
Abstract
Insufficient pancreatic β-cell mass or insulin-producing β-cells are implicated in all forms of diabetes mellitus. However, the molecular mechanisms underlying β-cell destruction are complex and not fully defined. Here we observed that activation of STAT3 is intensely and specifically inhibited in β-cells under hyperglycemic conditions. By knocking out STAT3 specifically in mouse β-cells, we found that the loss of STAT3 sensitized mice to three low doses of STZ stimulation resulting in hyperglycemia. Mechanistically, accumulating PTEN, induced by STAT3 deficiency, directly represses phosphorylation of AKT, which negatively modulates transcription factor activation, dysregulates β-cell function, positively promotes apoptotic signaling, and finally induces β-cell apoptosis. Notably, the defective secretion of insulin and β-cells apoptosis was completely rescued by PTEN ablation in STAT3-null islets or PTEN inhibitor bpv(phen) treatment. Thus our data suggest that STAT3 is a vital modulator of β-cell survival and function, highlighting a critical role for STAT3 in the negative regulation of PTEN-AKT signaling pathway associated with β-cell dysfunction and apoptosis.
Similar content being viewed by others


Introduction
Pancreatic β-cell destruction is the primary cause of type 1 diabetes (T1D) and a pivotal factor underlying β-cell mass reduction in type 2 diabetes (T2D) [1, 2]. Both T1D and T2D are characterized by impaired insulin secretion and resultant hyperglycemia. Though hyperglycemia can be controlled, no agent halts the disease progression by targeting the endogenous β-cells [3], which are exceptionally vulnerable to apoptotic damages under stress [4,5,6]. Thus preventing pancreatic β-cell apoptosis is an attractive intervention to conquer serious hyperglycemia. However, the molecular mechanisms of β-cell apoptosis remain elusive.
Signal transducer and activator of transcription 3 (STAT3), a member of the STAT protein family [7, 8], has been implicated in the regulation of apoptosis [9, 10]. STAT3 exerts its antiapoptotic action and facilitates malignant cell proliferation through induction of survival genes, repression of tumor-suppressor genes, inhibition of death receptors, or regulation of nuclear factor-κB pathway [11,12,13]. Intriguingly, several other studies show that STAT3 promotes apoptosis in distinct biological processes such as inducing apoptosis after major histocompatibility complex-1 ligation on T cells [14]. Inhibition of STAT3 suppresses epithelial apoptosis and delays mammary gland involution [15].
Previous studies have uncovered that STAT3 is highly related to metabolic diseases [16]. STAT3 in the hypothalamus [17] and liver [18] is essential for glucose homeostasis. Excessive STAT3 signaling aggravates the development of skeletal muscle insulin resistance [19] and STAT3 in T cells [20] is responsible for adipose tissue inflammation during diet-induced obesity causing insulin desensitization. Recently, STAT3 has been shown to be dispensable for pancreatic β-cell development and function [21], whereas STAT3 knockout (KO) mice express glucose intolerance and insulin secretion defects, likely due to altered pancreatic microvasculature rather than directly targeting β-cells [22]. In the infant pancreas, STAT3-activating mutation causes premature endocrine differentiation and hence pancreatic hypoplasia [23]. However, STAT3 activation after cytokine treatment induces α-to-β-cell reprogramming and regenerates β-cell mass in adult diabetic mice [24]. In chronic pancreatitis, STAT3 modulates cell cycle and protects β-cells from DNA damage [25], but others showed STAT3 is critical for transforming growth factor-β1-mediated β-cell epithelial–mesenchymal transition [26]. Although diverse functions of STAT3 have been reported in islet cells, the relationship between STAT3 and the regulation of β-cell survival has not been explored.
Here we show that pancreatic STAT3 activation is severely inhibited in β-cell-damaged models and conditional ablation of STAT3 remarkably sensitized β-cells to streptozocin (STZ)-induced apoptosis and hyperglycemia. Using STAT3/phosphatase and tensin homolog (PTEN) double conditional KO mice and applying pharmacological inhibitors of PTEN and AKT, our data further suggest that STAT3-PTEN signaling is critical for the progression of hyperglycemia by regulating AKT activity. Moreover, we also show that inactivating of PTEN by a small molecule inhibitor bpv(phen) protects β-cells against STZ-induced destruction in STAT3-deficient mice, which offers an opportunity for precision intervention in STAT3 mutation hyperglycemia.
Results
The activation of pancreatic STAT3 is inhibited in damaged β-cells
To explore whether STAT3 activation is correlated with β-cell insults, we exposed cultured INS-1 cells, a rat β-cell line, to a serial concentration of glucose mimicking hyperglycemic milieu [27]. Interestingly, phosphorylated STAT3 (pSTAT3) was remarkably decreased in cells treated with high glucose concentration (33.3 mM), while the phosphorylation of STAT1 and STAT6 remained unchanged (Fig. 1a). Similar to chronic effects of high glucose, pSTAT3 was downregulated after STZ-triggered DNA damage and hydrogen peroxide (H2O2)-induced acute oxidative stress (Fig. 1b, c), suggesting that the activation of STAT3 is inhibited in β-cells under stress.

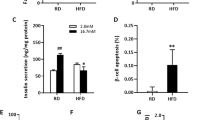
Signal transducer and activator of transcription 3 (STAT3) activation is inhibited in damaged β-cells. a–c INS-1 cells were exposed to normal (11.1 mM) and high glucose conditions (22.2 mM, 33.3 mM) glucose for 72 h (a) or 1 mM, 2 mM streptozocin (STZ) (b) or 100 μM, 200 μM H2O2 (c) in the presence of 11.1 mM or 33.3 mM glucose for 8 h. The protein level of STAT1, pSTAT1, STAT3, pSTAT3, STAT6, and pSTAT6 were detected by western blotting. Glc glucose. β-Actin or GAPDH was used as the loading control. d–f Ten-week-old male wild-type (WT) mice were injected with 150 mg kg−1 STZ or 130 mg kg−1 STZ (i.p.) or vehicle for single dose after fasting overnight (n = 10 mice/group). i.p., intraperitoneal. d Random blood glucose levels of vehicle and STZ-induced mice (n = 5 mice/group) 24 days after vehicle or STZ injection. Cont control (vehicle), H1 hyperglycemia (≥11.1 mM), H2 hyperglycemia (≥16.7 mM). e Immunohistochemistry and quantification for STAT3 and pSTAT3 in islet sections from control mice, as well as STZ-induced hyperglycemia (H1 and H2) mice at day 24. Scale bar, 200 μm. f Islets were isolated from vehicle mice and hyperglycemic mice (≥16.7 mM) at day 24. Western blotting was used to detect the protein expression (n = 5 mice/group). g, h Four-week-old male WT mice fed a normal chow diet or a high-fat diet (HFD) for 16 weeks. g Blood glucose levels of HFD-induced and chow-fed mice (n = 5 mice/group) performed at week 16. h Immunohistochemistry and quantification for STAT3 and pSTAT3 in islet sections from control mice and HFD-induced mice at week 16. Scale bar, 200 μm. Immunoreactivity was assessed by quantitative morphometry with automated image analysis (Image-Pro plus, Version6.0). Data represent the mean ± s.d., *P < 0.05, **P < 0.01; ###P < 0.001 (Student’s t test in g, h, one-way analysis of variance with Tukey’s multiple comparisons test in d, e)
The pSTAT3 reduction was further tested using STZ-induced β-cell destruction in vivo. The random glucose levels of STZ-induced mice were measured before sacrifice, and the mice were divided into two hyperglycemia (≥11.1 or 16.7 mM) groups (Fig. 1d). Notably, immunohistochemistry (IHC) staining results showed that the expression level of pSTAT3 in β-cells was gradually downregulated during disease progression (Fig. 1e). Meanwhile, the pSTAT3 expression in islets isolated from hyperglycemic mice exhibited a remarkable loss compared with normal mice, while neither pSTAT1 nor pSTAT6 was altered (Fig. 1f). However, no obvious difference in pSTAT3 expression was observed in high-fat diet (HFD)-fed mice comparing to the control mice treated with normal chow (Fig. 1g, h). Collectively, these results demonstrated inactivation of STAT3 in damaged β-cells.
