
Abstract
Oleate, the most abundant endogenous and dietary cis-unsaturated fatty acid, has the atypical property to cause the redistribution of microtubule-associated proteins 1A/1B light chain 3B (referred to as LC3) to the trans-Golgi network (TGN), as shown here. A genome-wide screen identified multiple, mostly Golgi transport-related genes specifically involved in the oleate-induced relocation of LC3 to the Golgi apparatus. Follow-up analyses revealed that oleate also caused the retention of secreted proteins in the TGN, as determined in two assays in which the secretion of proteins was synchronized, (i) an assay involving a thermosensitive vesicular stomatitis virus G (VSVG) protein that is retained in the endoplasmic reticulum (ER) until the temperature is lowered, and (ii) an isothermic assay involving the reversible retention of the protein of interest in the ER lumen and that was used both in vitro and in vivo. A pharmacological screen searching for agents that induce LC3 aggregation at the Golgi apparatus led to the identification of “oleate mimetics” that share the capacity to block conventional protein secretion. In conclusion, oleate represents a class of molecules that act on the Golgi apparatus to cause the recruitment of LC3 and to stall protein secretion.
Similar content being viewed by others


Introduction
Autophagy is characterized by the redistribution of microtubule-associated proteins 1A/1B light chain 3B (hereafter referred to as LC3) from a diffuse cytosolic pattern towards the membranes of autophagosomes and autolysosomes [1,2,3]. This process can be monitored by measuring the lipidation of LC3 (i.e., its conjugation to phosphatidylethanolamine that increases its electrophoretic mobility in immunoblot experiments) or more conveniently by visualizing the redistribution of LC3 into cytoplasmic puncta that can be detected by immunofluorescence or by conjugation of LC3 to fluorescent proteins [4, 5]. A myriad of different endogenous and exogenous factors can stimulate or suppress autophagy, with important implications for cellular physiology and organismal health. As a general pattern, autophagy is required for maintenance of cellular homeostasis, meaning that its inhibition precipitates the manifestation of degenerative diseases, while its activation can delay the aging process [6,7,8,9].
Fatty acids have a profound effect on autophagy. Oleic acid (IUPAC name: cis-9-octadecenoic acid) is the most abundant mono-unsaturated fatty acid in our body, as well as in our diet [10, 11]. As other medium and long-chain cis-unsaturated fatty acids, be they mono- or poly-unsaturated, oleate (the oleic acid anionic form) induces a non-canonical type of autophagy in which LC3 and other proteins from the LC3 family are conjugated to phosphatidylethanolamine and preferentially redistribute to the Golgi apparatus. This process is “non-canonical” in the sense that it does not require the activation of the Beclin-1 (BECN1)-phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3, best known as VPS34) complex [12]. In sharp contrast, saturated fatty acids, among which palmitic acid (IUPAC name: Hexadecanoic acid, the most abundant one), induce autophagy through a canonical pathway that requires both the ATG5/ATG7/ATG12 conjugation system (that conjugates LC3 to phosphatidylethanolamine) and the BECN1/PIK3C3 complex, resulting into the distribution of LC3 to cytoplasmic puncta without any preference for the Golgi apparatus [12]. Finally, trans-fatty acids such as elaidic acid (IUPAC name: trans-9-octadecenoic acid, an isomer of oleic acid) do not induce autophagy at all and rather inhibit the LC3 lipidation and redistribution induced by palmitic acid but not by oleic acid [13].
The aforementioned findings suggest that the physicochemical properties of different classes of fatty acids (saturated versus cis- or trans-unsaturated carbon chains) rather than their individual biochemical functions (e.g., in specific lipidation reactions) or their binding to specific G-protein-coupled receptor (which distinguish fatty acids of distinct lengths) [14,15,16] determine their effects on autophagy. Moreover, it is tempting to speculate that the well-described negative effects of trans-unsaturated fatty acids (contained in industrially processed hydrogenated oil) on human health are due to autophagy inhibition [16,17,18]. Similarly, the positive effect of cis-unsaturated fatty acids (contained in specific plant oils such as olive oil) [19] might be explained by the activation of autophagy-related circuitries [20].
Enticed by these perspectives, we decided to investigate the genetic basis of the oleate effect on human cells, starting with a genome-wide screen to identify the pathways that explain the specifics of autophagy induction by this mono-cis-unsaturated fatty acid.
Results
A genome-wide screen to explore oleate-induced LC3 puncta
Biosensor cell lines expressing a fusion protein composed by LC3 and green fluorescent protein (GFP) can be used to monitor autophagy-related processes [21]. Both oleate and palmitate induce GFP-LC3 puncta, though with different morphologies [12, 13], namely the formation of relatively few puncta, usually close to one pole of the nucleus, for oleate and a more speckled pattern in which multiple puncta distribute all over the cytoplasm for palmitate. To understand the genetic basis of the oleate-induced phenotype, we performed a robotized genome-wide small interfering (si)RNA-based phenotypic screen in which human osteosarcoma U2OS cells stably expressing GFP-LC3 were transfected with 18 120 distinct siRNAs targeting a majority of the known human mRNAs, then treated with either oleate or palmitate, and finally monitored to quantify cytoplasmic GFP-LC3 dots by automated high-content image analysis (Fig. 1a).

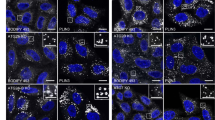
a–d Human osteosarcoma U2OS cells stably expressing GFP-LC3 were transfected with 18 120 distinct small-interfering RNAs (siRNAs) for 24 h. After transfection, cells were left untreated or treated with either 500 μM oleate or 500 μM palmitate for 6 h, before fixation, nuclei counterstaining with Hoechst 33342 and image acquisition by fluorescence microscopy. LC3 aggregation was evaluated by assessing the formation of GFP-LC3 puncta by fluorescence microscopy. a Representative images of untreated cells (CTR), cells treated with oleate (OL) or palmitate (PAL) and transfected with either an unrelated siRNA (siUNR), siATG5 (used as a control of autophagy inhibition) or siSTX5 (Golgi-related gene of interest) are depicted. The scale bar equals 10 µm. b The average area of GFP-LC3 puncta per cell was quantified, and data were normalized between negative (cells left untreated and transfected with siUNR) and positive controls (cells treated with OL and transfected with siUNR) and scaled by calculating Z-scores. GFP-LC3 dots surface scores were then computed by performing a sigmoid transformation on Z-scores and reported in a bi-parametric plot, showing the comparison between oleate versus palmitate treatment. The gray dots represent one hundred primarily selected oleate-specific hits. Autophagy-related and Golgi-related genes are represented in green and red respectively, and all other genes are represented as isopleths in a contour map. c Gene ontology (GO) relevant terms (excluding shared hits between oleate and palmitate) were reported in a bar chart showing the number of hits in each category. Colors represent the p value for each GO term. d Results from STRING analysis are displayed as a network interaction map and relevant GO terms (including shared hits between oleate and palmitate) are indicated. e–h U2OS cells stably co-expressing GALT1-GFP and RFP-LC3 were transfected with the indicated siRNAs and then treated with 500 μM oleate (OL) for 6 h. Upon treatment, cells were fixed, their nuclei counterstained with Hoechst 33342 and the phosphorylation of eIF2α (P-eIF2α) was assessed by means of immunofluorescence staining using a phosphoneoepitope-specific antibody recognizing eIF2α phosphorylation on serine 51, via fluorescence microscopy. RFP-LC3 dot aggregation (RFP-LC3), cell loss due to toxicity (TOX), Golgi surface (GOL), and P-eIF2α were quantified. e Representative images of GOL and RFP-LC3 of cells transfected with siUNR and siCOPE are depicted. The scale bar equals 10 µm. f For TOX, maps representing the cellular density per well of cells transfected with siUNR, siXBP1, siATF6, siATG5, siATG7, siCOPE are displayed and the scale bar represents 20 µm. g Representative images of P-eIF2α of cells transfected with siUNR, siEIF2AK4, siEEF2K, siATG5, siCOPB1, and siSTX3 are depicted and the scale bar equals 10 µm. h Each parameter was normalized to the unrelated (UNR) siRNA control, so that each value falls between 0 and 100%, the last value corresponding to the original effect, and the first to the maximum possible inhibition of the phenotype. SiRNAs depicted in green and red correspond to negative and positive controls respectively, while manually added genes are marked in pink.
Most of the siRNAs had no effect on the formation of puncta in response to either oleate or palmitate, while some targeting the central autophagic protein conjugation system (ATG5, ATG7, ATG12) reduced GFP-LC3 dots induced by both fatty acids. Other siRNAs participating in the Beclin 1 complex (such as ATG16L1 and BECN1) only interfered with palmitate, not oleate, -induced GFP-LC3 puncta, in accordance with a prior report [12]. In contrast, a small subset of semi-manually selected 85 genes appears to specifically interfere with the formation of puncta induced by oleate, but not palmitate (Fig. 1b). Among these siRNAs, two target subunits of the COPI complex (COPB1, COPB2) that is also targeted by brefeldin A, an inhibitor of ER-to-Golgi anterograde protein transport known to prevent oleate-induced relocation of GFP-LC3 to the Golgi apparatus without affecting palmitate-induced GFP-LC3 puncta [12]. Similarly, another protein involved in ER-to-Golgi transport, syntaxin-5 (STX5) [22], is required for the oleate, but not palmitate, -induced phenotype (Fig. 1a, b). Gene ontology (GO) analysis of the most prominent oleate-specific siRNAs revealed that most of the highly significant (p < 0.001) terms deal with Golgi-relevant transport processes (Fig. 1c), while the palmitate specific siRNAs did not relate to any significant biological process, even when including a wider set of genes.
