
Abstract
The amino acid antiporter system Xc− is important for the synthesis of glutathione (GSH) that functions to prevent lipid peroxidation and protect cells from nonapoptotic, iron-dependent death (i.e., ferroptosis). While the activity of system Xc− often positively correlates with the expression level of its light chain encoded by SLC7A11, inhibition of system Xc− activity by small molecules (e.g., erastin) causes a decrease in the intracellular GSH level, leading to ferroptotic cell death. How system Xc− is regulated during ferroptosis remains largely unknown. Here we report that activating transcription factor 3 (ATF3), a common stress sensor, can promote ferroptosis induced by erastin. ATF3 suppressed system Xc−, depleted intracellular GSH, and thereby promoted lipid peroxidation induced by erastin. ATF3 achieved this activity through binding to the SLC7A11 promoter and repressing SLC7A11 expression in a p53-independent manner. These findings thus add ATF3 to a short list of proteins that can regulate system Xc− and promote ferroptosis repressed by this antiporter.
Similar content being viewed by others

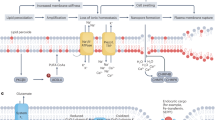

Introduction
System Xc− is an amino acid antiporter that mediates the exchange of extracellular cystine and intracellular glutamtate across the plasma membrane [1, 2]. Because cysteine reduced from cystine is the rate-limiting substrate for the synthesis of the antioxidant glutathione (GSH) [2], inhibition of system Xc−-mediated cystine import by small molecules (e.g., erastin) causes depletion of intracellular GSH and subsequent iron-dependent lipid peroxidation, which in turn leads to a form of nonapoptotic, iron-dependent cell death referred to as ferroptosis [3, 4]. Ferroptosis is regulated by a network revolving around glutathione peroxidase 4 (GPX4) that can reduce lipid peroxides at the expense of GSH, and thus GPX4 inhibitors (e.g., (1S, 3R)-RSL3, or RSL3 in short) are another major class of small molecules that can induce ferroptosis [5,6,7]. While ferroptosis has been linked to neurological disorders and ischemic injury in animal models [7], emerging evidence supports important roles that ferroptosis plays in tumor suppression and cancer resistance to therapy [8,9,10,11,12]. As the light chain encoded by SLC7A11 is a subunit unique to system Xc−, the SLC7A11 expression level often positively correlates with the activity of the antiporter [2]. Indeed, repression of SLC7A11 expression by the tumor suppressor p53 or the histone deubiquitinating enzyme BAP1 inhibits cystine uptake and promotes ferroptosis [8, 9]. However, the mechanism by which SLC7A11 expression is regulated remains largely unknown.
Activation transcription factor 3 (ATF3) is a member of the ATF/CREB family of transcription factors, and its expression is rapidly induced by a wide range of cellular stresses, including DNA damage, oxidative stress, and cell injury [13]. ATF3 contains a basic-region leucine-zipper (bZIP) domain that binds to the ATF/CREB cis-regulatory element or the AP-1 element [14], and as a result, can either repress or activate transcription depending on cell context. Although ATF3 can bind to more than 20,000 genome sites containing the ATF/CREB or the AP-1 element [14], only a small number of genes have been characterized so far as direct transcriptional targets of ATF3. In addition to its family members, ATF3 can also interact with other proteins (e.g., p53 and Tip60) and regulates their activities independent of its transcriptional activity [15, 16]. ATF3 has been shown to be involved in many pathological conditions such as cancer [17, 18], infections/inflammation [19,20,21,22], diabetes [23], and ischemic injury of hearts, livers, or brains [24, 25], highlighting the importance of understanding the exact role of ATF3 in the cellular stress response. Here we report that ATF3 could suppress system Xc− and predispose cells to ferroptosis by repressing SLC7A11 expression.
Materials and methods
Cell culture and reagents
HT1080 cells expressing ATF3 and empty vector were generated previously [26]. Retinal pigment epithelial (RPE) cells immortalized with hTERT were obtained from Dr. Todd Waldman [27]. To generate ATF3-knockout cells, a target sequence (5′-AAAATGATGCTTCAACACCC-3′) spanning the ATF3 start codon was inserted into pSpCas9(BB)-2A-puro, and used to transfect RPE and U2OS cells as described [28]. ATF3-knockout HCT116 cells were generated using rAAV-mediated homologous recombination as described previously [14]. These cells were cultured in media supplemented with 10% fetal bovine serum. We used DMEM for HT1080, RPE, DU145, and U2OS cells, RPMI 1640 for PC3 cells, and McCoy’s 5A for HCT116 cells. Erastin, RSL3, ferrostatin-1, BSO, Z-VAD-FMK, and FIN56 were purchased from Cayman Chemical. N-acetylcysteine (NAC) was purchased from Thermo Fisher Scientific. Other reagents were obtained from Sigma-Aldrich.
Cell viability assays
Cells treated in 24-well plates were trypsinized, and stained with 0.2% of Trypan blue. Viable cells were then counted using a hemocytometer under a microscope. In some experiments, cell viability was assessed by MTT assays as described [29].
SLC7A11 promoter cloning and mutagenesis
The human SLC7A11 promoter (−678 to +8) was PCR amplified from genomic DNA derived from HT1080 cells using a forward primer (5′-ATGGTACCTCTGGAGTCCTGGTGAATTTTG-3′) and a reverse primer (5′-TAACTCGAGACAAACCAGCTCAGCTTCCT-3′). After purification, the DNA fragment was digested with KpnI and XhoI, and cloned into the pGL3-Basic vector following a standard cloning protocol. To generate promoter constructs with BS-1 (m1) or BS-2 (m2) mutations, overlapping PCR was performed using the wild-type promoter as the template, and the forward/reverse primer coupled with a pair of complementary primers spanning the BS-1 or the BS-2 site. These primers were: m1-forward (5′-TTCTCATGTGGCGGGTGCAAACCTGG-3′), m1-reverse (5′-CCAGGTTTGCACCCGCCACATGAGAAG-3′); m2-forward (5′-CCTGGAGAATTTGCACCCTCATTTAGCTGTAG-3′), m2-reverse (5′-CTACAGCTAAATGAGGGTGCAAATTCTCCAGG); the mutated nucleosides were underlined. Double mutation were generated by using m1 as the template coupled with the m2 primer pair. The mutant promoter fragments were cloned into pGL3-Basic at KpnI and XhoI sites. To generate the promoter construct with a deletion of the predicted p53-BS, reverse PCR was performed using the wild-type promoter construct as the template and a forward primer (5′-TGCCTGTCACACCAACTTAC-3′) and a reverse primer (5′-ATGAGGAAGCTGAGCTGGTT-3′). The PCR product was digested with 5 units of DpnI, purified, and ligated with T4 DNA ligase for transformation. The sequences of the wild-type and mutant promoters were confirmed by DNA sequencing.