Ablation of STAT3 increases hyperglycemia and β-cell apoptosis induced by STZ
To understand the biological relevance of pancreatic STAT3 inhibition, we generated β-cell-specific STAT3 KO mice (β-STAT3KO) [28]. Loss of STAT3 expression in β-cells of β-STAT3KO mice indicated the effective disruption of STAT3 compared with either wild-type or Stat3flox/flox mice (Fig. S1a) and significantly increased body weight compared to wild-type (WT) mice (Fig. S1b). First, we tested the effect of STAT3-specific deletion on the development of HFD-induced obesity mice. We fed β-STAT3KO mice and their littermates a normal diet or an HFD for 16 weeks (Fig. S1c). Consequently, both weight gain and food intake (data not shown) in β-STAT3KO mice were comparable with those in WT mice during HFD (Fig. S1d). Moreover, both β-STAT3KO and WT mice exhibited slightly increased blood glucose and impaired glucose tolerance after HFD treatment (Fig. S1e, f), which accorded with C57BL/6J mice characteristics [29], with compensated islet mass, normal β-cell function, and integrated islets (Fig. S1g, h). These results suggest that STAT3 deficiency has no impact on β-cell compensatory proliferation during HFD-induced obesity.
Next, we sought to test whether STAT3 deletion, in turn, accelerates hyperglycemia pathogenesis. We challenged WT and β-STAT3KO mice both with a single high dose and multiple low doses (three times, 3MLD) of STZ and monitored plasma glucose levels every 3 days (Fig. 2a). Strikingly, 3MLD-STZ injections [30] induced persistent blood glucose elevation in β-STAT3KO mice, while WT mice remained unaffected (Fig. 2b). However, a single high dose of STZ treatment showed no difference in plasma glucose between β-STAT3KO and WT mice (Fig. S2a). Additionally, 3MLD-STZ-treated β-STAT3KO mice significantly lost more body weight than WT mice from day 12 (Fig. S2b). The fasting plasma insulin level in 10 min was higher in β-STAT3KO mice than in littermates (Fig. S2c). However, the 3MLD-STZ treatment delayed glucose clearance and impaired insulin secretion after glucose challenging in β-STAT3KO mice compared with the control group (Fig. 2c, d). Meanwhile, insulin tolerance tests (ITTs) showed similar blood glucose reduction after insulin injection between 3MLD-STZ-treated β-STAT3KO and WT mice (Fig. S2d), implying that the impaired glucose tolerance of β-STAT3KO mice was not due to changes in peripheral insulin sensitivity. To further assess the role of STAT3 in regulating β-cells, we harvested mice at day 24 and analyzed β-cell mass. Contrasting to a slight loss of β-cell mass in WT mice, β-STAT3KO mice suffered from a plunge in β-cell mass after 3MLD-STZ induction (Fig. 2e). Furthermore, IHC analysis suggested that, compared to WT mice, islets from 3MLD-STZ-treated β-STAT3KO mice were architecturally distorted, containing significantly lower insulin staining and a smaller β-cell fraction (Fig. 2f). These defects likely clarify the resulting deficiency in compensatory insulin secretion and developing hyperglycemia.

Signal transducer and activator of transcription 3 (STAT3)-deficient mice are more sensitive to streptozocin (STZ) treatment. a Ten-week-old male β-STAT3KO (KO) mice (n = 21) and their littermates (n = 22) were injected with 40 mg kg−1 STZ (i.p.) or vehicle for 3 consecutive days after fasting overnight. b Blood glucose levels of KO mice and their littermates at the indicated time points. i.p., intraperitoneal. c Glucose tolerance test (i.g.) was performed at day 21; blood glucose was measured at 0, 15, 30, 60, 90, 120, and 180 min. i.g., intragastric. d Plasma insulin levels were measured at 0 and 30 min after glucose gavage at day 23. *P < 0.05 (STZ-KO 0 min vs. STZ-WT 0 min); ##P < 0.01 (STZ-KO 30 min vs. STZ-WT 30 min). e β-Cell mass from KO mice and their littermates injected with vehicle or STZ at day 24. f Left, the pancreas of KO mice and their littermates injected with vehicle or STZ were at day 24 and immunostained for Insulin and Glucagon, respectively. Scale bars, 200 μm (inset), 1 mm (top), 100 μm (bottom). Right, quantification of the percentage of Insulin+ α-cells and Glucagon+ β-cells (n = 8 mice/group). g Western blotting was carried out to detect apoptosis in mouse islets from KO and WT mice. GAPDH, loading control. h Immunostaining of Cleaved Caspase 3 (CC3) with Insulin in the WT and KO mice pancreas at day 24. Scale bar, 100 μm. i Quantification of the percentage of CC3+ cells and Ki67+ cells among insulin+ β-cells at day 24 (n = 8 mice/group). j Quantitative reverse transcription PCR was carried out to analyze β-cell identity genes in pancreatic islets from 3MLD-STZ-induced or vehicle-treated KO mice and WT mice at day 24 (n = 5 mice/group). Gapdh, control. All values are expressed as mean ± s.d., *P < 0.05, **P < 0.01, ***P < 0.001; ##P < 0.01, ###P < 0.001 (Student’s t test in b–d, i and j, one-way analysis of variance with Tukey’s multiple comparisons test in e, f)
To elucidate how STAT3 deletion affects β-cell disruption, we evaluated apoptosis and proliferation in β-cells. As STAT3 modulates β-cell apoptosis under a β-cell toxic environment in vitro (Fig. S2e), we hypothesized that STAT3 deficiency may aggravate hyperglycemia progression in vivo. Results showed that WT mice exhibited slight cell apoptosis after STZ treatment, but β-STAT3KO mice revealed a remarkable apoptosis increase (Fig. 2g–i, Fig. S2f). The proliferation rate of β-cells was higher in 3MLD-STZ-treated WT mice than in saline-treated WT mice, indicating the compensatory capacity in response to physiological challenges. STAT3 KO significantly inhibited β-cell proliferation after 3MLD-STZ treatment (Fig. 2i, bottom, Fig. S2g). Compared to 3MLD-STZ-treated WT mice, β-STAT3KO mice exhibited much lower expression levels of β-cell-specific genes (Mafa, Pax6, Nkx6.1, and Pdx1) [31], which are critically engaged in the biosynthesis of β-cells (Fig. 2j). We next investigated whether immunoregulation [32] is involved in β-STAT3KO mice responses before/after STZ treatment and found equivalent levels of t helper type 1 (Th1) and Th17 cell populations from pancreatic lymph nodes (PLNs) between normal β-STAT3KO and control mice (Fig. S3a). Contrasting to classical multiple low doses (five times, 5MLD) of STZ-induced inflammation in WT mice [33], histological analysis showed that both 3MLD-STZ-injected WT and β-STAT3KO mice exhibited no inflammatory infiltrate in islets (Fig. S3b–d). Collectively, these data indicated that β-cell-specific deletion of STAT3 exacerbated progressive hyperglycemia in 3MLD-STZ-treated mice through destruction of functional β-cells and increasing apoptosis.
Depletion of PTEN alleviates β-cell dysfunction induced by STAT3 deficiency in 3MLD-STZ mice
In order to identify the regulatory genes responsible for β-cell apoptosis induced by STAT3 deficiency, we used quantitative reverse transcription PCR (qRT-PCR) analysis to validate well-known targets [34], such as glucose metabolism-related genes, insulin signaling pathway, and diabetes-regulated genes (Fig. 3a). Among them, some genes such as Mst1 [27] and Chop [5], critical in the progression of diabetes, exhibited higher expression in STZ-treated STAT3-deficient mice than that in WT mice, while STZ administration inhibited Mtor [35] and Glut2 gene expression, indicating impaired β-cell homeostasis and insulin processing. Although MyD88 [36] expression is obviously increased in β-STAT3KO mice than WT littermates, it was comparable between two groups after 3MLD-STZ treatment (Fig. 3a). Enzyme-linked immunosorbent assay (ELISA) and qRT-PCR analysis displayed no difference in MyD88-related inflammatory factors between STZ-induced WT and β-STAT3KO mice (Fig. S4a, b). Moreover, MyD88 deficiency had no effect on β-cell apoptosis induced by STAT3 knockdown and incubation with glucose (Fig. S4c). Interestingly, the tumor-suppressor Pten was the most significantly upregulated gene in 3MLD-STZ-treated β-STAT3KO mice.