A post-deconvolution bioinformatic STRING analysis that reconstructs documented protein-protein interactions among the oleate hits yielded a network of proteins involved in Golgi-related transport and ER stress with a central node that involves eukaryotic initiation factor 2α (EIF2A, best known as eIF2α) and several of its kinases (Fig. 1d) as part of the “integrated stress response” [23,24,25,26]. As a consequence, we decided to focus our study on these processes.
Validation of selected hits from the screen
In the next step, we retrieved all the siRNAs identified in the robotized screen as oleate-specific inhibitors of GFP-LC3 dots and tested them manually on U2OS cells co-expressing two fluorescent biosensors, namely a fusion protein composed by beta-1,4-galactosyltransferase 1 (GALT1) and GFP (for assessing Golgi morphology) and another chimeric protein composed by red fluorescent protein (RFP) and LC3 (for assessing RFP-LC3 puncta) (Fig. 1e). In addition, we measured the overall density of cells as a surrogate marker of viability (Fig. 1f), as well as the phosphorylation of eIF2α (on serine 51) by immunofluorescence staining with a phospho-neoepitope-specific antibody (Fig. 1g). Oleate-induced eIF2α phosphorylation was confirmed by immunoblot (Fig. S1). Moreover, the ER chaperone calreticulin, which is induced in the context of unfolded protein response (UPR) [27], was upregulated in response to oleate (Fig. S2A, B) and these oleate-induced effects occurred in a dose-dependent fashion (Fig. S2A–H).
Since oleate was previously described as an inducer of the UPR [13], we decided to add siRNAs targeting several UPR-related genes (ATF4, ATF6, XBP1, EIF2AK1 to 4, HSPA5, ERN1) to the validation experiments. Of note, many of these siRNAs (with the notable exception of ERN1), modulated the oleate effect (Fig. 1h). At least half of the siRNAs were validated with respect to the inhibition of oleate-induced RFP-LC3 puncta (including 3 out of the 4 eIF2α kinases), a reduction of eIF2α phosphorylation (including ATG5; eIF2α kinase 4, EIF2AK4; eukaryotic elongation factor 2 kinase, EEF2K; COPB1; XBP1) and attenuated the cytotoxicity of oleate (including ATG5, ATG7, EIF2AK4, as well as other ER stress-relevant genes such as ATF6 and XBP1; and 2 subunits of the COPI complex). A minority of the siRNAs also prevented the shrinkage of the Golgi apparatus induced by oleate (EIF2AK4; several COPI subunits) (Fig. 1h). These findings suggest an intimate link between the ATG5/7/12 protein conjugation system, ER stress, and COPI complex with respect to the oleate-triggered LC3 relocation to the Golgi apparatus.
To further explore this link, we used U2OS cells knockout for ATG5 confirming that oleate-induced GFP-LC3 dots entirely rely on this essential autophagy protein. Moreover, ATG5−/− cells showed a partial rescue from oleate cytotoxicity, a minor but significant reduction of eIF2α phosphorylation, but failure to interfere with Golgi shrinkage (which even was accentuated in the knockout cells) (Figs. 2a, b and S3A, C). A knock-in mutation by means of CRISPR/Cas9 technology on U2OS cells stably transduced with RFP-LC3 that renders eIF2α non phosphorylatable (by replacing serine 51 by an alanine residue: eIF2αS51A) [28] interfered with the induction of RFP-LC3 dots by oleate, enhanced oleate cell killing, yet again had no effects on the Golgi shrinkage (Figs. 2c, d and S3B, D). Similar results were obtained for mouse embryonic fibroblasts (MEF) subjected to a quadruple knockout (eIF2α4KO) to remove all four eIF2α kinases from the genome [29], showing that eIF2α phosphorylation is required for the formation of oleate-elicited RFP-LC3 puncta (Fig. S4A, B). These findings underscore the involvement of a complex cascade of molecular events that imply Golgi-related structural proteins, the COPI complex, and the “integrated stress response” for the oleate-induced phenotype.

Human osteosarcoma U2OS cells stably expressing GFP-LC3 that were either wild-type or knockout for ATG5 (ATG5−/−), as well as U2OS cells expressing non phosphorylatable eIF2α (eIF2αS51A) and RFP-LC3 were left untreated (CTR) or were treated with 5 μg/mL golgicide A (GCA), 500 μM oleate (OL), 2 μM staurosporine (STS), 0.3 μM torin 1 (TOR) or 3 μM thapsigargin (TP) for 6 h. For viability studies, cells were treated for additional 6 h and further co-stained with propidium iodide and Hoechst 33342, followed by immediate live-cell acquisition. For all other conditions, cells were fixed, their nuclei counterstained with Hoechst 33342 and images were acquired by fluorescence microscopy. GFP- (or RFP-) LC3 dots area, eIF2α phosphorylation (P-eIF2α) (assessed by immunofluorescence staining with a phosphoneoepitope-specific eIF2α antibody), Golgi morphology (Golgi surface detected by immunofluorescence staining with anti-B4GALT1 antibody) and cell death (assessed by the cell count of cells with Hoechstdim and PI- nuclei) were quantified. a, c Representative images of untreated cells (CTR), cells treated with OL or cells treated with the positive control for each assessed parameter (POS) (i.e., TOR for LC3 dots area, TP for P-eIF2α, GCA for Golgi disruption, and STS for cell death) are depicted for each knockout cell line (ATG5−/− and eIF2αS51A) together with the corresponding parental one (GFP-LC3 and RFP-LC3, respectively). Nuclei counterstained with Hoechst 33342 are represented in blue. The scale bar equals 10 μm. b, d For each assessed parameter and cell line, data were normalized to the untreated control, fold change (FC) means±SD are reported in bar charts for one representative experiment depicting the oleate effect together with appropriate internal controls. Each condition was compared to the untreated control (*) and to the parental cell line (#) by means of a Welch’s t-test (#p < 0.05, **/##p < 0.01, ***/###p < 0.001).
Oleate stalls protein secretion at the level of the Golgi
Intrigued by the partial colocalization of oleate-induced GFP-LC3 puncta with the Golgi marker GALT1 (Fig. 1e), we sought to more precisely map their subcellular localization using a quantitative immunofluorescence staining approach at higher resolution. For this, we subjected U2OS cells stably expressing GFP-LC3 to the induction of puncta by oleate or palmitate and then stained the cells with antibodies specific for the ER protein calreticulin (CALR), ER-Golgi intermediate compartment-53 (ERGIC-53 encoded by the LMAN1 gene), trans-Golgi network glycoprotein 46 (TGN46, official name: trans-Golgi network integral membrane protein 2, TGOLN2), 130 kDa cis-Golgi matrix protein (GM130, official name: Golgin subfamily A member 2, GOLGA2). Quantitative assessment of the colocalization of the fluorescent signals using a recently developed software [30] revealed that oleate (but not palmitate nor torin 1 and thapsigargin, which are “pure” activators of autophagy and ER stress, respectively) induced a selective and time-dependent colocalization of GFP-LC3 with the trans-Golgi marker TGN46, but not with the cis-Golgi network nor the ERGIC-53 or the ER (Figs. 3a-c and S5). This translocation of GFP-LC3 to the trans-Golgi was inhibited by brefeldin A and golgicide A (Fig. 3d–f).

Human osteosarcoma U2OS cells stably expressing GFP-LC3 were left untreated (CTR) or treated with 5 μg/mL brefeldin A (BFA), 5 μg/mL golgicide A (GCA), 500 μM oleate (OL), 500 μM palmitate (PAL), 2 μM staurosporine (STS), 0.3 μM torin 1 (TOR), 3 μM thapsigargin (TP), or treated with a combination of OL with BFA or GCA for 2 h, 4 h or 6 h. After fixation, nuclear counterstaining with Hoechst 33342 and antibody-mediated staining of trans-Golgi network glycoprotein 46 (TGN46), 130 kDa cis-Golgi matrix protein (GM130), ER-Golgi intermediate compartment-53 (LMAN1) and endoplasmic reticulum calreticulin (CALR), the images at high-resolution were acquired by confocal microscopy at 60X and analyzed to determine the co-occurrence of the fluorescent signals. a–c For each timepoint, representative images of untreated cells (CTR), cells treated with OL or PAL and stained with anti-TGN46 antibody are depicted. The background of the images has been removed by subtracting the Gaussian blur of each channel from the original image. Scale bar = 10 μm. d–f For each timepoint, the average surface overlap coefficient (SOC) between GFP-LC3 puncta and the different subcellular compartment per cell was calculated from the cleaned images and data are shown as boxplots with p values calculated using a Mann–Whitney U test (*p < 0.05, **p < 0.01, ***p < 0.001).
Intrigued by these observations, we wondered whether oleate might interfere with protein secretion via the trans-Golgi network. To evaluate this conjecture, we took advantage of cells transiently expressing a thermosensitive mutant of vesicular stomatitis virus-G (VSVG) protein [31, 32] that is fused to yellow fluorescent protein (YFP). This protein is retained in the ER lumen at high temperature (40 °C), yet undergoes a conformational change compatible with its traffic through the secretory pathway, transiting through the Golgi apparatus towards the plasma membrane, once the temperature is lowered to a permissive level (32 °C) (Fig. 4a, b). While classical autophagy induction by means of the mechanistic target of rapamycin (mTOR) inhibitor torin 1 failed to interfere with the membrane translocation of VSVG-YFP although clearly stimulating the formation of RFP-LC3 puncta, oleate prevented VSVG-YFP to move beyond a structure that resembles the Golgi apparatus in close proximity or overlap with RFP-LC3 puncta, thus hindering it from reaching the plasma membrane (Fig. 4c–e). These time-dependent changes were quantified and subjected to statistical analysis to document the capacity of oleate to retain VSVG-YFP within the cell, to enhance the colocalization of VSVG-YFP and RFP-LC3 and to reduce the dispersion of RFP-LC3 dots (Fig. 4f–i). Hence, oleate interferes with conventional protein secretion at the level of the Golgi apparatus, likely at the level of the trans-Golgi network.

