
Abstract
Cancer cells reprogram their energy metabolic system from the mitochondrial oxidative phosphorylation (OXPHOS) pathway to a glucose-dependent aerobic glycolysis pathway. This metabolic reprogramming phenomenon is known as the Warburg effect, a significant hallmark of cancer. However, the detailed mechanisms underlying this event or triggering this reprogramming remain largely unclear. Here, we found that histone H2B monoubiquitination (H2Bub1) negatively regulates the Warburg effect and tumorigenesis in human lung cancer cells (H1299 and A549 cell lines) likely through controlling the expression of multiple mitochondrial respiratory genes, which are essential for OXPHOS. Moreover, our work also suggested that pyruvate kinase M2 (PKM2), the rate-limiting enzyme of glycolysis, can directly interact with H2B in vivo and in vitro and negatively regulate the level of H2Bub1. The inhibition of cell proliferation and nude mice xenograft of human lung cancer cells induced by PKM2 knockdown can be partially rescued through lowering H2Bub1 levels, which indicates that the oncogenic function of PKM2 is achieved, at least partially, through the control of H2Bub1. Furthermore, PKM2 and H2Bub1 levels are negatively correlated in cancer specimens. Therefore, these findings not only provide a novel mechanism triggering the Warburg effect that is mediated through an epigenetic pathway (H2Bub1) but also reveal a novel metabolic regulator (PKM2) for the epigenetic mark H2Bub1. Thus, the PKM2-H2Bub1 axis may become a promising cancer therapeutic target.
Similar content being viewed by others

Introduction
Monoubiquitinated histone H2B (H2Bub1), catalyzed by the E2 ubiquitin-conjugating protein RAD6 (also named ubiquitin-conjugating enzyme E2A or E2B, UBE2A/B)-E3 ubiquitin-protein ligase BRE1 (also named ring finger protein 20 or 40, RNF20/40) ubiquitination machinery [1, 2], is a key epigenetic modification that plays significant roles in various biological processes. For instance, we and others found that H2Bub1 is essential for embryonic stem cell (ESCs) differentiation [3,4,5]. Recently, our group also determined that H2Bub1 is a key epigenetic switch in the regulation of autophagy activity [6, 7]. In addition, increasing studies indicated that H2Bub1 also plays significant roles in cancer development [8,9,10,11,12]. However, more investigations are still required to dissect the detailed mechanisms that how H2Bub1 participates in the control of tumorigenesis.
In the 1920s, Otto Warburg and colleagues found that unlike normal tissues, tumors show an elevated rate of glucose uptake and an increased amount of lactate production. In addition, even in the presence of oxygen, glucose can be fermented to produce lactate. This phenomenon is known as aerobic glycolysis or the Warburg effect [13, 14]. Compared with the amount of ATP generated through mitochondrial respiration, aerobic glycolysis is a less sufficient way to generate ATP. However, the Warburg effect benefits tumor cells in several aspects, such as results in rapid production of ATP [13, 15], contributes to biosynthetic processes [13, 16, 17], regulates tumor microenvironment [13, 18,19,20], and cancer cell signaling [13].
Pyruvate kinase, the final rate-limiting enzyme of glycolysis, catalyzes the conversion of phosphoenolpyruvate to pyruvate and yields one molecule of ATP [14]. There are four isoforms (M1, M2, L, and R) of pyruvate kinase in mammals, and they are differentially expressed in a spatial and temporal-specific manner. For instance, in humans, pyruvate kinase M2 (PKM2) is highly expressed in fetal tissues and progressively replaced by the other isoforms during development. In human cancer cells, the expression of PKM2 is substantially increased, indicating that PKM2 plays a key role in tumorigenesis [14]. In addition to PKM2’s canonical roles in glycolysis control, increasing evidence indicates that PKM2 may contribute to tumorigenesis through multiple nonmetabolic pathways. For example, PKM2 plays significant roles in the regulation of cancer-related gene expression [21, 22]. Moreover, recent studies showed that PKM2 likely possesses intrinsic protein kinase activity. The protein kinase activity of PKM2 may be essential for cancer development [14, 23,24,25,26,27], but this idea remains controversial [28, 29].
In the past decades, several hypotheses have been proposed to interpret the mechanisms underlying the Warburg effect. For example, Warburg originally proposed that mitochondrial dysfunction is the root of aerobic glycolysis and the primary cause of cancer [13, 30]. However, accumulating data suggested that multiple cancer cells possess normal mitochondria and show active respiratory capability [13]. In addition, others suspected that glycolysis may inhibit oxidative phosphorylation (OXPHOS) during cancer development [31,32,33,34]. However, lack of direct supports for this hypothesis. Therefore, although the Warburg effect has been extensively investigated, the detailed mechanisms triggering the Warburg effect during tumorigenesis remain unclear.
In this report, we show that H2Bub1 negatively regulates the Warburg effect and tumor cell growth in human lung cancer cells, likely through controlling the expression of a set of mitochondrial respiratory genes, which are essential for OXPHOS. In addition, PKM2, which contributes to the Warburg effect [14, 35, 36], negatively regulates the levels of H2Bub1. Detailed analyses revealed that PKM2 directly interacts with histone H2B and inhibits the access of the H2Bub1-specific ubiquitination machinery to H2B, resulting in the decrease of H2Bub1. Furthermore, our results also indicated that PKM2 likely controls the Warburg effect and tumorigenesis, at least partially, through the H2Bub1 pathway. In addition, the levels of PKM2 and H2Bub1 are negatively correlated in clinical specimens. The expression of PKM2 is significantly increased, whereas the levels of H2Bub1are strikingly decreased in cancerous tissues. Our results highlight a novel mechanism triggering the Warburg effect that is mediated through an epigenetic pathway (H2Bub1) and a novel metabolic regulator (PKM2) for the epigenetic mark H2Bub1. These findings have implications for better understanding of the role of epigenetic in Warburg effect. In addition, the PKM2-H2Bub1 axis may become a promising cancer therapeutic target.
Results
Loss of H2Bub1 promotes the Warburg effect and tumorigenesis
To understand whether histone modifications directly regulate the Warburg effect, we performed a small-scale screen with histone modification site-specific K to R mutants. Surprisingly, we found that the K120R mutant of histone H2B significantly affects the Warburg effect, suggesting that H2Bub1 may participate in the regulation of the Warburg effect and tumorigenesis. As expected, H2Bub1 was efficiently depleted with two methods, K120R mutation and RNF20 knockdown (Figs. 1a and S1a), and we found that loss of H2Bub1 results in a significant increase in lactate production, a typical indicator of the Warburg effect [13] (~0.4 fold increase in H2BK120R cells and approximately onefold (in H1299 cells) or ~0.25 fold (in A549 cells) increase in RNF20 knockdown cells, Figs. 1b Fig. S1b). Consistent with its effect on lactate production, depletion of H2Bub1 also results in an acidic environment (Figs. 1c and S1c).