Transfections and reporter assays
Transfections were carried out using Lipofectamine 2000 (Invitrogen) according to a protocol provided by the manufacturer. For reporter assays, cells seeded in 24-well plates were transfected with 100 ng of pGL3 or pGL3-pSLC7A11, 5 ng of pRL-CMV, and 400 ng of pCG-ATF3 or pCG, or 400 ng of p53. After 48 h, cells were lysed, and the reporter activity was measured using the Dual-Luciferase Reporter Assay System (Promega) following the recommendation.
Lentiviral infections
To generate the lentiviral vector expressing ATF3, the ATF3 cDNA was PCR amplified and cloned into pCDH-CMV-MCS-EF1-Puro at NheI and BamHI sites. The lentivectors (3 μg) were transfected into 293FT cells plated in a 100 mm dish along with 3 μg of pLP1, 3 μg of pLP2, and 3 μg of pLP/VSVG. Supernatants were harvested 2 days later and used to infect cells along with 4 μg/ml of polybrene. The SLC7A11-expressing lentiviral vector (pLOC-SLC7A11) and its control vector (pLOC-RFP) were previously described [30] and provided by Dr. Nicholas Clemons.
Chromatin immunoprecipitation assays
These were carried out as described previously [14]. We used an anti-ATF3 antibody (Santa Cruz, sc-188) or normal rabbit IgG to pull down sheared chromatin. Immunoprecipitated DNA was purified using the QIAquick PCR Purification Kit (Qiagen), and quantitated using real-time PCR. The primers used were as follows: SLC7A11, forward 5′-TTGAGCAACAAGCTCCTCCT-3′, reverse 5′-CAAACCAGCTCAGCTTCCTC-3′; ATF3, forward 5′-TGGCAACACGGAGTAAACGAC-3′, reverse 5′AGAGAAGAGAGCTGTGCAGTGC; RNF-ex, forward 5′-GGCTGGATTTTTGCAAGTTGA-3′, reverse 5′-TTGACGCCTCCAGCATCTG-3′.
DNA affinity precipitation assays
Nuclear extracts were prepared as described previously [31]. 100 μg of nuclear extracts or 0.25–1 μg of recombinant ATF3 [16] were incubated with 2 μg of annealed biotin-labeled or unlabeled oligonucleotide (biotin-5′-CATGTGGCTTGATGCAAACCTGGAGAATTTGCATCATCATTTAGC-3′) in a buffer containing 25 mM Tris-HCl, pH7.9, 2 mM EDTA, 1 mM EGTA, 0.1% NP-40, 100 mM NaCl, 1 mM DTT, and 5% glycerol at 4 °C for 1 h, and then at 4 °C for 2 h with addition of 15 μl of Dynabeads M-280. The beads were washed with the incubation buffer supplemented with 150 mM NaCl and 0.5% NP-40 for five times. Bound proteins were eluted by boiling in 20 μl of SDS loading buffer, and subjected to western blotting. For competition assays, ATF3 was incubated with 50 μg of unlabeled oligonucleotide along with 2 μg of labeled probe.
Glutamate release assays
The Glutamate-Glo Assay kit (Promega) was used to measure the amount of glutamate released into condition medium. Cells (2 × 105) plated in six-well plates were washed with PBS twice, and incubated in glutamine-free medium in the presence/absence of 10 μM of erastin for 1 h. To measure the glutamate level, 50 μl of condition medium was transferred to a 96-well plate, and mixed with 50 μl of a reaction mixture containing glutamate dehydrogenase, NAD, reductase, pro-luciferin, and luciferin detection solution following the manufacturer’s protocol. The plate was shaken for 30–60 s and incubated at room temperature for 1 h, and luminescence was measured with SpectraMax L (Molecular Devices). The glutamate level was first calculated in reference to a glutamate standard curve, and then normalized to the total cell number determined by MTT assays at the end of the experiment.
GSH assay
The Glutathione Assay Kit (Cayman Chemical) was used for these experiments. Cells (2 × 105) in six-well plates were treated with or without 10 μM of erastin for 5 h, scraped into 500 μl of 10 mM phosphate buffer containing 1 mM EGTA, and then lysed by sonication. After centrifugation, supernatants were deproteinated by incubating with 500 μl of 10% metaphosphoric acid at room temperature for 5 min, and then centrifuged for 3 min at 4000 rpm. The resulted supernatants were mixed with 50 μl of 4 M triethanolamine. 50 μl of each sample was then transferred to a 96-well plate, and incubated with 150 μl of Assay Cocktail containing the Ellman’s reagent (5,5′-dithio-bis- 2-(nitrobenzoic acid) or DTNB) at room temperature for 25 min. The absorbance at 405 nm was measured, and used to calculate the GSH amount in reference to a GSH standard curve.
Lipid peroxidation detection
Cells (2 × 105) in six-well plates were incubated with 1 ml of fresh medium containing 5 μM of BODIPY 581/591 C11 (Invitrogen) for 20 min. Cells were then trypsinized, washed, and resuspended in 0.5 ml of PBS for flow cytometry analysis. A minimum of 20,000 cells were analyzed per condition.
Quantitative RT-PCR
Total RNA was extracted from cells using TRIzol (Invitrogen). 2 μg of total RNA was then reverse transcribed using the iScript Advanced cDNA kit for RT-qPCR (Bio-Rad), and subjected to real-time PCR using the SYBR Green reagents (Bimake). The sequences of the primers used for real-time PCR were: SLC7A11, forward 5′-TCATTGGAGCAGGAATCTTCA-3′, reverse 5′-TTCAGCATAAGACAAAGCTCCA-3′; ATF3, forward 5′-GTGCCGAAACAAGAAGAAGG-3′, reverse 5′-TCTGAGCCTTCAGTTCAGCA-3′; CHAC1, forward 5′-GAACCCTGGTTACCTGGGC-3′, reverse 5′-CGCAGCAAGTATTCAAGGTTGT-3′; ASNS, forward 5′-CTGTGAAGAACAACCTCAGGATC-3′, reverse 5′-AACAGAGTGGCAGCAACCAAGC-3′; p21, forward, 5′-CTGGAGACTCTCAGGGTCGAAA-3′, reverse 5′-GATTAGGGCTTCCTCTTGGAGAA-3′; MMP-2, forward 5′-CCCACTGCGGTTTTCTCGAAT-3′, reverse 5′-CAAAGGGGTATCCATCGCCAT-3’; β-actin, forward 5′-TCCATCATGAAGTGTGACG-3′, reverse 5′-TACTCCTGCTTGCTGATCCAC-3′.