The oleate-induced inhibition of the trans-Golgi network (TGN)-to-plasma membrane (PM) protein trafficking was evaluated by means of a thermosensitive vesicular stomatitis virus G glycoprotein (VSVGtsO45) fused to yellow fluorescent protein (YFP) at its cytoplasmic tail (VSVG-YFP). a The principle of the VSVG-YFP assay is outlined. VSVG-YFP is retained in the endoplasmic reticulum (ER) at 40 °C and relocates from the ER, passing through the Golgi apparatus (GA) in ~0.5 h to the plasma membrane (PM) upon temperature reduction to 32 °C. b Human osteosarcoma U2OS cells stably expressing RFP-LC3 were transiently transfected with VSVG-YFP and were incubated at 40 °C for 16 h. Cells were left untreated (CTR) or treated with 0.3 μM torin 1 (TOR) or 500 μM oleate (OL) for 2 h at 40 °C, the temperature was then reduced to 32 °C followed by immediate live-cell acquisition with an image frequency acquisition of 10 min for 4 h. c–e Representative time-lapse images of VSVG-YFP and RFP-LC3 of untreated cells (CTR) and cells treated with TOR or OL are depicted. The scale bar corresponds to 10 μm. f–i Quantification of the relative VSVG-YFP dots surface, the relative RFP-LC3 surface, the surface overlap coefficient (SOC) between RFP-LC3 and VSVG-YFP, and the distance of LC3 dots to their centroid, is represented as means±SD throughout the time series for one representative experiment. The p value of each pairwise comparison was calculated using the Mann–Whitney U test (***p < 0.001, NS not significant).
Confirmation of oleate-inhibited protein secretion with the RUSH system in vitro
To further explore the protein trafficking, we turned to yet another approach, the retention using selective hooks (RUSH) system [33], in which streptavidin was targeted to the ER surface (by fusing it to CD74 in the N-terminus), where it retains a GFP-LC3 chimera fused to streptavidin-binding domain (SBP, placed in the C-terminus), especially in the absence of free biotin (which can be quenched by the addition of avidin to the culture medium) [34]. Upon addition of a molar excess of biotin (which has a much higher affinity for streptavidin, in the range of 10−14 M, than SBP, outcompeting the latter), the SBP-GFP-LC3 fusion protein is released from the streptavidin “hook” and can rapidly redistribute to other locations (Fig. S6B, C). When biotin was combined with torin 1, SBP-GFP-LC3 was diffusely present in the cytoplasm, as determined by time-lapse microscopy. Conversely, when oleate was introduced into the system together with biotin, it inhibited the biotin-induced dispersion of SBP-GFP-LC3 (Fig. S6A, D), which however was efficiently released from streptavidin, as indicated by the loss of the co-staining of GFP and streptavidin (Fig. S6E, G) and colocalized with the trans-Golgi marker TGN46 (Fig. S6F, G). Interestingly, even when avidin was added to the system after oleate treatment, SBP-GFP-LC3 still colocalized with TGN46, probably showing an irreversible perturbation in the distribution of this protein by the fatty acid. These results, substantiated by quantitative image analysis, confirm that oleate deviates the intracellular voyage of GFP-LC3 towards the trans-Golgi network, even in conditions in which protein movements are synchronized by the RUSH system.
In another RUSH assay [35], streptavidin was targeted to the ER lumen (by means of the classical ER retention signal KDEL placed in the N-terminus), where it retains GFP fused to a streptavidin-binding domain (SBP, placed in the C-terminus). Upon addition of biotin, the SBP-GFP fusion protein can be secreted via the Golgi apparatus into the culture supernatant (Fig. 5a). We then determined the effects of oleate on this protein release assay. As a positive control, brefeldin A caused full retention of SBP-GFP in the cell, in the proximity of the streptavidin hook, while the negative control torin 1 and palmitate failed to do so (Fig. 5b–f). Oleate caused a dose-dependent reduction in GFP secretion (Figs. 5b and S7A). In contrast, induction of the UPR by thapsigargin or tunicamycin failed to suppress GFP secretion in this system (Figs. 5b, c and S7A). Moreover, oleate did not affect protein synthesis (Fig. S7B, C), and inhibition of protein synthesis by cycloheximide did not interfere with the RUSH assay (Figs. 5b, c and S7B, C). Of note, instead of causing SBP-GFP to be withhold in the ER (as this would be observed with brefeldin A, Fig. 5d, e), oleate treatment led to a retention of GFP at the trans-Golgi network, as indicated by the colocalization of SBP-GFP and TGN46 (Fig. 5d, f).

The retention using selective hooks (RUSH) assay was used to evaluate protein secretion from the endoplasmic reticulum (ER) in vitro. a The principle of the RUSH-secretion assay is schematized. Human osteosarcoma U2OS cells stably express a GFP reporter coupled to a streptavidin binding peptide (SBP) that in absence of biotin is retained by a streptavidin (Str) expressing hook targeting via KDEL to the ER. Upon biotin addition, the SBP-GFP reporter exits the ER due to the binding of biotin to streptavidin and it is secreted via exocytosis. U2OS cells stably co-expressing Str-KDEL and SBP-GFP were treated for 2 h in the absence of biotin. Biotin was added without removing the culture medium and live time-lapse images were acquired every 20 min for 4 h. b The integrated GFP fluorescence intensity was quantified at each time point and kinetics of protein secretion are represented as means in conditions without biotin (full line) and with biotin (dashed line) of cells left untreated (CTR), or treated with 5 μg/mL brefeldin A (BFA) as positive control for conventional secretion inhibition, 50 μM cycloheximide (CHX), 3 μM thapsigargin (TP), 3 μM tunicamycin (TM), 0.3 μM torin 1 (TOR) or 500 μM palmitate (PAL). For oleate (OL), cells were treated with a concentration range (125 μM, 250 μM, 375 μM and 500 μM) and data are displayed as lines with increasing thickness corresponding to increasing doses. c Representative time-lapse images of U2OS Str-KDEL-SBP-GFP untreated cells (CTR) and cells treated with BFA, CHX, TP, TM, TOR, OL, or PAL, in the presence (+) or absence (−) of biotin are displayed. The scale bar equals 10 μm. d Cells were fixed in the presence of Hoechst 33342 and antibody-mediated staining of trans-Golgi network glycoprotein 46 (TGN46) and streptavidin (STR) was performed. High-resolution images of untreated cells (CTR), and cells treated with BFA, TOR, and OL in presence (+) or absence (−) of biotin are depicted. The background of the images has been removed by subtracting the Gaussian blur of each channel to the original image. The scale bar indicates 10 μm. The colocalization of SBP-GFP/STR (e) and SBP-GFP/STR (f) was quantified by means of the surface overlap coefficient (SOC), and data are displayed as mean±SD for one representative experiment with p values calculated using a family-wise Welch’s t-test against untreated control (fixed biotin treatment,*) or control without biotin (fixed drug, #) (*/#p < 0.05, **p < 0.01, ***/###p < 0.001).
In conclusion, oleate indeed blocks protein secretion at a step at which the protein of interest (here SBP-GFP) is trapped in the trans-Golgi network. Of note, the oleate-induced entrapment of SBP-GFP at the Golgi apparatus does not seem to require ATG5 and ATG7 (Fig. S8), meaning that LC3 translocation to the Golgi (which relies on ATG5 and ATG7, see above in Fig. 1) might not be required for the blockade of protein secretion induced by oleate.
Identification of other compounds that induce an oleate-like inhibition of protein secretion
We wondered whether the oleate-induced alteration in protein trafficking is a unique feature or whether other chemical entities might exert similar cellular effects. For this, we exposed U2OS cells stably co-expressing GALT1-GFP and RFP-LC3 cells to 1 280 compounds from the Prestwick chemical library (mostly composed by FDA-approved drugs) to which we added oleate, as well as LTX-401, because we had noted in the past that this agent induces the redistribution of LC3 towards the Golgi [36] and then measured RFP-LC3 puncta, shrinkage of the Golgi and effects on cellular viability. Hierarchical clustering yielded a series of oleate/LTX-401 “neighbors” that share similar cell biology effects, yet belong to rather different pharmacological classes including two antiprotozoal/antimalarial agents that target lysosomes (mefloquine and quinacrine), several serotonin reuptake inhibitors (paroxetine, sertraline, as well as indatraline that also acts on dopamine and norepinephrine reuptake), and the nonselective calcium channel blocker fendiline (Fig. 6a, b). Moreover, we performed a bioinformatic analysis of reported drug targets for these agents (extracted from the biochemical literature) to identify genes/proteins that interact with these “oleate mimetics”, as defined above as well as in a previous work [13] (Fig. S9A). Of note, in the target database generated during the genome-wide search for modulators of the oleate-induced GFP-LC3 phenotype (see above, Fig. 1), such genes were relatively enriched as positive modulators/contributors (meaning that the siRNAs targeting them attenuated the phenotype), and this tendency was borderline significant (p = 0.072, Kolmogorov–Smirnov test) (Fig. S9B, C). This suggests that the “oleate mimetics” act through a similar, at least partially overlapping, pathway to induce cytotoxicity and the GALT1-GFP/RFP-LC3 phenotype as oleate does.