Loss of H2Bub1 promotes the Warburg effect and tumor cell growth. a Left: H1299 cells were transfected with GFP-tagged wild-type H2B (GFP-H2BWT) or GFP-tagged K120R-mutated H2B (GFP-H2BK120R) for 48 h. Cells were then lysed and subjected to Western blot analysis with antibodies as indicated. Right: H1299 cells were transfected with control shRNA (shCont.) or RNF20-specific shRNA (shRNF20) for 48 h. Cells extracts were then prepared and subjected to Western blot analysis with antibodies as indicated. b H1299 cells transfected with GFP-tagged wild-type H2B plasmid (GFP-H2BWT) or GFP-tagged K120R-mutated H2B plasmid (GFP-H2BK120R) and control shRNA (shCont.) or RNF20-specific shRNA (shRNF20) were prepared. The cells (2 × 104) were then seeded into a 96-well plate, and the culture medium from each well was harvested to assess the levels of lactate production. c H1299 cells were transfected with HA-tagged wild-type H2B (H2BWT) or HA-tagged K120R-mutated H2B (H2BK120R) and control shRNA (shCont.) or RNF20-specific shRNA (shRNF20) for 72 h. The cells were then photographed, and the pH of the culture medium was determined. d H1299 cells (1 × 105) transfected with HA-tagged wild-type H2B plasmid (HA-H2BWT) or K120R-mutated H2B plasmid (HA-H2BK120R) were seeded into 96-well plates. After 12 h, oxygen consumption levels were assessed by the time-resolved fluorescence (TR-F) measurement. Values are expressed as the slope of RFU/min. e H1299 cells transfected with GFP-tagged wild-type H2B (GFP-H2BWT) or GFP-tagged K120R-mutated H2B (GFP-H2BK120R) were prepared. The cells (1 × 104) were then seeded into a 96-well plate in the presence or absence of 100 ng/ml oligomycin for 1 h, and the luminescence signals of each well were assessed to indicate the inhibition of intracellular ATP by oligomycin. Note that oligomycin, a specific inhibitor of mitochondrial ATP synthase, decreases oxidative phosphorylation. f Equal amounts (5 × 104) of H1299 cells transfected with GFP-tagged wild-type H2B (GFP-H2BWT), K120R-mutated H2B (GFP-H2BK120R), control siRNA (siCont.), or RNF20-specific siRNA (siRNF20) were plated and counted continuously for 4 days. g Soft agar anchorage-independent growth assay was performed upon H2Bub1 depletion in both A549 and H1299 cells (detailedly described in figure legends of Fig. S1k), and the relative number and diameter of the colonies were quantified. h In vivo tumor growth analysis was performed with H1299 cells stably transfected with GFP-tagged wild-type H2B (H2BWT) or GFP-tagged K120R-mutated H2B (H2BK120R) in nude mice. n = 7 mice in each group. The tumor weights are shown blow (one column only represents the weight of one tumor). The results of f are shown as mean ± S.D., and the others are shown as the mean with 95% CI. *p < 0.05; **p < 0.01; ***p < 0.001
As OXPHOS activity is usually impaired when the Warburg effect occurs. We therefore examined the levels of oxygen consumption upon H2Bub1 depletion, which is an indicator of the OXPHOS activity [37]. Consistently, we found that loss of H2Bub1 shows a statistically significant decrease in oxygen consumption (~30% decrease in H2BK120R cells, Figs. 1d and S1d). As mentioned above, the energy providing system is adapted from OXPHOS to glycolysis to promote rapid growth of cancer cells. Therefore, the dependency on OXPHOS pathway for ATP production is decreased when the Warburg effect occurs. We then analyzed the intracellular ATP concentration in the treatment of oligomycin, a specific inhibitor of mitochondrial ATP synthase. Our results suggested that treatment with oligmycin led to a significant decrease in the intracellular ATP concentration of control cells compared with that in H2Bub1-depleted cells (Fig. 1e). This result suggested that control cells rely more on OXPHOS for ATP generation, while H2Bub1-depleted cells rely less. In order to verify our results, we also repeated the above experiments including lactate production, glucose uptake, and inhibition of ATP production by oligomycin using the K120A and K120M mutants of H2B, and consistent results were obtained (Fig. S1e–i).
As the Warburg effect is a well-known hallmark of cancer [13, 16, 17], we therefore examined the effect of H2Bub1 on cancer cell growth. Our in vitro examination (cell counting assay and soft agar anchorage-independent growth assay) revealed that, consistent with its effect on the Warburg effect, loss of H2Bub1 significantly promotes the proliferative ability of H1299 and A549 cells (Figs. 1f, S1j and S2a, b), and the number and diameter of the colonies are both increased in H2Bub1-depleted cells (the number is nearly onefold increased in H2BK120R cells, Figs. 1g and S1k). We next performed xenograft assay with nude mice to determine the effect of H2Bub1 on tumorigenesis in vivo. Our results also showed that loss of H2Bub1 significantly promotes the abilities of cells to produce xenograft, which is similar as the in vitro data (Fig. 1h). Together, these observations indicated that H2Bub1 is likely a negative regulator of the Warburg effect and tumorigenesis.
H2Bub1 regulates the expression of a group of mitochondrial respiratory genes
To determine the biological roles of H2Bub1, we previously performed a microarray analysis following loss of H2Bub1 in H2BK120R mutated cells [38]. Gene Set Enrichment Assay (GSEA) of the reported microarray data indicated that a group of mitochondrial respiratory genes are enriched in H2Bub1-affected genes, and the expression of most of these genes is decreased in H2Bub1-depleted cells (H2BK120R) [38]. Here, we selected the affected mitochondrial respiratory genes and listed (Fig. 2a), and RT-PCR validation of these genes in response to H2Bub1 depletion was performed. We found that the expressions of most of the selected genes are decreased upon loss of H2Bub1, except that of OXA1L, SDHA, UQCRC1, NDUFS7, NDUFA9, UQCRB in H2BK120R cells and OXA1L, SDHA, UQCRC1, NDUFS7 in RNF20 knockdown cells, suggesting that not all the mitochondria respiratory genes are regulated by H2Bub1, and other mechanisms may be involved (Fig. 2b, c). We also selected some genes, and examined their protein levels upon loss of H2Bub1. Consistent with the effect on the mRNA levels, loss of H2Bub1 downregulates the protein levels of most of the selected mitochondria genes (Fig. 2d).

H2Bub1 regulates a group of mitochondrial respiratory genes. a Microarray analysis was previously reported in HEK293T cells transfected with Myc-tagged H2BK120R plasmid or with empty Myc plasmid (Cont.) to predict the downstream targets of H2Bub1 [38]. A group of genes encoding the mitochondrial respiratory chain were reported to be affected following H2BK120R overexpression [38]. The affected genes are listed. b, c Experimental validation (RT-PCR analysis) of the H2Bub1-affected genes listed in a. H1299 cells transfected with GFP-tagged wild-type H2B (H2BWT-GFP) or K120R-mutated H2B (H2BK120R-GFP) and control siRNA (siCont.) or RNF20-specific siRNA (siRNF20) were lysed and subjected to RT-PCR analysis. d Western Blot analysis of the selected H2Bub1-affected genes and the related quantitative analysis of protein expression levels. H1299 or A549 cells transfected with control siRNA (siCont.) or RNF20-specific siRNA (siRNF20) were lysed and subjected to western blot analysis with antibodies as indicated. e Chromatin immunoprecipitation (Ch-IP) analysis was performed with anti-H2Bub1 antibody or normal mouse IgG (NIgG). Two intergenic regions were used as negative controls. f Chromatin immunoprecipitation (Ch-IP) analysis was performed with anti-Pol-II antibody or normal rabbit IgG (NIgG). An intergenic region was used as a negative control. The results of d are shown the mean with 95% CI, and the others are shown as mean ± S.D. n = 3 per group. *p < 0.05; **p < 0.01; ***p < 0.001
Next, to determine whether H2Bub1 directly regulates the expression of these genes, a chromatin immunoprecipitation (Ch-IP) assay was performed with a H2Bub1 antibody. The result indicated that H2Bub1 is enriched in the regulatory regions (~300 bp downstream regions from transcriptional start site) of all the selected mitochondrial respiratory genes (Fig. 2e). Moreover, to demonstrate that these genes are transcriptionally activated, we also performed a Ch-IP assay with RNA polymerase II (Pol-II) antibodies. As expected, our results suggested that these genes are indeed transcriptionally activated in normal cells (Fig. 2f).