Western blotting
These were performed as described previously [15]. Briefly, cells were lysed in RIPA buffer containing 50 mM Tris-HCl, pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM NaF, 1 mM Na3VO4, and protease inhibitor cocktail (Roche) for 30 min, and subjected to SDS-PAGE for western blotting. The following antibodies were used: SLC7A11 (1:1000, #12691) from Cell Signaling, ATF3 (1:1000, sc-188) and p53 (1:1000, sc-6243) from Santa Cruz, and β-actin (1:10000, A5441) from Sigma.
Results
ATF3 promotes ferroptosis induced by erastin
A recent finding that the ATF3 mRNA level was increased during erastin-induced ferroptosis [4] prompted us to hypothesize that ATF3 may play a role in the regulation of this important form of cell death. Indeed, we found that erastin could induce ATF3 expression in as short as 1 h (Fig. 1a–c). The induction of ATF3 expression was transient in the immortalized normal RPE cell line (Fig. 1b–c), probably due to transcriptional autorepression [32]. NAC, the precursor of intracellular antioxidant GSH, could inhibit ATF3 induction by erastin (Fig. 1d, e), suggesting that the Nrf2-mediated antioxidant pathway activated by FIN-induced redox imbalance [20] might mediate ATF3 induction by these agents. Indeed, while ATF3 is a known oxidative stress-responsive gene [33], Nrf2 has been shown to bind to the ATF3 promoter and mediate ATF3 expression induced by reactive oxygen species (ROS) [34]. Similarly, while ATF3 expression can be induced by the endoplasmic reticulum (ER) stress via the PERK/ATF4-mediated pathway [35], ATF3 induction by erastin was impaired when the cells were pretreated with an PERK inhibitor GSK 2606414 (Fig. 1f), suggesting that erastin-induced ER stress [4] might contribute to ATF3 induction as well. However, although ATF3 was a p53 target gene in a certain contexts [36, 37], the observations that the p53 level was barely elevated by erastin (Fig. 1a, b, d, e) suggest that p53 was not involved in ATF3 induction by erastin.

Metabolic stress induces ATF3 expression. a, b HT1080 and RPE cells were treated with 5 µM of erastin for different time as indicated, and subjected to western blotting for ATF3 and p53 expression. c Indicated cells were treated with 5 μM of erastin for different time and subjected to qRT-PCR to measure the ATF3 mRNA level. The data are presented as mean ± SD. d, e HT1080 and RPE cells treated with 5 μM of erastin with or without 3 mM of NAC were subjected to western blotting. f RPE cells were pretreated with or without 1 µM of GSK2606414 (GSK′414) for 30 min, and then treated with erastin (5 µM) for western blotting
Ferroptosis inducers (FINs) can readily induce HT1080 cells to undergo ferroptosis—an event characterized by lipid peroxidation that can be alleviated by ferrostatin-1 (Ferr-1) [3]. Intriguingly, ATF3 expression sensitized HT1080 cells to erastin-induced death (Fig. 2a, b) that could be rescued by Ferr-1 but not Z-VAD-FAM (Fig. 2c), indicating that ATF3 promoted erastin-induced ferroptosis. Consistent with this effect, ATF3 expression promoted erastin-induced elevation of the lipid peroxide level in these cells (Fig. 2d). These effects were not caused by clonal variation, as we saw similar effects in an unselected cell population expressing ATF3 via lentiviral infections (Supplementary Fig. S1A). Although overexpression of an ectopic gene may trigger ROS production leading to cell death, it was highly unlikely that the observed ATF3-mediated effects were merely a consequence of gene overexpression as overexpression of an ATF3 mutant (ATF3-ΔBR) that lacks its DNA-binding domain (the basic region, or BR), and thus does not regulate gene transcription, failed to sensitize HT1080 cells to erastin (Supplementary Fig. S2). To corroborate these results, we generated ATF3-knockout (KO) RPE cells with the CRISPR/Cas9 tool (Fig. 2e). While the normal epithelial cells were sensitive to erastin (Fig. 2f), knockout of ATF3 expression dramatically suppressed erastin-induced ferroptosis and lipid peroxidation (Fig. 2f–h). ATF3 promoted erastin-induced ferroptosis in other cell lines (e.g, DU145 and U2OS) as well (Supplementary Fig. S1B, S1C). Taken together, our results indicate that ATF3 can promote ferroptosis induced by erastin. Of note, ATF3 expression promoted, while ATF3 knockout suppressed ferroptosis induced by the GPX4 inhibitor RSL3—a type 2 FIN—as well (Supplementary Fig. S3A, S3B). However, ATF3 had a negligible effect on cell sensitivity to FIN56, a type 3 FIN, which induces ferroptosis by depleting CoQ10 and GPX4 [7, 38] (Supplementary Fig. S3D, 3E).

ATF3 promotes ferroptosis induced by FINs. a HT1080 cells expressing ATF3 or transfected with the empty vector (EV) were treated with 2.5 μM of erastin for 24 h, and subjected to western blotting. b Viable HT1080 cells treated with erastin for 24 h were counted after stained with Trypan blue. c Viable HT1080 cells were counted after treatments with erastin (5 μM), Ferr-1 (2 μM), and/or Z-VAD-FMK (10 μg/ml) for 24 h. d HT1080 cells treated with/without erastin for 6 h were stained with C11-BODIPY, and subjected to flow cytometry analysis. e ATF3-knockout (KO) or -wild-type (WT) RPE cells were treated with 2.5 μM of erastin for 24 h for western blotting. f Viable RPE cells treated with erastin for 24 h were counted. g Viable RPE cells were counted after treated with erastin (5 μM), Ferr-1 (2 μM), and/or Z-VAD-FMK (10 μg/ml) for 24 h. h RPE cells treated with/without erastin for 6 h were stained with C11-BODIPY for flow cytometry analysis
ATF3 suppresses system Xc−
System Xc−-mediated cystine uptake crucial for GSH synthesis is a major mechanism that blocks lipid peroxidation and suppresses ferroptosis [5, 6]. To understand the mechanism by which ATF3 promotes ferroptosis, we determined the activity of system Xc− by measuring the amount of glutamate released into cultured media [4]. While erastin inhibited glutamate release as expected [4], ATF3 expression caused a decrease in the amount of released glutamate even in the absence of erastin (Fig. 3a). Conversely, ATF3 KO promoted glutamate release (Fig. 3b). The effects of ATF3 on glutamate release were consistent with the results that ATF3 expression increased while KO decreased the intracellular GSH level (Fig. 3c, d), arguing for the notion that ATF3 suppresses system Xc−. Thus, ATF3 might promote ferroptosis through suppressing system Xc−.

