex increases the stability of PGC-1α, a master regulator of mitochondrial biogenesis, and gluconeogenesis. PGC-1α interacts with HCF-1/OGT through its HBM motif, leading to its subsequent O-GlcNAcylation by OGT. O-GlcNAcylation of PGC-1α facilitates BAP1 recruitment, which in turn deubiquitinates PGC-1α, preventing its proteasomal degradation and thus promoting gluconeogenesis. BAP1/ASXLs/FOXKs complexes regulate several metabolic pathways. PGC-1α peroxisome proliferator-activated receptor-γ co-activator 1α.
Altogether, these results suggest that BAP1 orchestrates multiple cell metabolism pathways through its interaction with several protein partners. Given the multifaceted links between cell metabolism and cancer, it will be interesting to determine how BAP1 inactivation in cancer impacts cell metabolism and results in the rewiring of nutrient sensing and utilization pathways.
BAP1 regulation by interacting partners and post-translational modifications (PTMs)
The multitude of interactions between BAP1 and its partners suggest that this DUB could dynamically associate with specific factors to orchestrate cellular responses and signaling events. BAP1 requires ASXLs for enzymatic activation [2, 10, 11]. Importantly, depletions of ASXLs greatly reduce BAP1 protein levels indicating the importance of these factors in the regulation of BAP1 function [9]. Indeed, ASXLs can form mutually exclusive complexes with BAP1 and this can endow BAP1 complexes with distinct functions [9, 108]. Nonetheless, it is still unclear how BAP1 interactions with ASXLs are coordinated. On the other hand, BAP1 strongly associates with HCF-1 and this interaction is important for cell-cycle regulation [4,5,6]. HCF-1 is a major target of O-GlcNAcylation by OGT, which is required for HCF-1 maturation and cleavage, generating HCF-1N and HCF-1C fragments, both of which remain associated with the BAP1 complex along with OGT [109, 110] (Fig. 1A). While the significance of HCF-1 cleavage remains unknown, the HCF-1 (N and C)-OGT complex plays an important role in the regulation of PGC-1α by O-GlcNAcylation (see above). Interestingly, the lysine demethylase KDM1B (LSD2) was also identified in BAP1 complexes [3, 5, 6]. Through its newly discovered E3 ubiquitin ligase activity, LSD2 targets OGT for proteasomal degradation [111]. Thus, it will be interesting to determine whether BAP1 and LSD2 dynamically coordinate OGT ubiquitination state and function. Indeed, OGT was shown to be a substrate of BAP1 [33].
Several transcription factors are associated, at different stoichiometries, with BAP1 complexes and these appear to play important roles in coordinating its function at chromatin and gene regulatory regions. For instance, YY1 recruits BAP1 to chromatin for the transcriptional control of genes regulating cell growth and proliferation [6]. FOXK2 promotes BAP1 recruitment to chromatin to ensure deubiquitination of H2Aub and transcriptional control [8, 12, 47]. Overall, increasing evidence suggests that BAP1 could act as a hub bringing together several epigenetic writers and erasers for deposition or removal of histone modifications, respectively.
BAP1 is also regulated by PTMs. Indeed, BAP1 NLS is targeted by multi-monoubiquitination catalyzed by the E2/E3 hybrid enzyme UBE2O [7] (Fig. 5). This ubiquitination masks the NLS domain of BAP1 and sequesters the protein in the cytoplasm. Interestingly, BAP1 can auto-deubiquitinate its NLS motif, counteracting UBE2O ubiquitination, and promoting BAP1 nuclear entry. The importance of this mechanism for tumor suppression is emphasized by BAP1 cancer mutations K637-C638delinsN and E631-A634del that have increased interaction with UBE2O, which results in enhanced BAP1 NLS ubiquitination and subsequent retention in the cytoplasm (Fig. 5). As described earlier, BAP1 is phosphorylated during DNA damage on multiple sites including S592 and S276 within ATM/ATR SQ/TQ motifs and these events promote BAP1 recruitment to chromatin and enhance cell survival [65]. Although the exact mechanism remains unknown, these phosphorylation events do not directly target the DUB activity of BAP1. BAP1 is also phosphorylated on T493 and this regulates its interaction with FOXK1/2 [8] (Fig. 5). Thus, BAP1 recruitment to gene regulatory regions can be dynamically regulated by PTMs in response to cell-signaling events. On the other hand, BAP1 is modified by glutamylation at E651, a PTM that corresponds to an amide bond between the γ-carboxyl group of glutamic acid of the target protein and the amino group of glutamic acid [112]. Glutamylation is a reversible modification catalyzed by a group of tubulin tyrosine-ligase like enzymes and removed by cytosolic carboxypeptidase enzymes [113, 114] (Fig. 5). BAP1 glutamylation promotes its ubiquitination and proteasomal degradation, resulting in reduced self-renewal of long-term hematopoietic stem cells in mice. It would be of interest to determine if BAP1 glutamylation occurs in other cell types and how this PTM coordinates the transcriptional programs regulated by BAP1.