PKM2 negatively regulates H2Bub1 levels
Because PKM2 is the most important regulator of the Warburg effect [13, 14, 39,40,41,42] and directly phosphorylate histone H3 at T11 [14, 43], although it is still controversial [28, 29], we examined whether PKM2 regulates H2Bub1 levels in human cells. To our surprise, western blot analysis showed that overexpression of Flag-tagged PKM2 in multiple cell lines severely downregulates H2Bub1 levels (Fig. 3a). Consistent with the effect of PKM2 overexpression on H2Bub1 levels, depletion of PKM2 by PKM2-specific shRNA or siRNA [14, 22, 42] upregulates the levels of H2Bub1 (Fig. 3b). In addition, we overexpressed HA-tagged PKM1, another isoform of pyruvate kinase, at an increasing dosage in H1299 cells. However, there is no obvious effect on H2Bub1 levels, suggesting a PKM2-specific role in the control of H2Bub1 (Fig. 3c). Moreover, the pyruvate kinase activity of PKM2 is essential for its impact on H2Bub1; unlike wild-type (WT) PKM2, the pyruvate kinase-dead (KD) mutant of PKM2 (K367M) [14, 22] does not show obvious effects on H2Bubl levels (Fig. 3d). These data suggested that PKM2 is likely a novel negative regulator of H2Bub1 in human cells.

PKM2 negatively regulates the levels of H2Bub1. a H1299, HEK293T, and HeLa cells were transfected with or without (-) Flag-tagged PKM2 at an increasing dosage for 48 h. Western blot analysis was then performed. b H1299, A549, and HEK293T cells were transfected with control or PKM2-specific shRNA or siRNA as indicated [14, 22, 42] for 48 h, followed by western blot analysis. c H1299 cells were transfected with or without (-) HA-tagged PKM1 at an increasing dosage for 48 h. Western blot analysis was then performed with antibodies as indicated. d H1299 cells were transfected with Flag-tagged wild-type PKM2 (Flag-PKM2-WT), pyruvate kinase-dead mutant PKM2 (Flag-PKM2-KD (K367M)), or Flag empty vector (Cont.) for 48 h. Western blot analysis was then performed using antibodies as indicated. e H1299 cells were transfected with control (shCont.) or PKM2-specific (shPKM2) shRNA for 48 h, followed by western blot analysis. All the statistical analyses of western blot are presented by mean with 95% CI. *p < 0.05, **p < 0.01, ***p < 0.001
PKM2 is unlikely to affect the ubiquitination components essential for establishing H2Bub1, including RNF20, RNF40, and RAD6, as no obvious changes in the protein levels of these proteins occur following PKM2 depletion (Fig. 3e), indicating that other mechanisms may be involved in the regulation of H2Bub1 by PKM2.
PKM2 interacts with H2B in vivo and in vitro
To understand the mechanisms of how PKM2 regulates H2Bub1 levels, we first determined whether PKM2 interacts with H2B by a co-immunoprecipitation (Co-IP) assay. We expressed Flag-tagged PKM2 and GFP-tagged H2B in H1299 cells and performed Co-IP assays. To our surprise, we found that Flag-tagged PKM2 indeed associates with GFP-tagged H2B in vivo (Fig. 4a). Furthermore, Flag-tagged PKM2 and endogenous PKM2 all associate with endogenous H2B in human cells (Figs. 4b and S3a), but not with H2Bub1 (Fig. S3b). As histone H3 was reported to be a PKM2-binding histone [14], here H3 was used as positive control for our Co-IP assay (Figs. 4b and S3b). In addition, we purified GST-tagged H2B and His-tagged PKM2 in Escherichia coli (Fig. S3c, d) and performed GST pull-down analyses. Our result suggested that PKM2 directly interacts with H2B in vitro (Figs. 4c and S3e). Besides, to visualize the in situ subcellular interaction between them, Duolink proximity ligation assay was performed in H1299 cells and A549 cells. The results showed that PKM2 do interact with H2B and the signals of protein–protein interaction couples are almost in the nucleus in vivo. (Fig. 4d). To further support the interaction between PKM2 and H2B in the nucleus, we co-transfected H1299 cells with Flag-tagged PKM2 and GFP-tagged H2B and performed immunofluorescence (IF) analysis. Consistently, a weak PKM2 signal is observed in the nucleus where H2B is mainly localized (Fig. 4e). Consistent with the proximity ligation assay and IF results, a few amount of PKM2 indeed distributes in the nucleus, although most are distributed in the cytoplasm (Fig. 4f, left). However, the interaction between PKM2 and H2B occurs only in the nucleus (Fig. 4f, right). Moreover, the KD mutant of PKM2 (K367M) does not interact with H2B in human cells (Fig. 4g), consistent with the above observation that PKM2-KD does not affect H2Bub1 levels (Fig. 3d). In addition, PKM2 does not interact with the H2Bub1-specific ubiquitination components RAD6 and RNF20 in vivo (Fig. 4h), further supporting that the regulation of H2Bub1 by PKM2 may be achieved through the interaction between PKM2 and H2B.

PKM2 interacts with histone H2B. a H1299 cells were transfected with Flag-tagged PKM2 and GFP-tagged H2B for 48 h. The cells were lysed and subjected to co-immunoprecipitation (Co-IP) analysis. Normal IgG (NIgG) was used as a negative control. b H1299 cells transfected with (upper) or without (middle and lower) Flag-tagged PKM2 were lysed and subjected to Co-IP analysis with an antibody against Flag (upper), H2B (middle), or PKM2 (lower) followed by western blot analysis with antibodies as indicated. Normal IgG (NIgG) was used as a negative control. c His-tagged PKM2, GST-tagged H2B, and GST were purified and subjected to Ni-Bead pull-down (left) or GST pull-down (right) analysis. d Duolink proximity ligation assay using antibodies against H2B (mouse) and PKM2 (rabbit) was performed in H1299 and A549 cells. Red spots correspond protein–protein interaction couples. Blue: DAPI-stained nuclei. The control shown represents the omission of the primary antibody against PKM2. e H1299 cells were transfected with Flag-tagged PKM2 and GFP-tagged H2B for 48 h, followed by immunofluorescence (IF) analysis. f Cytoplasmic and nuclear fractions of H1299 cells were separately prepared. Western blot analysis was then performed with antibodies as indicated (left). H1299 cells were co-transfected with Flag-tagged PKM2 and GFP-tagged H2B for 48 h. Cytoplasmic and nuclear fractions were then prepared and subjected to Co-IP analysis with anti-GFP antibody followed by western blot analysis with antibodies as indicated (right). Normal IgG (NIgG) was used as a negative control. g H1299 cells co-transfected with Flag-tagged pyruvate kinase-dead mutant of PKM2 (Flag-PKM2-KD (K367M)) and GFP-tagged H2B were harvested and subjected to Co-IP analysis with an antibody against Flag followed by western blot analysis with antibodies as indicated. Normal IgG (NIgG) was used as a negative control. h H1299 cells transfected with Flag-tagged PKM2 were lysed and subjected to Co-IP analysis with an antibody against Flag followed by western blot analysis with antibodies as indicated. Normal IgG (NIgG) was used as a negative control
To further support the interaction between PKM2 and H2B, we performed molecular docking analysis with the structures of PKM2 (PDB#: 4FXJ) and H2B (PDB#: 2RVQ). Our analysis indicated the presence of a groove in the PKM2 protein that nicely accommodates the N-terminal region (amino acids about 9–20) of the H2B protein (Fig. 5a). In addition, a bud jutting out the PKM2 protein likely blocks the K120 site of H2B, suggesting a potential mechanism for the regulation of H2Bub1 by PKM2 (Fig. 5a). Molecular docking analysis showing the association between PKM2 and H2B under nucleosome conditions is shown in Fig. 5b. Upon focusing the interface of PKM2 and H2B binding region, we found that there is a “U-shaped” groove containing three acidic amino acids (E396, E397, and E418) in the groove region of PKM2, while the N-terminal region of H2B contains four basic amino acid (K11, K12, K15, and K16). These two groups of amino acids are close to each other and may form a lump of electrostatic interaction that may well account for the structure basis for the PKM2–H2B interaction (Fig. 5c). In addition, we found that the K367 site of PKM2 locates nearly to the “U-shaped” groove (Fig. 5c). Therefore, the K367M mutation of PKM2 may affect the structure of PKM2-H2B interaction region, providing a possible explanation for why PKM2-KD mutant does not interact with H2B (Fig. 4g).

