
Abstract
The BH3-only class of Bcl-2 family proteins triggers mitochondrial apoptosis. Several mechanisms are used to restrain the pro-apoptotic activity of these proteins. Dynein light chain (DYNLL) 1 and 2 has been proposed to negatively regulate the activity of Bim and Bmf, respectively, and the Bim–DYNLL1 interaction leads to the formation of large protein complexes on mitochondria. Here we found that Bim and Bmf interact with both isoforms of DYNLL (DYNLL1 and DYNLL2). DYNLL1/2 not only induced homo-dimerization of Bim and Bmf but also led to the formation of ternary complexes (Bim–DYNLL–Bmf), both in cell-free and in cellular systems. DYNLL-induced oligomerization stabilized Bmf in cultured cells and inhibited its degradation by the ubiquitin-independent 20S proteasome in a cell-free system. Surprisingly, overexpression of wild-type Bmf but not of a DYNLL-binding-deficient mutant induced degradation of endogenous Bim in different cell lines, but both variants sensitized to apoptosis. Mutant Bmf incapable of interacting with anti-apoptotic Bcl-2 proteins and of inducing apoptosis still caused Bim degradation. These results suggest that Bmf overexpression-induced Bim degradation is not due to the displacement of Bim from anti-apoptotic Bcl-2 proteins but a direct consequence of the modulation of Bim–DYNLL association. A peptide derived from the DYNLL-binding domain of Bim also led to the degradation of Bim as well as of its preferred binding partner Mcl-1. Thus DYNLL regulates the mitochondrial pathway of apoptosis by determining the stability of Bmf, Bim, and Mcl-1 proteins.
Similar content being viewed by others


Introduction
Mitochondrial apoptosis is regulated by the Bcl-2 protein family. This family encompasses three functional groups of proteins: two effector molecules (Bax and Bak) can permeabilize the outer mitochondrial membrane, release cytochrome c and thereby activate caspases. A second pro-apoptotic group, known as BH3-only proteins, can activate Bax/Bak. Some BH3-only proteins can directly bind to Bax/Bak, while all of them can at least enhance apoptosis by inactivating anti-apoptotic Bcl-2 proteins. The anti-apoptotic Bcl-2 proteins can bind both Bax/Bak and BH3-only proteins and thereby inhibit apoptosis (reviewed in [1,2,3]).
How the Bcl-2 protein family integrates upstream signals to initiate mitochondrial permeabilization is still an area of considerable uncertainty. BH3-only proteins may be transcriptionally upregulated, for instance, by p53-activity [3]. There are, however, also instances where no changes in the expression levels of Bcl-2 family members are seen [4], suggesting posttranslational regulation. The BH3-only protein Bim that is important in the immune system [5] as well as for apoptosis in epithelial tumors [6] is a case in point. Bcl-2-modifying factor (Bmf) is another important BH3-only protein that has been implicated in B cell development [7], anoikis [8, 9], and in apoptosis induction during inhibition of CAP-dependent protein synthesis [10].
Studies of the isolated BH3 domains have reported that Bim is a more potent direct activator of Bax and Bak [11, 12] when compared to Bmf [13]. Bim is also stronger in terms of indirect activation of Bax/Bak, which occurs through the inactivation of anti-apoptotic Bcl-2 proteins [11, 14].
Intriguingly, similar functions of Bim and Bmf have been observed in a number of experimental systems. In neurons, both Bim [15, 16] and Bmf [17, 18] are involved in the regulation of apoptosis during kinase signaling. Extracellular signal–regulated kinase signaling has substantial effects on the activity of Bim [19] as well as on Bmf [20]. A HDAC4-RelB-p52 complex repressed both Bmf and Bim gene expression [21]. Both Bim and Bmf have been found to regulate B cell and hematopoietic stem cell survival [7, 22,23,24]. Molecularly, the two BH3-only proteins share another salient feature, namely, the ability to bind dynein light chain (DYNLL). By yeast-two-hybrid screening, Bim isolated DYNLL1 while Bmf preferentially isolated DYNLL2 clones [25, 26]. However, in pull-down assays using GFP-DYNLL1/2 and truncated Bim or Bmf, no binding preference was observed [27], which is not surprising since the two isoforms of DYNLL, DYNLL1 and DYNLL2, are almost identical. It has been initially suggested that DYNLL1 and 2 sequester Bim and Bmf to the microtubule and actin cytoskeleton-associated motor complexes, respectively. However, Bim localizes to mitochondria upon synthesis [28] and is largely found in the mitochondrial fraction [29]. The situation has not been as thoroughly investigated for Bmf although epitope-tagged Bmf was also found at mitochondria [10]. The view that DYNLL proteins act as cargo adaptor for the cytoskeleton-associated motor complexes is no longer considered correct; rather, DYNLL1/2 can bind two identical protein molecules and dimerize them [30, 31].
Nonetheless, DYNLL appears to play a role in regulating the activity of Bim and Bmf. The isoform of Bim (BimS) lacking the DYNLL-binding domain (DBD) has been shown to be more active [26]. Intriguingly, Bmf isoforms that lacked a functional BH3 domain but retained DYNLL-binding enhanced colony-forming potential of HeLa cells upon overexpression [32], suggesting an activity other than interaction with Bcl-2 protein families. Furthermore, conditional loss of the DYNLL1-regulating transcription factor ASCIZ or DYNLL1 leads to B cell lymphopenia due to enhanced Bim-dependent apoptosis [33, 34].
We have recently analyzed the Bim–DYNLL interaction at the molecular level [35]. However, the actual molecular activity of Bmf has not been investigated beyond the binding activity of its BH3 domain within the Bcl-2 family. In this study, we tested biophysical and biochemical characteristics of Bmf–DYNLL interaction in a cell-free system. In intact cells, we investigated the consequences of Bmf–DYNLL interaction in regulation of its turnover and its pro-apoptotic activity. Our data show that the role of DYNLL goes beyond homo-oligomerization of Bim and Bmf and identify ternary complexes of Bim–DYNLL–Bmf, extending the interactions in the Bcl-2 family to complexes of two different BH3-only proteins. Furthermore, we find that DYNLL-induced oligomerization is essential for the posttranslational stability of Bim and Bmf. Overexpressed Bmf destabilized Bim, probably by occupying both DYNLL-binding grooves.
Materials and methods
Cell culture
Bax−/− Bak−/− mouse embryonic fibroblasts (MEFs) immortalized with SV40 large-T antigen (provided by the lab of Dr. David Huang, Walter and Eliza Hall Institute, Melbourne) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), antibiotics (100 IU/mL penicillin G, 100 μg/mL streptomycin sulfate), and 50 μM β-mercaptoethanol. SV40 Immortalized Bmf−/− MEFs, 293FT, Phoenix and HeLa cells were cultured in DMEM supplemented with 10% FCS and antibiotics (100 IU/mL penicillin G, 100 μg/mL streptomycin sulfate). The different Hoxb8 neutrophil progenitor cells (wild type (wt), Bim−/−, Noxa−/−, Bim−/− Noxa−/−) were derived from bone marrow of C57BL/6-deficient mice as described earlier [36]. Hoxb8 neutrophil progenitor cells (Hoxb8 NPCs) were established by retroviral transduction of ER-Hoxb8 and cells were selected in the presence of stem cell factor. The transduced cell lines were cultured in Optimem medium (Invitrogen) supplemented with 10% FCS (Gibco), 30 mM β-mercaptoethanol (Sigma), antibiotics (100 IU/mL penicillin G and 100 µg/mL streptomycin sulfate), 1% supernatant from stem cell factor (SCF)-producing Chinese hamster ovary cells (gift from Dr. Hans Haecker, Memphis), and 1 μM β-estradiol (Sigma). The SCF withdrawal or differentiation assays with Hoxb8 NPCs were performed by either culturing the cells in a medium devoid of SCF or β-estradiol, respectively. All the cells were incubated in a CO2 incubator (37 °C at 5% CO2). The Mcl-1 inhibitor S63845 (SelleckChem) was dissolved in dimethyl sulfoxide and further diluted in the culture medium to the desired concentration.