a, b Human osteosarcoma U2OS cells stably co-expressing RFP-LC3 and GALT1-GFP were left untreated (CTR), were treated with 500 μM oleate (OL), 5 μg/mL LTX-401 or were treated with the compounds from the Prestwick chemical library at 1 or 10 μM for 6 h. After fixation and nuclear counterstaining with Hoechst 33342, images were acquired by fluorescence microscopy and analyzed. a For RFP-LC3 dots surface (LC3), Golgi surface (GOL), and cell viability (VIAB), Z-scores were computed and reported in a hierarchically-ranked heatmap. Oleate neighboring is displayed as an expanded view below heatmap. b Representative images from the phenotypic screen of untreated cells (CTR) or cells treated with OL, LTX-401 as well as fendiline (FEN) are displayed. The scale bar represents 10 μm. c–i Human osteosarcoma U2OS cells stably expressing RFP-LC3 were transiently transfected with the thermosensitive vesicular stomatitis virus G glycoprotein (VSVGtsO45) fused to a fluorescent protein YFP at its cytoplasmic tail (VSVG-YFP) to evaluate the secretory pathway. Cells were left untreated (CTR) or treated with 5 μg/mL LTX-401, 10 μM FEN or 10 μM sertraline (SER), pharmacological compounds mimicking the phenotypic effect of oleate, as well as 0.3 μM torin 1 (TOR). c–e Representative time-lapse images of cells treated with LTX-401, FEN, and SER are depicted. The scale bar equals 10 μm. f–i The quantification of the relative VSVG-YFP dots surface, the relative RFP-LC3 dots surface, the surface overlap coefficient (SOC) between RFP-LC3 and VSVG-YFP and the RFP-LC3 dots distance to centroid are represented for the time series and in boxplots for each condition for one representative experiment. The p value of each pairwise comparison against control (CTR) (*) or torin 1 (TOR) (#) was calculated using a Mann–Whitney U test (#p < 0.05, **/##p < 0.01, ***/###p < 0.001).
Using a more direct approach, in another series of experiments, we confirmed that sertraline and fendiline acted similarly to oleate and LTX-401 to suppress protein secretion in the VSVG-YFP assay discussed above (Fig. 6c–i). Thus, pharmacological agents that induce alterations in Golgi morphology and the subcellular distribution of LC3 that resemble those induced by oleate are also inhibitors of conventional protein secretion.
Validation of inhibition of protein secretion by oleate in vitro and in vivo
To demonstrate that oleate can inhibit protein secretion in more physiological settings, we measured the release of tumor necrosis factor-α (TNF-α) from primary bone marrow-derived macrophages in response to bacterial lipopolysaccharide (LPS) in vitro (Fig. S10A). Like BFA, oleate (but not palmitate, nor torin 1 or thapsigargin) inhibited the ELISA-detectable release of TNF-α into the supernatant (Fig. S10B), as it caused the retention of immunofluorescence detectable TNF-α in the cytoplasm of the macrophages (Fig. S10C, D). In addition, we tested the capacity of oleate to inhibit protein secretion in vivo, in mice. For this, we created a RUSH system in which the ER-sessile streptavidin hook retains an SBP-tagged Gaussia luciferase as a bate (Fig. 7a, b). Upon hydrodynamic injection of the two plasmids (the hook in threefold molar excess over the bate), this RUSH system is expressed in hepatocytes and responds to the intraperitoneal injection of biotin by a surge of luciferase in the blood plasma that can be easily quantified by means of a chemiluminescence assay (Fig. 7c, d). Pre-treatment of mice with oleate (but not palmitate) at doses that cause LC3 lipidation [12] 2 h before biotin injection induced a significant reduction in luciferase secretion in this system (Fig. 7e, f). These results demonstrate that oleate can cause a partial inhibition of protein secretion in vivo.

The in vivo retention using selective hooks (RUSH) assay was employed to evaluate protein secretion in mice. a Vectors encoding the KDEL-Streptavidin hook and the secretory reporter streptavidin-binding peptide (SBP) coupled with the naturally secretable Gaussia princeps luciferase (Gluc) are displayed. b The principle of the RUSH-secretion assay is schematized. In presence of biotin, that outcompetes with streptavidin (Str), the SBP-Gluc bate is released from the Str-KDEL hook and secreted from the cell. c, d The hook and reporter vectors were co-expressed in hepatocytes by hydrodynamic intravenous (i.v.) injection through the tail vein. Seventy-two hours later, mice were treated by systemic (intraperitoneal, i.p.) injection for 2 h before i.p. injection of biotin (T0), causing the release of the Gluc into the circulation. Before biotin injection, and every 30/60 min, blood was sampled to assess the Gluc chemiluminescence activity. e Seventy-two hours after hydrodynamic i.v. injection of plasmids, mice were treated either with vehicle (10% BSA in endotoxin-free DPBS) or 100 mg/kg oleate (OL) for 2 h, followed by i.p. injection of biotin (+Biotin) or equivalent vehicle solution. Luciferase luminescence was assessed in serum samples and it was quantified throughout time, normalized to the control at the initial time and data are displayed as fold change (FC) mean±SEM (at least 6 mice per group are represented). f In an independent experiment, 72 h after hydrodynamic i.v. injection of plasmids, mice were treated either with vehicle (BSA), 100 mg/kg oleate (OL) or 100 mg/kg palmitate (PAL) for 2 h. Biotin was then injected i.p. and the luminescence FC 30 min after biotin injection is represented as boxplots with individual points. Outlier mice were excluded and p values were calculated using Mann–Whitney U test against the vehicle control (*p < 0.05, **p < 0.01, NS not significant).
Discussion
Oleate, which is the most abundant endogenous and exogenous cis-unsaturated fatty acid, is clearly distinct from palmitate, the most abundant saturated fatty acid, with respect to its cellular effects, at multiple levels. Indeed, in contrast to palmitate, oleate activates a pathway that leads to the translocation of LC3 to, and its retention at, the trans-Golgi network. We have shown in the past that this oleate-induced process requires ATG genes involved in the conjugation of LC3 family proteins to phosphatidylethanolamine (such as ATG5, ATG7, and ATG12) but does not require a series of other ATG genes involved in phosphatidylinositol phosphorylation (such as ATG14, BECN1, and PIK3C3/VPS34) [12]. This led us to postulate that oleate induces a “non-canonical” pathway of autophagy. Here, we extend and refine this notion, based on a genome-wide screen for genes required for oleate to induce the redistribution of LC3 from a diffuse towards an oligo-punctiform pattern. Indeed, this screen led to the discovery that multiple genes involved in Golgi-related transport processes are specifically required for this LC3 relocation induced by oleate but not by palmitate.
Following up on this finding, we investigated the capacity of oleate to interfere with canonical (Golgi-dependent, brefeldin A-inhibitable) protein secretion, using two experimental systems in which the movement of proteins through the Golgi apparatus can be synchronized. The first (classical) system is based on a YFP-coupled thermosensitive VSVG protein that is retained in the ER until the temperature is switched to a low permissive range, while the second RUSH system is performed at isothermic conditions (excluding temperature effects on membrane fluidity) and involves the reversible (biotin-inhibitable) retention of GFP fused to SBP in the ER lumen by streptavidin. In both systems, oleate had similar effects. Oleate did not affect the rapid move of the protein of interest (VSVG-YFP or SBP-GFP) from the ER to the Golgi, yet caused its retention in the Golgi, in or at the same compartment that accumulates LC3, thus blocking protein secretion. Inhibition of protein secretion by oleate was also demonstrated in vitro, for LPS-inducible TNF-α release from macrophages, as well as in vivo, using a tailor-made RUSH system measuring the secretion of luciferase from hepatocytes. These findings suggest that oleate may influence protein secretion at physiologically relevant concentrations. Whether such levels of oleate may also be obtained by dietary ingestion of oleate-rich food items (such as olive oil) remains to be investigated.
Mechanistically, oleate acts differently from brefeldin A, which causes dispersion of the Golgi apparatus. Oleate caused partial shrinkage of the Golgi, while stimulating the retention of LC3 and secreted proteins at and within the trans-Golgi compartment, respectively. Brefeldin A (or knockdown of subunits of the COPI complex, its pharmacological target) did not cause the redistribution of LC3 to puncta to the same extend as oleate but rather prevented the oleate-induced relocation of LC3 to the Golgi, supporting the contention that brefeldin A and oleate block protein secretion through rather distinct mechanisms. Of note, ATG5 and ATG7 were not required for the inhibition of protein secretion by oleate, supporting the idea that LC3 distribution to the Golgi (which relies on ATG5 and ATG7) can be uncoupled from the effects of oleate on protein secretion. Similarly, it appears that the oleate-induced redistribution of LC3 to the Golgi requires inhibition of mTOR [12], but mTOR inhibition by torin 1 did not interfere with protein secretion, underscoring that both Golgi-related processes, LC3 aggregation and secreted protein retention, are separable from each other. Finally, oleate induced the phosphorylation of eIF2α (as do most autophagy inducers) [37,38,39] is a requirement for LC3 recruitment but not for Golgi shrinkage, again dissociating the two phenomena. At this point, it is not clear through which molecular mechanisms oleate affects the function of the trans-Golgi network. It can be speculated that oleate selectively affects the particular membrane properties of this Golgi compartment, which differs in its biochemical composition and biophysical features from other cellular endomembranes [40,41,42].