BAP1 is subjected to several post-translational modifications that regulate its functions and tumor suppressor activity. Following DNA damage, ATM and ATR phosphorylate BAP1 to increase its recruitment on the site of DNA damage and allow proper DNA repair. BAP1 was also found to be targeted by glutamylation on E651. This modification is catalyzed by TTL5/7 enzymes and removed by CPP3. BAP1 glutamylation was found to be important for normal HSCs maintenance and self-renewal. Phosphorylation of T493 of BAP1 is important for its interaction with the FOXK1 and FOXK2 transcription factors. The NLS motif of BAP1 is modified by multi-monoubiquitination by the atypical E2/E3 hybrid enzyme UBE2O. NLS ubiquitination results in the retention of BAP1 in the cytoplasm. Interestingly, BAP1 deubiquitinates its own NLS domain and counteracts the effect of UBE2O. CCP3 cytosolic carboxypeptidase, TTLL5/7 tubulin tyrosine-ligase like.
BAP1 in cancer pathogenesis
BAP1 inactivating mutations were initially identified in non-small cell lung carcinoma cells [1], and subsequently in numerous cancers (Fig. 6A). BAP1 was found to be mutated in ~40% of patients with UM [115]. Interestingly, UM is characterized by prevalent activating mutations of GNAQ or GNA11, likely corresponding to the initiating mutational events of this cancer. Thus, constitutive activation of G protein-coupled receptor signaling in conjunction with BAP1 inactivation play a critical role in the pathogenesis of UM. Interestingly, in metastatic tumors originated from UM, BAP1 mutations ramp up to 80%, suggesting that its inactivation is an important feature for disease progression [14, 24]. Indeed, UM tumors with decreased BAP1 show higher microvascular density and infiltration of immune cells, suggesting that BAP1 could also influence tumor angiogenesis and microenvironment [116].

A Repartition of BAP1 mutations across human cancers. B Most BAP1 cancer mutations cause truncations resulting in the loss of the NLS motif, which ultimately sequesters BAP1 in the cytoplasm. C Point mutations of BAP1 can disrupt its DUB activity toward its substrates in the nucleus, notably H2AK119ub. IP3R3 receptor was recently found to be a substrate for BAP1 and disruption of BAP1 DUB activity by mutations could also alter its function in the cytoplasm. D BAP1H666-R669del mutation that specifically disrupts interaction with the ASXLs proteins required for H2AK119ub deubiquitination by BAP1. E BAP1K637-C638delinsN and BAP1E631-A634del mutants have increased interaction with UBE2O protein and, as a result, increased multi monoubiquitination of BAP1 NLS causing an overall retention of BAP1 in the cytoplasm. UM uveal melanoma, TETs thymic epithelial tumor, MPM malignant pleural mesothelioma, MPeM malignant peritoneal mesothelioma, HC hepatocellular carcinoma, ccRCC clear cell renal cell carcinoma, PDA pancreatic ductal adenocarcinomas, CUBI composite ubiquitin-binding interface.
MPM, a cancer often associated with asbestos exposure, presents near 60% of BAP1 mutations. Interestingly, frequent mutations of CDKN2A and NF1 tumor suppressors are also observed in MPM tumors and combined mutations of BAP1, NF2, and CDKN2A are observed in about 34% MPM, indicating the relative importance of these tumor suppressors in preventing MPM development [21, 22, 117, 118]. Indeed, combined deletion of Bap1, Nf2, and Cdkn2a genes causes rapid disease onset in mice [38, 39]. While the tumors show some characteristics of the human disease [39], it will be interesting to model in mouse cancer models, the sequential series of genetic and molecular events that lead to MPM development. Of note, BAP1 was also recently found to be frequently mutated in malignant peritoneal mesothelioma (MPeM), a rare cancer usually not associated with asbestos exposure [119]. BAP1 mutations are found in about 25% of cases along with mutations of other chromatin modifiers indicating the importance of epigenomic regulation in MPeM pathogenesis. Interestingly, BAP1-mutated tumors have a distinct inflammatory tumor microenvironment associated with an increased expression of immune checkpoint receptors, perhaps identifying a vulnerability for MPeM treatment with immune checkpoint inhibitors [119].
Clear cell renal cell carcinoma (ccRCC), a common renal cancer characterized by mutations of the tumor suppressor VHL, is also associated with frequent BAP1 mutations (10–15% of the cases) [16, 120]. Interestingly, PBRM1 and SETD2, encoding chromatin-remodeling/modifying factors, are also mutated in ccRCC at 30–41% and 7–15%, respectively, and these genes are all located in the 3p chromosomal region, which is a major target of loss of heterozygosity in ccRCC [20, 121,122,123]. This mutational profile also provides evidence for the importance of epigenetic regulation in ccRCC development. Moreover, conditional ablation of Vhl, along with one allele of Bap1, in nephron progenitor cells resulted in renal cell carcinoma in mice with features that resemble those observed in the human disease [34]. These include kidney tumors at different stages, cysts with multiple levels of epithelium stratification, and neoplastic nodules. At the cellular level, cancer cells show enlarged nuclei, a severe chromatin reorganization, and clear or eosinophilic cytoplasm, which are typically observed in ccRCC [34].
Frequent BAP1 mutations are also found in other cancers including intrahepatic cholangiocarcinoma (25%) [17, 19], hepatocellular carcinoma (14–17%) [23, 124], and thymic epithelial tumors (6–8%) [125]. Finally, BAP1 mutations are also observed, although at very low levels, in a wide range of other cancers such as breast cancer [126], colorectal cancer [127], pancreatic cancer [128], and bladder cancer [129]. Interestingly, heterozygous loss of BAP1 is found in 25% of pancreatic ductal adenocarcinomas and 40% of acinar cell carcinoma suggesting that BAP1 is a haploinsufficient tumor suppressor [40]. Moreover, the loss of BAP1 is associated with a history of chronic pancreatitis and this can be recapitulated following Bap1 inactivation in mice [40].