Generation of plasmids, viral vectors, and cell lines
The genes coding for murine Bmf_short (referred to as Bmf here) were amplified by PCR and subcloned into pENTR/SD/D-TOPO Gateway vector (Life Technologies). Addition of N-terminal tags (HA-tag or V5-tag) to the Bmf constructs were achieved by overlap extension PCR followed by sub-cloning into pENTR/SD/D-TOPO. Mutations in the DYNLL binding (BmfT70A Q71A / BmfAA) or in the BH3 domain (Bmf I134E L138E I141E F145E/Bmf-BH3ΔΔ) of Bmf were done with the Q5® Site-Directed Mutagenesis Kit (NEB). The constructs were then sub-cloned into desired expression vectors via Gateway (GW) LR recombinase reaction (Life Technologies). The Lenti (pF5xUAS-SV40-Puro, pFhEF1α) or the retrovirus particles (pMIG-GW) were generated by transfecting 293FT or Phoenix cells, respectively, with specific packaging vectors. The culture supernatant containing the virus particles were collected and used for generation of different cell lines. For the constitutive expression in Bax−/− Bak−/− MEFs, the cells were transduced with pMIG-GW-retroviral vectors carrying different Bmf constructs (HA-Bmf/HA-BmfAA/HA-Bmf-BH3ΔΔ or HA-BmfAA-BH3ΔΔ). For the study of Bmf–Bmf or Bmf–Bim interactions, these cells were further transduced with lentiviral vectors carrying different V5-tagged Bmf (pFhEF1α-GW-SV40-puro-V5Bmf/BmfAA) or Bim mutants (pFhEF1α-GW-SV40-puro-V5BimEL/BimELAA). The tamoxifen (4-HT)-inducible V5-tagged Bmf/BmfAA cells were generated by transducing the Bmf−/− MEFs with pFU-G147EV16-PGK-Hygro and then with the lentiviral vector pF5xUAS-GW-SV40-Puro encoding the transgenes. Selection of cells was done using 800 µg/mL hygromycin (Invitrogen) and 5 μg/mL puromycin (InVivoGen). The Bmf constructs were induced by treating the cells with 400 nM 4-HT (Sigma) for the indicated periods of time. The Hoxb8 NPCs (wt, Noxa−/−, and Bim−/− Noxa−/−) stably expressing different Bmf constructs were transduced by the pMIG-GW retrovirus by centrifuging 100 × 103 cells in 1 mL of virus supernatant (supplemented with SCF, β-estradiol, and polybrene) at 1800 rpm for 90 min at 30 °C in a 12-well plate. Thereafter, 1 mL of fresh culture medium was added and cells were incubated in a CO2 incubator. The green fluorescent protein (GFP)-positive cells were then sorted after 1 week of culture and used for further experiments. For GFP-DBD/DBD-AA construct, GFP was amplified from pEGFP-N1 vector using GFP-specific forward primer. The coding sequences for the DBD or DBDAA are included in the reverse primers. 293FT cells were transfected using FuGENE® HD transfection reagent (Promega) following the manufacturer’s protocol. The sequences of the primer are enlisted in Table 1.
Immunoprecipitation (IP)
Anti-HA (anti-HA affinity matrix; Roche) or anti-V5 IP (anti-V5 agarose affinity matrix; Sigma) was performed with 500 µg of whole-cell lysates (lysis buffer: 20 mM Tris/HCl at pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, protease inhibitor mix [Roche]) of Bax−/− Bak−/− MEFs expressing the indicated Bmf or Bim constructs. To test the interaction between endogenous Bim with Bmf, anti-Bim and anti-Bmf IP were performed with 1.5 mg of whole-cell lysates from HeLa cells. Protein G-Agarose beads (Roche, Cat. no. 11719416001) were incubated with anti-Bim (C34C5, Cell Signaling) or anti-Bmf (ab181148, Abcam) antibodies along with cell lysate for 4 h at 4 °C. Anti-rabbit IgG (Cell Signaling) IP was taken as isotype control. Induction of Bim–Bmf complexes by DYNLL was determined by incubating 0.8 mM large unilamellar vesicles (LUVs) with different combinations of recombinant BimL (50 nM), Bmf (50 nM), and DYNLL1 or DYNLL2 (100 nM) for 30 min at 30 °C. Protein complexes were then solubilized in 1% digitonin buffer (20 mM Tris/HCl pH 7.4, 50 mM NaCl, 1% digitonin) for 15 min. Afterwards, samples were centrifuged and the supernatant was used to perform anti-Bim IP. Interaction between recombinant Bmf and DYNLL1 or DYNLL2 was tested by incubating 25 nM Bmf with 50 nM DYNNL1 or DYNLL2 in 1% digitonin buffer for 30 min at 30 °C. Samples were then used to perform anti-Bmf IP. All IP samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by western blotting.
Gel electrophoresis and western blotting
The cells were harvested by trypsinization, washed once with phosphate-buffered saline (PBS) and lysed in lysis buffer (20 mM Tris/HCl at pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, protease inhibitor mix [Roche]) for 30 min on ice. After the incubation, cells were centrifuged at 4800 rpm for 10 min. The supernatant were collected and proteins in the lysates were quantified using Bradford assay (Bio-Rad). The proteins were then denatured with 1× Laemmli buffer and by heating at 95 °C for 5 min. Equal amounts of protein samples were separated by SDS-PAGE and blotted on a polyvinylidene difluoride (PVDF) membrane using wet transfer. The membranes were blocked in 5% milk in TBS-T and probed with different primary antibodies overnight at 4 °C. Incubation with secondary antibody was done for 2 h at room temperature (RT).
For BN-PAGE with isolated mitochondria, the harvested cells were washed once with PBS and resuspended in MB-EDTA buffer for 15 min prior to fractionation as described before [28]. Mitochondrial fractions were prepared, and proteins were quantified by Bradford assay. Equal protein amounts were solubilized with BN-PAGE lysis buffer (20 mM Tris/HCl at pH 7.4, 50 mM NaCl, 0.1 mM EDTA, 1% digitonin, 1 mM PMSF) for 15 min on ice. The samples were then centrifuged at 10,000 × g and the supernatant was collected in a separate tube with 1× BN-PAGE loading dye. Samples were separated on a 6–16.5% polyacrylamide gradient gel followed by wet transfer to a PVDF membrane. The blotted proteins were then detected as described above.
Antibodies against Bim (Cell Signaling, no. C34C5), Bmf (Enzo Life Sciences, no. ALX-804–508, Abcam, no. ab181148 or clone 12E10, a gift from A. Strasser and L. O’Reilly), Bcl-XL (Cell Signaling, no. 54H6), Bcl-2 (Cell Signaling, no. 2870) Bax (Cell Signaling, no. 2772), tubulin (Sigma, no. T9026), DYNLL1 (Abcam, no. EP1660Y, Enzo, ALX-804–340), DYNLL2 (Merck, No. ABT140), Mcl-1 (Cell Signaling, no. 94296), Noxa (Abcam, ab13654), cytochrome c (BD, no. 556432), HA (Cell Signaling, no. C29F4), and V5 (Invitrogen, no. r960–25) were used as suggested by the manufacturers.
The primary antibodies were detected using horseradish peroxidase-conjugated secondary antibodies against anti-rabbit (Sigma), anti-mouse (Dianova), or anti-hamster (Dianova). The signals were detected using enhanced chemiluminescence reagent (GE Healthcare or Thermo Scientific) on an Intas Imaging system.
In situ proximity ligation assay (PLA)
PLA was performed with the Duolink In Situ Fluorescence Kit (OLINK). Briefly, 25,000 cells were seeded on coverslips in 24-well cell culture plates. After 24 h of culture, the cells were washed and fixed with 4% paraformaldehyde (PFA) for 10 min. After washing thrice with PBS, the cells were permeabilized with 0.1% Triton X-100/PBS, and blocked with 5% bovine serum albumin (BSA)/PBS. The cells were then incubated with the primary antibodies (V5 antibody 1:400; and α-HA antibody 1:1500) in 5% BSA/0.5% saponin/PBS for 1 h. Incubation with anti-mouse and anti-rabbit probe conjugated secondary antibodies and detection were performed according to the manufacturer’s instructions. Cells were analyzed with the appropriate filters in a Keyence BZ-9000 fluorescence microscope. The interaction spots were counted using the ImageJ program.
Cytochrome c release with isolated mitochondria
Mitochondria were isolated as described above and the cytochrome c release assay was performed with recombinant Bmf, BimL, and Bax as described earlier [28]. The recombinant human Bax protein was purified using a published protocol [37] and pTYB1-Bax as an expression vector. Purification strategy for full-length BimL has been described earlier [35]. Recombinant Bmf was purified following a similar strategy as described for BimL.
Labeling of recombinant proteins with fluorophore
Full-length recombinant Bmf (Atto488 or Atto655), BimL (Alexa488 or Atto655), and DYNLL1/2 (Atto655) were labeled by incubating the proteins overnight with ten-fold molar excess of the respective reactive dyes at 4 °C. The free dye and the salt were separated using a PD-10 desalting column packed with Sephadex G-25 resin (GE Healthcare). The fluorophores emitting in the green (Atto488 and Alexa488) and red channel (Atto655 and Alexa647) are denoted here with the letter “G” and “R,” respectively, after the name of the labeled proteins.