Molecular docking analysis of the interaction between PKM2 and H2B. a Molecular docking analysis of PKM2-H2B interaction. The structures of PKM2 (PDB#:4FXJ) and H2B (PDB#:2RVQ) were used for molecular docking analysis, and the image was visualized with PyMOL software. b Molecular docking analyses of PKM2-H2B interaction under nucleosome conditions are shown. c Detailed analysis of the interface region of PKM2–H2B interaction
PKM2 disrupts the association of the RNF20-RAD6 ubiquitination machinery with H2B
To understand the detailed mechanism of how PKM2 regulates H2Bub1, we next tested whether PKM2 serves as a protein kinase for histone H2B, As previous studies reported that PKM2 possesses protein kinase activity [14, 23,24,25,26,27, 43] although it remains controversial [28, 29]. However, our in vitro phosphorylation assay indicated that PKM2 failed to directly phosphorylate H2B (Fig. S4a, b).
Together with the findings that PKM2 directly interacts with H2B (Figs. 4c and S3e), and may structurally block K120 site of H2B (Fig. 5a), we wondered whether the binding of PKM2 to H2B disrupts the association of H2Bub1-specific ubiquitination machinery with H2B. As expected, PKM2 overexpression results in a decreased association of the RNF20-RAD6 ubiquitination machinery with H2B (Fig. 6a, left and middle). By contrast, depletion of PKM2 leads to an increased association between the RNF20-RAD6 machinery and H2B (Fig. 6b). However, PKM2 is unlikely to affect the intrinsic organization of the H2Bub1-specific ubiquitination machinery as PKM2 overexpression shows no obvious effect on the association between RNF20 and RAD6 (Fig. 6a, middle). Together, these findings suggested that the regulation of H2Bub1 by PKM2 is likely achieved through limiting the access of the H2Bub1-specific ubiquitination machinery to histone H2B. In addition, the KD mutant of PKM2 (K367M) overexpression failed to change the association of the RNF20-RAD6 ubiquitination machinery with H2B (Fig. 6a, right), consistent with the above observation that PKM2-KD does not interact with H2B and affect H2Bub1 levels (Figs. 3d and 4g).

PKM2 disrupts the access of the RNF20-RAD6 ubiquitination machinery to H2B. a GFP-tagged H2B transfected H1299 cells were co-transfected with or without (−) Flag-tagged PKM2 (left and middle) or Flag-tagged KD mutant of PKM2 (K367M) (right) at an increasing dosage for 48 h. The cells were then lysed and subjected to Co-IP analysis. Normal IgG (NIgG) was used as a negative control. b H1299 and A549 cells were transfected with control or PKM2-specific shRNA or siRNA as indicated for 48 h. The cells were then lysed and subjected to Co-IP analysis with an antibody against H2B followed by western blot analysis with antibodies as indicated. Normal IgG (NIgG) was used as a negative control. c H1299 cells were co-transfected with Flag-tagged PKM2 and GFP-tagged H2B for 48 h. The cells were then treated with 100 ng/mL EGF for the indicated times. Co-IP analysis was performed with anti-GFP antibody followed by western blot analysis with antibodies as indicated. d H1299 cells were treated with 100 ng/mL EGF for the indicated times. The cells were then harvested, and western blot analysis was performed with antibodies as indicated. e H1299 cells transfected with control siRNA (siCont.) or PKM2-specific siRNA (siPKM2) were treated with 100 ng/mL EGF for the indicated times. The cells were then harvested, and western blot analysis was performed with antibodies as indicated. f GFP-H2B transfected H1299 cells were treated with 100 ng/mL EGF for the indicated times. The cells were then lysed and subjected to Co-IP analysis with anti-GFP antibody, and western blot analysis was performed with antibodies as indicated. Normal IgG (NIgG) was used as a negative control
Moreover, consistent with the effect of PKM2 on the levels of H2Bub1, epidermal growth factor (EGF), a reported stimulator of PKM2 nuclear translocation [22, 42], promotes the interaction between PKM2 and H2B in vivo (Fig. 6c). Furthermore, EGF treatment results in a significant decrease in the levels of H2Bub1 in human cells (Fig. 6d), which depends on the existence of PKM2 (Fig. 6e). We next tested whether EGF further affects the association of H2B with its ubiquitination machinery. As expected, our Co-IP result revealed that EGF treatment indeed disrupts the interaction between H2B and its ubiquitination machinery in vivo (Fig. 6f). Together, these results indicated that the regulation of H2Bub1 by PKM2 occurs under relatively physiological conditions.
PKM2 regulates the expression of the same set of mitochondrial respiratory genes and the Warburg effect through H2Bub1
As we observed that PKM2 is a negative regulator of H2Bub1, we examined whether PKM2 regulates the expression of the H2Bub1-regulated mitochondrial respiratory genes. As expected, similar to the effect of loss of H2Bub1, overexpression of PKM2 also decreases the expression of the selected mitochondrial respiratory genes, except that of NDUFS7 and UQCRB (Fig. 7a), suggesting that not all the mitochondria respiratory genes are regulated by PKM2, and other mechanisms may be involved. In addition, our Ch-IP result revealed that overexpression of PKM2 decreases the enrichment of H2Bub1 in the regulatory regions of the selected mitochondrial respiratory genes (Fig. 7b).

PKM2-H2Bub1 axis regulates the Warburg effect. a H1299 cells were transfected with empty Flag plasmid (Cont.) or Flag-tagged PKM2 plasmid for 48 h. The cells were then lysed and subjected to RT-PCR analysis. b H1299 cells transfected with empty Flag plasmid (Cont.) or Flag-tagged PKM2 plasmid were subjected to Ch-IP analysis with an antibody against H2Bub1. n = 3 per group in a, b. c H1299 cells transfected with GFP-tagged wild-type H2B combined with control shRNA (GFP-H2BWT + shCont.), GFP-tagged wild-type H2B together with PKM2-specific shRNA (GFP-H2BWT + shPKM2), or GFP-tagged K120R-mutated H2B together with PKM2-specific shRNA (GFP-H2BK120R + shPKM2) were prepared. Lactate production was assessed. d H1299 cells transfected with HA-tagged wild-type H2B combined with control shRNA (HA-H2BWT + shCont.), HA-tagged wild-type H2B together with PKM2-specific shRNA (HA-H2BWT + shPKM2), or HA-tagged K120R-mutated H2B together with PKM2-specific shRNA (HA-H2BK120R + shPKM2) were subjected to oxygen consumption analysis. e Inhibition of intracellular ATP concentrations by oligomycin was examined in H1299 cells transfected with GFP-tagged wild-type H2B combined with control siRNA (GFP-H2BWT + siCont.), GFP-tagged wild-type H2B together with PKM2-specific siRNA (GFP-H2BWT + siPKM2), or GFP-tagged K120R-mutated H2B together with PKM2-specific siRNA (GFP-H2BK120R + siPKM2). f H1299 cells transfected with HA-tagged wild-type H2B combined with control shRNA (HA-H2BWT + shCont.), HA-tagged wild-type H2B together with PKM2-specific shRNA (HA-H2BWT + shPKM2), or HA-tagged K120R-mutated H2B together with PKM2-specific shRNA (HA-H2BK120R + shPKM2) were prepared. The cells (2 × 104) were seeded into a 96-well plate to assess the levels of glucose uptake. g Working model. We speculated that PKM2 likely negatively regulates the expression of mitochondrial respiratory genes through a H2Bub1-mediated epigenetic pathway and further results in impairment of OXPHOS. Thus, glycolysis may be enhanced to compensate for the defect in OXPHOS, eventually resulting in the Warburg effect. The results of a and b are shown as mean ± S.D., and the others are shown as the mean with 95% CI *p < 0.05; **p < 0.01; ***p < 0.001
We next asked whether PKM2 regulates the Warburg effect via the H2Bub1 pathway. PKM2 has been reported as a positive regulator of the Warburg effect and promotes the production of lactate in human cells [37, 42, 44, 45]. Consistently, we found that the depletion of PKM2 in H1299 cells results in a decreased lactate production (~20% decrease in PKM2-depleted cells, Figs. 7c and S5a, b), and, intriguingly, the inhibition of lactate production by depletion of PKM2 is almost fully rescued by loss of H2Bub1 (Figs. 7c and S5a, b), suggesting that the regulation of lactate production by PKM2 may be achieved through a H2Bub1-mediated pathway. In addition, depletion of PKM2 significantly increases oxygen consumption (~30% increase in PKM2-depleted cells), and this upregulated oxygen consumption can be fully rescued by loss of H2Bub1, suggesting that the regulation of oxygen consumption by PKM2 is achieved via H2Bub1 (Fig. 7d). Moreover, depletion of PKM2 results in a striking increase in the levels of OXPHOS-produced ATP (approximately twofold increase in PKM2-depleted cells), and this effect of PKM2 is also rescued by loss of H2Bub1 (Figs. 7e and S5c). Consistent with the above results, depletion of PKM2 decreases the activity of glucose uptake (~40% decrease in PKM2-depleted cells), and this inhibition is also rescued by loss of H2Bub1 (Fig. 7f). We also performed the related analyses in A549 cells including lactate production and oxygen consumption, and similar conclusions were obtained (Fig. S5d–f). Together, these data indicated that regulation of the Warburg effect by PKM2 is at least partially achieved through H2Bub1.