Depletion of phosphatase and tensin homolog (PTEN) alleviates β-cell dysfunction induced by signal transducer and activator of transcription 3 (STAT3) deficiency in 3MLD-STZ mice. a Ten-week-old male knockout (KO) mice and their littermates were injected with 40 mg kg−1 streptozocin (STZ, i.p.) for 3 consecutive days after fasting overnight. i.p., intraperitoneal. Quantitative reverse transcription PCR (qRT-PCR) quantification showing the expression of selected genes in pancreatic islets (n = 5 mice/group). Gapdh, control. b, c INS-1 cells were transfected with siSTAT3 #1 or control siRNA (siNC) and treated with or without 33.3 mM glucose for 48 h. 11.1 mM, normal condition as control; 33.3 mM, high glucose. qRT-PCR (b) and western blotting (c) were used to detect STAT3 and PTEN. GAPDH, control. ***P < 0.001 (11.1 mM + siNC vs. 11.1 mM + siSTAT3 #1); ##P < 0.01, ###P < 0.001 (33.3 mM + siNC vs. 33.3 mM + siSTAT3 #1). d Islets were isolated from different groups (wild-type (WT), KO, STZ-induced WT, and STZ-induced KO mice) at day 24, the expression of STAT3 and PTEN were examined by western blotting. e Representative immunostaining image and quantitative analysis for Insulin (red) and PTEN (green) at day 24 (n = 8 mice/group). Scale bar, 100 μm. f–h Ten-week-old male β-STAT3-PTENDKO (DKO) mice, KO mice, and their littermates were injected with 40 mg kg−1 STZ (n = 10, 15, 10) or vehicle (n = 10 mice/group) for 3 consecutive days after fasting overnight. f Random blood glucose was checked at the indicated times. g Glucose tolerance test experiment was performed at day 21 to detect the blood glucose at 0, 15, 30, 60, 90, 120, and 180 min. h Immunostaining (left) and quantification (right) of the percentage of Insulin (red) and Glucagon (green) or CC3 (green) from KO and DKO mice at day 27 (n = 5 mice/group). Scale bar, 100 μm. All values are expressed as mean ± s.d., **P < 0.01, ***P < 0.001; ##P < 0.01, ###P < 0.001 (Student’s t test in b, e–g, one-way analysis of variance with Tukey’s multiple comparisons test in a, h)
PTEN, a regulator of cell cycle and apoptosis, is an important negative modulator of insulin signaling [37]. Hence, we asked whether PTEN could override islet function due to STAT3 deficiency. We employed small interfering RNA (siRNA) against STAT3 and observed a remarkable PTEN accumulation at both transcriptional and translational levels (Fig. 3b, c, Fig. S5a). Consistent with our results in vitro, STAT3 KO caused elevated PTEN levels in β-cells of β-STAT3KO mice and further accumulated after STZ treatment in vivo (Fig. 3d, e).
We next explored whether PTEN deficiency could reverse glucose intolerance and β-cell apoptosis in β-STAT3KO mice. Cumulative expression of PTEN by deletion of STAT3 was abolished in islets of Ins2CrePtenflox/floxStat3flox/flox mice [38] (hereafter referred to as β-STAT3-PTENDKO) (Fig. S5b, c). As expected, random plasma glucose levels (Fig. 3f) and glucose intolerance (Fig. 3g) in 3MLD-STZ-induced β-STAT3-PTENDKO mice were largely reversed by genetic disruption of PTEN comparing to β-STAT3KO mice. In addition, β-STAT3KO mice had architecturally distorted islets and apoptotic β-cells with high PTEN expression in most CC3+ β-cells (Fig. S5d). PTEN deficiency could completely prevent β-cell apoptosis and structural destruction of pancreas islets in β-STAT3-PTENDKO mice (Fig. 3h). Therefore, PTEN deletion could reverse impaired glucose intolerance and β-cell apoptosis induced by STAT3 deficiency upon 3MLD-STZ treatment, demonstrating that PTEN is a vital downstream effector of STAT3 in the regulation of β-cell survival and function.
AKT mediates the alleviation of β-cell dysfunction and glucose intolerance by depleting PTEN in β-STAT3KO mice
Next, we examined the effects of PTEN on regulatory elements of islet-enriched PDX1 and MAFA in 293FT cells. Luciferase reporter assays showed that STAT3 knockdown resulted in attenuated promoter activity of PDX1 and MAFA, whereas the repressive activity was significantly diminished by inhibition of PTEN with bpv(phen) [39] (Fig. 4a, Fig. S6a). Additionally, no difference in the expression of β-cell-specific transcription factors was observed between islets from β-PTENKO mice treated with STZ or vehicle (Fig. S6b). However, STAT3-PTEN double knockout (DKO) protected mice from STZ treatment and displayed an obvious mRNA increase of β-cell-related transcription factors compared to β-STAT3KO mice (Fig. 4b), further confirming that STAT3 could regulate gene expression of β-cell-specific genes through PTEN. Consistent with our previous observation, 3MLD-STZ-treated β-STAT3KO mice exhibited a decreased nuclear localization of PDX1 in β-cells, while its proportion was re-established in 3MLD-STZ-treated β-STAT3-PTENDKO mice (Fig. 4c). Therefore, β-cell-specific genes were dictated by STAT3-PTEN signaling both in vitro and in vivo.

Signal transducer and activator of transcription 3 (STAT3)-phosphatase and tensin homolog (PTEN) regulates β-cell survival and function through AKT activation. a 293FT cells were transfected with luciferase reporters driven by PDX1 or MAFA promoter together with siNC or siSTAT3 and treated with/without bpv(phen) as indicated for 24 h. b–e Ten-week-old male DKO mice, knockout (KO) mice, and their littermates were injected with 40 mg kg−1 streptozocin (STZ) or vehicle for 3 consecutive days after fasting overnight (n = 10 mice/group). b Quantitative reverse transcription PCR was performed to analyze β-cell identity genes in islets from DKO mice, KO mice and their littermates at day 24 (n = 5 mice/group). Gapdh, control. Values are expressed as mean ± s.e. c Representative images of immunolabeling (left) and quantification (right) for PDX1 (green) and Insulin (red) from DKO mice, KO mice and their littermates at day 24 (n = 5 mice/group). Scale bar, 100 μm. d Pancreatic islet cells isolated from the above mice were subjected to immunoblotting against STAT3, PTEN, AKT, pAKT, PDX1, Nkx6.1, Cleaved Caspase 3, and Cleaved poly ADP-ribose polymerase (PARP) 27 days after last STZ. GAPDH, loading control. e Immunohistochemistry for pAKT in sections from mouse pancreases at day 24. Scale bar, 200 μm. f,g Ten-week old-male DKO mice (n = 5), KO mice (n = 6), and their littermates (n = 5) were injected with 40 mg kg−1 STZ or vehicle for 3 consecutive days after fasting overnight, then treated with AZD5363 (i.g., 200 mg kg−1 daily) from day 11 to day 27, and sacrificed at day 27. f Blood glucose levels of randomly fed of DKO, KO mice, and the relative wild-type littermates with or without AZD5363 treatment. The arrow represents the first day of AZD5363 administration. g Blood glucose levels were measured at day 21 by glucose tolerance test (i.g.) at 0, 15, 30, 60, 90, 120, and 180 min. Immunoreactivity was assessed by quantitative morphometry with automated image analysis (Image-Pro plus, Version6.0). i.g., intragastric. All values are expressed as mean ± s.d., *P < 0.05, **P < 0.01, ***P < 0.001; #P < 0.05, ###P < 0.001 (Student’s t test in a, f, g, one-way analysis of variance with Tukey’s multiple comparisons test in b, c)
Given that PTEN could antagonize survival/proliferative paradigm and consequently decrease the activity of downstream AKT, which is critical for β-cell survival [40, 41], we hypothesized that the dysfunction of β-cells in 3MLD-STZ-treated β-STAT3KO mice is mediated by PTEN-AKT signaling. Western blotting analysis showed that STZ treatment led to PTEN protein build-up and AKT inactivation after STAT3 KO, with increased Cleaved Caspase 3/Cleaved poly ADP-ribose polymerase (PARP) and decreased PDX1/Nkx6.1. Notably, PTEN deficiency reversed AKT activity and protected β-cells from apoptosis (Fig. 4d). IHC results also proved pAKT signal disappearance in islets of 3MLD-STZ-treated β-STAT3KO mice and increased pAKT expression after PTEN KO (Fig. 4e). To further validate the importance of AKT in β-cell function regulated by STAT3 and PTEN, we used the AKT-specific inhibitor AZD5363 [42], which suppressed AKT phosphorylation over time (Fig. S6c). The results showed that all 3MLD-treated animals exhibited elevated glucose levels and glucose intolerance after AZD5363 administration (Fig. 4f, g). Moreover, AZD5363-treated hyperglycemic β-STAT3KO mice exhibited much graver β-cell apoptosis compared to the WT mice after STZ injection, and PTEN depletion in β-STAT3KO mice largely reversed β-cell apoptosis (Fig. S6d). qRT-PCR data revealed that AZD5363 treatment slightly decreased the expression of β-cell-specific genes (Mafa, Pax6, Nkx6.1, and Pdx1) in STZ-treated WT mice. Furthermore, hyperglycemic STAT3-null mice exhibited a significant decrease in transcriptional activities after AZD5363 treatment (Fig. S6e). Together, these results demonstrate that AKT is downstream of PTEN, mediating β-cell impaired functions and apoptosis in 3MLD-STZ-induced β-STAT3KO mice.