ATF3-mediated suppression of system Xc− contributes to the promotion of ferroptosis. a, b HT1080 and RPE cells were treated with or without 10 μM of erastin for 1 h, and the amount of glutamate released into culture medium was measured. c, d Indicated cells treated with 10 μM of erastin for 5 h were lysed, and the intracellular GSH level was measured. e Viable HT1080 cells after treatments with erastin (1 μM) and/or 2-ME (20 μM) were counted. f Viable RPE cells treated with erastin (5 μM) and/or 2-ME (20 μM) were counted. g, h Cells were treated with erastin and/or NAC (3 mM), and subjected to MTT assays. The data are presented as mean ± SD. *p < 0.05; ***p < 0.001, Student’s t-test
ATF3 promotes ferroptosis through regulating system Xc–
2-Mercaptoethanol (2-ME) can reduce cystine to cysteine extracellularly, and thus bypass system Xc− to supply cysteine for GSH synthesis. If suppression of system Xc− is the main mechanism by which ATF3 promotes ferroptosis, bypassing system Xc− by treating cells with 2-ME would alleviate the effects of ATF3 on ferroptosis. Indeed, 2-ME rescued ferroptosis induced by erastin in ATF3-expressing HT1080 cells (Fig. 3e). Similarly, erastin-induced ferroptosis in RPE cells was also rescued (Fig. 3f). Of note, it is unlikely that the general/nonspecific reducing activity of 2-ME resulted in the observed rescuing effect, as 2-ME did not recue ferroptosis induced by cystine deprivation (Supplementary Fig. S4). We also treated the cells with NAC, which can convert into cysteine, thereby bypassing system Xc−. Again, bypassing system Xc− using NAC abolished ferroptosis induced by erastin in both HT1080 and RPE cells (Fig. 3g, h). These results thus support our hypothesis whereby ATF3 promotes erastin-induced ferroptosis through suppressing system Xc−.
ATF3 binds to and represses the SLC7A11 promoter
As the SLC7A11 expression level positively correlates with the system Xc− activity [2], we tested if ATF3 suppresses system Xc− by downregulating SLC7A11 expression. Intriguingly, our genome-wide ChIP-seq analysis [14] revealed a very strong ATF3-binding peak in a SLC7A11 promoter region proximal to the transcription start site (TSS) (Fig. 4a, arrow). This ATF3-binding peak was also found in almost all of the cell lines examined by the ENCODE project, including human embryonic stem cells (Fig. 4a), suggesting that ATF3 likely strongly binds to the SLC7A11 promoter and regulates SLC7A11 transcription. It is important to note that the SLC7A11 promoter is on the list of top ten promoters with highest ATF3-binding scores (Fig. 4b). As a comparison, the binding score of DDIT3 or CHOP—a gene well-characterized as an ATF3 target [39]—was much lower than that of SLC7A11 (Fig. 4b). The ATF3-binding peak in the SLC7A11 promoter spans a region containing two adjacent sites (BS-1 and BS-2) that are in opposite orientation but have the same sequence (5′-TGATGCAA-3′) as the –20 site that was previously characterized as a functional ATF3-binding site in the ATF3 promoter and known to be responsible for its autorepression [32] (Fig. 4c), suggesting that ATF3 likely binds to the SLC7A11 promoter at these two sites. Indeed, we carried out ChIP assays using HT080 cells, and confirmed that ATF3 strongly bound to this region in spite of the very low basal ATF3 level in the cells (Fig. 4d). We used the ATF3-20 site (ATF3) and a region spanning a RNF43 exon (RNF-ex) as the positive and the negative control, respectively, based on our previous report [14]. Overexpressing ATF3 in HT080 cells significantly increased the amount of ATF3 bound to the SLC7A11 promoter (Fig. 4e), further demonstrating the specificity of the binding. We also carried out DNA affinity precipitation assays, and confirmed that ATF3 bound to an oligonucleotide spanning this region (Fig. 4f). Moreover, while recombinant ATF3 also bound to the oligonucleotide (Fig. 4g), such binding was competitively blocked by an excess amount of unlabeled oligonucleotide (Fig. 4h), suggesting that ATF3 can directly bind to the SLC7A11 promoter.

ATF3 binds to and represses the SLC7A11 promoter. a A UCSC genome browser view of the ATF3-binding peak in the human SLC7A11 promoter. The ChIP-seq data previously reported [14] were reanalyzed and compared with the data from the ENCODE project. WT and KO, ATF3-wildtype and -knockout cells, respectively. b Peak scores of ATF3-binding peaks localized in promoters (defined as regions +/− 1 kb relative to transcription start sites) are depicted. Insert indicates top promoters with highest peak scores. c The sequence of the center of the ATF3-binding peak in the human SLC7A11 promoter is shown. The locations of BS-1, BS-2, and p53-BS relative to the transcription start site are indicated. d, e HT1080 cells were subjected to ChIP assays with an ATF3-specific antibody, and the amount of precipitated DNA was quantitated by real-time PCR. f A biotin-labeled oligonucleotide containing the BS-1 and BS-2 sites (biotin-5′-CATGTGGCTTGATGCAAACCTGGAGAATTTGCATCATCATTTAGC-3′) was incubated with nuclear extracts prepared from HT1080-ATF3 cells. Bound proteins were pulled down using Dynabeads, washed, and subjected to western blotting. g The biotin-labeled and unlabeled oligonucleotides were incubated with recombinant ATF3 for pulldown assays as in f. h The biotin-labeled oligonucleotide were incubated with recombinant ATF3 with or without an excess amount of unlabeled oligonucleotide for pulldown assays. i The human SLC7A11 promoter (pSLC7A11) was cloned into pGL3, and transfected into HT1080 cells for dual luciferase assays. j HT1080 cells were transfected with the indicated reporters with or without increasing amounts of ATF3 for dual luciferase assays. k The SLC7A11 promoters with point mutations in the BS-1 (5′-TTGCATCA-3′ to 5′-TTGCACCC-3′) and/or BS-2 site (5’-TGATGCAA-3’ to 5′-GGGTGCAA-3′) were transfected with/without ATF3 into HT080 cells for dual luciferase activity assays
To determine whether ATF3 binding to the SLC7A11 promoter is functional, we cloned the human SLC7A11 promoter for luciferase reporter activity assays. While the SLC7A11 promoter exhibited a dramatically-reduced activity in HT1080 cells stably expressing ATF3 (Fig. 4i), transient ATF3 expression also significantly decreased the SLC7A11 promoter activity in a dose-dependent manner (Fig. 4j). Importantly, mutating either BS-1 or BS-2 dramatically alleviated ATF3-mediated repression of the SLC7A11 promoter (Fig. 4k), indicating that ATF3 bound to both BS-1 and BS-2 sites to repress the SLC7A11 promoter. Of note, mutating either BS-1 or BS-2 site largely decreased the basal activity of the promoter (Fig. 4k)—a result arguing for a model whereby ATF3 represses the SLC7A11 promoter by competing for BS-1/BS-2 binding with transcriptional activators (see more in “Discussion”).