Solution fluorescence cross-correlation spectroscopy (FCCS)
For solution FCCS experiments, 40 nM of Bmf and BimL and 80 nM DYNLL1/2 were incubated in 200 μL of PBS (pH 7.4) for 30 min in a well of pre-blocked (1% BSA) Lab-Tek chambers (Nunc). Images were acquired on a LSM710 confocal microscope and each acquisition lasted 20 s (>10,000 times longer than the diffusion time of the labeled proteins) to ensure accumulation of sufficient data points for the generation of autocorrelation curves. The cross-correlation (CC) was calculated assuming three-dimensional (3D) diffusion of the proteins as described [38].
Calcein release assay
In order to perform calcein release experiments, we extruded LUVs as described above using a buffer containing 10 mM Tris pH 7.4, 0.5 mM EDTA, and 70 mM calcein (Sigma Aldrich). To remove excessive calcein, extruded LUVs were applied to a chromatographic column (Bio-Rad) loaded with CL-4B sepharose beads (Sigma Aldrich). Using an elution buffer containing 10 mM Tris pH 7.4, 0.5 mM EDTA, and 100 mM NaCl, they were then eluted in 0.5 mL fractions. Collected fractions were tested for release of the fluorophore by loading 50 µL of LUVs plus 150 µL of elution buffer with or without Triton X-100 on a pre-blocked (2 h with 5% BSA at RT) black 96-well plate (Thermofischer Scientific). Using a fluorescence plate reader (Tecan), samples were excited at 485 nm and analyzed with the Magellan software (Tecan), and fractions displaying at least 5× difference in signal strength between full release (Triton X-100 containing samples) and control state were selected for further experiments.
To assess the Bax-activating capacity of recombinant BimL and Bmf, calcein-loaded LUVs were incubated with a combination of 100 nM Bax and either 10 nM BimL or different amounts of Bmf (20, 50, and 100 nM). Release of calcein was measured every 2 min for a time period of 1 h, and every time point was displayed as a percentage of the fluorophore release caused by the 1% Triton X-100-containing buffer (full release).
Results
DYNLL binding causes dimerization and complex formation of Bmf
DYNLL1 and 2 are near-identical proteins (over 93% sequence identity), yet have been proposed to perform non-redundant functions in vivo. We first analyzed the interaction of Bmf with DYNLL1 or DYNLL2 in solution by FCCS. We produced full-length mouse Bmf, DYNLL1, and DYNLL2 in Escherichia coli using an expression and purification protocol described earlier [35]. Bmf labeled with Atto488 showed equal CC (%) with both Atto655-labeled DYNLL1 and DYNLL2, indicating a similar extent of complex formation. This interaction was not seen when Bmf with an inactivating mutation in the DYNLL interaction domain (BmfT70AQ71A/BmfAA) was used (Fig. 1a, Fig. S1). Since DYNLL1 causes Bim dimerization [35, 39], we tested whether the same may be the case for Bmf. As shown in Fig. 1b, a clear CC signal of Bmf labeled with two different dyes was obtained in the presence but not in the absence of recombinant DYNLL2, which indicates that DYNLL2 promotes Bmf dimerization. Again, Bmf homo-dimerization induced by DYNLL2 depended on the integrity of its DYNLL interaction domain (Fig. 1b). Changes in diffusion time (ζD) of the soluble proteins measured by FCCS also suggest complex formation. Indeed, addition of DYNLL2 significantly increased the diffusion time of Bmf but not of BmfAA (Fig. 1c). As expected, DYNLL1 also induced dimerization of Bmf in FCCS experiments (Fig. S2). We also performed IP experiments with the unlabeled recombinant proteins. Again, both isoforms of DYNLLL interacted with Bmf (Fig. S3). These results show that Bmf can interact with DYNLL1 and DYNLL2 in solution and that DYNLL can induce the formation of Bmf dimers.

Bmf forms homomeric complexes through interactions with either isoform of dynein light chain (DYNLL). a Quantification of cross-correlation (CC) between Bmf and DYNLL1 or DYNLL2. Solution FCCS experiments were performed with Bmf-Atto488 (Bmf-Green/Bmf-G) or BmfAA-Atto488 (BmfAA-G) with the two isoforms of DYNLL-Atto655 (DYNLL1-Red or DYNLL2-Red/DYNLL1/2-R). Bmf showed equal CC with both the isoforms of DYNLL. Mutation in the DYNLL-binding domain (Bmf-AA) abrogated this interaction. The results are calculated from three independent experiments (at least five runs per experiment) and shown as mean ± SD. Statistical significance were analyzed by two-way analysis of variance (ANOVA) using GraphPad. ****p < 0.0001; p > 0.05; ns: non-significant. Symbols show individual measurements. b, c Quantification of CC and diffusion time (ζD) of Bmf from solution FCCS experiments. b Percentage of CC between Bmf-G and Bmf-Red (Bmf-R) or BmfAA-R was calculated in the presence or absence of DYNLL2. Addition of DYNLL2 significantly enhanced the CC between Bmf-G and Bmf-R but had no effect on the CC between Bmf-G and BmfAA-R. c Diffusion time (ζD) of Bmf was analyzed in the presence or absence of DYNLL2 for the red channel. Diffusion time of Bmf-R but not of the BmfAA-R increased in the presence of DYNLL2, suggesting its oligomerization in the presence of DYNLL2. The data are calculated from three independent experiments with at least five runs per experiment. The data are shown as mean ± SD. ****p < 0.0001, one way ANOVA using GraphPad. Symbols show individual measurements. d–f DYNLL-dependent oligomerization of Bmf in mouse embryonic fibroblasts (MEFs). Anti-HA immunoprecipitation (IP) (d) or in situ proximity ligation assay (PLA) (e, f) was performed with Bax−/− Bak−/− MEFs constitutively expressing HA-tagged Bmf with either V5-tagged Bmf or Bmf-AA. d The IP fraction shows the co-precipitation of V5-Bmf with HA-Bmf but not of V5-BmfAA. Data are representative of three independent experiments. e Fluorescence microscopic images showing the proximity of Bmf molecules using PLA. Bax−/− Bak−/− MEFs constitutively expressing HA-tagged Bmf with either V5-tagged Bmf or Bmf-AA were probed with anti-HA and anti-V5 antibodies, followed by labeling with secondary antibodies and visualization. Cells expressing HA-Bmf only acted as a control for the assay. Yellow dots represent interaction spots. Images were acquired under identical conditions of magnification (×40) and exposure times and are representative of three independent experiments. Bars, 20 μm. f Quantification of interaction spots from the microscopic images. Interaction spots were counted from 300 cells from three independent PLA experiments using ImageJ. The fold change in the number of interaction spots was calculated (interaction spots in V5-Bmf-overexpressing cells/V5-BmfAA). The results are represented as mean ± SD. **p < 0.01, two-tailed unpaired Student’s t test. (wt: wild type V5-tagged Bmf; mut: mutant V5-tagged Bmf-expressing cells). g, h DYNLL binding leads to the formation of large Bmf complexes on synthetic membranes (g) and on mitochondria (h). g Recombinant murine Bmf or BmfAA (50 nM) were incubated with large unilamellar vesicles (LUVs) in the absence or presence of DYNLL1 (100 nM) for 30 min at 30 °C. LUVs were pelleted by centrifugation and solubilized in 1% digitonin buffer, loaded onto a 6–16.5% gradient BN gel, and probed for Bmf. Data are representative of three independent experiments. h BN-PAGE was performed with solubilized mitochondrial fractions (1% digitonin buffer) from Bax−/− Bak−/− MEFs overexpressing HA-tagged Bmf or BmfAA. The complexes in wild-type Bmf-expressing cells ran >150 kDa while mutant Bmf formed relatively lower-order complexes (<66 kDa)
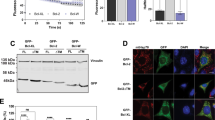
To test for such interaction in intact cells, we co-expressed HA-tagged Bmf with V5-tagged Bmf or BmfAA in MEFs (Bax−/− Bak−/− cells were used to prevent apoptosis). IP of HA-Bmf showed clear co-precipitation of V5-tagged Bmf but not of BmfAA and vice versa (Fig. 1d, Fig. S4). A considerably higher amount of DYNLL was co-precipitated with HA-Bmf in the cells co-expressing V5-tagged wt Bmf. To probe for DYNLL in western blots, we used an antibody raised against DYNLL1. However, experiments with recombinant DYNLL1 and DYNLL2 proteins suggested that, although antibodies raised against either isoform are available, they lack specificity in western blot experiments (Fig. S5). Therefore, we cannot be certain of the isoform precipitated from these cells. In a complementary approach, we also assessed the proximity of two Bmf molecules by microscopy using in situ proximity ligation assay (PLA). A strong PLA signal (higher number of interaction spots) was observed in cells expressing HA and V5-tagged wt Bmf as compared to cells expressing V5-BmfAA (Fig. 1e, f). The data indicate that Bmf forms homomeric complexes both in solution and in intact cells through the interaction with DYNLL1/2.