Based on these data, we speculated that PKM2 likely negatively regulates the expression of mitochondrial respiratory genes through a H2Bub1-mediated epigenetic pathway and further results in impairment of OXPHOS. Thus, glycolysis may be enhanced to compensate for the defect in OXPHOS, eventually resulting in the Warburg effect (Fig. 7g).
PKM2-H2Bub1 axis regulates tumor cell growth
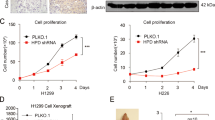
We next examined the significance of the PKM2-H2Bub1 axis in tumor cell growth both in vitro (cell-counting assay) and in vivo (xenograft assay). Our results revealed that depletion of PKM2 decreases the growth of H1299 and A549 cells both in vitro and in vivo, and this inhibition is rescued by loss of H2Bub1 (Figs. 8a, b, S5g and S2c, d), suggesting that the regulation of cell growth by PKM2 may also be achieved at least partially through H2Bub1.

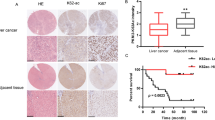
PKM2-H2Bub1 axis regulates tumor cell growth. a Equal amounts (5 × 104) of H1299 cells transfected with GFP-tagged wild-type H2B combined with control shRNA (H2BWT + shCont.), GFP-tagged wild-type H2B together with PKM2-specific shRNA (H2BWT + shPKM2), or GFP-tagged K120R-mutated H2B together with PKM2-specific shRNA (H2BK120R + shPKM2) were plated and counted continuously for 4 days (left). Equal amounts (5 × 104) of H1299 cells transfected with control siRNA (siCont.), PKM2-specific siRNA (siPKM2), or siPKM2 together with RNF20-specific siRNA (siPKM2 + siRNF20) were plated and counted continuously for 4 days (right). The results are shown as mean ± S.D. b In vivo tumor growth analysis was performed with H1299 cells stably transfected with GFP-tagged wild-type H2B combined with control shRNA (shCont. + H2BWT), GFP-tagged wild-type H2B together with PKM2-specific shRNA (shPKM2 + H2BWT), or GFP-tagged K120R-mutated H2B together with PKM2-specific shRNA (shPKM2 + H2BK120R) in nude mice. n = 7 mice in each group. Tumor weights are shown (right, one column only represents the weight of one tumor). c IHC staining with the indicated antibodies was performed on 75 pairs of nonsmall cell lung cancer specimens. Representative photos are shown, and quantification data is shown on the left. d IF (n = 4) and western blot (n = 8) analyses with the indicated antibodies were performed on 12 pairs of breast cancer specimens. Representative photos of IF staining and western blot are shown. e Working model. In addition to the canonical pathway of PKM2 in the regulation of the Warburg effect through functioning as a rate-limiting enzyme of glycolysis, our work demonstrates that a H2Bub1-mediated epigenetic pathway is also employed by PKM2 to control reprogrammed energy metabolism during cancer development
We next performed immunohistochemical (IHC) analysis to examine the in vivo correlation between PKM2 and H2Bub1 in human lung cancer specimens. The results show that the levels of PKM2 and H2Bub1 are negatively correlated. PKM2 is upregulated in nonsmall cell lung cancer (NSCLC) tissues, whereas the levels of H2Bub1 are downregulated (Fig. 8c). Similar results were also observed in human breast cancer tissues as we found that the levels of PKM2 are significantly increased in cancerous tissues, consistent with previous reports [46, 47], whereas the levels of H2Bub1 are strongly decreased (Fig. 8d), suggesting that the interplay of PKM2 and H2Bub1 may be a general event in multiple cancers. Together, these results suggested that the PKM2-H2Bub1 axis likely plays significant roles in tumor development.
Discussion
Typically, the following main hypotheses are proposed to illustrate the underlying mechanisms triggering the Warburg effect: (1) irreversible mitochondrial defects occurring during tumorigenesis and (2) inhibition of the mitochondrial OXPHOS process by aerobic glycolysis in cancer cells [31,32,33,34, 48]. However, imperfections remain in both hypotheses; for example, mitochondrial functions are normal, OXPHOS normally occurs in many cancers, and there is a lack of direct experimental evidence for the second hypothesis [13]. Therefore, the mechanisms triggering the adaption of energy metabolism during tumorigenesis remain poorly understood. In this study, we describe an important and previously unknown mechanism underlying the Warburg effect through a H2Bub1-mediated epigenetic pathway, and this pathway is tightly involved in the function of PKM2 in controlling the Warburg effect and tumorigenesis.
We found that H2Bub1 is required for the expression of mitochondrial respiratory genes (Fig. 2) that are essential for the OXPHOS pathway, the main energy providing system in normal cells. However, the levels of H2Bub1 are decreased in cancer cells (Fig. 8c, d). Therefore, loss of H2Bub1 may epigenetically inactive the expression of mitochondrial respiratory genes, further resulting the inhibition of the OXPHOS pathway. Then, the aerobic glycolysis pathway may be compensatorily employed by cancer cells to produce enough energy to support the survival of tumor cells. In addition, this study indicated that PKM2, a well-known key regulator of glycolysis and the Warburg effect, is a novel negative regulator of H2Bub1 (Fig. 3a, b). Though there is no previous report on the interaction between PKM2 and H2B, our results show that they can interact directly (Figs. 4a–f and S3). In addition, we also predicted the possible structural basis that PKM2 regulates H2Bub1 through molecular docking analysis, and found that PKM2 may structurally block the K120 site of H2B (Fig. 5). Moreover, our Co-IP experiments also confirmed this prediction that PKM2 overexpression indeed inhibits the association of RAD6-BRE1 ubiquitination machinery with H2B (Fig. 6a, b). Therefore, this study provides a possible molecular mechanism for PKM2 to affect the H2Bub1 levels. Consistent to its effect on the levels of H2Bub1, PKM2 also inhibits the expression of mitochondrial respiratory genes likely through a H2Bub1-mediated epigenetic pathway (Fig. 7a, b). Furthermore, our results indicated that the effect of PKM2 on the Warburg effect is also achieved through H2Bub1, at least partially (Fig. 7c–f). Therefore, our present results likely provide significant support for the second hypothesis that key regulators of glycolysis (such as PKM2) indeed show an inhibitory effect on the OXPHOS pathway in human cells. In addition to the canonical roles of PKM2 in regulation of glycolysis and the Warburg effect, our findings together with those of other studies [14, 21,22,23] revealed that epigenetics, as a nuclear event, is also employed by PKM2 in regulation of the Warburg effect and tumorigenesis (Fig. 8e).