PTEN inhibitor rescues β-cell survival and disrupted function by 3MLD-STZ treatment in β-STAT3KO mice
As PTEN depletion combats β-cell apoptosis and restores β-cell function in 3MLD-STZ-induced β-STAT3KO mice, PTEN inhibitors may suppress the hyperglycemia progression. Hence, we began exploring the effect of bpv(phen) on classical β-cell damaged models both induced by a large dose and 5MLD of STZ in WT mice. Since the severely elevated blood glucose levels were not mitigated by bpv(phen) injection (Fig. S7a, b), bpv(phen) was used when 3MLD-STZ-induced β-STAT3KO mice started hyperglycemia (Fig. 5a, Fig. S7c). Interestingly, β-STAT3KO mice without bpv(phen) injection developed fasting hyperglycemia, while bpv(phen) treatment markedly decreased plasma glucose levels, similar to WT mice (Fig. 5b). Additionally, bpv(phen) treatment reversed the body weight loss of β-STAT3KO mice during persistent hyperglycemia (Fig. 5c). As expected, bpv(phen) treatment could restore the level of both blood glucose and plasma insulin to normality in 3MLD-STZ-injected β-STAT3KO mice (Fig. 5d, e). Consistently, bpv(phen)-treated mice prevented β-cell mass reduction, islet architecture disruption, abnormal β-cell fraction, and decreased insulin secretion (Fig. 5f, g). Moreover, western blotting (Fig. 5h) and immunofluorescence (Fig. 5i, Fig. S7d) analysis confirmed the extent of β-cell survival and proliferation, accompanied by markedly reduced β-cell apoptosis, increased Nkx6.1/PDX1 expression (Fig. 5h), and over four-fold Ki67+ cells in islets of bpv(phen)-treated hyperglycemic β-STAT3KO mice vs. those of untreated β-STAT3KO littermates (Fig. 5i). Importantly, AKT activation was rescued after bpv(phen) injection in contrast to the robust inactivation seen in STZ-induced β-STAT3KO mice (Fig. 5j), according to the results in β-STAT3-PTENDKO mice. The percentage of PDX1 in β-cells was also restored by bpv(phen) treatment (Fig. 5k, l). To confirm this finding, we examined the levels of β-cell critical transcription factors such as Mafa, Pax6, Nkx6.1, and Pdx1, which were attenuated by STZ injection and exhibited normal or higher expression in pancreatic islets of bpv(phen)-treated β-STAT3KO mice (Fig. 5m). Overall, these results suggested that the PTEN inhibitor bpv(phen) effectively lowers blood glucose levels and ameliorates hyperglycemia symptoms in 3MLD-STZ-treated β-STAT3KO mice by restoring β-cell survival and function.

bpv(phen) rescues β-cell survival and function disrupted by 3MLD-STZ-treatment in β-STAT3KO mice. a Ten-week-old male knockout (KO) mice (n = 16) and their littermates (n = 15) were injected with 40 mg kg−1 streptozocin (STZ, i.p.) or vehicle for 3 consecutive days after fasting overnight, daily injected with 2 mg kg−1 bpv(phen, i.p.) or vehicle from day 11 to day 27. i.p., intraperitoneal. b Blood glucose levels were checked at the indicated times of KO mice or wild-type (WT) littermates. The arrow represents the first day of bpv(phen) administration. c Body weight monitored every 3 days. d Blood glucose was analyzed after 0, 15, 30, 60, 90, 120, and 180 min in glucose tolerance test (i.g.) experiment at day 21. e Insulin secretion was measured before (0 min) and 30 min after glucose gavage at day 23. ***P < 0.001 (STZ-KO 0 min vs. STZ-KO-bpv 0 min); ###P < 0.001 (STZ-KO 30 min vs. STZ-KO-bpv 30 min). f β-cells mass from KO mice and their littermates at day 27. g Representative images (left) of α-cell (green) and β-cell (red) staining in islets from different groups and quantification (right) at day 27 (n = 5 mice/group). Scale bar, 100 μm. h Islets were isolated from WT and KO male mice treated with STZ or combined with bpv(phen) (n = 5 mice/group) at day 27. Western blotting was used to detect Nkx6.1, PDX1, Cleaved Caspase 3, and Cleaved poly ADP-ribose polymerase (PARP). GAPDH, loading control. i Quantification of the percentage of Ki67+ pancreatic β-cells from KO and WT mice (n = 5 mice/group). j Immunohistochemical staining of pAKT in pancreatic sections. Scale bar, 200 μm. k, l Representative images (k) of PDX1 (green) and Insulin (red) staining in islets from different groups and quantification analysis (l) (n = 5 mice/group). Scale bar, 100 μm. m Quantitative reverse transcription PCR analysis of β-cell identity gene expression in islets from KO mice, bpv(phen)-injected KO mice, and their control mice (n = 5 mice/group) at day 27. Gapdh, control. Immunoreactivity was assessed by quantitative morphometry with automated image analysis (Image-Pro plus, Version6.0). All values are expressed as mean ± s.d., *P < 0.05, **P < 0.01, ***P < 0.001; #P < 0.05, ##P < 0.01, ###P < 0.001 (Student’s t test in b–e, one-way analysis of variance with Tukey’s multiple comparisons test in f, g, i, l, m)
In order to explore whether bpv(phen) alleviates STZ-induced β-cell apoptosis through AKT signaling in β-STAT3KO mice, we used AKT inhibitor AZD5363 to arrest this function. AZD5363 treatment blocked the rescue of blood glucose and glucose intolerance by bpv(phen) treatment in 3MLD-STZ-induced β-STAT3KO mice (Fig. 6a, b). AZD5363 also induced disruptions in islet structure where α-cells invaded the interior and consequently increased β-cell apoptosis (Fig. 6c, d). Additionally, AZD5363 abolished the normalization of PDX1 levels by bpv(phen) treatment in the β-cells of 3MLD-STZ-treated β-STAT3KO mice (Fig. 6e), suggesting that bpv(phen) relieves hyperglycemia through activation of AKT in STAT3-depleted mice.