In the case of Bim, binding to DYNLL1 induces the formation of larger-order complexes. We tested the same for Bmf and found that indeed DYNLL1 induced the formation of large molecular weight Bmf complexes on LUV as analyzed by blue native PAGE (BN-PAGE) (Fig. 1g). In mitochondrial fractions isolated from Bax−/− Bak−/− MEFs, also comparatively large Bmf complexes (≈200 kDa) were seen in wt Bmf-overexpressing cells when compared to BmfAA (Fig. 1h). Unexpectedly, complexes of around 500 kDa were noticed in both Bmf and BmfAA-overexpressing cells. Therefore, it appears that in intact cells BmfAA still has some residual binding to DYNLL or it gets recruited to the large complexes indirectly via interaction with other binding partners, most likely other Bcl-2 family proteins.
DYNLL induces the formation of ternary complexes containing Bim and Bmf
Since DYNLL1-induced homo-dimerization both of Bim [35] and Bmf, it seemed possible that DYNLL binding could also induce hetero-dimers containing both Bim and Bmf. Hence, we first established that Bim bound not only DYNLL1 but also DYNLL2. As expected, similar FCCS signals were obtained between BimL and either DYNLL1 or DYNLL2 (Fig. 2a). When tested by FCCS, hetero-dimerization of Bim and Bmf was very clearly seen in the presence of DYNLL2 (Fig. 2b) as well as with DYNLL1 (data not shown). Consistently, IP performed with untagged recombinant proteins also showed DYNLL1- or 2-induced Bim–Bmf hetero-dimerization (Fig. S6). Co-IP experiments from Bax−/− Bak−/− MEFs gave similar results. Overexpressed HA-Bmf pulled down wt V5-tagged BimEL but not BimELAA and vice versa (Fig. 2c, Fig. S7). Overexpressed HA-tagged wt Bmf but not BmfAA co-precipitated endogenous Bim in Bax−/− Bak−/− MEFs (Fig. 2d). In addition, Bmf co-precipitated endogenous BimEL in thymic extracts from BCL-2 transgenic mice (Fig. 2e) and Bmf IP–liquid chromatography/mass spectrometric analysis using Ba/F3 cells overexpressing Bmf plus Bcl-2 identified in addition to DYNLL1/2-derived peptides also peptides covering Bim (Table S1). PLA in MEFs also detected significantly higher number of interaction spots between HA-Bmf and V5-BimEL as compared to BimELAA (Fig. 2f, g). We further performed IP experiments in Bmf−/− MEFs reconstituted with V5-Bmf/BmfAA under a tamoxifen-inducible promoter. Again, endogenous Bim was co-precipitated with wt but not with BmfAA-expressing cells (Fig. 2h). Finally, in HeLa cells, endogenous Bim co-precipitated endogenous Bmf and vice versa (Fig. 2i). To rule out the role of BH3 domain in Bim–Bmf hetero-dimerization, we performed IP experiments with Bax−/− Bak−/− MEFs overexpressing Bmf with four inactivating mutations in its BH3 domain (Bmf-BH3ΔΔ or BmfAA-BH3ΔΔ). Consistently, Bim was co-precipitated in cells expressing Bmf with intact DBD (Fig. S8). Overall, these results identify the existence of complexes containing both Bim and Bmf, which require the binding to DYNLL. They further confirm that Bim and Bmf do not show any specificity in binding to the two isoforms of DYNLL, at least in vitro.

Dynein light chain (DYNLL) binding leads to the formation of hetero-oligomeric complexes of Bim and Bmf in solution and in intact cells. a BimL interacts with both DYNLL isoforms. Dot plot showing the quantification of cross-correlation (CC) from solution FCCS experiments with BimL-Alexa488 (Bim-Green/BimL-G) and Atto655-labeled DYNLL molecules (DYNLL1-R or DYNLL2-R). No significant difference was observed in the interaction of BimL with DYNLL1 or DYNLL2. The results are calculated from three independent experiments (at least five runs per experiment) and shown as mean ± SD. (ns: non-significant) p > 0.05, two-tailed paired Student’s t test. Symbols show individual measurements. b Dot plot showing the quantification of cross-correlation from solution FCCS experiments with BimL-Alexa647 (BimL-R) and Atto488-labeled Bmf (Bmf-G) in the presence or absence of unlabeled DYNLL2. The CC between Bim and Bmf increased significantly after the addition of DYNLL2 indicating DYNLL-dependent hetero-oligomerization of the two proteins. The results are calculated from three independent experiments (at least five runs per experiment) and shown here as mean ± SD. ****p < 0.0001, two-tailed paired Student’s t test. Symbols show individual measurements. c Anti-HA IP was performed with the whole-cell lysates of Bax−/− Bak−/− mouse embryonic fibroblasts (MEFs) constitutively expressing HA-tagged Bmf with V5-tagged BimEL or BimELAA. The immunoprecipitated (IP) fraction shows that only V5-BimEL but not the V5-BimELAA was co-precipitated with HA-Bmf. Data are representative of three independent experiments. d Anti-HA IP was performed with whole-cell lysates of Bax−/− Bak−/− MEFs constitutively expressing either HA-tagged Bmf or BmfAA. The IP fraction shows the co-precipitation of endogenous Bim and DYNLL1/2 with HA-Bmf but not with HA-BmfAA. Data are representative of three independent experiments. e Anti-Bmf IP was performed with thymic lysates from a vav-Bcl-2 transgenic mouse. The IP fraction shows the co-precipitation of endogenous Bim with Bmf. Anti-IgG IP was used as an isotype control. f Fluorescence microscopic images showing the in situ interaction of Bim and Bmf by proximity ligation assay (PLA). PLA was performed in Bax−/− Bak−/− MEFs constitutively expressing HA-tagged Bmf with either V5-tagged BimEL or BimELAA. Cells were probed with anti-HA and anti-V5 antibodies and PLA was performed as per the manufacturer’s protocol. HA-Bmf-only-expressing cells stained with anti-HA and anti-V5 antibodies were used as a negative control. The yellow dots represent the interaction spots. Images were acquired under identical conditions of magnification (×40) and exposure times and are representative of three independent experiments. Bars, 20 μm. g Quantification of interaction spots from the microscopy images of PLA. Interaction spots were counted from 300 cells from three independent experiments using ImageJ program. The fold change in the number of interaction spots was calculated (average interaction spots per cell in V5-BimEL-overexpressing cells/V5-BimELAA). (wt: wild type V5-tagged BimEL; mut: mutant V5-tagged BimEL expressing cells). The results are represented as mean ± SD. **p < 0.01, two-tailed unpaired Student’s t test. h Anti-V5 IP was performed with the whole-cell lysates of Bmf−/− MEFs reconstituted with V5 tagged-Bmf or BmfAA under the control of a tamoxifen (4-HT)-inducible promoter. Cells were treated with 4-HT (400 nM for 24 h) for the induction of Bmf. The IP fraction shows the co-precipitation of endogenous Bim with HA-Bmf but not with HA-BmfAA. The input and unbound fractions are 10% of the IP fraction. i Anti-Bim or Bmf IP was performed with the whole-cell lysates of HeLa cells to determine the interaction between endogenous Bim and Bmf. The IP fraction shows the co-precipitation of endogenous Bim with Bmf and vice versa. Anti-IgG IP was used as an isotype control. The input and unbound fractions are 10% of the IP fraction
DYNLL was initially proposed to induce formation of ternary complexes with two structurally different binding partners. However, later studies questioned this model based on the reduced thermostability of such complexes. Interestingly, our results here show existence of DYNLL-dependent complexes containing both Bim and Bmf. These two proteins belong to the BH3-only subfamily of Bcl-2 proteins and they share structural elements beyond the DBD. Therefore, it seems possible that Bim and Bmf can occupy the two opposing binding grooves of DYNLL without destabilizing the complex.