Of note, oleate is not unique in its capacity to induce the redistribution of LC3 to the Golgi and to simultaneously stall protein secretion at this level. Indeed, systematic screening of 1 280 pharmacological agents revealed the existence of other, chemically unrelated (though mostly lipophilic) compounds that induce the redistribution of LC3 towards the Golgi, as shown here, and that share with oleate the capacity to block protein secretion [35], stalling their transport at the trans-Golgi network. At this point, it is difficult to understand which might be the shared mode of action among these “oleate mimetics”, although one common mode of action might be a perturbation of intracellular trafficking processes.
Mefloquine (an antimalarial agent) and quinacrine (an antiprotozoal and anthelmintic) share structural features, cause lysosomal alkalization [43, 44] and are both used for the treatment of systemic autoimmunity, especially in patients with cutaneous lupus erythematosus [45, 46]. It is possible, yet remains to be determined, that the inhibition of pro-inflammatory cytokine secretion by mefloquine or quinacrine in vivo [47, 48] is mediated by blocking protein secretion in the trans-Golgi compartment.
Several of the “oleate mimetics” act as reuptake inhibitors of neurotransmitters, as this applies to the antidepressants paroxetine, sertraline and indatraline [49, 50]. Such antidepressants are administered at a much lower dose (by one log) than mefloquine and quinacrine, suggesting that clinically useful concentrations will not lead to general protein secretion. However, it is possible that their accidental or intentional (suicidal) overdosing might induce such effects in vivo. Irrespective of these uncertainties, it appears that several pharmacological agents can affect cellular physiology in a similar fashion as oleate does.
In conclusion, oleate represents a class of molecules that cause the accumulation of LC3 at the Golgi apparatus as a sign of a local perturbation that culminates in the blockade of protein transport at the level of the trans-Golgi network.
Materials and methods
Cell lines
Human osteosarcoma U2OS wild-type (WT) cells were purchased from ATCC. U2OS cells stably expressing GFP-LC3, RFP-LC3, co-expressing RFP-LC3 and GALT1-GFP, co-expressing SBP-GFP and Str-KDEL, co-expressing SBP-GFP-LC3 and Str-CD74, U2OS ATG5 knockout (ATG5−/−) expressing GFP-LC3 and U2OS bearing a mutant non phosphorylatable version of eIF2α (U2OS eIF2αS51A) and co-expressing RFP-LC3 were generated by our group in the past [28, 34,35,36, 38, 51, 52]. Mouse embryonic fibroblasts MEF WT and knockout (KO) for the four eIF2α kinases (MEF eIF2α4KO) were provided by Pr. Seeichi Oyadomari from Tokushima University, Tokushima, Japan. Both MEF WT RFP-LC3 and eIF2α4KO RFP-LC3 were constructed from the aforementioned cell lines which were transduced using LentiBrite™ RFP-LC3 Lentiviral Biosensor viral particles (Merck Millipore, Burlington, MA, USA) according to the manufacturer’s instructions [39].
Cell culture
Human osteosarcoma U2OS cells and mouse embryonic fibroblasts (MEF) were cultured in Dulbecco’s Modified Eagle Medium (Thermo Fisher Scientific, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco® Thermo Fisher Scientific), 1% nonessential amino acids (Thermo Fisher Scientific), 1% HEPES (Thermo Fisher Scientific) in a humidified incubator with 5% CO2 at 37 °C. For U2OS cells co-expressing SBP-GFP and Str-KDEL, 0.25 mg/mL hygromycin (Invivogen, San Diego, CA, USA) and 0.5 mg/mL G418 (Invivogen) were added to the culture medium. For U2OS cells co-expressing SBP-GFP-LC3 and Str-CD74 culture medium was supplemented with 0.25 mg/mL hygromycin, 0.5 mg/mL G418, and 40 μM biotin (Sigma Aldrich, St Louis, MO, USA). Plastic supplies were purchased from Corning (Corning, NY, USA) and Greiner Bio-One (Kremsmünster, Austria).
Compounds and reagents
The Prestwick chemical library was obtained from Prestwick Chemical (Illkirch, France) and used at 1 and 10 μM. Fendiline and sertraline were aliquoted from the Prestwick chemical library and both used at 10 μM. LTX-401 was provided by Lytix Biopharma (Tromsø, Norway) and used at 5 μg/mL. Avidin (A9275) used at 1 μM, biotin (B4501) used at 40 μM, brefeldin A (B6542) used at 5 μg/mL, staurosporine (S4400) used at 2 μM, thapsigargin (T9033) used at 3 μM, tunicamycin (T7765) used at 3 μM and cycloheximide (01810) used at 50 μM were purchased from Sigma-Aldrich (St Louis, MO, USA). Golgicide A (3584) used at 5 μg/mL and torin 1 (4247) used at 0.3 μM were purchased from Tocris Bioscience (Bristol, UK). Oleate (10-1801-13) and palmitate (10-1600-13), both used at 500 μM, were purchased from Larodan (Malmö, Sweden). Sodium oleate (O7501) and sodium palmitate (P9767) used at 100 mg/kg for in vivo experimentation were purchased from Sigma-Aldrich. Hoechst 33342 (H3570) and Lipofectamine® 2000 reagent were purchased from Thermo Fisher Scientific. Propidium iodide (P4864), formaldehyde (F8775), and Triton X-100 (T8787) were purchased from Sigma Aldrich (St. Louis, MO, USA).
Antibodies
Rabbit monoclonal antibodies against phospho-eIF2α (Ser51) (ab32157) used 1/500 for U2OS cells, LMAN1 (ab125006) used 1/150, GM130 (ab52649) used 1/300 and rabbit polyclonal antibody against calreticulin (ab2907) used 1/300 were purchased from Abcam (Cambridge, UK). Rabbit monoclonal antibody against phospho-eIF2α (Ser51) (119A11) used 1/500 for MEF cells was purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit polyclonal antibody against TGN46 (13573-1-AP) used 1/150 was purchased from Proteintech (Rosemont, IL, USA). Rabbit polyclonal antibody against B4GALT1 (PAB20512) used 1/500 was purchased from Abnova (Taipei, Taiwan). Mouse monoclonal antibody against streptavidin (sc52234) used 1/500 was purchased from Sant Cruz Biotechnology (Dallas, TX, USA). Anti-rabbit and anti-mouse AlexaFluor 488-, 568- and 647-labeled antibodies used 1/1000 were purchased from Thermo Fisher Scientific. For immunoblotting, LC3B (2775 S, Cell Signaling Technologies), PeIF2a (ab32157, Abcam), and eIF2α (9722 S, Cell Signaling Technologies) were used 1/1000, β-actin (ab6276, clone AC-15, Abcam) was diluted 1/10000.
Fluorescence microscopy, image acquisition and analysis
One day before treatment, U2OS WT or ATG5−/−cells stably expressing GFP-LC3 or co-expressing RFP-LC3 and GALT1-GFP, U2OS eIF2αS51A expressing RFP-LC3 and MEF or MEF eIF2α4KO cells stably expressing RFP-LC3 were seeded in 384-well μClear imaging plates (Greiner Bio-One) and let adhere for 24 h. The next day, cells were treated to assess LC3 aggregation and Golgi morphology at 6 h, co-occurrence of LC3 puncta and subcellular structures at 2, 4, and 6 h, eIF2α phosphorylation (P-eIF2α) at 6 h and viability at 12 h. For viability studies, cells were further co-stained with 0.5 μg/mL propidium iodide and 2.5 μg/mL Hoechst 33342 for 20 min followed by immediate live-cell acquisition. For all other conditions, cells were fixed with 3.7% formaldehyde containing 1 μg/mL Hoechst 33342 for 1 h at room temperature. When measuring the formation of LC3 puncta and the morphology of the Golgi, the fixative was exchanged to PBS and the plates were analyzed by automated microscopy. To assess P-eIF2α and co-occurrence of LC3 puncta and subcellular structures, an additional immunostaining was performed. Cells were permeabilized with 0.1% Triton X-100 for 10 min (except for P-eIF2α in U2OS cells) and unspecific antibody binding was blocked by incubation with 2% BSA for 1 h at room temperature. After overnight incubation at 4 °C with the primary antibody, cells were repeatedly washed with PBS, stained with AlexaFluor-568 (or 488, or 647)-coupled secondary antibody for 2 h at room temperature and then washed with PBS before acquisition. For automated fluorescence microscopy, a robot-assisted Molecular Devices IXM XL BioImager and a Molecular Devices IXM-C (Molecular Devices, Sunnyvale, CA, USA) equipped with either a SpectraX or an Aura II light source (Lumencor, Beaverton, OR, USA), adequate excitation and emission filters (Semrock, Rochester, NY, USA) and a 16-bit monochromes sCMOS PCO.edge 5.5 camera (PCO Kelheim, Germany) or an Andor Zyla camera (Belfast, Northern Ireland) and a 20X or 60X PlanAPO objective (Nikon, Tokyo, Japan) were used to acquire a minimum of four view fields per well. For live cellular imaging, the atmospheric environment was controlled by means of an Ibidi gas mixer (Gräfelfing, Germany). The images were segmented and analyzed with the freely available software R (https://www.r-project.org), integrated with the EBImage package from the Bioconductor repository (https://www.bioconductor.org), the MetaxpR package (https://github.com/kroemerlab/MetaxpR), the RBioFormats package (https://github.com/aoles/RBioFormats), as well as the MorphR package (https://github.com/kroemerlab/MorphR). The primary region of interest (ROI) was defined by a polygon mask around the nucleus allowing for the enumeration of cells, the detection of morphological alterations of the nucleus and nuclear fluorescence intensity. Secondary cytoplasmic ROIs were used for the quantification of P-eIF2α intensity. To quantify GFP- (or RFP-) LC3 aggregation, a segmentation mask of high-intensity dots was generated in the cytoplasm of cells. For co-occurrence analysis, images were analyzed using the ColocalizR software [30] calculating the surface overlap coefficient (SOC) between fluorophores of interest. Golgi morphology was evaluated by quantifying the Golgi surface. For viability studies, the number of nuclei was measured in fixed cells, while in live-imaged cells dead cells (PI+) were quantified compared to live cells (Hoechstdim and PI-). After exclusion of cellular debris and dead cells, parameters of interest were normalized, statistically evaluated, and graphically depicted by using R software. Images were extracted, pixel intensities scaled to be visible (in the same extent for all images of a given experiment) and the background of the images has been removed by subtracting the Gaussian blur of each channel to the original image.