Many studies have shown that H2Bub1 is downregulated in cancerous tissues, and the downregulation may favor tumorigenesis [8,9,10,11,12]. The change of H2Bub1 levels in cancer is now mainly explained by the mutation, low expression of E3 ubiquitin–ligase complex RNF20/RNF40 or the overexpression of multiple-deubiquitylating enzymes, including USP22, USP44, and others. Our research may provide a new possibility that the overexpression of PKM2 in cancer may lead to the downregulation of H2Bub1 levels. Furthermore, our clinic-based studies suggested that the levels of PKM2 and H2Bub1 are strongly negatively correlated in human specimens (Fig. 8c, d). Loss of H2Bub1 promotes tumor cell growth both in vitro and in vivo (Fig. 1f–h, and S1j, k), further supporting the tumor suppressor function of H2Bub1. Together, these data indicated that loss of H2Bub1 may be a significant epigenetic mark of tumorigenesis, and the PKM2-H2Bub1 axis may become a promising cancer therapeutic target in the future. In addition, we report in previous articles [6, 7] that starvation-induced downregulation of H2Bub1 levels will induce autophagy, and whether the changes in H2Bub1 levels associated with PKM2 can also induce the changes of autophagy in tumor cells deserve further studies.
Materials and methods
Cell lines culture, RNAi, and vectors
H1299 and A549 human lung cancer cells, HEK293T human embryonic kidney cell and HeLa human cervical carcinoma cells were purchased from Center of Cell Source, Shanghai Institute of Biological Sciences, CAS, China. All the cells were confirmed to have no mycoplasma contamination by the Mycoplasma Detection Kit (Lonza; #LT07-705) once per month. H1299, HEK293T and HeLa cell lines were authenticated using Short Tandem Repeat (STR) analysis by Genetic Testing Biotechnology Corporation (Suzhou, China). These cells were cultured at 37 °C in DMEM (Gibco, #11960-044) supplemented with 10% fetal bovine serum (Gibco, #10099141) and 1% penicillin and streptomycin (Gibco, #15070-063) in a 5% CO2 incubator.
The shRNA against PKM2 [14] was a kind gift from Dr Weiwei Yang from the Shanghai Institute of Biochemistry and Cell Biology, CAS. The GV298 PKM2 shRNA was generated with CATCTACCACTTGCAATTA oligonucleotide. The GV298 RNF20 shRNA was generated with GTGCTGTAACATGCGTAAA oligonucleotide. The siRNA against RNF20 (5′-GCGGCACAAUCACUAUCAATT-3′ and 5′-UUGAUAGUGAUUGUGCCGCTT-3′) and siRNA against PKM2 (5′-GGCUGGACUACAAGAACAUTT-3′ and 5′-AUGUUCUUGUAGUCCAGCCTT-3′) were designed and synthesized by the GenePharm Company (Shanghai, China). The transfection of shRNA or siRNA into cultured cells was performed using Lipofectamine 2000 (Invitrogen, #11668-019) according to the manufacturer’s protocol.
The Flag-tagged PKM2, Flag-tagged PKM2 K367M, HA-tagged PKM1, and His-tagged PKM2 [14] all were kind gifts from Dr Weiwei Yang from the Shanghai Institute of Biochemistry and Cell Biology, CAS. The GFP-tagged H2BWT, GFP-tagged H2BK120R, GFP-tagged H2BK120A, and GFP-tagged H2BK120M were respectively ligated into pEGFP-N1 vector. The GST-tagged H2BWT was ligated into pET-42a(+) vector. The HA-tagged H2BWT and HA-tagged H2BK120R were ligated into pLVX-IRES-ZsGreen1 vector. The transient transfection of the constructs into the cells was performed using Lipofectamine 2000 (Invitrogen, #11668-019), according to the manufacturer’s standard protocol. For creating stable cell lines, HA-tagged H2BWT, HA-tagged H2BK120R (pLVX-IRES-ZsGreen1 vector), shPKM2 (GV298 vector), and the control vector were packaged into Lentivirus by co-transfection with helper vectors (VSV-G, delta8.9) in 293FT cells. Virus titers were measured and stored at ultra-freezer (−80 °C) as aliquots. H1299 cells were infected by the virus and the positive cells were picked up by FACS using ZsGreen or mCherry.
Co-immunoprecipitation (Co-IP)
Cells were transfected with the indicated constructs using Lipofectamine 2000 (Invitrogen, #11668-019). After 48 h, the cells were harvested, washed with ice-cold PBS three times, resuspended in ATM lysis buffer (containing 100 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.2 mM EDTA, 20% glycerol, 0.4% NP-40, 2% Tween-20, and 0.2 mM PMSF) and sonicated on ice ten times (3 s each), with 20% efficiency. Cell lysates were incubated with normal mouse IgG (Santa Cruz Biotechnology, #sc-2025) or normal rabbit IgG (Cell Signaling Technology, #2729 S) as a negative control or the indicated antibodies for immunoprecipitation at 4 °C overnight. Protein A/G agarose beads (Santa Cruz Biotechnology, #sc-2003) were subsequently added and incubated for another 3 h. The solution was centrifuged to harvest the agarose beads after they were washed five times with lysis buffer. Precipitated proteins were released by boiling in loading buffer and resolved via SDS-PAGE. Western blot analyses were performed using related antibodies.
Antibodies and western blot analysis
The anti-H2Bub1 antibody was purchased from Medimabs (#MM-0029). The antibodies against PKM2 (#4053), RAD6 (#4944), RNF20 (#11974), RNF40 (#12187), H2B (#2934), and Pol-II (#14958) were purchased from Cell Signaling Technology. The antibodies against Flag (#M20008), His (#M20001), GST (#M20007), GFP (#M20004), and Actin (#M20011) were purchased from Abmart (Shanghai, China). The antibodies against H2B (#ab52484), H3 (#ab1791), NDUFS1 (#ab169540), NDUFAB1 (#ab181021), NDUFS4 (#ab137064), NDUFA1(#ab176563), NDUFS2 (#ab192022), UQCRH (#ab134949), and SDHA (#ab137040) were purchased from Abcam.
Cells were lysed in ATM lysis buffer (containing 100 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.2 mM EDTA, 20% glycerol, 0.4% NP-40, 2% Tween-20, and 0.2 mM PMSF). The protein concentration in the supernatant was measured using a BCA Assay Kit (Novagen, #71285-3). Next, samples were loaded into a 10% or 15% gel to resolve proteins. Different amounts of total protein were loaded in each experiment to facilitate the detection of different target proteins. After electrophoresis, the proteins were transferred to PVDF membranes (Amersham, #10600021) and hybridized with primary antibodies at a dilution of 1:2000. HRP-labeled secondary antibodies (Zhongshan Golden Bridge, #ZDR-5306 and #ZDR-5307) were applied at a dilution of 1:4000. An ECL detection system (Calbiochem, #345818) was used to detect the signals on the membranes.
All the western blot analyses in this work were repeated more than three times.
RT-PCR assay
Cells were lysed to isolate total RNA using TRIzol reagent (Invitrogen, #15596-026), according to the manufacturer’s instructions. Reverse transcription was performed using a reverse transcription kit (Takara, #2641A). Briefly, total RNA (5 μg) was reverse transcribed to synthesize cDNA in a volume of 20 μL using M-MLV reverse transcriptase. In each 25-μL PCR mixture, 1 μL of cDNA was used for real-time PCR or semiquantitative PCR analyses. For semiquantitative PCR, PCR products were loaded onto a 2% agarose gel, stained with ethidium bromide, imaged, and quantified. Real-time PCR was performed with SYBR Advantage qPCR Premix (Clontech, #639676) on an iQ5 Real-Time PCR System (Bio-Rad). Fold differences in gene expression levels were calculated and normalized against the internal control GAPDH. Primers used were listed in Table S1.
Chromatin immunoprecipitation (Ch-IP) assay
Ch-IP was performed according to the published protocols from Upstate and our previous work [3, 6, 49]. Primers used were listed in Table S2.