P53 is not required for ATF3-mediated repression of SLC7A11 promoter
Previously, p53 was shown to repress SLC7A11 transcription by binding to a consensus site (p53-BS) proximal to BS-1 and BS-2 sites (Fig. 4c) [8]. As ATF3 can interact with p53 and colocalize with p53 at promoters/enhancers to regulate transcription [14, 15], it is probable that ATF3 might regulate the SLC7A11 promoter through interacting with p53. However, ATF3 retained its SLC7A11-repressing activity in p53-null PC3 cells (Fig. 5a). In line with these results, while the amount of ATF3 bound to the promoter of an ATF3- and p53-coregulated gene MSI2 [14] was decreased, knockout of p53 did not impair ATF3 binding to the SLC7A11 promoter (Supplementary Fig. S5). Moreover, ATF3 repressed a mutated SLC7A11 promoter where p53-BS was deleted (mtp53BS) to the same extent as the wild-type promoter (Fig. 5b). Surprisingly, p53 also repressed the mutant promoter (Fig. 5b). While p53 may repress transcription without binding to a consensus site [40, 41], it is worth noting that, unlike in the mouse promoter [8], two of the four nucleotides crucial for p53 binding (marked in red) in the human SLC7A11 promoter don’t match that of the consensus site (Fig. 5c).

ATF3 represses the SLC7A11 promoter independent of p53. a p53-null PC3 cells were transfected with the SLC7A11 promoter reporter with or without ATF3/p53 for dual luciferase activity assays. b The p53-binding site (p53-BS) was deleted from the SLC7A11 promoter. The mutant promoter (mtp53BS) was transfected into PC3 cells with or without ATF3 or p53 for dual luciferase activity assays. c The sequence of the predicted p53-binding site in the human SLC7A11 promoter was compared with that in the mouse promoter and the consensus p53-binding sequence. Red color marks the four nucleotides crucial for p53 binding. The nucleotides that do not match the consensus sequence are underlined. The data are presented as mean ± SD
ATF3 represses SLC7A11 expression
Consistent with the results that ATF3 repressed the SLC7A11 promoter, both the SLC7A11 mRNA and protein levels were dramatically decreased in the ATF3-expressing cells compared to the control HT1080 cells (Fig. 6a) - an effect unlikely caused by clonal variation as lentiviral-mediated ATF3 expression repressed SLC7A11 expression in HT1080 (Fig. 6b) and RPE cells (Fig. 6c) as well. ATF3 also decreased the SLC7A11 level in p53-null PC3 cells (Fig. 6d), in line with the notion that ATF3 represses SLC7A11 expression independent of p53. Conversely, knockout of ATF3 expression in RPE cells caused an increase in the SLC7A11 level (Fig. 6e). This effect was not due to nonspecific gene targeting by the ATF3-sgRNA, as knockout of ATF3 expression using a distinct, rAAV-based approach [14] also increased the SLC7A11 expression level (Fig. 6f). To further support the notion that ATF3 represses SLC7A11 expression, we analyzed publicly available gene expression datasets and found that the ATF3 expression level inversely correlated with the SLC7A11 level in two cohorts of glioma patients (Fig. 6g). This correlation is of significance given that SLC7A11 overexpression found in glioma cells has been shown to promote the growth of the brain tumors [42]. As the SLC7A11 protein had a relatively long half-life, it was not surprising that ATF3 did not affect the stability of this protein (Fig. 6h).

ATF3 represses SLC7A11 expression. a HT1080 cells were subjected to qRT-PCR and western blotting for SLC7A11 expression. b, c HT1080 (b) and RPE (c) cells were infected with LentiATF3 or control viruses for qRT-PCR assays. d PC3 cells infected with LentiATF3 was subjected to qRT-PCR and western blotting for SLC7A11 expression. e The SLC7A11 expression level was determined in ATF3-wiltype (WT) and -knockout (KO) RPE cells. f ATF3-knockout (KO) HCT116 cells generated via rAAV-mediated gene targeting were subjected to qRT-PCR and western blotting for SLC7A11 expression. g The correlation between ATF3 and SLC7A11 expression in two cohorts of glioma patients was analyzed using the R2 Genomics Analysis and Visualization Platform. h HT1080 cells were treated with cycloheximide (CHX) for western blotting.The data are presented as mean ± SD
Stress-induced ATF3 does not regulate SLC7A11 expression
Erastin can induce SLC7A11 expression as an adaptive response to system Xc− inhibition [4]. Indeed, erastin induced a slight increase of the SLC7A11 mRNA level (Fig. 7a), but profound accumulation of the SLC7A11 protein (Fig. 7b), in RPE cells, suggesting that erastin might induce an adaptive cellular response (e.g., inducing expression of the heat shock protein HSPA5 [10]) to protect SLC7A11 from degradation. To our surprise, knockout of ATF3 expression in RPE cells caused a decrease in the SLC7A11 mRNA level after erastin treatments (Fig. 7a). However, because erastin only slightly increased the SLC7A11 mRNA level (Fig. 7a) and since the half-life of SLC7A11 protein was relatively long (Fig. 7b), the SLC7A11 protein level remained higher in the knockout cells as compared to the wild-type cells at all of the time points during erastin treatments (Fig. 7b), which would ensure higher system Xc− activity in the KO cells. Intriguingly, while ATF3 could bind to the CHAC1 and the ASNS promoter (Fig. 4b), ATF3 promoted erastin-induced expression of these two genes [4] (Fig. 7c, d). While the role of neither CHAC1 nor ASNS in the regulation of ferroptosis has been established [4], our results suggest that ATF3 more likely controls the basal SLC7A11 level and predispose cells to a state sensitive to ferroptosis.