Disruption of AKT abolishes alleviated hyperglycemia by deleted phosphatase and tensin homolog (PTEN) in knockout (KO) mice. a–e Ten-week old-male KO mice and their littermates were injected with 40 mg kg−1 streptozocin (STZ, i.p.) for 3 consecutive days after fasting overnight, then treated with bpv(phen) (i.p., 2 mg kg−1 once daily) alone or with AZD5363 (i.g., 200 mg kg−1 once daily) and at day 27 (n = 9 mice/group). a Random-fed blood glucose was monitored every 3 days of KO mice or the respective wild-type (WT) littermates. The arrow represents the first day of bpv(phen) administration or with AZD5363. b Plasma glucose was measured after 0, 15, 30, 60, 90, 120, and 180 min in glucose tolerance test (i.g.) experiment at day 21. c Representative staining and quantification analysis for Insulin (red) and Glucagon (green) in islets from different groups at day 27 (n = 5 mice/group). Scale bar, 100 μm. d Representative images and statistics of anti-CC3 (green) and anti-Insulin (red) immunostaining (n = 5 mice/group). Scale bar, 100 μm. e Immunofluorescence and percentage of anti-PDX1 (green) and anti-Insulin (red) immunostaining (n = 5 mice/group). Scale bar, 100 μm. f Left, in the normal milieu, activated signal transducer and activator of transcription 3 (STAT3) inhibits PTEN and promotes AKT phosphorylation, thereby prevents β-cells from apoptosis and triggers β-cell-related gene transcriptions, which is involved in β-cell function. Right, the proposed model for the role of STAT3-PTEN signaling axis in β-cells. In hyperglycemic stimuli or β-cells suffering from oxidative/DNA damage, which inhibits STAT3 activation, PTEN is heavily accumulated, and AKT activation is impaired that then triggers Cleavage of Caspase 3 and acceleration of β-cell death. β-Cell-related gene transcription factors are also impaired, further disturbing β-cell survival and function. i.p., intraperitoneal; i.g., intragastric. All values are expressed as mean ± s.d., ***P < 0.001 (Student’s t test)
Discussion
Our results demonstrate that STAT3 dysfunction profoundly contributes to defective insulin secretion and β-cell apoptosis under stress. Inactivation of PTEN by genetic deletion or pharmacological inhibition preserves β-cell function and combats STZ insults in STAT3-deficient mice through its regulation of AKT and downstream effects.
It is critical to illuminate molecular underpinnings of β-cell apoptosis for its role as a vital culprit underlying hyperglycemia progression. Our work shows that pancreatic STAT3 serves as a negative modulator for severe hyperglycemia by using 3MLD-STZ-induced mouse model, which is different from previous studies examining glucose metabolism in normal β-STAT3KO mice [17, 21]. Also notably, we only observed the effect of pancreatic STAT3 ablation on hyperglycemia susceptibility after 3MLD-STZ injection but not with a single high dosage, suggesting that STAT3 is more sensitive to progressive and constant stimulation [43] rather than acute toxicity with serious β-cell damage [44]. Intriguingly, 3MLD-STZ-treated WT mice recruited few inflammatory cells and exhibited proliferative capacity to resist β-cell apoptosis without suffering from T1D or even hyperglycemia [30]. Consistent with previous studies [45, 46], 5MLD-STZ-induced mice showed severe invasive insulitis and β-cell destruction and failed to proliferate and expand islets [47], ultimately leading to hyperglycemia (Figs. S3b, c and S8a–d). As we know, β-cells can compensate the reduced efficiency of insulin action and keep normal blood glucose in diet-induced obesity [48], which is consistent with our results (Fig. S1g) and partly explains that STAT3 is not directly involved in β-cell proliferation. In conclusion, we showed that pancreatic STAT3 plays a critical role in protecting β-cells from external stimulation-induced apoptosis and further explained why depletion of STAT3 in β-cells only affects STZ-induced hyperglycemia rather than HFD-induced obesity.
Knocking out STAT3 gene using an Ins2/RIP-Cre transgene led to mild obesity and glucose intolerance (Fig. S1b, f), which is likely connected to the resulting STAT3 deletion from neurons expressing the leptin receptor [17]. β-STAT3KO mice have similar or slightly decreased insulin level compared to WT mice, which, however, showed hyperinsulinemia after glucose gavage (Fig. S2c). We noticed that β-STAT3KO mice were glucose intolerant compared to controls in chow diet, but this effect was significantly increased by treatment with the β-cell-specific cytotoxic STZ (Fig. 2c), suggesting that STAT3 plays a critical role in β-cell apoptosis. Furthermore, we showed that STAT3 knockdown caused remarkable apoptosis increases upon STZ exposure in vitro (Fig. S2e), which could partially explain the specific function of STAT3 in β-cell survival and apoptosis in vivo. Additionally, Ins2/RIP-Cre mice are widely used to study the function of certain genes for β-cells [35, 49, 50]. In conclusion, our results showed that STAT3 mainly acts in β-cells rather than leptin receptor-positive neurons under apoptotic damages.
In major insulin-related organs, PTEN deficiency improves glucose metabolism and induces resistance to diabetes in patients and rodents [51,52,53,54]. We found that PTEN is obviously accumulated in 3MLD-STZ-induced β-STAT3KO mice, consistent with recent studies, suggesting that a similar STAT3-PTEN regulatory relation exists in cancer [55, 56] and other diseases [57, 58]. However, such relationship in β-cell destruction-induced hyperglycemia is poorly understood. We corroborated that the cumulative increase of PTEN is the main reason for rising blood glucose and glucose intolerance when suffering from STZ treatment in β-STAT3KO mice, and PTEN blockage would prevent β-cell apoptosis and restore cell function. Because PTEN inhibitors are not as efficient as genetic KO, PTEN KO exhibited lower plasma glucose than pharmacological inhibition of PTEN in STZ-induced β-STAT3KO mice. The vital consequence of PTEN accumulation leads to the inactivation of AKT signaling, which upregulates insulin transcription, insulin secretion, β-cell growth, and survival [59]. Because of different AKT activation states among STZ-induced mice, AZD5363-injected β-STAT3KO mice displayed much more severe hyperglycemia, poor glucose tolerance, and numerous apoptotic β-cells while β-STAT3-PTENDKO mice presented minimal phenotypic changes. Consistently, recent studies have suggested crosstalk between AKT and islet-enriched transcription factors [60, 61] and also implicated AKT in orchestrating apoptosis [34, 62]. Together, our findings demonstrate that STAT3 regulates β-cell apoptosis by disrupting PTEN-AKT pathway under multiple stresses.
Despite the potentially huge clinical benefits of functional β-cells, none of the currently used hypoglycemic agents target maintenance of endogenous β-cells [3]. Some small molecule compounds were reported to stimulate β-cell proliferation, improve glucose production, and regulate islet autoimmunity [63,64,65]. However, it is quite common for patients to be insensitive or have a serious adverse reaction to clinical therapeutics [66, 67]. Therefore, novel precision medicines for a special population with higher risks for hyperglycemia are more clinically beneficial. From human single-cell transcriptomes, we found that STAT3 expression in β-cells presents large individual difference between human beings [68] that enlightened us on exploring preferable strategies for effective control of hyperglycemic patients in whose β-cells inactivating STAT3 mutation occur. To examine the impact of reducing PTEN signaling in the pathways above, we utilized a PTEN inhibitor bpv(phen) to assess its effects on hyperglycemia induced by 3MLD-STZ and found that bpv(phen) could reverse 3MLD-STZ-induced β-STAT3KO mice β-cell apoptosis, promote β-cell proliferation, and improve glycemia control, without affecting normal hyperglycemic mice or control mice. We speculate that PTEN overaccumulation is required for bpv(phen) treatment which suggests that it can accurately be used for inactivating STAT3-mutant patients with little side effect of hypoglycemia.
In summary, our findings uncovered that dysfunctional STAT3-PTEN-AKT pathway is a critical mediator for defective β-cell function and apoptosis in STZ-induced hyperglycemia, and pharmacological interventions targeting this signaling pathway, such as inhibition of PTEN activity, may be a potential strategy to develop new drugs to achieve precision treatment for potential inactivating STAT3-mutant patients.