DYNLL binding determines interaction of Bmf with anti-apoptotic Bcl-2 proteins
We next analyzed the functional consequences of Bmf–DYNLL interaction. In the case of Bim, one consequence of its interaction with DYNLL1 is the much higher binding efficiency of Bim to anti-apoptotic Bcl-2 family proteins [35]. Indeed, we also observed that overexpressed Bmf was more efficient in co-immunoprecipitating endogenous Bcl-XL, Bcl-2, Bcl-w, and Mcl-1 than BmfAA (Fig. 3a, b). Since we cannot be certain from this experiment whether the binding to anti-apoptotic proteins was direct to Bmf or through the association of Bmf with Bim, we performed the same experiment with cells overexpressing Bmf with inactivating mutations in its BH3 domain (Bmf-BH3ΔΔ or BmfAA-BH3ΔΔ). These mutations disrupted the binding of Bmf to anti-apoptotic Bcl-2- proteins while the binding to Bim through DYNLL remained intact (Fig. S8). Bmf thus binds to anti-apoptotic Bcl-2 family proteins directly, while being part of a complex with Bim.

Bmf–dynein light chain (DYNLL) interaction increases the efficiency of co-precipitation of anti-apoptotic Bcl-2 proteins with Bmf. a, b Anti-HA immunoprecipitation (IP) was performed from whole-cell lysates of Bax−/− Bak−/− mouse embryonic fibroblasts stably expressing either HA-tagged Bmf or BmfAA. The IP fraction compares the co-precipitation of endogenous Bcl-XL, Bcl-2, Bcl-w, DYNLL1/2 (a), and Mcl-1 (b) between HA-Bmf- and HA-BmfAA-expressing cells. Data are representative of three independent experiments
DYNLL-induced oligomerization of Bmf determines its stability
Structural analysis of Bmf suggests that it contains intrinsically unstructured/disordered areas. Many intrinsically disordered proteins are recognized and degraded in an ubiquitin-independent pathway by the 20S rather than the 26S proteasome [40]. The two intrinsically disordered Bcl-2 proteins, Bim and Mcl-1, have previously been shown to be degraded via the 20S proteasome [41, 42]. We have also reported that loss of DYNLL1 induces proteasomal degradation of Bim and Mcl-1 [35]. This suggested the possibility that DYNLL binding could also determine the stability of Bmf. We tested this first by directly comparing the turnover of stably overexpressed Bmf and BmfAA in MEFs. At steady state, no obvious difference in the cellular levels of Bmf and BmfAA was noticed (Fig. 4a, b). However, the cellular levels of BmfAA were substantially reduced when compared to wt Bmf at 6 h of cycloheximide blockade of protein synthesis indicating increased stability of Bmf through complex formation.

Dynein light chain (DYNLL)-binding determines the stability of Bmf protein in cellular and in cell-free assays. a Western blot comparing the turnover of HA-Bmf and HA-BmfAA in the presence of translation inhibitor cycloheximide. Cell lysates were prepared from Bax−/− Bak−/− mouse embryonic fibroblasts constitutively expressing either HA-tagged Bmf or BmfAA upon treatment with cycloheximide (CHX, 15 µg/mL) for 2, 4, or 6 h. b Quantification of band densities from three independent western blotting experiments (as in Fig. 4a) showing almost eight-fold higher HA-Bmf as compared to HA-BmfAA after 6 h of CHX treatment. The band densities were quantified using ImageJ and normalized to the loading control GAPDH. Bar diagram represents fold change (mean ± SD) in the normalized band densities of HA-Bmf in comparison to HA-BmfAA. **p < 0.01, two-tailed unpaired Student’s t test. c, d Western blot comparing the degradation of recombinant wild-type and DYNLL-binding mutant Bmf in an in vitro 20S proteasome degradation assay. Recombinant full-length murine Bmf or BmfAA (25 nM) were incubated with 20S proteasome (1 µL) either in the absence or presence of recombinant DYNLL2 (100 nM) at 37 °C for the indicated periods of time. Samples were denatured by addition of Laemmli buffer and heated for 5 min at 95 °C. c In the absence of DYNLL2, Bmf and BmfAA were equally susceptible to degradation by 20S proteasome. d Addition of DYNLL2 inhibited the degradation of Bmf but not of BmfAA
We further confirmed the role of DYNLL in Bmf stability by in vitro 20S proteasome-mediated protein degradation assay. Interestingly, recombinant Bmf and BmfAA were equally susceptible to degradation by recombinant 20S proteasome in vitro (Fig. 4c). The addition of DYNLL2, however, inhibited degradation of wt Bmf by 20S proteasome but no such effect was seen for BmfAA (Fig. 4d). This indicates that the DYNLL-induced complex formation indeed plays a role in determining the turnover of Bmf.
Bmf can regulate the expression levels of Bim by a mechanism depending on DYNLL binding
These and previous [35] results show that DYNLL1/2-induced oligomerization of Bmf and Bim determines their posttranslational stability. Since Bmf and Bim can be present in the same complex, we hypothesized that these proteins might negatively regulate the cellular levels of each other in a DYNLL-binding-dependent manner. This would also suggest that apoptosis regulated by either BH3-only protein may be affected by the levels of the other. When we tested for protein levels, we made the surprising observation that the constitutive overexpression of Bmf but not of BmfAA led to a clear reduction in endogenous BimEL protein levels in Bax−/− Bak−/− MEFs and in wt Hoxb8 NPCs (Fig. 5a, b). To further confirm this, we reconstituted Bmf−/− MEFs with either V5-tagged Bmf or BmfAA under the control of a tamoxifen-inducible promoter. We have previously noticed that tamoxifen treatment induces endogenous Bim expression in MEFs (unpublished observation). As shown in Fig. 5c, tamoxifen treatment caused induction of Bmf along with the endogenous Bim in these cells. However, again the levels of endogenous Bim were lower in Bmf-expressing cells as compared to control or BmfAA-expressing cells. Proteasome inhibition partially restored Bim levels in Bmf-overexpressing cells, confirming that the loss of Bim is primarily due to enhanced proteasomal degradation (Fig. 5c).

Bmf expression induces the degradation of BimEL in a dynein light chain-binding-dependent manner. a, b Western blots comparing the endogenous Bim levels in a Bax−/− Bak−/− mouse embryonic fibroblasts (MEFs) or in b wild-type Hoxb8 neutrophil progenitor cells (NPCs) constitutively expressing HA-tagged or untagged Bmf variants, as indicated. Bmf but not of BmfAA expression led to the loss of endogenous BimEL. Uninfected Bax−/− Bak−/− MEFs or wild-type Hoxb8 NPCs served as a control for the assay. Data are representative of three independent experiments. Tubulin served as a loading control. c Bim destabilization induced by Bmf expression is reduced by the proteasome inhibitor MG-132. Western blots were performed with the whole-cell lysates of Bmf−/− MEFs expressing V5-tagged Bmf or BmfAA under the control of a tamoxifen (4-HT)-inducible promoter. Uninfected Bmf−/− parental cells were used as control. Cells were treated with 4-HT (400 nM) for 24 h to induce Bmf expression. Note that treatment with 4-HT also induces the expression of endogenous Bim in these cells. Levels of BimEL in uninfected control and in BmfAA-expressing cells were higher compared to Bmf-expressing cells. Inhibition of the proteasome with MG-132 stabilized Bim in these cells, suggesting a role of the proteasome in Bim loss
Interaction of Bim with anti-apoptotic Bcl-2 proteins has been proposed to stabilize Bim [43, 44]. Bmf-induced Bim loss, therefore, could be due to the displacement of Bim from anti-apoptotic Bcl-2 proteins. We tested this possibility by expressing a Bmf mutant with four inactivating mutations in its BH3 domain in Noxa−/− Hoxb8 NPCs (Bmf-BH3ΔΔ). As expected, these mutations almost completely abolished the binding of Bmf to anti-apoptotic Bcl-2 proteins in Bax−/− Bak−/− MEFs (Fig. S8). We chose Noxa−/− cells because in these cells SCF withdrawal-driven apoptosis is strongly Bim dependent, making it possible to determine the role of overexpressed Bmf on the activity of Bim.
We then compared the effect of Bmf or Bmf-BH3ΔΔ expression on endogenous BimEL levels in Noxa−/− Hoxb8 NPCs. Both Bmf and Bmf-BH3ΔΔ destabilized BimEL in a DYNLL-binding-dependent mechanism (Fig. 6a, b). However, the loss of BimEL was higher in cells overexpressing Bmf as compared to Bmf-BH3ΔΔ-expressing cells (Fig. 6c, d). When we directly compared the Bmf levels, we observed that Bmf-BH3ΔΔ was expressed at substantially lower levels compared to Bmf/BmfAA (Fig. 6d). This suggests that the observed difference in reducing Bim levels between Bmf and Bmf-BH3ΔΔ was probably due to the unequal expression levels of these proteins. The difference in Bmf levels may be due to enhanced stability of intact Bmf through binding to anti-apoptotic Bcl-2 proteins.