In addition to somatic mutations, germline mutations of BAP1 have also been identified, predisposing patients to multiple cancers, defining the BAP1 cancer syndrome, which is associated with a high risk of developing UM, MPM, ccRCC, and other cancers [15, 25, 26, 30, 130,131,132,133]. Individuals with inherited BAP1 mutations have a highly increased risk of cancer development and tumors, notably UM, are often more aggressive and with metastatic behavior. Some individuals even develop UM and MPM in their lifetime, which is predicted to be of an extremely low incidence if the pathogenesis of these cancers was independent of BAP1. Consistently, mice carrying heterozygous Bap1 mutations are more susceptible to asbestos-induced MPM than wild-type mice [35, 36]. These findings also raised a note of caution that exposure of individuals with germline BAP1 mutations to even minimal doses of carcinogens should be prevented. Thus, at least for some cancers, BAP1 constitutes an interesting example for gene–environment interactions in carcinogenesis.
At the molecular level, BAP1 is targeted by mutations, along its genomic sequence, with no hotspot region [14,15,16,17,18,19,20,21,22,23,24,25, 117, 132, 134]. Most of these BAP1 mutations produce protein truncations without the NLS, which explains why an important number of cancers lose nuclear BAP1 staining (Fig. 6B). Missense mutations inside the UCH domain, the NORS, or the CTD are also observed. These point mutations have been instrumental in understanding the tumor suppressor function of BAP1. For instance, mutations in the UCH domain, including the catalytic cysteine (C91), indicate the importance of DUB activity for tumor suppression [14, 16, 17, 19, 22, 66, 71, 135] (Fig. 6C). Of note, several mutations outside the UCH domain, e.g., R666-H669del (in the CTD domain) also cause inactivation of BAP1 DUB activity [9]. This is due to the intramolecular interactions between the UCH domain, the CTD domain and the DEUBAD domain of ASXLs, which creates a composite ubiquitin-binding interface that enables a BAP1 conformation suitable for catalysis and regulation by ASXLs. Interestingly, the protein encoded by the BAP1R666-H669del mutant specifically loses interaction with ASXLs proteins without perturbing interaction with the other partners [9] (Fig. 6D). Other mutations of BAP1 that target the C-terminal region (BAP1K637-C638delinsN, BAP1E631-A634del) result in increased interaction of mutated BAP1 with UBE2O and enhanced cytoplasmic localization [7] (Fig. 6E). In summary, ample evidence indicated that BAP1 mutations in cancer are inactivating and result in a loss of its nuclear function and DUB activity. It will be interesting to further determine how BAP1 cancer mutations might reveal additional co-factors, interacting partners, and signaling pathways that could be critical for its tumor suppressor function. Finally, loss of BAP1 protein expression in several cancers can also be observed in the absence of mutations [16, 22, 136]. For instance, a proportion of MPM tumors with no BAP1 gene mutation and with normal mRNA expression are negative for BAP1 protein staining, suggesting that post-translational events regulating its stability might also be responsible for BAP1 loss-of-function [22].
Although the prevalent model indicates that BAP1 is a tumor suppressor, recent data also suggest that gain-of-function and oncogenic properties might also be attributed to this DUB [78, 134, 137, 138]. For instance, ASXL1 is frequently mutated in acute myeloid leukemia, MDS, chronic myelomonocytic leukemia, myeloproliferative neoplasms, and other cancers [139,140,141,142,143,144]. Several studies reported that expression of a truncated form of ASXL1, found in hematological malignancies, leads to a gain-of-function phenotype [137, 145,146,147]. While the mutated form of ASXL1 (N-terminal fragment containing the DEUBAD) could promote BAP1 DUB activity and gene derepression, other mechanisms of chromatin regulation have also been proposed [137, 145,146,147]. Altogether, while these observations suggest that BAP1 might also act as an oncogene, additional studies using genuine mouse models, with protein expression levels that resemble disease conditions, are necessary to ascertain potential oncogenic properties of BAP1.
Concluding remarks and potential future directions
Recent progress indicated that BAP1 regulates several cellular processes that orchestrate cell fate decisions and cell differentiation, cell-cycle progression, cell death, and cell metabolism. Nonetheless, several questions remain unresolved on (i) how BAP1 is recruited to chromatin by transcription factors and how BAP1 chromatin localization influences transcriptional regulation? (ii) Does BAP1 regulate additional processes in the cytoplasm, in addition to ER-associated Ca2+ signaling? (iii) What are the gene expression programs controlled by the multiple BAP1 complexes? (iv) How PTMs of BAP1 and associated factors orchestrate the functions of the BAP1 complexes. On the other hand, studies are needed to determine the ordered molecular events that lead to malignant transformation following BAP1 inactivation. It is still largely unclear why the loss of BAP1 results in transformation only in some cell types and tissues. Finally, a deep understanding of the rewiring of signaling events responsible for the maintenance of BAP1-deficient tumors could result in the identification of actionable vulnerabilities to treat BAP1-associated cancers. Indeed, recent studies suggested that the loss of BAP1 could sensitize cells and/or predict patient response to several agents and therapies [44, 65, 88, 148,149,150], revealing a new space for the discovery of therapies targeting BAP1-associated cancers.
References
Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16:1097–112.
Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465:243–7.
Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403.