Methods
Experimental animals
All animal models were on a C57BL/6J background. Homozygous STAT3 floxed mice (Stat3flox/flox) were crossed with Ins2-Cre mice to generate heterozygous Ins2-Cre-STAT3 floxed mice (Ins2creStat3flox/+), which were then intercrossed with Stat3flox/flox mice to generate β-STAT3KO (Ins2creStat3flox/flox) mice. A similar mating strategy was used for generating β-cell-specific STAT3-PTEN DKO mice (Ins2creStat3flox/floxPtenflox/flox, referred to β-STAT3-PTENDKO) and β-cell-specific PTEN KO mice (Ins2crePtenflox/flox, referred to β-PTENKO). Stat3flox/flox and Ins2-Cre mice were obtained from the Model Animal Research Center of Nanjing University, and Ptenflox/flox mice were obtained from Jackson Laboratory. All animal care and experimental procedures were conducted in accordance with the rules and regulations of the Institutional Animal Care &Use Committee (IACUC) of Zhejiang University, with ethical approval numbers IACUC-16003 and IACUC-17023.
For MLD-STZ experiments, 10-week-old male β-STAT3KO mice, β-PTENKO mice, DKO mice, their WT littermates, or WT mice were fasted for 12 h and injected with STZ (40 mg kg−1, Sigma) freshly dissolved in 50 mM sodium citrate buffer (pH 4.5) for 3 or 5 consecutive days. For STZ single injection, 10-week-old male WT mice were fasted for 12 h and injected with a single dose of STZ at 130 or 150 mg kg−1. For the HFD experiments, 4-week-old β-STAT3KO mice and their WT littermates were fed a standard laboratory chow or an HFD [27, 69] (fat, carbohydrate, and protein content was 58, 26, and 16 kcal%, respectively) for 16 weeks, and body weight and blood glucose levels were recorded weekly. For both models, random blood was obtained from the tail vein of nonfasted mice, and glucose was measured through a Glucometer (Bayer).
Glucose tolerance test (GTT) and ITT and measurement of insulin secretion
GTTs and ITTs were performed after 12-h fast. Briefly, the fasted mice were infused with glucose (Sinopharm Chemical Reagent Co., Ltd, 2 g kg−1) by oral gavage or subcutaneously injected with insulin (Novolin, Novo Nordisk, 0.5 U kg−1) for GTT and ITT, respectively. For GTT, blood samples were taken at time points 0, 15, 30, 60, 90, 120, and 180 min for glucose measurements. For ITT, blood samples were measured before (0 min) and after (15 and 30 min) injection of insulin. Plasma insulin levels were measured before (0 min) and after (5, 10, 30 min) glucose gavage and tested using the Mouse Insulin Elisa Kit (EMD Millipore).
Islet isolation, cell culture, and transfection
Islets from β-STAT3KO mice, β-STAT3-PTENDKO mice, and their WT littermates were isolated as described previously [70]. Briefly, pancreases were perfused with collagenase P (Sigma, 0.5 mg ml−1) via an indwelling needle, followed by digestion at 38 °C for 10 min. The digested pancreases were filtered with a 600-μm cell strainer, followed by washing and density gradient separation method. INS-1 cells (Institute of Basic Medicine, Chinese Academy of Medical Sciences) were cultured in complete RPMI-1640 medium (Gibco) supplemented with 10 mM HEPES (Gibco), 1 mM sodium pyruvate (Gibco), and 50 μM β-mercaptoethanol (Sigma). HEK 293FT cells (Cyagen Biosciences Inc) were cultured using Dulbecco’s modified Eagle’s medium (Gibco). All media included 1% penicillin–streptomycin and 10% fetal bovine serum (Gemini). The cell lines used for experiments had been passaged no more than 20 times and cells were monitored for mycoplasma contamination every 6 months.
All siRNA (Genepharma) transfection was performed using jet medium and jet Primer (Polyplus). HEK 293FT cells were transiently transfected according to the manufacturer’s protocols. INS-1 cells were electroporated with the Letrans Kit for Cell Lines (LAIZEE; cat#LZT010) through the Nucleofector II Device (Lonza) according to the manufacturer’s instruction. Cells were harvested at 24–48 h after transfection. The sequences used were as follows: Rat siSTAT3 #1 (sense: 5′-CCCGCCAACAAAUUAAGAATT-3′; anti-sense: 5′-UUCUUAAUUUGUUGG CGGGTT-3′), #2 (sense: 5′-GCAGGAUCUAGAACAGAAATT-3′; anti-sense: 5′-UUUCUGUUCUAGAUCCUGCTT-3′), Rat siMyD88 (sense: 5′-GGACUUUCC UAGUAUCCUATT-3′; anti-sense: 5′-UAGGAUACUAGGAAAGUCCTT-3′).
Compound intervention
bpv (phen, Santa Cruz) was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 4 mM (1000× stock). Glucose was dissolved in 1640 medium at a concentration of 33.3 M (1000× stock). H2O2 (Sinopharm Chemical Reagent Co., Ltd) was dissolved in 1640 medium at a concentration of 200 mM (1000× stock). STZ was dissolved in 1640 medium immediately at a concentration of 1 mM or 2 mM before use in vitro.
bpv(phen) was dissolved in 0.9% NaCl solution and AZD5363 was dissolved in special solvent (10% DMSO 25% w/v Kleptose HPB). For mice, the corresponding vehicle (0.9% NaCl) and bpv (phen, 2 mg kg−1) were given intraperitoneally, and AZD5363 (TargetMol, 200 mg kg−1) and vehicle (10% DMSO 25% w/v Kleptose HPB) were administrated by oral gavages.
Histology and islet morphometry
Mouse pancreases were fixed in 4% paraformaldehyde and embedded in paraffin or embedded in O.C.T. compound (Tissue-Tek). Pancreas sections (4 μm) were deparaffinized, rehydrated, and incubated overnight at 4 °C with antibodies for IHC [71]. Pancreas cryosections (8 μm) were incubated with blocking buffer (phosphate-buffered saline with 5% normal goat serum and 0.3% Triton-100) for 1 h for immunofluorescence. The primary antibodies were as follows: anti-Insulin (ab6995, 1:200), anti-Glucagon (ab10988, 1:100), anti-Ki67 (ab16667, 1:50), anti-PTEN (ab79156, 1:100), anti-pSer473-AKT (ab81283, 1:500), and anti-CD3 (ab5690, 1:100) antibodies from Abcam; anti-STAT3 (9139, 1:100), anti-STAT3 (12640, 1:100), anti-pTyr705-STAT3 (9145, 1:500), anti-pTyr705-STAT3 (4113, 1:500), anti-Cleaved Caspase 3 (9661, 1:100), anti-PTEN (9559, 1:100), anti-PDX1 (5679, 1:100), anti-Insulin (3014, 1:200), and anti-Glucagon (2760, 1:500) antibodies from Cell Signaling Technology (CST). The secondary antibodies were as follows: anti-rabbit secondary antibody (Beijing Zhongshan Biotechnology, PV-6001), anti-mouse secondary antibody (Beijing Zhongshan Biotechnology, PV-6002), Alexa Fluor® 488 anti-rabbit IgG secondary antibody (Invitrogen, A-11008), Alexa Fluor® 568 anti-rabbit IgG secondary antibody (Invitrogen, A-10042), Alexa Fluor® 488 anti-mouse IgG secondary antibody (Invitrogen, A-21202), and Alexa Fluor® 568 anti-mouse IgG secondary antibody (Invitrogen, A-10037). According to the previously reported methods [34, 72], at least 8 evenly 20 mm apart sections throughout the entire pancreas were taken from each mouse. Mean percentage of β-cell fraction per pancreas was calculated as the ratio of insulin+ area to the whole pancreatic tissue area. β-Cell mass was obtained by multiplying the β-cell ratio by the pancreas weight. Images were acquired with a Leica DMI3000 fluorescence microscope or Zeiss LSMS 10 Meta microscope or Leica DM2500 microscope.