High-throughput siRNA interference
U2OS cells stably expressing GFP-LC3 were seeded in 384-well μClear imaging plates and let adhere for 24 h. The day after, cells were transfected with the siGenome genome-wide small-interfering RNA (siRNA) library (Dharmacon, Lafayette, CO, USA) targeting 18 120 genes of the human genome (one gene represented as a SMARTpool of 4 individual siRNAs in one well) for 24 h at a final concentration of 30 nM by means of DharmaFECT™ transfection reagent (Dharmacon) according to the manufacturer’s instructions. A siUNR (UAAGGCUAUGAAGAGAUAC) was used as a non-targeting control. Medium was discarded, cells let adapt for additional 24 h and then treated for 6 h with oleate or palmitate or left untreated as control prior to fixation and image acquisition as previously described. The average surface of GFP-LC3 puncta per cell was quantified. Before scaling data by calculating Z-scores, the data was normalized between negative (untreated cells and cells transfected with siUNR) and positive controls (cells treated with oleate and transfected with siUNR). A sigmoid transformation was performed on Z-scores to generate smoothened GFP-LC3 dots surface scores [53]. A bi-parametric plot was generated for the comparison of oleate- and palmitate-specific GFP-LC3 dots surface scores and 100 oleate-specific hits were semi-automatically determined and 15 non-expressed genes in the U2OS cell line were excluded. Briefly, 2 thresholds were defined by the 5% percentile of oleate and palmitate GFP-LC3 dots surface scores, thus defining a quadrant of oleate-specific hits, inside of which a manual gating was applied. Corresponding genes plus control siRNAs and manually added siRNAs were used to transfect U2OS cells stably expressing RFP-LC3 and GALT1-GFP for 24 h, the medium was then discarded, cells let adapt for additional 24 h before a 6 h treatment with oleate, fixation, and image acquisition as previously described. RFP-LC3 aggregation, toxicity, Golgi surface, and P-eIF2α intensity were measured as explained above. Each of the latter parameter was normalized to the unrelated (UNR) siRNA control and for plotting the heatmap, each value was ranged between 0 and 100% of oleate-induced effect, the first value corresponding to maximum inhibition of the phenotype and the last value corresponding to the original effect.
Bioinformatic analysis on oleate-specific hits
Genes modulating oleate-induced effects were subjected to Gene Ontology (GO) analysis by means of the clusterProfiler package (https://www.bioconductor.org). Relevant enriched terms from the “biological process” category were kept based on their associated p value (<0.1). In addition, proteins encoded by the identified target genes were further analyzed using the STRING tool (https://www.string-db.org), indicating confirmed and putative protein-protein interactions.
VSVG trafficking experiments
One day prior to transfection, U2OS cells stably expressing RFP-LC3 were seeded in 96-well μClear imaging plates (Greiner Bio-One) and let adhere for 24 h. The following day, transient transfection with ts045 VSVG-YFP plasmid was performed using Lipofectamine® 2000 according to the manufacturer’s protocol (Thermo Fisher Scientific). The plasmid was a kind gift from Dr. Frank Perez from Institut Curie, Paris, France. On the next day, cells were incubated for 16 h at 40 °C to maintain VSVG-YFP in the endoplasmic reticulum. Cells were then treated or left untreated for 2 h at 40 °C. Upon temperature shift to 32 °C, cells were observed by live-cell microscopy for 4 h with a frequency of image acquisition of one image every 10 min. The images were segmented and analyzed with R using the EBImage package. The relative VSVG-YFP and RFP-LC3 dots surfaces, the surface overlap coefficient (SOC) between VSVG and LC3 and the LC3 average dots distance to centroid (indicating the degree of dots spread) were quantified throughout the time series.
Retention using selective hooks (RUSH) system to evaluate protein trafficking in vitro
U2OS cells stably co-expressing Str-KDEL and SBP-GFP or co-expressing Str-CD74 and SBP-GFP-LC3 were seeded in 384-well μClear imaging plates in the absence and presence of biotin respectively and let adapt for 24 h. U2OS Str-CD74-SBP-GFP-LC3 cells were incubated additional 12 h in the presence of avidin. Cells were then treated or left untreated in the absence of biotin and in the presence of avidin respectively for 2 h and observed by live-cell microscopy for 2 h with a frequency of image acquisition of one image every 30 min. Next, 40 μM biotin was added and images were acquired every 20 min for 4 h. For U2OS Str-KDEL-SBP-GFP, cell integrated cytoplasmic GFP intensity was quantified in the entire image (after background correction by gaussian blur subtraction) for each timepoint. For U2OS Str-CD74-SBP-GFP-LC3, the dispersion of LC3 was quantified as dots distance to centroid. For both cell lines, co-occurrence between GFP and streptavidin, and between GFP and TGN46 was quantified using ColocalizR software. For siRNA interference, U2OS Str-KDEL-SBP-GFP were transfected for 24 h at a final concentration of 30 nM by means of DharmaFECT™ transfection reagent (Dharmacon) according to the manufacturer’s instructions. Medium was discarded, cells let adapt for additional 48 h and then left untreated or treated in absence of biotin for 2 h prior to biotin addition and live imaging acquisition as previously described. Fluorescence loss was quantified on an image-basis, by calculating the sum of integrated intensities differences over time, divided by the initial value.
Phenotypic screen of oleate mimetics
U2OS cells stably expressing RFP-LC3 and GALT1-GFP were seeded in 384-well μClear imaging plates and let adhere for 24 h. The following day, cells were treated with compounds from the Prestwick chemical library, oleate, and LTX-401 for 6 h. Cells were fixed, imaged, segmented, and analyzed as previously described. Data were submitted to a min-max normalization using appropriate controls for each assessed parameter (untreated control/oleate for LC3 aggregation, untreated control/golgicide A for Golgi disruption, untreated control alone for cellular viability), and then scaled to unit variance. Thereafter, hierarchical clustering was performed on single drugs by selecting for each agent the concentration that induced maximal LC3 aggregation.
Statistical analysis
Statistics were performed using freely available software R. Unless mentioned, all depicted barcharts are showing mean±SD from triplicate instances. Each experiment was conducted independently at least three times, with one being represented. Unless mentioned, significance levels were assessed by means of a Welch’s t-test (when comparing means) or a Mann–Whitney U test (when comparing medians) against appropriate control (*p < 0.05, **p < 0.01, ***p < 0.001).
Immunoblotting
After treatment, cells were washed with cold PBS, harvested with pre-warmed 0.25% trypsin/EDTA and lysed with RIPA buffer (89901, Thermo Fisher Scientific) supplemented with Pierce protease and phosphatase inhibitors (A32955 and A32957, Thermo Fisher Scientific) on ice. Samples were sonicated and centrifuged at 12000 g for 15 min at 4 °C, followed by the quantification of protein content by means of the Bio-Rad laboratory DC Protein Assay (500-0113, 500-0114, 500-0115, BioRad, CA, USA) following the manufacturer’s instructions. Next, 20 μg of protein were dissolved in NuPAGE® LDS sample buffer 4X (NP0007, Invitrogen) and NuPAGE® sample reducing agent 10X (NP0009, Invitrogen) and denaturized at 100 °C for 10 min. Protein samples were separated on pre-cast 4-12% polyacrylamide NuPAGE™ Bis-Tris gels (NP0336, Thermo Fisher Scientific) in NuPAGE® MOPS SDS Running buffer (NP0001, Thermo Fisher Scientific) and electro-transferred to EtOH-activated PVDF membranes (88518, Thermo Fisher Scientific) in transfer buffer (25 mM Tris, 190 mM glycine, 20% ethanol in H2O) (EU0550, Euromedex, Souffelweyersheim, France). Membranes were incubated in Tris-buffered saline with Tween®20 detergent (TBST, 20 mM Tris, pH7.5, 150 mM NaCL, 0.1% Tween®20 in H2O) (P1379, Sigma Aldrich; ET220-B, Euromedex) supplemented with 5% BSA (04-100-812, Euromedex) for 1 h in order to saturate unspecific binding sites. Thereafter, membranes were probed with primary antibodies specific to LC3B, PeIF2a, and eIF2α, diluted 1/1000 in 5% BSA in TBST overnight at 4 °C on a rocking shaker. Equal loading was verified by means of β-actin diluted 1/10000 in 5% BSA in TBST. The day after, membranes were extensively washed with TBST and then incubated with goat anti-rabbit IgG(H + L) horseradish peroxidase (HRP)-labeled secondary antibody (4050-05, Southern Biotech, Birmingham, AL, USA) diluted 1/5000 in 5% BSA in TBST for 1 h at room temperature. Immunoreactive bands were visualized by means of the ECL™ Prime western blotting detection reagents (RPN2236, Cytiva, Marlborough, MA, USA) and chemiluminescence images were acquired with an ImageQuant LAS4000 digital imaging system (GE Healthcare, Little Chalfont, UK).