Misaghi S, Ottosen S, Izrael-Tomasevic A, Arnott D, Lamkanfi M, Lee J, et al. Association of C-terminal ubiquitin hydrolase BRCA1-associated protein 1 with cell cycle regulator host cell factor 1. Mol Cell Biol. 2009;29:2181–92.
Machida YJ, Machida Y, Vashisht AA, Wohlschlegel JA, Dutta A. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. J Biol Chem. 2009;284:34179–88.
Yu H, Mashtalir N, Daou S, Hammond-Martel I, Ross J, Sui G, et al. The ubiquitin carboxyl hydrolase BAP1 forms a ternary complex with YY1 and HCF-1 and is a critical regulator of gene expression. Mol Cell Biol. 2010;30:5071–85.
Mashtalir N, Daou S, Barbour H, Sen NN, Gagnon J, Hammond-Martel I, et al. Autodeubiquitination protects the tumor suppressor BAP1 from cytoplasmic sequestration mediated by the atypical ubiquitin ligase UBE2O. Mol Cell. 2014;54:392–406.
Okino Y, Machida Y, Frankland-Searby S, Machida YJ. BRCA1-associated protein 1 (BAP1) deubiquitinase antagonizes the ubiquitin-mediated activation of FoxK2 target genes. J Biol Chem. 2015;290:1580–91.
Daou S, Hammond-Martel I, Mashtalir N, Barbour H, Gagnon J, Iannantuono NV, et al. The BAP1/ASXL2 histone H2A deubiquitinase complex regulates cell proliferation and is disrupted in cancer. J Biol Chem. 2015;290:28643–63.
Daou S, Barbour H, Ahmed O, Masclef L, Baril C, Sen Nkwe N, et al. Monoubiquitination of ASXLs controls the deubiquitinase activity of the tumor suppressor BAP1. Nat Commun. 2018;9:4385.
Sahtoe DD, van Dijk WJ, Ekkebus R, Ovaa H, Sixma TK. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat Commun. 2016;7:10292.
Ji Z, Mohammed H, Webber A, Ridsdale J, Han N, Carroll JS, et al. The forkhead transcription factor FOXK2 acts as a chromatin targeting factor for the BAP1-containing histone deubiquitinase complex. Nucleic Acids Res. 2014;42:6232–42.
Bononi A, Giorgi C, Patergnani S, Larson D, Verbruggen K, Tanji M, et al. BAP1 regulates IP3R3-mediated Ca(2+) flux to mitochondria suppressing cell transformation. Nature. 2017;546:549–53.
Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–3.
Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43:1022–5.
Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012;44:751–9.
Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet. 2013;45:1470–3.
Goldstein AM. Germline BAP1 mutations and tumor susceptibility. Nat Genet. 2011;43:925–6.
Chan-On W, Nairismagi ML, Ong CK, Lim WK, Dima S, Pairojkul C, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet. 2013;45:1474–8.
Kovac M, Navas C, Horswell S, Salm M, Bardella C, Rowan A, et al. Recurrent chromosomal gains and heterogeneous driver mutations characterise papillary renal cancer evolution. Nat Commun. 2015;6:6336.
Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48:407–16.
Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet. 2011;43:668–72.
Woo HG, Choi JH, Yoon S, Jee BA, Cho EJ, Lee JH, et al. Integrative analysis of genomic and epigenomic regulation of the transcriptome in liver cancer. Nat Commun. 2017;8:839.
Karlsson J, Nilsson LM, Mitra S, Alsen S, Shelke GV, Sah VR, et al. Molecular profiling of driver events in metastatic uveal melanoma. Nat Commun. 2020;11:1894.
Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43:1018–21.
Abdel-Rahman MH, Pilarski R, Cebulla CM, Massengill JB, Christopher BN, Boru G, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48:856–9.
Carbone M, Ferris LK, Baumann F, Napolitano A, Lum CA, Flores EG, et al. BAP1 cancer syndrome: malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J Transl Med. 2012;10:179.
Njauw CN, Kim I, Piris A, Gabree M, Taylor M, Lane AM, et al. Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneous-ocular melanoma families. PLoS ONE. 2012;7:e35295.
Farley MN, Schmidt LS, Mester JL, Pena-Llopis S, Pavia-Jimenez A, Christie A, et al. A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res. 2013;11:1061–71.
Popova T, Hebert L, Jacquemin V, Gad S, Caux-Moncoutier V, Dubois-d’Enghien C, et al. Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet. 2013;92:974–80.
Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer. 2013;13:153–9.
Carbone M, Harbour JW, Brugarolas J, Bononi A, Pagano I, Dey A, et al. Biological mechanisms and clinical significance of BAP1 mutations in human cancer. Cancer Discov. 2020;10:1103–20.
Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012;337:1541–6.
Wang SS, Gu YF, Wolff N, Stefanius K, Christie A, Dey A, et al. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proc Natl Acad Sci USA. 2014;111:16538–43.
Napolitano A, Pellegrini L, Dey A, Larson D, Tanji M, Flores EG, et al. Minimal asbestos exposure in germline BAP1 heterozygous mice is associated with deregulated inflammatory response and increased risk of mesothelioma. Oncogene. 2016;35:1996–2002.
Kadariya Y, Cheung M, Xu J, Pei J, Sementino E, Menges CW, et al. Bap1 is a bona fide tumor suppressor: genetic evidence from mouse models carrying heterozygous germline Bap1 mutations. Cancer Res. 2016;76:2836–44.