Bmf-overexpression-induced BimEL degradation is not due to enhanced interaction of Bmf with anti-apoptotic Bcl-2 proteins. a–d Western blotting was performed with lysates of Noxa−/− Hoxb8 NPCs constitutively expressing the HA-tagged Bmf/BmfAA without (a) or with inactivating mutations in the BH3 domain (b, labeled as Bmf-BH3ΔΔ). Substantial loss of endogenous BimEL was noticed only in Bmf-overexpressing cells (Bmf or Bmf-BH3ΔΔ) but not in control or in dynein light chain (DYNLL)-binding mutant Bmf (BmfAA or BmfAA-BH3ΔΔ). The cellular levels of the anti-apoptotic proteins Bcl-2 or Mcl-1 were not affected by Bmf overexpression. Data are representative of three independent experiments. Shown are the four amino acid residues (highlighted in red) in the BH3 domain of Bmf that were mutated to inhibit Bmf interaction with other Bcl-2 family members (BH3ΔΔ). c Normalized band intensities of BimEL against tubulin from western blotting experiments (as in Fig. 6d) using ImageJ program. Data are represented as mean ± SD of four independent experiments and statistical significance was analyzed by two-way analysis of variance (ANOVA) using GraphPad. ****p < 0.0001, **p < 0.01. d Cellular levels of Bmf and Bim in Noxa−/− Hoxb8 NPCs constitutively expressing different Bmf mutants, as indicated. e–g Transient overexpression of a 17 amino acid residue peptide harboring the Bim DYNLL-binding domain (DBD) fused to the carboxy terminus of green fluorescent protein (GFP) (GFP-DBD) binds to DYNLL and induces Bim and Mcl-1 degradation. e Anti-GFP immunoprecipitation (IP) was performed from lysates of HEK cells transiently expressing GFP-DBD or GFP-DBDAA. The image in the inset shows the sequence of DBD and DBDAA. The conserved residues for DYNLL binding are marked by the blue box while the two amino acid residues substituted in the mutant are highlighted in red color. Co-precipitation of DYNLL1/2 with GFP-DBD confirmed that the GFP-fused sequence interacts with DYNLL1/2. f Input samples for the IP show loss of endogenous Bim and Mcl-1 only in GFP-DBD transfected but not in GFP or in GFP-DBDAA-transfected cells. g Band intensities (normalized against tubulin) of BimEL and Mcl-1 from western blotting experiments using ImageJ program. Data are shown as the mean ± SD of three independent experiments and statistical significance was analyzed by two-way ANOVA using GraphPad. **p < 0.01, *p < 0.05, (ns) p > 0.05
The destabilization of Bim by Bmf therefore requires Bmf recruitment into DYNLL-coordinated complexes but seems independent of Bmf binding to anti-apoptotic Bcl-2 proteins. Finally, when transiently expressed, the DBD of Bim (17 amino acids fused to carboxy-terminus of GFP; GFP-DBD) in 293FT cells was sufficient to interact with DYNLL1/2 and to induce BimEL degradation (Fig. 6e–g). We also observed destabilization of Mcl-1 in GFP-DBD expressing cells (Fig. 6f, g). Overall, these results support our hypothesis that DYNLL-induced complex formation is a mechanism to stabilize Bim and Bmf. The data are consistent with the interpretation that, when Bmf outcompetes Bim in DYNLL-coordinated complexes, Bim is degraded by the proteasome.
Bmf overexpression sensitizes cells to apoptotic stimuli through a DYNLL-binding-independent mechanism
We used the cellular system of Hoxb8 NPCs to test for the pro-apoptotic potential of Bmf and the requirement for DYNLL binding as well as for binding to anti-apoptotic Bcl-2 proteins. These cells depend on the presence of SCF and die rapidly when it is withdrawn (20–30% cell death after 6 h, around 90% after 24 h, Fig. 7a). Overexpression of Bmf in wt Hoxb8 NPCs showed a non-significant trend to faster cell death; this difference was slightly greater and significant (at 6 h) when BmfAA was expressed (Fig. 7a). Deficiency of both Bim and Noxa provides good protection against this form of cell death [36] (Fig. 7b; expression levels are shown in Fig. S9). In Noxa-deficient cells, both Bmf and BmfAA enhanced sensitivity to SCF-withdrawal-induced cell death to the same degree (Fig. 7c). Bmf also sensitized to Mcl-1-inhibition and to cell death induced during differentiation but only minor differences between cells expressing Bmf and BmfAA were observed (Fig. S10). Several studies have shown that loss of DYNLL function (either by altering its cellular levels or by targeting its binding groove) sensitizes cells to apoptosis. DYNLL is a hub protein that regulates several vital cellular functions. Therefore, it is still possible that loss of DYNLL leads to the activation of upstream signaling pathways that stabilizes Bim/Bmf in the absence of complex formation. Indeed, c-Jun N-terminal kinase-mediated phosphorylation of Bim (also Bmf) has been proposed to disrupt Bim–DYNLL interaction as well as to regulate its turnover [45, 46].

Bmf or BmfAA expression sensitizes Hoxb8 neutrophil progenitor cells (NPCs) to stem cell factor (SCF)-withdrawal-induced apoptosis to a similar extent. Apoptosis was induced by SCF withdrawal in wild-type (a), Bim−/− Noxa−/− (b), or Noxa−/− (c) Hoxb8 NPCs constitutively expressing either untagged or HA-tagged Bmf/BmfAA, as indicated. Cells were stained with propidium iodide (PI) and analyzed by flow cytometry after 6 and 24 h of SCF withdrawal. d Bmf-induced cell death in Noxa−/− cells require its intact BH3 domain. Apoptosis was induced by SCF withdrawal in Noxa−/− Hoxb8 NPCs constitutively expressing the BH3 domain mutant Bmf (Bmf-BH3ΔΔ) or BmfAA (BmfAA-BH3ΔΔ). No significant difference in cell death was noticed between cells overexpressing Bmf-BH3ΔΔ or BmfAA-BH3ΔΔ cells when compared to the vector control cells. Data are the mean ± SD of three independent experiments and statistical significance was analyzed by two-way analysis of variance using GraphPad. ****p < 0.0001, ***p < 0.001, **p < 0.01, (ns) p > 0.05. e, f Bmf is unable to directly activate Bax. e Release of cytochrome c through Bax activation by Bmf on isolated mitochondria. Mitochondria isolated from Bax−/− Bak−/− mouse embryonic fibroblasts were incubated with recombinant Bax (100 nM) with the indicated concentrations of Bmf at 30 °C for 30 min. Mitochondria were pelleted by centrifugation. Pellet and the supernatant fractions were subjected to western blotting. Cytochrome c was detected in pellet (retained in mitochondria) and supernatant (released) fractions. Incubation with BimL and Bax served as a positive control. Results are representative of three independent experiments. f Inability of Bmf to permeabilize liposomes through direct activation of Bax. Calcein-loaded large unilamellar vesicles (LUVs) were incubated with recombinant Bax (100 nM) together with increasing concentrations of Bmf (20, 50, 100 nM) or with BimL (10 nM). Membrane permeabilization was monitored as the increase in fluorescence due to the release of quenched calcein from the LUVs over time. Data (mean ± SD) are represented as the percentage of the calcein released by the positive control (Triton X-100-treated LUVs)
Only Bmf but not BmfAA has an effect on Bim levels. Since the cells expressing either Bmf variant showed the same cell death sensitivity, this suggested that Bmf itself exerted the additional killing effect. To test this further, we expressed Bmf or BmfAA with inactivating mutations in the BH3 domain (Bmf-BH3ΔΔ) in Noxa-deficient cells. Indeed, these mutants of Bmf showed no sensitizing effect (Fig. 7d). To test directly for the killing activity of Bmf, we expressed Bmf or BmfAA in Bim/Noxa-double-deficient Hoxb8 NPCs. Intriguingly, at least in the absence of Noxa, Bmf had even stronger sensitizing activity than Bim (Fig. 7b; compare results of Bmf in Fig. 7b with the control cells in Fig. 7c). Again, no difference between Bmf and BmfAA was seen.