In silico gene target analysis of oleate-mimetics
Gene targets of oleate mimetics, enriched with neighbors stemming from a colocalization analysis performed in a previous work [30], were retrieved using the Power User Gateway interface of PubChem to query the BioAssay database [54], and relevant genes were kept when at least two drugs had associated records, with at least one of them described as active. The list of genes was further curated by excluding non-expressed genes in the U2OS cell line, as reported in the protein atlas (https://www.proteinatlas.org). An enrichment in low scores was investigated by comparing the distribution of the obtained targets versus whole-genome gene set LC3 aggregation scores by performing a Kolmogorov–Smirnov test.
AHA incorporation to assess translation
Translation was studied by evaluating the incorporation of L-azidohomoalanine (AHA), a labeled amino acid analog of methionine containing an azide moiety, by means of the Click-iT™ chemistry according to the manufacturer’s instructions (C10289, Thermo Fisher Scientific). In short, U2OS wild-type cells were seeded in 384-well μClear imaging plates and let adapt for 24 h. The next day, cells were treated or left untreated in complete medium for 4 h and multiple PBS washing steps were performed. Cells were then treated 30 min in the presence of methionine-free medium (21013024, Thermo Fisher Scientific), followed by additional 1.5 h in methionine-free medium supplemented with 25 μM L-azidohomoalanine (AHA). Afterward, cells were fixed with 3.7% formaldehyde containing 1 μg/mL Hoechst 33342 for 1 h at room temperature, permeabilized with 0.1% Triton X-100 for 10 min and unspecific antibody binding was blocked by incubation with 2% BSA. After incubation with AlexaFluor 488-coupled azide for 2 h at room temperature, cells were imaged as previously described. The cytoplasmic GFP intensity (AHA) was quantified with the software R and data was normalized to the control.
In vivo experimentation
Wild-type C57BL/6 mice were purchased from Envigo (HARLAN FRANCE, Gannat, France) and were housed in the animal facility of the Gustave Roussy Cancer Campus with 12 h light and dark cycles, in a pathogen-free, temperature-controlled environment where they received water and food ad libitum. Animal experiments were conducted in compliance with the EU Directive 63/2010/EU from the European Parliament and were approved by the local Ethics Committee of the Gustave Roussy Cancer Campus (CEEA IRCIV/IGR no. 26, registered at the French Ministry of Research, with the protocol number 23146 / 2019112800493053). “BiostaTGV” software was used to calculate the number of animals to be included in each group in order to reach statistical significance, based on expected results.
In vivo RUSH assay
The RUSH plasmids pCDH-streptavidin (Str)-KDEL [35] and pCDH-ss-SBP-Gluc were injected into the tail vein of C57Bl/6 10 weeks old female mice via hydrodynamic injection as described previously [55]. pCDH-ss-SBP-Gluc was generated by replacing GFP in pCDH-ss-SBP-EGFP [35] with the coding sequence of secretable Gaussia princeps luciferase (Gluc) synthesized by Eurofins Genomics (Ebersberg, Germany). In short, mice were injected with 90 µg pCDH-streptavidin (Str)-KDEL plus 30 µg pCDH-ss-SBP-Gluc diluted in 37 °C pre-warmed sterile saline solution. DNA injection was performed with 5 mL BD Luer-Lok syringes (309646, BD Biosciences) equipped with 25 G needles. The entire volume was injected within 5-8 s under continuous pressure.
Seventy-two hours after plasmid delivery, mice were randomly treated via a single intraperitoneal injection (i.p.) of 100 mg/kg oleate or 100 mg/kg palmitate both diluted in endotoxin-free DPBS supplemented with 10% BSA or equivalent vehicle solution. Two hours later, mice were treated with 15 mg/kg biotin i.p. or equivalent vehicle solution for 3 h. Before biotin injection, 30 min and every hour after that, 10 μL of blood was withdrawn through the mandibular vein and immediately mixed with 2 μL of 50 mM EDTA anticoagulant, centrifuged at 6000 × g for 2 min at 4 °C and plasma was frozen at −20 °C.
Bioluminescence was assessed with 100 μM coelenterazine (sc-205904, Santa Cruz Biotechonolgy) prior diluted in PBS supplemented with 5 mM NaCl, at pH 7.2 and stabilized at room temperature for 30 min. In short, 5 μL serum per sample were transferred into Nunc™ F96 MicroWell™ white polystyrene plates (236105, Thermo Fisher Scientific), 100 μL/well of 100 μM coelenterazine were added and the Gluc chemiluminescence activity was immediately measured using a FluorStar Optima Plate Reader. Data were analyzed using R as follows: first, each individual was normalized to its initial value after biotin injection, and outliers were excluded timewise when its absolute scaled value (to unit variance) was higher than 2. Means and SEM of at least six mice per group were thereafter computed for plotting purposes, and time series or endpoint groups were compared by means of a pairwise Mann–Whitney U-test.
Measurement of extracellular TNF-α by sandwich ELISA
The ELISA max™ Deluxe Set for mouse TNF-α (430904, BioLegend®) was employed to measure TNF-α released in cell culture supernatants. Bone marrow‐derived macrophages (BMDM) were generated from femurs and tibias of dissected legs from 8 to 10-week-old C57Bl/6 mice. Bone marrow was extracted from bones by cutting joints with a scalpel and flushing the exposed bone marrow with a 10 mL syringe filled with 1% BSA in PBS. Clumps were dissociated by pipetting and red blood cells were lysed with cell lysis buffer (0.01 M Tris, 0.83% NH4Cl in Milli‐Q water). The cell suspension was passed through a 70 μm cell strainer and cells were seeded into 15 cm Petri-dishes in BMDM culture medium composed of RPMI 1640 medium supplemented with 10% FBS, 200 mM L-Glutamine (35050061, Thermo Fisher Scientific), 100 U/ml penicillin (15140122, Thermo Fisher Scientific), 1 M HEPES (15630056, Thermo Fisher Scientific), 1% MEM Non‐Essential Amino Acids Solutions (11140035, Thermo Fisher Scientific), and 50 μM β‐mercaptoethanol (31350010, Thermo Fisher Scientific) supplemented with 50 ng/ml recombinant mouse M‐CSF (416-ML-050, R&D Systems, MI, USA).
On day 3, the supernatant was removed and BMDM growth medium freshly supplemented with M-CSF was added. BMDM were harvested on day 6 and seeded in 24-well cell culture plates (3524, Corning) in BMDM growth medium freshly supplemented with M-CSF. The following day, BMDM were activated with 100 ng/mL lipopolysaccharide (LPS) (L4391, Sigma Aldrich) for 16 h. Next, BMDM were washed twice with PBS and treated for 2 h with selected compounds in combination or absence of LPS. Supernatant was collected and centrifuged. ELISA was performed as described in the provider’s protocol. Briefly, high binding 96-well plates (3361, Corning) were coated with anti-TNF-α capture antibody overnight at 4 °C. After multiple washing with washing buffer, blocking buffer was added to block unspecific binding for 1 h at RT on a rocking shaker. 100 μL of standards and samples diluted in 1% BSA in PBS were added in each coated-well for 2 h at RT with shaking. After washing, the anti-TNF-α detection antibody was added for 1 h with shaking. Plates were then rinsed and incubated with avidin-HRP-conjugated secondary antibody. After extensive washing, 100 μL of TMB substrate solution (34029, Thermo Fisher Scientific) was added in all wells and incubated for 15 min at RT in the dark until colorizing. Immediately after, stop solution (2 M H2SO4) was added and the absorbance at 450 nm was assessed with a SpectraMax I3 multi‐mode microplate reader (Molecular Devices). TNF-α concentration was quantified with software R by establishing an absorbance-concentration model using a mixed polynomial-exponential fit.
Assessment of intracellular TNF-α by flow cytometry
BMDM were collected, differentiated, activated, and treated as described above. Cells were detached with cell lifters, centrifuged, and washed twice with PBS. Firstly, BMDM were stained with LIVE/DEAD™ Fixable Yellow Dead Cell Stain Kit (L34968, Invitrogen) and incubated with purified anti-mouse CD16/32 antibody (101302, BioLegend®) to block Fc receptors and non-specific antibody binding for 20 min, at 4 °C. BMDM were then stained with F4/80 monoclonal antibody (BM8) coupled to AlexaFluor 488 (53-4801-82, Invitrogen) at 1 μg/mL in 1% BSA in PBS for 30 min at 4 °C covered in foil. Fixation and permeabilization for intracellular staining were performed by means of the Cyto-Fast™ Fix/Perm Buffer Set (426803, BioLegend®) according to manufacturer’s protocol. BMDM were then stained with 0.125 μg of anti-mouse TNF-α monoclonal antibody (MP6-XT22) coupled to APC (17-7321-82, Invitrogen). In control samples, the isotype control antibody rat IgG1 kappa isotype control (eBRG1) coupled to APC (17-4301-82, Invitrogen) was included. After multiple washes, samples were run on a BD LSR Fortessa flow cytometer, and data were acquired using BD FACS Diva software (BD Biosciences). Intracellular TNF-α was assessed by quantifying the ratio of live F4/80+ APC+ BMDM among the total amount of live F4/80+ BMDM using R software, combined with the flowCore package (available on https://www.bioconductor.org/ repository).
References
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8.
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. The EMBO journal. 2017;36:1811–36.
Mizushima N. The ATG conjugation systems in autophagy. Current Opinion Cell Biol. 2019;63:1–10.
Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26.
Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222.
Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–80.
Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19:579–93.
Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42.
Morishita H, Mizushima N. Diverse cellular roles of autophagy. Annual Rev Cell Dev Biol. 2019;35:453–75.
Sales-Campos H, Souza PR, Peghini BC, da Silva JS, Cardoso CR. An overview of the modulatory effects of oleic acid in health and disease. Mini Rev Med Chem. 2013;13:201–10.