Gu YF, Cohn S, Christie A, McKenzie T, Wolff N, Do QN, et al. Modeling renal cell carcinoma in mice: Bap1 and Pbrm1 inactivation drive tumor grade. Cancer Discov. 2017;7:900–17.
Kukuyan AM, Sementino E, Kadariya Y, Menges CW, Cheung M, Tan Y, et al. Inactivation of Bap1 cooperates with losses of Nf2 and Cdkn2a to drive the development of pleural malignant mesothelioma in conditional mouse models. Cancer Res. 2019;79:4113–23.
Badhai J, Pandey GK, Song JY, Krijgsman O, Bhaskaran R, Chandrasekaran G, et al. Combined deletion of Bap1, Nf2, and Cdkn2ab causes rapid onset of malignant mesothelioma in mice. J Exp Med. 2020;217:e20191257.
Perkail S, Andricovich J, Kai Y, Tzatsos A. BAP1 is a haploinsufficient tumor suppressor linking chronic pancreatitis to pancreatic cancer in mice. Nat Commun. 2020;11:3018.
Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–8.
de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell. 2004;7:663–76.
Pengelly AR, Kalb R, Finkl K, Muller J. Transcriptional repression by PRC1 in the absence of H2A monoubiquitylation. Genes Dev. 2015;29:1487–92.
Wang L, Zhao Z, Ozark PA, Fantini D, Marshall SA, Rendleman EJ, et al. Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat Med. 2018;24:758–69.
Campagne A, Lee MK, Zielinski D, Michaud A, Le Corre S, Dingli F, et al. BAP1 complex promotes transcription by opposing PRC1-mediated H2A ubiquitylation. Nat Commun. 2019;10:348.
He M, Chaurushiya MS, Webster JD, Kummerfeld S, Reja R, Chaudhuri S, et al. Intrinsic apoptosis shapes the tumor spectrum linked to inactivation of the deubiquitinase BAP1. Science. 2019;364:283–5.
Kolovos P, Nishimura K, Sankar A, Sidoli S, Cloos PA, Helin K, et al. PR-DUB maintains expression of critical genes through FOXK1/2 and ASXL1/2/3-dependent recruitment to chromatin and H2AK119ub1 deubiquitination. Genome Res. 2020;30:1119–30.
Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev. 2003;17:896–911.
Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–49.
Narayanan A, Ruyechan WT, Kristie TM. The coactivator host cell factor-1 mediates Set1 and MLL1 H3K4 trimethylation at herpesvirus immediate early promoters for initiation of infection. Proc Natl Acad Sci USA. 2007;104:10835–40.
Julien E, Herr W. A switch in mitotic histone H4 lysine 20 methylation status is linked to M phase defects upon loss of HCF-1. Mol Cell. 2004;14:713–25.
Tyagi S, Chabes AL, Wysocka J, Herr W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol Cell. 2007;27:107–19.
Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med. 2009;15:1312–7.
Liu W, Tanasa B, Tyurina OV, Zhou TY, Gassmann R, Liu WT, et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466:508–12.
Peng H, Nogueira ML, Vogel JL, Kristie TM. Transcriptional coactivator HCF-1 couples the histone chaperone Asf1b to HSV-1 DNA replication components. Proc Natl Acad Sci USA. 2010;107:2461–6.
Revenko AS, Kalashnikova EV, Gemo AT, Zou JX, Chen HW. Chromatin loading of E2F-MLL complex by cancer-associated coregulator ANCCA via reading a specific histone mark. Mol Cell Biol. 2010;30:5260–72.
Yu L, Jearawiriyapaisarn N, Lee MP, Hosoya T, Wu Q, Myers G, et al. BAP1 regulation of the key adaptor protein NCoR1 is critical for gamma-globin gene repression. Genes Dev. 2018;32:1537–49.
Dai F, Lee H, Zhang Y, Zhuang L, Yao H, Xi Y, et al. BAP1 inhibits the ER stress gene regulatory network and modulates metabolic stress response. Proc Natl Acad Sci USA. 2017;114:3192–7.
Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20:1181–92.
Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell. 2013;49:795–807.
Soria G, Polo SE, Almouzni G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell. 2012;46:722–34.
Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204.
van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009;19:207–17.
Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45.
Yu H, Pak H, Hammond-Martel I, Ghram M, Rodrigue A, Daou S, et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci USA. 2014;111:285–90.
Ismail IH, Davidson R, Gagne JP, Xu ZZ, Poirier GG, Hendzel MJ. Germline mutations in BAP1 impair its function in DNA double-strand break repair. Cancer Res. 2014;74:4282–94.
Lee HS, Lee SA, Hur SK, Seo JW, Kwon J. Stabilization and targeting of INO80 to replication forks by BAP1 during normal DNA synthesis. Nat Commun. 2014;5:5128.
Lee HS, Seo HR, Lee SA, Choi S, Kang D, Kwon J. BAP1 promotes stalled fork restart and cell survival via INO80 in response to replication stress. Biochem J. 2019;476:3053–66.
Watanabe S, Peterson CL. The INO80 family of chromatin-remodeling enzymes: regulators of histone variant dynamics. Cold Spring Harb Symp Quant Biol. 2010;75:35–42.
Gerhold CB, Gasser SM. INO80 and SWR complexes: relating structure to function in chromatin remodeling. Trends Cell Biol. 2014;24:619–31.