Recombinant Bmf protein can activate Bax but only indirectly
Bmf on its own thus had clear pro-apoptotic activity, which required its BH3 domain. As discussed above, it is not clear whether full-length Bmf can cause apoptosis only through inactivation of anti-apoptotic Bcl-2 proteins or through direct activation of Bax/Bak. Here we tested this by assessing membrane permeabilization using recombinant Bmf and Bax proteins. Cytochrome c release was tested on mitochondria isolated from Bax−/− Bak−/− MEFs. Addition of 40 or 80 nM of recombinant Bmf together with a constant concentration of Bax caused the release of a minor fraction of cytochrome c (Fig. 7e). Lower amounts of Bim (10 nM) were sufficient to cause complete release of cytochrome c (Fig. 7e). When calcein release from LUVs was tested, Bim efficiently activated recombinant Bax, as measured by the release of calcein from LUVs. No Bax-activating activity of Bmf was detected in this assay, even at ten-fold higher concentrations than those required for Bim to achieve near-complete calcein release (Fig. 7f). The main difference between the two systems is the presence of anti-apoptotic proteins on mitochondria but not on LUVs. The results therefore suggest that Bmf can activate Bax indirectly, through inactivation of anti-apoptotic Bcl-2 proteins, but not directly.
Discussion
Our analysis of Bmf–DYNLL interaction provides insight into the roles of DYNLL in the regulation of mitochondrial apoptosis. The DYNLL-binding activity of Bmf has long been known [25, 27], and the recent description of Bim complex formation by DYNLL1 suggested that Bmf might be subject to regulation similar to Bim. The finding of Bim–DYNLL–Bmf ternary complexes extends our understanding of complexes within the Bcl-2 protein family.
DYNLL1 and DYNLL2 are virtually identical proteins, with DYNLL1 usually expressed at much higher levels [33, 47]. As predicted from nuclear magnetic resonance (NMR) studies of truncated Bmf (and Bim) [27], we found no difference in binding of DYNLL1 or DYNLL2 to Bim or Bmf. Therefore, it appears that the in vivo function of DYNLL1 and DYNLL2 mainly depends on their spatio-temporal expression patterns. Intriguingly, Bmf was identified as interacting with Bim through binding to DYNLL, indicating that it is incorporated into Bim complexes in intact cells. DYNLL was initially proposed to induce the formation of ternary complexes with two structurally different binding partners. However, later studies questioned the stability of such complexes, and the current view is that DYNLL only induces the formation of stable homo-dimers. Bim and Bmf are similar proteins, and it may therefore be possible that DYNLL can induce hetero-dimerization of these proteins. We found that IP against BmfAA could not isolate Bim, and an anti-BimAA IP could not precipitate Bmf. The competition for DYNLL binding suggested by the Bmf-induced loss of Bim (which depended on an intact DYNLL-binding site) also argues that both DYNLL-binding sites are required: this suggests that indeed ternary complexes of Bim, Bmf, and DYNLL are formed. We cannot, however, formally exclude the possibility that complex formation occurs through additional sites outside the DBD. A detailed structural analysis of complexes containing Bim and Bmf, for instance, by NMR, will be necessary to exclude this possibility completely.
Similar to Bim, binding to DYNLL led to the dimerization and formation of large Bmf complexes on mitochondria. Several Bcl-2 family proteins (Bim, Bmf, Noxa [48], Mcl-1) have long stretches of intrinsically unstructured region. The stability of such disordered proteins is typically modulated through degradation by the 20S proteasome [40]. Binding partners of intrinsically disordered proteins are known to induce spatial structure and thereby modulate their function as well as posttranslational stability. Interestingly, the presence of intrinsically disordered region is also an essential feature of DYNLL-binding partners [35].
The binding of Bmf to DYNLL probably induces spatial structure in disordered regions of Bmf, which would explain the increased affinity of Bmf for anti-apoptotic Bcl-2 proteins. Disruption of large Bim complexes by targeting DYNLL led to degradation of Bim as well as of its preferred binding partner Mcl-1 [35]. Both Bim and Mcl-1 have been shown to be degraded via ubiquitin-independent 20S proteasome. In the present study also, DYNLL-induced complex formation stabilized Bmf. Therefore, it is plausible that DYNLL-induced complex formation is relevant in regulating proteasome-mediated degradation of its binding partners. DYNLL thus regulates the stability of Bmf, Bim, and Mcl-1 proteins and therefore probably apoptosis.
A striking observation was the regulation of Bim levels by Bmf in a DYNLL-dependent but Bcl-2-binding-independent mechanism. It is conceivable that there is a limited availability of DYNLL and that the overexpressed Bmf outcompetes Bim from the complexes and subsequently Bim is degraded. Interestingly, isoforms of Bmf lacking the BH3 domain but retaining the DYNLL-binding domain enhanced colony-forming potential of HeLa cells [32]. Similarly, we also found that expression of a conserved DBD is sufficient to induce Bim degradation. Therefore, it is a likely possibility that Bmf splice variants lacking the BH3 domain can exert their pro-survival effects by regulating the levels of Bim.
Bmf has mostly been considered a BH3-only protein of limited pro-apoptotic activity; other than in B cells, there are few examples where the loss of Bmf had a clear anti-apoptotic effect. This may be a consequence of low expression of Bmf in other cell types. When we expressed it in Hoxb8 NPCs, Bmf had a strong pro-apoptotic effect in that it sensitized the cells to SCF withdrawal, to the inhibition of Mcl-1, and to the stress of differentiation. We, however, did not observe any effect of DYNLL binding on the pro-apoptotic activity of Bmf.
A number of studies have investigated the affinity of BH3-only proteins for anti-apoptotic Bcl-2 proteins (recently reviewed in [49]). Most of these studies (and all studies investigating Bmf) have used synthetic peptides of the BH3 domain rather than full-length proteins, which may generate inherent inaccuracies. In these studies, the Bmf BH3 domain was found to bind well to Bcl-2, Bcl-XL, and Bcl-w but poorly (with the exception of one study that used a shorter peptide [49]) to Mcl-1 and negligibly to A1/Bfl-1. In the studies available, the Bmf BH3 peptide was unable to directly activate Bax. When Bmf was, however, tested as a recombinant chimera, where the BH3 domain of Bid was replaced with the BH3 domain of Bmf, this protein could directly activate Bax although the activity was considerably smaller than in the case of the Bim BH3 domain [50]. Despite cytochrome c-releasing activity and although Bmf had substantial pro-apoptotic activity in vivo, we failed to find direct Bax-activating activity of recombinant Bmf. Bmf therefore seems not to be able directly to activate Bax/Bak, but at least in the Hoxb8 model this did not affect its pro-apoptotic activity.
Many models still are built on the one-to-one interactions of BH3-only proteins and anti-apoptotic Bcl-2 proteins. The identification of DYNLL-mediated Bim-containing [35] and now Bmf–Bim complexes further extends the possibilities of regulation. Through the sequestration into Bim-containing complexes, the recruitment of anti-apoptotic Bcl-2 proteins, and the regulation of Bim abundance, Bmf likely has functions beyond the immediate neutralization of a subgroup of anti-apoptotic Bcl-2 proteins. Mitochondrial apoptosis, with all its biological and medical ramifications, thus is subject to regulation by the non-Bcl-2 family protein DYNLL.
References
Adams JM, Cory S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018;25:27–36.
Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63.
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310.
Parish IA, Rao S, Smyth GK, Juelich T, Denyer GS, Davey GM, et al. The molecular signature of CD8+ T cells undergoing deletional tolerance. Blood. 2009;113:4575–85.
Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–8.
Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, et al. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell. 2005;7:227–38.
Labi V, Woess C, Tuzlak S, Erlacher M, Bouillet P, Strasser A, et al. Deregulated cell death and lymphocyte homeostasis cause premature lethality in mice lacking the BH3-only proteins Bim and Bmf. Blood. 2014;123:2652–62.
Hornsveld M, Tenhagen M, van de Ven RA, Smits AM, van Triest MH, van Amersfoort M, et al. Restraining FOXO3-dependent transcriptional BMF activation underpins tumour growth and metastasis of E-cadherin-negative breast cancer. Cell Death Differ. 2016;23:1483–92.
Hausmann M, Leucht K, Ploner C, Kiessling S, Villunger A, Becker H, et al. BCL-2 modifying factor (BMF) is a central regulator of anoikis in human intestinal epithelial cells. J Biol Chem. 2011;286:26533–40.
Grespi F, Soratroi C, Krumschnabel G, Sohm B, Ploner C, Geley S, et al. BH3-only protein Bmf mediates apoptosis upon inhibition of CAP-dependent protein synthesis. Cell Death Differ. 2010;17:1672–83.
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92.
Leshchiner ES, Braun CR, Bird GH, Walensky LD. Direct activation of full-length proapoptotic BAK. Proc Natl Acad Sci USA. 2013;110:E986–995.
Du H, Wolf J, Schafer B, Moldoveanu T, Chipuk JE, Kuwana T. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem. 2011;286:491–501.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403.
Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29:629–43.
Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162:613–22.
Akhter R, Saleem S, Saha A, Biswas SC. The pro-apoptotic protein Bmf co-operates with Bim and Puma in neuron death induced by beta-amyloid or NGF deprivation. Mol Cell Neurosci. 2018;88:249–57.
Pfeiffer S, Anilkumar U, Chen G, Ramirez-Peinado S, Galindo-Moreno J, Munoz-Pinedo C, et al. Analysis of BH3-only proteins upregulated in response to oxygen/glucose deprivation in cortical neurons identifies Bmf but not Noxa as potential mediator of neuronal injury. Cell Death Dis. 2014;5:e1456.
Ley R, Ewings KE, Hadfield K, Cook SJ. Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell Death Differ. 2005;12:1008–14.
Sale MJ, Cook SJ. The BH3 mimetic ABT-263 synergizes with the MEK1/2 inhibitor selumetinib/AZD6244 to promote BIM-dependent tumour cell death and inhibit acquired resistance. Biochem J. 2013;450:285–94.
Vallabhapurapu SD, Noothi SK, Pullum DA, Lawrie CH, Pallapati R, Potluri V, et al. Transcriptional repression by the HDAC4-RelB-p52 complex regulates multiple myeloma survival and growth. Nat Commun. 2015;6:8428.
Woess C, Tuzlak S, Labi V, Drach M, Bertele D, Schneider P, et al. Combined loss of the BH3-only proteins Bim and Bmf restores B-cell development and function in TACI-Ig transgenic mice. Cell Death Differ. 2015;22:1477–88.
Labi V, Bertele D, Woess C, Tischner D, Bock FJ, Schwemmers S, et al. Haematopoietic stem cell survival and transplantation efficacy is limited by the BH3-only proteins Bim and Bmf. EMBO Mol Med. 2013;5:122–36.
Frenzel A, Labi V, Chmelewskij W, Ploner C, Geley S, Fiegl H, et al. Suppression of B-cell lymphomagenesis by the BH3-only proteins Bmf and Bad. Blood. 2010;115:995–1005.
Puthalakath H, Villunger A, O’Reilly LA, Beaumont JG, Coultas L, Cheney RE, et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–32.
Puthalakath H, Huang DC, O’Reilly LA, King SM, Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3:287–96.
Day CL, Puthalakath H, Skea G, Strasser A, Barsukov I, Lian LY, et al. Localization of dynein light chains 1 and 2 and their pro-apoptotic ligands. Biochem J. 2004;377(Pt 3):597–605.
Wilfling F, Weber A, Potthoff S, Vogtle FN, Meisinger C, Paschen SA, et al. BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death Differ. 2012;19:1328–36.
Gomez-Bougie P, Bataille R, Amiot M. Endogenous association of Bim BH3-only protein with Mcl-1, Bcl-xL and Bcl-2 on mitochondria in human B cells. Eur J Immunol. 2005;35:971–6.
Singh PK, Weber A, Hacker G. The established and the predicted roles of dynein light chain in the regulation of mitochondrial apoptosis. Cell Cycle. 2018;17:1037–47.
Rapali P, Szenes A, Radnai L, Bakos A, Pal G, Nyitray L. DYNLL/LC8: a light chain subunit of the dynein motor complex and beyond. FEBS J. 2011;278:2980–96.
Morales AA, Olsson A, Celsing F, Osterborg A, Jondal M, Osorio LM. Expression and transcriptional regulation of functionally distinct Bmf isoforms in B-chronic lymphocytic leukemia cells. Leukemia. 2004;18:41–47.
Jurado S, Gleeson K, O’Donnell K, Izon DJ, Walkley CR, Strasser A, et al. The Zinc-finger protein ASCIZ regulates B cell development via DYNLL1 and Bim. J Exp Med. 2012;209:1629–39.
King A, Li L, Wong DM, Liu R, Bamford R, Strasser A, et al. Dynein light chain regulates adaptive and innate B cell development by distinctive genetic mechanisms. PLoS Genet. 2017;13:e1007010.
Singh PK, Roukounakis A, Frank DO, Kirschnek S, Das KK, Neumann S, et al. Dynein light chain 1 induces assembly of large Bim complexes on mitochondria that stabilize Mcl-1 and regulate apoptosis. Genes Dev. 2017;31:1754–69.
Kirschnek S, Vier J, Gautam S, Frankenberg T, Rangelova S, Eitz-Ferrer P, et al. Molecular analysis of neutrophil spontaneous apoptosis reveals a strong role for the pro-apoptotic BH3-only protein Noxa. Cell Death Differ. 2011;18:1805–14.
Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–54.
Garcia-Saez AJ, Ries J, Orzaez M, Perez-Paya E, Schwille P. Membrane promotes tBID interaction with BCL(XL). Nat Struct Mol Biol. 2009;16:1178–85.
Fan J, Zhang Q, Tochio H, Li M, Zhang M. Structural basis of diverse sequence-dependent target recognition by the 8 kDa dynein light chain. J Mol Biol. 2001;306:97–108.
Ben-Nissan G, Sharon M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules. 2014;4:862–84.
Wiggins CM, Tsvetkov P, Johnson M, Joyce CL, Lamb CA, Bryant NJ, et al. BIM(EL), an intrinsically disordered protein, is degraded by 20S proteasomes in the absence of poly-ubiquitylation. J Cell Sci. 2011;124(Pt 6):969–77.
Stewart DP, Koss B, Bathina M, Perciavalle RM, Bisanz K, Opferman JT. Ubiquitin-independent degradation of antiapoptotic MCL-1. Mol Cell Biol. 2010;30:3099–110.
Jorgensen TN, McKee A, Wang M, Kushnir E, White J, Refaeli Y, et al. Bim and Bcl-2 mutually affect the expression of the other in T cells. J Immunol. 2007;179:3417–24.
Wuilleme-Toumi S, Trichet V, Gomez-Bougie P, Gratas C, Bataille R, Amiot M. Reciprocal protection of Mcl-1 and Bim from ubiquitin-proteasome degradation. Biochem Biophys Res Commun. 2007;361:865–9.
Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA. 2003;100:2432–7.
Geissler A, Haun F, Frank DO, Wieland K, Simon MM, Idzko M, et al. Apoptosis induced by the fungal pathogen gliotoxin requires a triple phosphorylation of Bim by JNK. Cell Death Differ. 2013;20:1317–29.
Zaytseva O, Tenis N, Mitchell N, Kanno S, Yasui A, Heierhorst J, et al. The novel zinc finger protein dASCIZ regulates mitosis in Drosophila via an essential role in dynein light-chain expression. Genetics. 2014;196:443–53.
Craxton A, Butterworth M, Harper N, Fairall L, Schwabe J, Ciechanover A, et al. NOXA, a sensor of proteasome integrity, is degraded by 26S proteasomes by an ubiquitin-independent pathway that is blocked by MCL-1. Cell Death Differ. 2012;19:1424–34.
Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25:65–80.
Hockings C, Anwari K, Ninnis RL, Brouwer J, O’Hely M, Evangelista M, et al. Bid chimeras indicate that most BH3-only proteins can directly activate Bak and Bax, and show no preference for Bak versus Bax. Cell Death Dis. 2015;6:e1735.
Acknowledgements
This study was supported by the German Research Council (DFG, FOR2036 to GH and AJG-S) and the Austrian Science Fund (FWF, project # I3271 to AV).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by D. Vaux
Supplementary information
41418_2019_365_MOESM10_ESM.ai
Bmf overexpression sensitizes Hoxb8 neutrophil progenitor cells to apoptosis induced by Mcl-1 inhibitor and differentiation
Rights and permissions
About this article

Cite this article
Singh, P.K., Roukounakis, A., Weber, A. et al. Dynein light chain binding determines complex formation and posttranslational stability of the Bcl-2 family members Bmf and Bim. Cell Death Differ 27, 434–450 (2020). https://doi.org/10.1038/s41418-019-0365-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-019-0365-y
This article is cited by
-
Isovincathicine from Catharanthus roseus induces apoptosis in A549 cells
Journal of Natural Medicines (2024)
-
Weighted gene co-expression network indicates that the DYNLL2 is an important regulator of chicken breast muscle development and is regulated by miR-148a-3p
BMC Genomics (2022)
-
Non-canonical phosphorylation of Bmf by p38 MAPK promotes its apoptotic activity in anoikis
Cell Death & Differentiation (2022)
-
FOXM1 repression increases mitotic death upon antimitotic chemotherapy through BMF upregulation
Cell Death & Disease (2021)
-
Serine and one-carbon metabolisms bring new therapeutic venues in prostate cancer
Discover Oncology (2021)