Widmer RJ, Flammer AJ, Lerman LO, Lerman A. The Mediterranean diet, its components, and cardiovascular disease. Am J Med. 2015;128:229–38.
Niso-Santano M, Malik SA, Pietrocola F, Bravo-San Pedro JM, Marino G, Cianfanelli V, et al. Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 2015;34:1025–41.
Sauvat A, Chen G, Muller K, Tong M, Aprahamian F, Durand S, et al. Trans-fats inhibit autophagy induced by saturated fatty acids. EBioMed. 2018;30:261–72.
Sundqvist M, Christenson K, Holdfeldt A, Gabl M, Martensson J, Bjorkman L, et al. Similarities and differences between the responses induced in human phagocytes through activation of the medium chain fatty acid receptor GPR84 and the short chain fatty acid receptor FFA2R. Biochim et biophys acta Mol Cell Res. 2018;1865:695–708.
Bolognini D, Barki N, Butcher AJ, Hudson BD, Sergeev E, Molloy C, et al. Chemogenetics defines receptor-mediated functions of short chain free fatty acids. Nat Chem Biol. 2019;15:489–98.
Luckmann M, Trauelsen M, Frimurer TM, Schwartz TW. Structural basis for GPCR signaling by small polar versus large lipid metabolites-discovery of non-metabolite ligands. Current Opinion Cell Biol. 2020;63:38–48.
de Souza RJ, Mente A, Maroleanu A, Cozma AI, Ha V, Kishibe T, et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: systematic review and meta-analysis of observational studies. Bmj. 2015;351:h3978.
Wang DD, Hu FB. Dietary fat and risk of cardiovascular disease: recent controversies and advances. Annual Rev Nutr. 2017;37:423–46.
Corella D, Coltell O, Macian F, Ordovas JM. Advances in understanding the molecular basis of the mediterranean diet effect. Annual Rev Food Sci Techn. 2018;9:227–49.
Enot DP, Niso-Santano M, Durand S, Chery A, Pietrocola F, Vacchelli E, et al. Metabolomic analyses reveal that anti-aging metabolites are depleted by palmitate but increased by oleate in vivo. Cell Cycle. 2015;14:2399–407.
Bravo-San Pedro JM, Pietrocola F, Sica V, Izzo V, Sauvat A, Kepp O, et al. High-throughput quantification of GFP-LC3(+) dots by automated fluorescence microscopy. Methods in enzymology. 2017;587:71–86.
Linders PT, Horst CV, Beest MT, van den Bogaart G. Stx5-Mediated ER-Golgi transport in mammals and yeast. Cells. 2019;8.
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–33.
Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, et al. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004;23:169–79.
Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol cell. 2010;40:280–93.
Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17:1374–95.
Heal R, McGivan J. Induction of calreticulin expression in response to amino acid deprivation in Chinese hamster ovary cells. Biochem J. 1998;329(Pt 2):389–94.
Humeau J, Sauvat A, Cerrato G, Xie W, Loos F, Iannantuoni F, et al. Inhibition of transcription by dactinomycin reveals a new characteristic of immunogenic cell stress. EMBO Mol Med. 2020;12:e11622.
Taniuchi S, Miyake M, Tsugawa K, Oyadomari M, Oyadomari S. Integrated stress response of vertebrates is regulated by four eIF2alpha kinases. Sci Rep. 2016;6:32886.
Sauvat A, Leduc M, Muller K, Kepp O, Kroemer G. ColocalizR: An open-source application for cell-based high-throughput colocalization analysis. Comp Biol Med. 2019;107:227–34.
Lafay F. Envelope proteins of vesicular stomatitis virus: effect of temperature-sensitive mutations in complementation groups III and V. J Virol. 1974;14:1220–8.
Coria AS, Masseroni ML, Diaz, Anel AM. Regulation of PKD1-mediated Golgi to cell surface trafficking by Galphaq subunits. Biol Cell. 2014;106:30–43.
Boncompain G, Divoux S, Gareil N, de Forges H, Lescure A, Latreche L, et al. Synchronization of secretory protein traffic in populations of cells. Nat Meth. 2012;9:493–8.
Loos F, Xie W, Sica V, Bravo-San Pedro JM, Souquere S, Pierron G, et al. Artificial tethering of LC3 or p62 to organelles is not sufficient to trigger autophagy. Cell Death Dis. 2019;10:771.
Zhao L, Liu P, Boncompain G, Loos F, Lachkar S, Bezu L, et al. Identification of pharmacological inhibitors of conventional protein secretion. Sci Rep. 2018;8:14966.
Zhou H, Sauvat A, Gomes-da-Silva LC, Durand S, Forveille S, Iribarren K, et al. The oncolytic compound LTX-401 targets the Golgi apparatus. Cell Death Differ. 2018;25:227–8.
Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Nat Acad Sci USA. 2002;99:190–5.
Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell. 2012;48:667–80.
Humeau J, Leduc M, Cerrato G, Loos F, Kepp O, Kroemer G. Phosphorylation of eukaryotic initiation factor-2α (eIF2α) in autophagy. 2020;11:433.
Geatti O, Shapiro B, Barillari B. Scintigraphic depiction of an insulinoma by I-131 metaiodobenzylguanidine. Clinical nuclear medicine. 1989;14:903–5.
Ha KD, Clarke BA, Brown WJ. Regulation of the Golgi complex by phospholipid remodeling enzymes. Biochimica et biophysica acta. 2012;1821:1078–88.
Casares D, Escriba PV, Rossello CA. Membrane lipid composition: effect on membrane and organelle structure, function and compartmentalization and therapeutic avenues. Int J Mol Sci. 2019;20.
Glaumann H, Motakefi AM, Jansson H. Intracellular distribution and effect of the antimalarial drug mefloquine on lysosomes of rat liver. Liver. 1992;12(4 Pt 1):183–90.
Parks A, Charest-Morin X, Boivin-Welch M, Bouthillier J, Marceau F. Autophagic flux inhibition and lysosomogenesis ensuing cellular capture and retention of the cationic drug quinacrine in murine models. PeerJ. 2015;3:e1314.
Hamaguchi M, Suzuki K, Fujita H, Uzuka T, Matsuda H, Shishido-Hara Y, et al. Successful treatment of non-HIV progressive multifocal leukoencephalopathy: case report and literature review. J Neurol. 2020;267:731–8.
Ugarte A, Danza A, Ruiz-Irastorza G. Glucocorticoids and antimalarials in systemic lupus erythematosus: an update and future directions. Current Opinion Rheumatol. 2018;30:482–9.
Ram A, Mabalirajan U, Singh SK, Singh VP, Ghosh B. Mepacrine alleviates airway hyperresponsiveness and airway inflammation in a mouse model of asthma. Int Immunopharmacol. 2008;8:893–9.
Alves P, Bashir MM, Wysocka M, Zeidi M, Feng R, Werth VP. Quinacrine suppresses tumor necrosis factor-alpha and IFN-alpha in Dermatomyositis and cutaneous lupus erythematosus. J Invest Dermatol Symp Proc. 2017;18:S57–S63.
Thase ME, Denko T. Pharmacotherapy of mood disorders. Annual Rev Clin Psych. 2008;4:53–91.
Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet. 2018;391:1357–66.
Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30:1147–58.
Chen G, Xie W, Nah J, Sauvat A, Liu P, Pietrocola F, et al. 3,4-Dimethoxychalcone induces autophagy through activation of the transcription factors TFE3 and TFEB. EMBO Mol Med. 2019;11:e10469.
Sauvat A, Cerrato G. Whole Genome Screenings results. figshare. Dataset. 2020. https://doi.org/10.6084/m9.figshare.13227686.
Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Zhou Z, et al. PubChem’s BioAssay Database. Nucleic Acids Res. 2012;40:D400–12.
Liu P, Zhao L, Loos F, Marty C, Xie W, Martins I, et al. Immunosuppression by mutated calreticulin released from malignant cells. Mol Cell. 2020;77:748–60 e9.
Acknowledgements
GC is supported by a scholarship of the Fondation pour la Recherche Médicale (FRM FDT202001011060). GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR)—Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancellerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085 and GDW20181100051), Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM).
Author information
Authors and Affiliations
Contributions
Conceptualization, GK, AS, and GC; Methodology, GK, AS, GC, JH, ML, and PL; Investigation, GC, KM, PL, LZ, WX, and SZ; Formal analysis, AS, GC, and ML; Writing—Original Draft, GK, GC, and AS; Writing—Review & Editing, GK, GC, OK, and JH; Funding acquisition, GK; Supervision, OK and GK.
Corresponding authors
Ethics declarations
Conflict of interest
GK and OK are cofounders of Samsara Therapeutics. GK is a founder of everImmune and Therafast Bio.
Ethics
Animal experiments were conducted in compliance with the EU Directive 63/2010/EU from the European Parliament and were approved by the local Ethics Committee of the Gustave Roussy Cancer Campus (CEEA IRCIV/IGR no. 26, registered at the French Ministry of Research.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Piacentini
Supplementary information
Rights and permissions
About this article

Cite this article
Cerrato, G., Leduc, M., Müller, K. et al. Oleate-induced aggregation of LC3 at the trans-Golgi network is linked to a protein trafficking blockade. Cell Death Differ 28, 1733–1752 (2021). https://doi.org/10.1038/s41418-020-00699-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00699-3
This article is cited by
-
A non-canonical role of ATG8 in Golgi recovery from heat stress in plants
Nature Plants (2023)
-
Everolimus and plicamycin specifically target chemoresistant colorectal cancer cells of the CMS4 subtype
Cell Death & Disease (2021)