Ventii KH, Devi NS, Friedrich KL, Chernova TA, Tighiouart M, Van Meir EG, et al. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res. 2008;68:6953–62.
Arenzana TL, Lianoglou S, Seki A, Eidenschenk C, Cheung T, Seshasayee D, et al. Tumor suppressor BAP1 is essential for thymic development and proliferative responses of T lymphocytes. Sci Immunol. 2018;3:eaal1953.
Pan H, Jia R, Zhang L, Xu S, Wu Q, Song X, et al. BAP1 regulates cell cycle progression through E2F1 target genes and mediates transcriptional silencing via H2A monoubiquitination in uveal melanoma cells. Int J Biochem Cell Biol. 2015;60:176–84.
van Ginkel PR, Hsiao KM, Schjerven H, Farnham PJ. E2F-mediated growth regulation requires transcription factor cooperation. J Biol Chem. 1997;272:18367–74.
Schlisio S, Halperin T, Vidal M, Nevins JR. Interaction of YY1 with E2Fs, mediated by RYBP, provides a mechanism for specificity of E2F function. EMBO J. 2002;21:5775–86.
Affar el B, Gay F, Shi Y, Liu H, Huarte M, Wu S, et al. Essential dosage-dependent functions of the transcription factor yin yang 1 in late embryonic development and cell cycle progression. Mol Cell Biol. 2006;26:3565–81.
Xu J, Kadariya Y, Cheung M, Pei J, Talarchek J, Sementino E, et al. Germline mutation of Bap1 accelerates development of asbestos-induced malignant mesothelioma. Cancer Res. 2014;74:4388–97.
Qin J, Zhou Z, Chen W, Wang C, Zhang H, Ge G, et al. BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nat Commun. 2015;6:8471.
Chen XX, Yin Y, Cheng JW, Huang A, Hu B, Zhang X, et al. BAP1 acts as a tumor suppressor in intrahepatic cholangiocarcinoma by modulating the ERK1/2 and JNK/c-Jun pathways. Cell Death Dis. 2018;9:1036.
Lee HJ, Pham T, Chang MT, Barnes D, Cai AG, Noubade R, et al. The tumor suppressor BAP1 regulates the hippo pathway in pancreatic ductal adenocarcinoma. Cancer Res. 2020;80:1656–68.
Deng R, Guo Y, Li L, He J, Qiang Z, Zhang H, et al. BAP1 suppresses prostate cancer progression by deubiquitinating and stabilizing PTEN. Mol Oncol. 2020;15:279–98.
Blanpain C, Daley GQ, Hochedlinger K, Passegue E, Rossant J, Yamanaka S. Stem cells assessed. Nat Rev Mol Cell Biol. 2012;13:471–6.
Laurenti E, Gottgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 2018;553:418–26.
Heidstra R, Sabatini S. Plant and animal stem cells: similar yet different. Nat Rev Mol Cell Biol. 2014;15:301–12.
Goodell MA, Rando TA. Stem cells and healthy aging. Science. 2015;350:1199–204.
Wu J, Izpisua Belmonte JC. Stem cells: a renaissance in human biology research. Cell. 2016;165:1572–85.
Baughman JM, Rose CM, Kolumam G, Webster JD, Wilkerson EM, Merrill AE, et al. NeuCode proteomics reveals Bap1 regulation of metabolism. Cell Rep. 2016;16:583–95.
LaFave LM, Beguelin W, Koche R, Teater M, Spitzer B, Chramiec A, et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2015;21:1344–9.
Kuznetsov JN, Aguero TH, Owens DA, Kurtenbach S, Field MG, Durante MA, et al. BAP1 regulates epigenetic switch from pluripotency to differentiation in developmental lineages giving rise to BAP1-mutant cancers. Sci Adv. 2019;5:eaax1738.
Danese A, Patergnani S, Bonora M, Wieckowski MR, Previati M, Giorgi C, et al. Calcium regulates cell death in cancer: roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim Biophys Acta Bioenerg. 2017;1858:615–27.
Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62–72.
Stefan CJ. Endoplasmic reticulum-plasma membrane contacts: Principals of phosphoinositide and calcium signaling. Curr Opin Cell Biol. 2020;63:125–34.
Bhosale G, Sharpe JA, Sundier SY, Duchen MR. Calcium signaling as a mediator of cell energy demand and a trigger to cell death. Ann N Y Acad Sci. 2015;1350:107–16.
Bauer TM, Murphy E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ Res. 2020;126:280–93.
Sime W, Niu Q, Abassi Y, Masoumi KC, Zarrizi R, Kohler JB, et al. BAP1 induces cell death via interaction with 14-3-3 in neuroblastoma. Cell Death Dis. 2018;9:458.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Affar EB, Carbone M. BAP1 regulates different mechanisms of cell death. Cell Death Dis. 2018;9:1151.
Lei P, Bai T, Sun Y. Mechanisms of ferroptosis and relations with regulated cell death: a review. Front Physiol. 2019;10:139.
Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. Obesity, inflammation, and cancer. Annu Rev Pathol. 2016;11:421–49.
Goodpaster BH, Sparks LM. Metabolic flexibility in health and disease. Cell Metab. 2017;25:1027–36.
Bantug GR, Galluzzi L, Kroemer G, Hess C. The spectrum of T cell metabolism in health and disease. Nat Rev Immunol. 2018;18:19–34.
Liu G, Summer R. Cellular metabolism in lung health and disease. Annu Rev Physiol. 2019;81:403–28.