Erastin-induced ATF3 does not repress SLC7A11 induction but promote CHAC1 and ASNS induction. a, b RPE cells treated with 5 μM of erastin were subjected to qRT-PCR (A) and western blotting (B). c, d CHAC1 and ASNS expression levels in ATF3-wildtype (WT) and –KO RPE cells were determined by qRT-PCR
SLC7A11 expression antagonizes ATF3-mediated promotion of ferroptosis
The results described above argue for that ATF3 represses SLC7A11 expression to suppress system Xc− and subsequently promote ferroptosis. If this hypothesis is true, restoring SLC7A11 expression in ATF3-expressing cells would restore system Xc− activity and consequently impair the capability of ATF3 to promote ferroptosis. Indeed, SLC7A11 overexpression via lentiviral infections (Fig. 8a) restored the system Xc− activity (evidenced by increased glutamate release) in ATF3-expressing cells (Fig. 8b). The reason why the SLC7A11 level in ATF3-expressing cells was lower than that in the empty-vector control cells after lentiviral infections was unknown, but it might be attributable to the capability of ATF3 to suppress viral integration [43]. Nevertheless, while SLC7A11 expression conferred resistance to erastin in both ATF3-expressing and control cells as expected, the difference in erastin sensitivity between these cells was diminished (Fig. 8c), indicating that SLC7A11 expression counteracted ATF3-mediated ferroptosis induced by erastin. Our results thus demonstrate that ATF3 can promote erastin-induced ferroptosis by repressing SLC7A11 expression and suppressing system Xc−.

SLC7A11 overexpression antagonizes ATF3-promoted ferroptosis. a HT1080 cells were infected with SLC7A11-expressing lentiviruses or control viruses (RFP) for western blotting. b The amount of glutamate in the condition media from the infected HT1080 cells treated with or without erastin (10 μM) was measured. c Infected cells were treated with erastin, and cell viability was measured by MTT assays. d Schematic presentation of a model whereby ATF3 represses SLC7A11 expression to suppress system Xc-, and thereby predispose cells to a state prone to ferroptosis
Discussion
Ferroptosis is a nonapoptotic form of cell death that can be induced by metabolic stress such as GSH depletion [7]. While system Xc− mediated uptake of cystine is crucial for the maintenance of redox balance and the blockage of lipid peroxidation, how the system Xc− activity is regulated remains largely unknown. Previously, CD44v/MUC1-C and BECN1 was shown to interact with SLC7A11 and regulate the stability of system Xc− [44, 45]. While Nrf2 and ATF4 can bind to the SLC7A11 promoter and activate its expression [46,47,48], binding of p53 to the SLC7A11 promoter or BAP1-mediated epigenetic modifications of the promoter represses SLC7A11 transcription [8, 9]. In this report, we have demonstrated that ATF3 is another major SLC7A11 repressor. ATF3 strongly bound to the SLC7A11 promoter, suppressed system Xc−, and promoted ferroptosis induced by erastin (Fig. 8d). Although ATF3 expression was induced by erastin, it is intriguing that ATF3 induction was more likely a mere response to redox imbalance caused by system Xc− inhibition (Fig. 1d, e). In this regard, while both the ATF3 and the SLC7A11 promoter contain binding sites for Nrf2 that is activated by oxidative stress [34, 46], erastin-induced ATF3 did not appear to repress SLC7A11 expression induced by redox imbalance. Rather, ATF3 contributed to erastin-induced CHAC1 expression (Fig. 7c). Although CHAC1 was shown to mediate GSH degradation [49, 50], its role in ferroptosis remains controversial [4, 51]. Therefore, it is more likely that ATF3 predisposes cells to a state prone to ferroptosis through repressing SLC7A11 expression. Our study thus adds ATF3 to a very short list of proteins that can regulate ferroptosis by suppressing system Xc−. Given that emerging evidence links ferroptosis to various pathological conditions (e.g., cancer and neural injury) [7] often involving induction or aberrant expression of ATF3 [13, 17], our finding suggests that ATF3 may regulate these events by promoting ferroptosis. It is worth noting that ATF3 is only structurally similar to ATF4 in the bZIP domain. In contrast to ATF3, ATF4 suppresses ferroptosis by transactivating SLC7A11 and HSPA5 [10, 48]. As Atf3−/− mice, unlike Atf4−/− mice, are developmentally normal [52], drugging ATF3 would be a better strategy than targeting ATF4 for harnessing ferroptosis to treat human diseases including cancer.
Intriguingly, ATF3 also appeared to promote RSL3-induced ferroptosis (Supplementary Fig. S3A, S3B). While ATF3-mediated suppression of system Xc− resulted in a decrease in the intracellular GSH level (Fig. 3c), which could in turn sensitize the cells to RSL3-mediated GPX4 inhibition, bypassing system Xc− with 2-ME or NAC indeed countered the effects of ATF3 on RSL3 sensitivity (Supplementary Fig. S3C). However, 2-ME/NAC only partly antagonized ATF3’s effects, suggesting that ATF3 could also promote RSL3-mediated ferroptosis via additional, unknown mechanism(s). Indeed, like ATF4, ATF3 might regulate expression of HSPA5, which encodes for a heat shock protein bound to GPX4 and protecting the latter from degradation [10].
ATF3 repressed SLC7A11 expression, but transactivated CHAC1 and ASNS expression in response to erastin. These results were not unexpected as binding of ATF3 to DNA can result in either transcriptional repression or activation [53]. ATF3 can form homodimer, or bind to its family members as well as many other transcription factors (e.g., p53, Sp1, NF-κB) [15, 54, 55], leading to distinct transcriptional outcomes. While a prevailing model proposes that ATF3 recruits and/or stabilizes transcriptional repressors/corepressors (e.g., HDAC1) to repress promoters [56, 57], our results that mutating the ATF3-binding sites decreased the activity of SLC7A11 promoter (Fig. 4k) suggest that ATF3 more likely repressed SLC7A11 expression through competing with transcriptional activators/coactivators for promoter binding. The BS-1/BS-2 sequence matches the consensus sequence of the C/EBP-ATF response elements, and thus these sites could be bound by both the C/EBP and the ATF/CREB family of transcription factors [58, 59], many of which are transcriptional activators. BS-1 and BS-2 sites are adjacent, separated by only 9 bp, and in different orientation (Fig. 4c). This unique feature might facilitate homodimerization of DNA-bound ATF3 and stabilize the ATF3-DNA interaction, thereby shielding these sites from access to transcriptional activators. Intriguingly, in addition to the –20 site, ATF3 appears to bind to its own promoter at another site proximal to the transcription start site [14]. Thus, ATF3 might repress its own promoter [32] with a mechanism similar to that for SLC7A11. In contrast, neither the CHAC1 nor the ASNS promoter contains two adjacent ATF3-binding sites for promoting ATF3 to bind to DNA.