Subcellular fractionation
Cytoplasmic and nuclear proteins were extracted using a Nuclear and Cytoplasmic Protein Extraction Kit from Beyotime Biotechnology (#P0027, Jiangsu, China) according to the manufacturer’s instructions.
GST pull-down
GST pull-down analysis was performed as previously described [50]. Briefly, bacterial-expressed His-tagged PKM2 samples were individually mixed with GST-tagged H2B or control GST bound to glutathione-Sepharose 4B for 2 h at 4 °C. After extensive washes, the beads were collected, boiled in SDS sample buffer and separated by SDS-PAGE. Interactions were analyzed by Coomassie brilliant blue staining and western blot analysis.
Immunofluorescence (IF) analysis
IF analysis was performed as previously described [6, 49]. Briefly, cells grown on culture slides (the slides were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature) or paraffin-embedded tissue slides (the slides were boiled at 96 °C for 20 min in sodium citrate for antigen retrieval) were blocked by 2% BSA in PBS and incubated with anti-Flag mouse antibody (1:1000 dilution, Abmart), anti-H2Bub1 rabbit antibody (1:800 dilution, Cell Signaling Technology) or anti-PKM2 rabbit antibody (1:800 dilution, Cell Signaling Technology) overnight at 4 °C. Cy3-coupled or FITC-coupled secondary antibodies (Invitrogen) were applied at a dilution of 1:500, and the DNA-staining marker, DAPI, was used at a concentration of 5 μg/mL. The slides were visualized with an inverted fluorescence microscope (Leica DMI4000B). All studies concerning human tumor specimens were approved by the Ethics Committee of Tongji University School of Medicine and informed consent was obtained from all patients. Our human-subjects study abides by the declaration of Helsinki principles.
Immunohistochemical (IHC) analysis
IHC analysis was performed by Shanghai Outdo Biotech Co., Ltd (HLug-NSCLC150PT-01). Briefly, human NSCLC and adjacent noncancerous tissues were fixed and prepared for staining. Paraffin-embedded tissue sections were deparaffinized, rehydrated, and boiled at 96 °C for 20 min in sodium citrate for antigen retrieval. Endogenous peroxidase activity was blocked by incubation for 5 min in 3% hydrogen peroxide solution. Next, the slides were stained with related antibodies overnight at 4 °C. Diaminobenzidine (DAB) was used to visualize the IHC, and hematoxylin was used as a counterstain.
Duolink Proximity Ligation Assay
The in situ proximity ligation assay was performed using the Duolink® In Situ Orange Starter Kit (Sigma, #DUO92102). In brief, cells were seeded on cover slides. The next day, cells were fixed with 4% Paraformaldehyde, permeabilized with 0.2% Triton X-100, blocked, incubated with primary antibodies against H2B (Abcam, #ab52484), and PKM2 (Cell Signaling Technology, #4053) at 4 °C overnight, and then processed according to the manufacturer’s instructions. The slides were visualized with an inverted fluorescence microscope (Leica DMI4000B). Red spots correspond protein–protein interaction couples.
Cell growth assay
A total of 5 × 104 cells per well were seeded into a six-well plate in DMEM with 10% bovine calf serum and counted continuously for 4 days after seeding.
Soft agar anchorage-independent growth assay
Cells (2 × 103 per well) were mixed with 0.3% agarose in growth medium, plated on top of a solidified layer of 0.6% agarose in growth medium, in a six-well plate, and fed every 3 day with growth medium. After 2 weeks, the colonies were dyed with Cristal Violet (0.005%) for 1 h in 37 °C, washed with PBS, and imaged with a digital camera (olympus E-M5II). The photos were analyzed by photoshop software.
Xenograft studies
Similar age and weight nude mice (BALB/C; male; 5 weeks old) were grouped randomly and related H1299 cell lines were injected subcutaneously into nude mice. Briefly, cells were suspended in PBS containing 10% matrigel (CORNING, #354230) and adjusted the concentration to 1 × 108/ml, then injected cells subcutaneously into nude mice (0.1 ml per mouse). Approximately, 6 weeks after injection, the tumors were dissected and weighed. Sample sizes (n = 7) represent the number of animal and the sizes were selected based on effect size and availability as per usual standard. The investigator was blinded to the group allocation during the experiment. This study was approved by the Ethics Committee of Tongji University School of Medicine and the authorization number is TJLAC-018-035.
EdU staining assay
EdU were added into cell culture with final concentration to 10 μM and incubated for 90 min. Then the cells were collected and analyzed with the Click-IT Plus EdU flow cytometry assay (Invitrogen, #C10635) according to the manufacturer’s protocol. DNA was labeled with propidium iodide. Flow cytometry was performed on FACS verse and the data was analyzed using FlowJo software.
In vitro kinase assays
Kinase reactions were performed as previously described [14]. Mass spectrometry analysis was then performed by PTM BioLabs, Inc. (Zhejiang, China) as previously described [51].
Glucose uptake and lactate production
The levels of glucose uptake by tumor cells were measured with a Glucose Uptake Colorimetric Assay Kit (BioVision, Milpitas, CA, USA). The cells were seeded in 96-well plates at a density of 1 × 104 cells per well. After 24 h, glucose uptake assays were performed according to the manufacturer’s instructions. Lactate production was measured using a commercially available colorimetric/fluorometric assay kit (BioVision, Milpitas, CA, USA). The cells were seeded into 96-well plates at a concentration of 2 × 104 cells per well with phenol red-free complete medium and cultured at 37 °C for 14 h. Aliquots of media from each well were assessed to determine the amount of lactate present. Glucose uptake and lactate production were normalized to cell numbers.
Oxygen consumption rate and intracellular ATP assays
The oxygen consumption rate was measured by an Oxygen Consumption Rate Assay Kit (MitoXpressXtra HS Method) (Cayman). Cells (1 × 105) were cultured in 96-well plates. After MitoXpressXtra was added to each well, we applied a layer of mineral oil to restrict back diffusion of atmospheric oxygen. Oxygen consumption levels were assessed by the time-resolved fluorescence (TR-F) measurement. Intracellular ATP concentrations were measured with a CellTiter-Glo Luminescent Cell Viability Assay (Promega). Cells (1 × 104) were seeded into 96-well plates and treated with or without oligomycin (100 ng/ml) for 1 h. The luminescence signals of each well were measured.
Statistics
Statistical testing was performed using the unpaired two-tailed Student’s t test. P values of 0.05 were considered to be statistically significant. Sample sizes, as described in figure legends, were selected based on effect size and availability as per usual standard. Our data all meet the assumptions of the two-tailed Student’s t tests. In addition, the variation was assessed and we take two as the TYPE parameter in the T test when the variance is similar between the groups, take three as the TYPE parameter when the variance is significant different. Results are expressed as scatter plots or histogram. Data are shown as mean with 95% CI or mean ± S.D. of at least three independent experiments.
References
Kim J, Hake SB, Roeder RG. The human homolog of yeast BRE1 functions as a transcriptional coactivator through direct activator interactions. Mol Cell. 2005;20:759–70.
Kim J, Guermah M, McGinty RK, Lee JS, Tang Z, Milne TA, et al. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell. 2009;137:459–71.
Chen S, Li J, Wang DL, Sun FL. Histone H2B lysine 120 monoubiquitination is required for embryonic stem cell differentiation. Cell Res. 2012;22:1402–5.
Fuchs G, Shema E, Vesterman R, Kotler E, Wolchinsky Z, Wilder S, et al. RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Mol Cell. 2012;46:662–73.
Karpiuk O, Najafova Z, Kramer F, Hennion M, Galonska C, König A, et al. The histone H2B monoubiquitination regulatory pathway is required for differentiation of multipotent stem cells. Mol Cell. 2012;46:705–13.
Chen S, Jing Y, Kang X, Yang L, Wang DL, Zhang W, et al. Histone H2B monoubiquitination is a critical epigenetic switch for the regulation of autophagy. Nucleic Acids Res. 2017;45:1144–58.