Bononi A, Yang H, Giorgi C, Patergnani S, Pellegrini L, Su M, et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ. 2017;24:1694–704.
Ruan HB, Han X, Li MD, Singh JP, Qian K, Azarhoush S, et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab. 2012;16:226–37.
Izawa T, Rohatgi N, Fukunaga T, Wang QT, Silva MJ, Gardner MJ, et al. ASXL2 regulates glucose, lipid, and skeletal homeostasis. Cell Rep. 2015;11:1625–37.
Yang C, Ding H, Yang Y, Yang L, Yang Y, Fang M, et al. BAP1 regulates AMPK-mTOR signalling pathway through deubiquitinating and stabilizing tumour-suppressor LKB1. Biochem Biophys Res Commun. 2020;529:1025–32.
Hauri S, Comoglio F, Seimiya M, Gerstung M, Glatter T, Hansen K, et al. A high-density map for navigating the human polycomb complexome. Cell Rep. 2016;17:583–95.
Daou S, Mashtalir N, Hammond-Martel I, Pak H, Yu H, Sui G, et al. Crosstalk between O-GlcNAcylation and proteolytic cleavage regulates the host cell factor-1 maturation pathway. Proc Natl Acad Sci U S A. 2011;108:2747–52.
Capotosti F, Guernier S, Lammers F, Waridel P, Cai Y, Jin J, et al. O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell. 2011;144:376–88.
Yang Y, Yin X, Yang H, Xu Y. Histone demethylase LSD2 acts as an E3 ubiquitin ligase and inhibits cancer cell growth through promoting proteasomal degradation of OGT. Mol Cell. 2015;58:47–59.
Xiong Z, Xia P, Zhu X, Geng J, Wang S, Ye B, et al. Glutamylation of deubiquitinase BAP1 controls self-renewal of hematopoietic stem cells and hematopoiesis. J Exp Med. 2020;217:e20190974.
Janke C, Rogowski K, Wloga D, Regnard C, Kajava AV, Strub JM, et al. Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science. 2005;308:1758–62.
Rogowski K, van Dijk J, Magiera MM, Bosc C, Deloulme JC, Bosson A, et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell. 2010;143:564–78.
Field MG, Durante MA, Anbunathan H, Cai LZ, Decatur CL, Bowcock AM, et al. Punctuated evolution of canonical genomic aberrations in uveal melanoma. Nat Commun. 2018;9:116.
Brouwer NJ, Gezgin G, Wierenga APA, Bronkhorst IHG, Marinkovic M, Luyten GPM, et al. Tumour angiogenesis in uveal melanoma is related to genetic evolution. Cancers. 2019;11:979.
Yoshikawa Y, Emi M, Hashimoto-Tamaoki T, Ohmuraya M, Sato A, Tsujimura T, et al. High-density array-CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc Natl Acad Sci USA. 2016;113:13432–7.
Hmeljak J, Sanchez-Vega F, Hoadley KA, Shih J, Stewart C, Heiman D, et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. 2018;8:1548–65.
Shrestha R, Nabavi N, Lin YY, Mo F, Anderson S, Volik S, et al. BAP1 haploinsufficiency predicts a distinct immunogenic class of malignant peritoneal mesothelioma. Genome Med. 2019;11:8.
Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–33.
Liao L, Liu ZZ, Langbein L, Cai W, Cho EA, Na J, et al. Multiple tumor suppressors regulate a HIF-dependent negative feedback loop via ISGF3 in human clear cell renal cancer. Elife. 2018;7:e37925.
Huang Y, Wang J, Jia P, Li X, Pei G, Wang C, et al. Clonal architectures predict clinical outcome in clear cell renal cell carcinoma. Nat Commun. 2019;10:1245.
Chen YB, Xu J, Skanderup AJ, Dong Y, Brannon AR, Wang L, et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun. 2016;7:13131.
Mosbeh A, Halfawy K, Abdel-Mageed WS, Sweed D, Rahman MHA. Nuclear BAP1 loss is common in intrahepatic cholangiocarcinoma and a subtype of hepatocellular carcinoma but rare in pancreatic ductal adenocarcinoma. Cancer Genet. 2018;224-5:21–8.
Wang Y, Thomas A, Lau C, Rajan A, Zhu Y, Killian JK, et al. Mutations of epigenetic regulatory genes are common in thymic carcinomas. Sci Rep. 2014;4:7336.
Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–4.
Je EM, Lee SH, Yoo NJ. Somatic mutation of a tumor suppressor gene BAP1 is rare in breast, prostate, gastric and colorectal cancers. APMIS. 2012;120:855–6.
Tayao M, Andrici J, Farzin M, Clarkson A, Sioson L, Watson N, et al. Loss of BAP1 expression is very rare in pancreatic ductal adenocarcinoma. PLoS ONE. 2016;11:e0150338.
Tesch ME, Pater JA, Vandekerkhove G, Wang G, Binnington K, So AI, et al. Concurrent germline and somatic pathogenic BAP1 variants in a patient with metastatic bladder cancer. NPJ Genom Med. 2020;5:12.
Carbone M, Flores EG, Emi M, Johnson TA, Tsunoda T, Behner D, et al. Combined genetic and genealogic studies uncover a large BAP1 cancer syndrome kindred tracing back nine generations to a common ancestor from the 1700s. PLoS Genet. 2015;11:e1005633.