Another important finding from this study was that ATF3 repressed SLC7A11 expression and promote ferroptosis independent of p53. While p53 can induce ATF3 expression [36, 37], ATF3 binds to p53 and stabilizes the tumor suppressor in response to DNA damage [15, 26, 60]. Although ATF3 was shown to reduce the p53 mRNA level in umbilical vein endothelial cells and keratinocytes [61, 62], we did not find that ATF3 expression decreased the p53 level in all of the cells that we tested. Intriguingly, while erastin slightly increased the p53 level (Fig. 1a, b), ATF3 appeared to be required for this induction (Fig. 1b). As the predicted p53-binding site (p53BS) in the human SLC7A11 promoter was dispensable for p53-mediated repression of SLC7A11 expression (Fig. 5b), our results raise an important question as to whether p53 represses SLC7A11 promoter through interacting with ATF3. It is worth noting that p53 appears to play opposite roles in the regulation of ferroptosis. On one hand, p53 can promote oxidative stress-induced ferroptosis by repressing SLC7A11 while activating SAT1 expression [8, 63]. On the other hand, p53 suppresses erastin-induced ferroptosis by inhibiting DPP4 in colorectal cancer (CRC) cells or inducing p21 expression in non-CRC cells [64, 65]. However, p53-mediated suppression of ferroptosis requires preactivation of p53 [65]. As ATF3 could directly bind to the SLC7A11 promoter and since SLC7A11 overexpression could abolish ATF3-mediated ferroptosis, we concluded that ATF3-mediated repression of SLC7A11 expression and suppression of system Xc− might be the major mechanism by which ATF3 promotes ferroptosis induced by metabolic stress.
References
Bridges R, Natale N, Patel S. System Xc− cystine/glutamate antiporter: an update on molecylar pharmacology and roles within the CNS. Br J Pharmacol. 2012;165:20–34.
Lewerenz J, Hewett S, Huang Y, Lambros M, Gout P, Kalivas P, et al. The cystine/glutamate antiporter system Xc− in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18:522–55.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Dixon S, Patel D, Welsch M, Skouta R, Lee E, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79.
Wang WS, Stockwell BR. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016;26:165–76.
Stockwell B, Friedmann Angeli J, Bayir H, Bush A, Conrad M, Dixon S, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20:1181–92.
Zhu S, Zhang Q, Sun X, Zeh H 3rd, Lotze M, Kang R, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77:2064–77.
Viswanathan V, Ryan M, Dhruv H, Gill S, Eichhoff O, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–7.
Hangauer M, Viswanathan V, Ryan M, Bole D, Eaton J, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–50.
Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U. ATF3 and stress responses. Gene Expr. 1999;7:321–5.
Zhao J, Li X, Guo M, Yu J, Yan C. The common stress responsive transcription factor ATF3 binds genomic sites enriched with p300 and H3k27ac for transcriptional regulation. BMC Genom. 2016;17:335.
Yan C, Lu D, Hai T, Boyd D. Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. EMBO J. 2005;24:2425–35.
Cui H, Guo M, Xu D, Ding Z-C, Zhou G, Ding H-F, et al. The stress-responsive gene ATF3 regulates the histone acetyltransferase Tip60. Nat Commun. 2015;6:6752.
Wang Z, He Y, Deng W, Lang L, Yang H, Jin B, et al. Atf3 deficiency promotes genome instability and spontaneous tumorigenesis in mice. Oncogene. 2018;37:18–27.
Gargiulo G, Cesaroni M, Serresi M, de Vries N, Hulsman D, Bruggeman SW, et al. In vivo RNAi screen for BMI1 targets identifies TGF-β/BMP-ER stress pathways as key regulators of neural- and malignant glioma-stem cell homeostasis. Cancer Cell. 2013;23:660–76.
Rosenberger C, Clark A, Treuting P, Johnson C, Aderem A. ATF3 regulates MCMV infection in mice by modulating IFN-γ expression in natural killer cells. Proc Natl Acad Sci USA. 2008;105:2544–9.
Hoetzenecker W, Echtenacher B, Guenova E, Hoetzenecker K, Woelbing F, Bruck J, et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat Med. 2012;18:128–34.
de Nardo D, Labzin LI, Kono H, Seki E, Schmidt SV, Beyer M, et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol. 2014;15:152–60.
Bambouskova M, Gorvel L, Lampropooulou V, Sergushichev A, Loginicheva E, Johnson K, et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ-ATF3 inflammatory axis. Nature. 2018;556:501–4.
Zmuda EJ, Qi L, Zhu MX, Mirmira RG, Montminy MR, Hai T. The roles of ATF3, an adaptive-response gene, in high-fat-diet-induced diabetes and pancreatic beta-cell dysfunction. Mol Endocrinol. 2010;24:1423–33.
Huang C, Chen J, Wu J, Tsai H, Lin H, Yan Y, et al. Novel link of anti-apoptotic ATF3 with pro-apoptotic CTMP in the ischemic brain. Mol Neurobiol. 2015;51:543–57.
Seijiffers R, Mills C, Woolf C. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci. 2007;27:7911–20.
Yan C, Wang H, Boyd D. ATF3 represses 72-kDa type IV collagenase (MMP-2) expression by antagonizing p53-dependent trans-activation of the collagenase promoter. J Biol Chem. 2002;277:10804–12.
Kim J-S, Bonifant C, BunZ F, Lane W, Waldman T. Epitope tagging of endogenous genes in diverse human cell lines. Nucleic Acids Res. 2008;36:e127.
Ran F, Hsu P, Wright J, Agarwala V, Scott D, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308.
Lv G, Sun D, Zhang J, Xie X, Wu X, Fang W, et al. Lx2-32c, a novel semi-synthetic taxane, exerts antitumor activity against prostate cancer cells in vitro and in vivo. Acta Pharm Sin B. 2017;7:52–8.
Liu D, Duong C, Haupt S, Montgomery K, HOuse C, Azar W, et al. Inhibiting the system Xc−/glutathone axis selectively targets cancers with mutant-p53 accumulation. Nat Commun. 2017;8:14844.
Yan C, Wang H, Boyd D. KiSS-1 represses 92-kDa type IV collagenase expression by down-regulating NF-κB binding to the promoter as a consequence of IκBα-induced block of p65/p50 nuclear translocation. J Biol Chem. 2001;276:1164–72.
Wolfgang CD, Liang G, Okamoto Y, Allen AE, Hai T. Transcriptional autorepression of the stress-inducible gene ATF3. J Biol Chem. 2000;275:16865–70.
Okamoto A, Iwamoto Y, Maru Y. Oxidative stress-responsive transcription factor ATF3 potentially mediates diabetic angiopathy. Mol Cell Biol. 2006;26:1087–97.
Kim K-H, Jeong J-Y, Surh Y-J, Kim K-W. Expression of stress-response ATF3 is mediated by Nrf2 in astrocytes. Nucleic Acids Res. 2010;38:48–59.
Jiang H-Y, Wek S, McGrath B, Lu D, Hai T, Harding H, et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol. 2004;24:1365–77.