Huang X, Yang L, Cai FF, Wang Y, Chen P, Mi J, et al. Autophagy-related protein ATG5 regulates histone H2B mono-ubiquitylation by translational control of RNF20. J Genet Genomics. 2017;44:503–6.
Spolverini A, Fuchs G, Bublik DR, Oren M. let-7b and let-7c microRNAs promote histone H2B ubiquitylation and inhibit cell migration by targeting multiple components of the H2B deubiquitylation machinery. Oncogene. 2017;36:5819–28.
Zhang K, Wang J, Tong TR, Wu X, Nelson R, Yuan YC, et al. Loss of H2B monoubiquitination is associated with poor-differentiation and enhanced malignancy of lung adenocarcinoma. Int J Cancer. 2017;141:766–77.
Tarcic O, Granit RZ, Pateras IS, Masury H, Maly B, Zwang Y, et al. RNF20 and histone H2B ubiquitylation exert opposing effects in Basal-Like versus luminal breast cancer. Cell Death Differ. 2017;24:694–704.
Tarcic O, Pateras IS, Cooks T, Shema E, Kanterman J, Ashkenazi H, et al. RNF20 links histone H2B ubiquitylation with inflammation and inflammation-associated cancer. Cell Rep. 2016;14:1462–76.
Prenzel T, Begus-Nahrmann Y, Kramer F, Hennion M, Hsu C, Gorsler T, et al. Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res. 2011;71:5739–53.
Liberti MV, Locasale JW. The Warburg effect: how doesit benefit cancer cells? Trends Biochem Sci. 2016;41:211–8.
Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–96.
Shestov AA, Liu X, Ser Z, Cluntun AA, Hung YP, Huang L, et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife. 2014;3:e03342.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20.
Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73:1524–35.
Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–63.
Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–70.
Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor1. Cell. 2011;145:732–44.
Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011;480:118–22.
Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609.
Jiang Y, Li X, Yang W, Hawke DH, Zheng Y, Xia Y, et al. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell. 2014;53:75–87.
Jiang Y, Wang Y, Wang T, Hawke DH, Zheng Y, Li X, et al. PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells. Nat Commun. 2014;5:5566.
He CL, Bian YY, Xue Y, Liu ZX, Zhou KQ, Yao CF, et al. Pyruvate kinase M2 activates mTORC1 by phosphorylating AKT1S1. Sci Rep. 2016;6:21524.
Wei Y, Wang D, Jin F, Bian Z, Li L, Liang H, et al. Pyruvate kinase type M2 promotes tumour cell exosome release via phosphorylating synaptosome-associated protein 23. Nat Commun. 2017;8:14041.
Hosios AM, Fiske BP, Gui DY, Vander Heiden MG. Lack of evidence for PKM2 protein kinase activity. Mol Cell. 2015;59:850–7.
Riscal R, Schrepfer E, Arena G, Cissé MY, Bellvert F, Heuillet M, et al. Chromatin-bound MDM2 regulates serine metabolism and redox homeostasis independently of p53. Mol Cell. 2016;62:890–902.
Warburg O. On the origin of cancer cells. Science. 1956;123:309–14.
Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrialphysiology, and tumor maintenance. Cancer Cell. 2006;9:425–34.
Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274:1393–418.
Bellance N, Benard G, Furt F, Begueret H, Smolková K, Passerieux E, et al. Bioenergetics of lung tumors: alteration of mitochondrial biogenesis and respiratory capacity. Int J Biochem Cell Biol. 2009;41:2566–77.
Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochim Biophys Acta. 2011;1807:552–61.
Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–3.
Mellati AA, Yücel M, Altinörs N, Gündüz U. Regulation of M2-type pyruvate kinase from human meningioma by allosteric effectors fructose 1,6 diphosphate and L-alanine. Cancer Biochem Biophys. 1992;13:33–41.
Chen Z, Wang Z, Guo W, Zhang Z, Zhao F, Zhao Y, et al. TRIM35 Interacts with pyruvate kinase isoform M2 to suppress the Warburg effect and tumorigenicity in hepatocellular carcinoma. Oncogene. 2015;34:3946–56.
Wu Y, Chen P, Jing Y, Wang C, Men YL, Zhan W, et al. Microarray analysis reveals potential biological functions of histone H2B monoubiquitination. PLoS One. 2015;10:e0133444.
Tekade RK, Sun X. The Warburg effect and glucose-derived cancer theranostics. Drug Disco Today. 2017;22:1637–53.
Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–6.
Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–83.
Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14:1295–304.
Li S, Swanson SK, Gogol M, Florens L, Washburn MP, Workman JL, et al. Serine and SAM responsive complex SESAME regulates histone modification crosstalk by sensing cellular metabolism. Mol Cell. 2015;60:408–21.
Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73.
Liang J, Cao R, Zhang Y, Xia Y, Zheng Y, Li X, et al. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat Commun. 2016;7:12431.
Chao TK, Huang TS, Liao YP, Huang RL, Su PH, Shen HY, et al. Pyruvate kinase M2 is a poor prognostic marker of and a therapeutic target in ovarian cancer. PLoS One. 2017;12:e0182166.
Wang C, Jiang J, Ji J, Cai Q, Chen X, Yu Y, et al. PKM2 promotes cell migration and inhibits autophagy by mediating PI3K/AKT activation and contributes to the malignant development of gastric cancer. Sci Rep. 2017;7:2886.
Smolková K, Plecitá-Hlavatá L, Bellance N, Benard G, Rossignol R, Ježek P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int J Biochem Cell Biol. 2011;43:950–68.
Chen S, Wang DL, Liu Y, Zhao L, Sun FL. RAD6 regulates the dosage of p53 by a combination of transcriptional and posttranscriptional mechanisms. Mol Cell Biol. 2012;32:576–87.
Chen S, Wei HM, Lv WW, Wang DL, Sun FL. E2 ligase dRad6 regulates DMP53 turnover in Drosophila. J Biol Chem. 2011;286:9020–30.
An H, Yang L, Wang C, Gan Z, Gu H, Zhang T. et al. Interactome analysis reveals a novel role for RAD6 in the regulation of proteasome activity and localization in response to DNA damage. Mol Cell Biol. 2017;37:e00419–16.
Acknowledgements
We thank Dr Weiwei Yang from the Shanghai Institute of Biochemistry and Cell Biology (SIBS, CAS) for kindly providing the PKM2-related plasmids. This work was supported by the National Key Research and Development Program of China (Grant No.: 2017YFA0103301 and 2016YFA0100403), the 973 program of the Ministry of Science and Technology of China (Grant No.: 2015CB856204, 2015CB964802), the National Natural Science Foundation of China (Grant No.: 91419304, 81773009, 81972650, 31330043, and 31271534), the Fundamental Research Funds for the Central Universities (Xi’an Jiao Tong University, Grant No.: 2017qngz13), and the China Postdoctoral Science Foundation (Grant No.: 2017M613149 and 2018T111038).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Piacentini
Supplementary information
Rights and permissions
About this article

Cite this article
Jing, YY., Cai, FF., Zhang, L. et al. Epigenetic regulation of the Warburg effect by H2B monoubiquitination. Cell Death Differ 27, 1660–1676 (2020). https://doi.org/10.1038/s41418-019-0450-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-019-0450-2
This article is cited by
-
Metformin Increases the Response of Cholangiocarcinoma Cells to Gemcitabine by Suppressing Pyruvate Kinase M2 to Activate Mitochondrial Apoptosis
Digestive Diseases and Sciences (2024)
-
Long non-coding RNAs affecting cell metabolism in cancer
Biology Direct (2022)
-
Gankyrin modulated non-small cell lung cancer progression via glycolysis metabolism in a YAP1-dependent manner
Cell Death Discovery (2022)
-
Novel circular RNA circSLIT2 facilitates the aerobic glycolysis of pancreatic ductal adenocarcinoma via miR-510-5p/c-Myc/LDHA axis
Cell Death & Disease (2021)
-
Serine and one-carbon metabolisms bring new therapeutic venues in prostate cancer
Discover Oncology (2021)