Walpole S, Pritchard AL, Cebulla CM, Pilarski R, Stautberg M, Davidorf FH, et al. Comprehensive study of the clinical phenotype of germline BAP1 variant-carrying families worldwide. J Natl Cancer Inst. 2018;110:1328–41.
Ohar JA, Cheung M, Talarchek J, Howard SE, Howard TD, Hesdorffer M, et al. Germline BAP1 mutational landscape of asbestos-exposed malignant mesothelioma patients with family history of cancer. Cancer Res. 2016;76:206–15.
Rawson RV, Watson GF, Maher AM, McCarthy SW, Thompson JF, Scolyer RA. Germline BAP1 mutations also predispose to cutaneous squamous cell carcinoma. Pathology. 2017;49:539–42.
Bhattacharya S, Hanpude P, Maiti TK. Cancer associated missense mutations in BAP1 catalytic domain induce amyloidogenic aggregation: a new insight in enzymatic inactivation. Sci Rep. 2015;5:18462.
Guo G, Chmielecki J, Goparaju C, Heguy A, Dolgalev I, Carbone M, et al. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res. 2015;75:264–9.
Farquhar N, Thornton S, Coupland SE, Coulson JM, Sacco JJ, Krishna Y, et al. Patterns of BAP1 protein expression provide insights into prognostic significance and the biology of uveal melanoma. J Pathol Clin Res. 2018;4:26–38.
Asada S, Goyama S, Inoue D, Shikata S, Takeda R, Fukushima T, et al. Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat Commun. 2018;9:2733.
Steurer S, Schwemmer L, Hube-Magg C, Buscheck F, Hoflmayer D, Tsourlakis MC, et al. Nuclear up regulation of the BRCA1-associated ubiquitinase BAP1 is associated with tumor aggressiveness in prostate cancers lacking the TMPRSS2:ERG fusion. Oncotarget. 2019;10:7096–111.
Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145:788–800.
Boultwood J, Perry J, Pellagatti A, Fernandez-Mercado M, Fernandez-Santamaria C, Calasanz MJ, et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010;24:1062–5.
Fisher CL, Pineault N, Brookes C, Helgason CD, Ohta H, Bodner C, et al. Loss-of-function additional sex combs like 1 mutations disrupt hematopoiesis but do not cause severe myelodysplasia or leukemia. Blood. 2010;115:38–46.
Gelsi-Boyer V, Trouplin V, Roquain J, Adelaide J, Carbuccia N, Esterni B, et al. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br J Haematol. 2010;151:365–75.
Katoh M. Functional and cancer genomics of ASXL family members. Br J Cancer. 2013;109:299–306.
Micol JB, Abdel-Wahab O. The role of additional sex combs-like proteins in cancer. Cold Spring Harb Perspect Med. 2016;6:a026526.
Balasubramani A, Larjo A, Bassein JA, Chang X, Hastie RB, Togher SM, et al. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat Commun. 2015;6:7307.
Yang H, Kurtenbach S, Guo Y, Lohse I, Durante MA, Li J, et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood. 2018;131:328–41.
Nagase R, Inoue D, Pastore A, Fujino T, Hou HA, Yamasaki N, et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J Exp Med. 2018;215:1729–47.
Kolluri KK, Alifrangis C, Kumar N, Ishii Y, Price S, Michaut M, et al. Loss of functional BAP1 augments sensitivity to TRAIL in cancer cells. Elife. 2018;7:e30224.
Ladanyi M, Sanchez Vega F, Zauderer M. Loss of BAP1 as a candidate predictive biomarker for immunotherapy of mesothelioma. Genome Med. 2019;11:18.
Figueiredo CR, Kalirai H, Sacco JJ, Azevedo RA, Duckworth A, Slupsky JR, et al. Loss of BAP1 expression is associated with an immunosuppressive microenvironment in uveal melanoma, with implications for immunotherapy development. J Pathol. 2020;250:420–39.
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research (2018–2023) to EBA. EBA is a senior scholar of the Fonds de la Recherche du Québec-Santé (FRQ-S). LM holds a PhD scholarship from the FRQ-S. AO holds PhD scholarships from the FRQ-S and the Cole Foundation. Certain forms and shapes displayed in figures were taken and modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. http://smart.servier.com/.
Author information
Authors and Affiliations
Contributions
EBA, ML, AO, and BE collected the references and wrote the manuscript. ML, AO, BE, and EBA realized the illustration. BL, NL, and AN contributed to the general improvement of the writing and critical review of the figures.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by F. Pentimalli
Rights and permissions
About this article

Cite this article
Masclef, L., Ahmed, O., Estavoyer, B. et al. Roles and mechanisms of BAP1 deubiquitinase in tumor suppression. Cell Death Differ 28, 606–625 (2021). https://doi.org/10.1038/s41418-020-00709-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00709-4
This article is cited by
-
Circ_0087851 suppresses colorectal cancer malignant progression through triggering miR-593-3p/BAP1-mediated ferroptosis
Journal of Cancer Research and Clinical Oncology (2024)
-
Emerging roles of deubiquitinating enzymes in actin cytoskeleton and tumor metastasis
Cellular Oncology (2024)
-
Targeting BAP1 with small compound inhibitor for colon cancer treatment
Scientific Reports (2023)
-
BAP1 as a guardian of genome stability: implications in human cancer
Experimental & Molecular Medicine (2023)
-
Deficiency of BAP1 inhibits neuroblastoma tumorigenesis through destabilization of MYCN
Cell Death & Disease (2023)