Zhang C, Gao C, Kawauchi J, Hashimoto Y, Tsuchida N, Kitajima S. Transcriptional activation of the human stress-inducible transcriptional repressor ATF3 gene promoter by p53. Biochem Biophys Res Commun. 2002;297:1302–10.
Fan F, Jin S, Amundson SA, Tong T, Fan W, Zhao H, et al. ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene. 2002;21:7488–96.
Shimada K, Skouta R, Kaplan A, Yang W, Hayano M, Dixon S, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503.
Wolfgang C, Chen B, Martindale J, Holbrook N, Hai T. Gadd153/Chop10, a potential target gene of the transcriptional repressor ATF3. Mol Cell Biol. 1997;17:6700–7.
Gridasova A, Henry R. The p53 tumor suppressor protein represses human snRNA gene transcription by RNA polymerases II and III independently of sequence-specific DNA binding. Mol Cell Biol. 2005;25:3247–60.
Vousden KH, Prives C. Blinded by the light: the growing complexicty of p53. Cell. 2009;137:413–31.
Chung W, Lyons S, Nelson G, Hamza H, Gladson C, Gillespie G, et al. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25:7101–10.
Rosenberger CM, Clark AE, Treuting PM, Johnson CD, Aderem A. ATF3 regulates MCMV infection in mice by modulating IFN-{gamma} expression in natural killer cells. Proc Natl Acad Sci USA. 2008;105:2544–9.
Hasegawa M, Takahashi H, Rajabi H, Alam M, Suzuki Y, Yin L, et al. Functional interactions of the cystine/glutamate antiporter, CD44v, and MUC1-C oncoprotein in triple-negative breast cancer cells. Oncotarget. 2016;7:11756–69.
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc− activity. Curr Biol. 2018;28:2388–99.
Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, et al. Electrophile response element-mediated induction of the cystine/glutamate excahnge transporter gene expression. J Biol Chem. 2002;277:44765–71.
Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, et al. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol. 2014;34:3421–34.
Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu I, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 2017;36:5593–608.
Kumar A, Tikoo S, Maity S, Sengupta S, Sengupta S, Kaur A, et al. Mammalian proapoptotic factor ChaC1 and its homologues function as γ-glutamyl cyclotransferases acting specifically on glutathione. EMBO Rep. 2012;13:1095–101.
Crawford R, Prescott E, Sylvester C, Higdon A, Shan J, Kilberg MS, et al. Human CHAC1 protein degrades glutathione, and mRNA induction is regulated by the transcription factors ATF4 and ATF3 and a bipartite ATF/CRE regulatory element. J Biol Chem. 2015;290:15878–91.
Chen M, Wang S, Hsu C, Yin P, Yeh T, Lee H, et al. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget. 2017;8:114588–602.
Hartman MG, Lu D, Kim ML, Kociba GJ, Shukri T, Buteau J, et al. Role for activating transcription factor 3 in stress-induced β-cell apoptosis. Mol Cell Biol. 2004;24:5721–32.
Thompson MR, Xu D, Williams BR. ATF3 transcription factor and its emerging role in immunity and cancer. J Mol Med. 2009;87:1053–60.
Xue Y, Lim S, Yang Y, Wang Z, Jensen LDE, Hedlund E-M, et al. PDGF-BB modulates hematopoiesis and tumor angiognesis by inducing erythropoietin production in stromal cells. Nat Med. 2012;18:100–10.
Hua B, Tamamori-Adachi M, Luo Y, Tamura K, Morioka M, Fukuda M, et al. A splice variant of stress response gene ATF3 counteracts NF-κB-dependent anti-apoptosis through inhibiting recruitment of CREB-binding protein/p300 coactivator. J Biol Chem. 2006;281:1620–9.
Chen BPC, Liang G, Whelan J, Hai T. ATF3 and ATF3ΔZip: transcriptional repression versus activation by alternatively spliced isoforms. J Biol Chem. 1994;269:15819–26.
Gilchrist M, Thorsson V, Li B, Rust A, Korb M, Roach J, et al. Systems biology approaches identify ATF3 as a negative regulator of toll-like receptor 4. Nature. 2006;441:173–8.
Brasses-Lagnel C, Lavoinne A, Husson A. Contrl of mammlian gene expression by amino acids, especially glutamine. FEBS J. 2009;276:1826–44.
Kilberg M, Shan J, Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trend Endocrinol Metab. 2009;29:436–43.
Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, et al. A human protein-protein interaction network: a resource for annnotating the proteome. Cell. 2005;122:957–68.
Kawauchi J, Zhang C, Nobori K, Hashimoto Y, Adachi MT, Noda A, et al. Transcriptional repressor activating transcription factor 3 protects human umbilical vein endothelial cells from tumor necrosis factor-α-induced apoptosis through down-regulation of p53 transcription. J Biol Chem. 2002;277:39025–34.
Wu X, Nguyen B, Dziunycz P, Chang S, Brooks Y, Lefort K, et al. Opposing roles for calcineurin and ATF3 in squamous skin cancer. Nature. 2010;465:368–72.
Ou Y, Wang S, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci USA. 2016;113:E6806–12.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 cctivity. Cell Rep. 2017;20:1692–704.
Tarangelo A, Magtanong L, Bieging-Rolett K, Li Y, Ye J, Attardi L, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018;22:569–75.
Acknowledgements
This work was supported by the National Institutes of Health grants (R01CA139107, R01CA190429). We thank Dr. Nicholas J. Clemons for providing the SLC7A11 expression vector.
Author information
Authors and Affiliations
Contributions
LW, YL, HY, TD, and LL conducted the experiments; H-FD, JZ, HW, and XC analyzed the data, and provided insights into manuscript writing. CY designed the experiments, performed data analysis, and interpreted the data. CY and LW wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by E. Baehrecke
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., Liu, Y., Du, T. et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc–. Cell Death Differ 27, 662–675 (2020). https://doi.org/10.1038/s41418-019-0380-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-019-0380-z
This article is cited by
-
GPR68-ATF4 signaling is a novel prosurvival pathway in glioblastoma activated by acidic extracellular microenvironment
Experimental Hematology & Oncology (2024)
-
Irisin attenuates type 1 diabetic cardiomyopathy by anti-ferroptosis via SIRT1-mediated deacetylation of p53
Cardiovascular Diabetology (2024)
-
Mitochondria-mediated ferroptosis induced by CARD9 ablation prevents MDSCs-dependent antifungal immunity
Cell Communication and Signaling (2024)
-
Puerarin attenuates valproate-induced features of ASD in male mice via regulating Slc7a11-dependent ferroptosis
Neuropsychopharmacology (2024)
-
TRIM21/USP15 balances ACSL4 stability and the imatinib resistance of gastrointestinal stromal tumors
British Journal of Cancer (2024)