Immunoblotting
Immunoblotting analysis of proteins in cell lysates was performed as previously described [71]. The primary antibodies were as follows: anti-STAT1 (9172, 1:1000), anti-pTyr701-STAT1 (9167, 1:1000), anti-STAT3 (9139, 1:1000), anti-pTry641-STAT6 (9361, 1:1000) antibodies, anti-pTyr705-STAT3 (9145, 1:1000), anti-pSer473-AKT (4060, 1:1000), anti-Cleaved Caspase 3 (9661, 1:1000), anti-Cleaved PARP (5625, 1:1000), anti-PTEN (9559, 1:1000) and anti-PDX1 (5679, 1:1000) antibodies were from CST; anti-STAT6 (ab54461, 1:1000) and anti-Nkx.6.1 (ab221544, 1:1000) antibodies were from Abcam; anti-AKT (sc-8312, 1:1000), anti-MyD88 (sc-74532, 1:1000), anti-α-Tubulin (sc-53646, 1:1000), anti-β-Actin (sc-1615, 1:1000) and anti-GAPDH (sc-25778, 1:1000) antibodies were from Santa Cruz. Appropriate secondary antibodies and ECL were performed to visualize chemiluminescence (ECL, Amersham Biosciences, Castle Hill, Australia). For the quantification, CC3+ cells were counted as apoptotic islet cells if they co-localized with Insulin or were located within the Insulin+ islet area [73]. The vertical coordinates were shown as “% CC3+ cells of total islets nuclei” according to a previous study [74] (20 islets were counted from each animal).
Flow cytometric analysis
Single-cell suspensions from PLNs were obtained by mechanical disruption and incubated for 30 min at 4 °C with anti-CD4 (BD Biosciences, 5233919) for staining of surface markers. For intracellular staining of cytokines, cells were stained with anti-CD4, followed by staining with anti-IFN-γ, anti-IL-17A (Biolegend, B213720, B201753) and incubation for 30 min at 4 °C. After staining, cells were washed and analyzed in a BD FACS-Calibur cytometer (Becton Dickinson, San Jose, CA).
RNA extraction and RT-PCR
Total RNA was isolated from tissues and cells using Trizol reagent (Invitrogen) according to the manufacturer’s protocols, and cDNA synthesis was performed by using the TransScript Kit (TransGen Biotech). qRT-PCR was carried out as described previously [75] with ABI Fast 7500 Real-time PCR instrument (Perkin-Elmer Applied Biosystems), and the relative mRNA levels were normalized to internal control such as Gapdh. The PCR primer sequences are available upon request.
Construction of the luciferase reporter vector and luciferase assay
The promoter regions of Pdx1 from −1865 to −1033 bp and Mafa from −1961 to −1132 bp of the transcription start site were amplified via PCR by using KOD DNA Polymerase (Toyobo, Japan). Amplicons were inserted into the pGL4.14 [LUC2/Hygro] Vector (Promega, USA) at the 5′ side of the LUC2 sequence. HEK 293FT cells were transfected with luciferase reporters siSTAT3 and pRL-TK renilla luciferase vector (Promega) and assayed 24 h posttransfection for luciferase activities by using a Dual-Luciferase Reporter Assay Kit (Promega) according to the manufacturer’s instructions. The luciferase intensity was detected using Varioskan Flash (BD). Results were normalized to the Renilla luciferase control and expressed relative to the average value of the controls.
Statistical analysis
Data analysis was performed using the GraphPad Prism Software Version 5.0a (GraphPad, San Diego, CA) and SigmaPlot.v10.0 (Systat Software Inc). Quantitative image analysis of IHC was performed using the Image-pro Plus 6.0 analysis software (Media Cybernetics). Data were presented as mean ± s.d. All experiments were repeated at least three times with representative data shown. Statistical significance was determined by unpaired two-side Student’s t test or one-way analysis of variance. 0.01 < *P < 0.05, 0.01 < **P < 0.001, and ***P < 0.001 represents a significant difference in all statistical comparisons.
Data availability
All relevant data are available from the authors on request.
References
Wilcox NS, Rui J, Hebrok M, Herold KC. Life and death of β cells in Type 1 diabetes: a comprehensive review. J Autoimmun. 2016;71:51–8.
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–10.
Stadler LK, Farooqi IS. A new drug target for type 2 diabetes. Cell. 2017;170:12–4.
Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–6.
Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest. 2008;118:3378–89.
Robertson RP, Harmon J, Tran POT, Poitout V. β-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53(suppl 1):S119–24.
Levy David E, Darnell JJr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62.
Darnell JE. STATs and gene regulation. Science. 1997;277:1630–5.
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809.
Yu H, Kortylewski M, Pardoll D. Tumour immunology: crosstalk between cancer and immune cells: role of STAT3 in the tumor microenvironment. Nat Rev Immunol. 2007;7:41–51.
Siddiquee KAZ, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008;18:254–67.
Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–60.
Okuma A, Hoshino K, Ohba T, Fukushi S, Aiba S, Akira S, et al. Enhanced apoptosis by disruption of the STAT3-IκB-ζ signaling pathway in epithelial cells induces Sjögren’s syndrome-like autoimmune disease. Immunity. 2013;38:450–60.
Skov S, Nielsen M, Bregenholt S, Ødum N, Claesson MH. Activation of Stat-3 is involved in the induction of apoptosis after ligation of major histocompatibility complex class I molecules on human Jurkat T cells. Blood. 1998;91:3566–73.
Chapman RS, Lourenco PC, Tonner E, Flint DJ, Selbert S, Takeda K, et al. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Gene Dev. 1999;13:2604–16.
Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signaling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736–46.
Cui Y, Huang L, Elefteriou F, Yang G, Shelton JM, Giles JE, et al. Essential role of STAT3 in body weight and glucose homeostasis. Mol Cell Biol. 2004;24:258–69.
Inoue H, Ogawa W, Asakawa A, Okamoto Y, Nishizawa A, Matsumoto M, et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 2006;3:267–75.
Mashili F, Chibalin AV, Krook A, Zierath JR. Constitutive STAT3 phosphorylation contributes to skeletal muscle insulin resistance in type 2 diabetes. Diabetes. 2012;62:457–65.
Priceman SJ, Kujawski M, Shen S, Cherryholmes GA, Lee H, Zhang C, et al. Regulation of adipose tissue T cell subsets by Stat3 is crucial for diet-induced obesity and insulin resistance. PNAS. 2013;110:13079–84.
Lee J-Y, Hennighausen L. The transcription factor Stat3 is dispensable for pancreatic β-cell development and function. Biochem Biophys Res Commun. 2005;334:764–8.
Kostromina E, Wang X, Han W. Altered islet morphology but normal islet secretory function in vitro in a mouse model with microvascular alterations in the pancreas. PLoS ONE. 2013;8:e71277.
Saarimäki-Vire J, Balboa D, Russell MA, Saarikettu J, Kinnunen M, Keskitalo S, et al. An activating STAT3 mutation causes neonatal diabetes through premature induction of pancreatic differentiation. Cell Rep. 2017;19:281–94.
Baeyens L, Lemper M, Leuckx G, De Groef S, Bonfanti P, Stangé G, et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol. 2014;32:76–83.
De Groef S, Renmans D, Cai Y, Leuckx G, Roels S, Staels W, et al. STAT3 modulates β-cell cycling in injured mouse pancreas and protects against DNA damage. Cell Death Dis. 2016;7:e2272.
Xiao X, Fischbach S, Zhang T, Chen C, Sheng Q, Zimmerman R, et al. SMAD3/Stat3 signaling mediates β-cell epithelial-mesenchymal transition in chronic pancreatitis-related diabetes. Diabetes. 2017;66:2646–58.
Ardestani A, Paroni F, Azizi Z, Kaur S, Khobragade V, Yuan T, et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat Med. 2014;20:385.
Li X, Cheng KK, Liu Z, Yang J-K, Wang B, Jiang X, et al. The MDM2–p53–pyruvate carboxylase signaling axis couples mitochondrial metabolism to glucose-stimulated insulin secretion in pancreatic β-cells. Nat Commun. 2016;7:11740.
Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, et al. Animal models of obesity and diabetes mellitus. Nat Rev Endocrinol. 2018;14:140–62.
Cai EP, Casimir M, Schroer SA, Luk CT, Shi SY, Choi D, et al. In vivo role of focal adhesion kinase in regulating pancreatic β-cell mass and function through insulin signaling, actin dynamics, and granule trafficking. Diabetes. 2012;61:1708–18.
Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J Clin Invest. 2013;123:3305–16.
Costa FR, Françozo MC, De Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med. 2016;213:1223–39.
Mellado-Gil J, Rosa TC, Demirci C, Gonzalez-Pertusa JA, Velazquez-Garcia S, Ernst S, et al. Disruption of hepatocyte growth factor/c-Met signaling enhances pancreatic β-cell death and accelerates the onset of diabetes. Diabetes. 2011;60:525–36.
Belgardt B-F, Ahmed K, Spranger M, Latreille M, Denzler R, Kondratiuk N, et al. The microRNA-200 family regulates pancreatic beta cell survival in type 2 diabetes. Nat Med. 2015;21:619–30.
Blandino-Rosano M, Barbaresso R, Jimenez-Palomares M, Bozadjieva N, Werneck-de-Castro JP, Hatanaka M, et al. Loss of mTORC1 signaling impairs β-cell homeostasis and insulin processing. Nat Commun. 2017;8:16014.
Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–13.
Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumor suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96.
Zhu H, Xiao F, Wang G, Wei X, Jiang L, Chen Y, et al. STAT3 regulates self-renewal of adult muscle satellite cells during injury-induced muscle regeneration. Cell Rep. 2016;16:2102–15.
Spinelli L, Lindsay YE, Leslie NR. PTEN inhibitors: an evaluation of current compounds. Adv Biol Regul. 2015;57:102–11.
Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405.
Tong Z, Fan Y, Zhang W, Xu J, Cheng J, Ding M, et al. Pancreas-specific Pten deficiency causes partial resistance to diabetes and elevated hepatic AKT signaling. Cell Res. 2009;19:710–9.
Davies BR, Greenwood H, Dudley P, Crafter C, Yu D-H, Zhang J, et al. Preclinical pharmacology of AZD5363, an orally bioavailable inhibitor of AKT: pharmacodynamics, antitumor activity and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012;11:873–87.
Chia JSJ, McRae JL, Thomas H, Fynch S, Elkerbout L, Hill P, et al. The protective effects of CD39 over-expression in multiple low dose streptozotocin-induced diabetes in mice. Diabetes. 2013;62:2026–35.
Paik S-g, Fleischer N, Shin S-i, et al. diabetes mellitus induced by subdiabetogenic doses of streptozotocin: obligatory role of cell-mediated autoimmune processes. PNAS. 1980;77:6129–33.
Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest. 2017;127:2881–91.
Wang M, Yang L, Sheng X, Chen W, Tang H, Sheng H, et al. T-cell vaccination leads to suppression of intrapancreatic Th17 cells through Stat3-mediated RORγt inhibition in autoimmune diabetes. Cell Res. 2011;21:1358.
Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–6.
Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–6.
Adam J, Ramracheya R, Chibalina MV, Ternette N, Hamilton A, Tarasov AI, et al. Fumarate hydratase deletion in pancreatic β cells leads to progressive diabetes. Cell Rep. 2017;20:3135–48.
Tang T, Abbott MJ, Ahmadian M, Lopes AB, Wang Y, Sul HS. Desnutrin/ATGL activates PPARδ to promote mitochondrial function for insulin secretion in islet β cells. Cell Metab. 2013;18:883–95.
Pal A, Barber TM, Van de Bunt M, Rudge SA, Zhang Q, Lachlan KL, et al. PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N Engl J Med. 2012;367:1002–11.
Wang L, Luk CT, Cai EP, Schroer SA, Allister EM, Shi SY, et al. PTEN deletion in pancreatic α cells protects against high fat diet-induced hyperglucagonemia and insulin resistance. Diabetes. 2014;64:147–57.
Wang L, Opland D, Tsai S, Luk CT, Schroer SA, Allison MB, et al. Pten deletion in RIP-Cre neurons protects against type 2 diabetes by activating the anti-inflammatory reflex. Nat Med. 2014;20:484–92.
Morley TS, Xia JY, Scherer PE. Selective enhancement of insulin sensitivity in the mature adipocyte is sufficient for systemic metabolic improvements. Nat Commun. 2015;6:7906.
Aghazadeh S, Yazdanparast R. Activation of STAT3/HIF-1alpha/Hes-1 axis promotes trastuzumab resistance in HER2-overexpressing breast cancer cells via down-regulation of PTEN. Biochim Biophys Acta. 2017;1861:1970–80.
de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, et al. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008;22:449–62.
Xue R, Lei S, Xia ZY, Wu Y, Meng Q, Zhan L, et al. Selective inhibition of PTEN preserves ischaemic post-conditioning cardioprotection in STZ-induced Type 1 diabetic rats: role of the PI3K/Akt and JAK2/STAT3 pathways. Clin Sci (Lond). 2016;130:377–92.
Sun S, Steinberg BM. PTEN is a negative regulator of STAT3 activation in human papillomavirus-infected cells. J Gen Virol. 2002;83:1651–8.
Tuttle RL, Gill NS, Pugh W, Lee J-P, Koeberlein B, Furth EE, et al. Regulation of pancreatic β-cell growth and survival by the serine/threonine protein kinase Akt1/PKBα. Nat Med. 2001;7:1133–7.
Humphrey RK, Yu S-M, Flores LE, Jhala US. Glucose regulates steady-state levels of PDX1 via the reciprocal actions of GSK3 and AKT kinases. J Biol Chem. 2010;285:3406–16.
Han SI, Aramata S, Yasuda K, Kataoka K. MafA stability in pancreatic beta cells is regulated by glucose and is dependent on its constitutive phosphorylation at multiple sites by glycogen synthase kinase 3. Mol Cell Biol. 2007;27:6593–605.
Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813:1978–86.
El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou J-Y, Shirakawa J, et al. SerpinB1 promotes pancreatic β cell proliferation. Cell Metab. 2016;23:194–205.
Axelsson AS, Tubbs E, Mecham B, Chacko S, Nenonen HA, Tang Y, et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci Transl Med. 2017;9:eaah4477.
Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang YQ, et al. Inhibition of DYRK1A and GSK3B induces human beta-cell proliferation. Nat Commun. 2015;6:8372.
Stein SA, Lamos EM, Davis SN. A review of the efficacy and safety of oral antidiabetic drugs. Expert Opin Drug Saf. 2012;12:153–75.
Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–43.
Wang YJ, Schug J, Won K-J, Liu C, Naji A, Avrahami D, et al. Single-cell transcriptomics of the human endocrine pancreas. Diabetes. 2016;65:3028–38.
Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163–7.
Maedler K, Spinas G, Dyntar D, Moritz W, Kaiser N, Donath MY. Distinct effects of saturated and monounsaturated fatty acids on β-cell turnover and function. Diabetes. 2001;50:69–76.
Weng Q, Wang J, Wang J, Tan B, Wang J, Wang H, et al. Folate metabolism regulates oligodendrocyte survival and differentiation by modulating AMPKalpha activity. Sci Rep. 2017;7:1705.
Ni Q, Gu Y, Xie Y, Yin Q, Zhang H, Nie A, et al. Raptor regulates functional maturation of murine beta cells. Nat Commun. 2017;8:15755.
Villa SR, Priyadarshini M, Fuller MH, Bhardwaj T, Brodsky MR, Angueira AR, et al. Loss of free fatty acid receptor 2 leads to impaired islet mass and beta cell survival. Sci Rep. 2016;6:28159.
Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic β cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014;19:872–82.
Weng Q, Wang J, Wang J, Wang J, Sattar F, Zhang Z, et al. Lenalidomide regulates CNS autoimmunity by promoting M2 macrophages polarization. Cell Death Dis. 2018;9:251.
Acknowledgements
We thank Dr. Ji Cao for comments and suggestions. We thank Dr. Berry Kalen for linguistic modification. This work was supported by the National Natural Science Foundation for Distinguished Young Scholar of China (No. 81625024) to BY and the National Natural Science Foundation (No. 81741172) to QW and Zhejiang Provincial Natural Science Foundation (No. LGF18H310001) to QW.
Author information
Authors and Affiliations
Contributions
QJW, MDY, and QJH conceived and designed the study. MTZ, JHZ, LJY, ZJX, ZKZ, and JCW performed the experiments. QJW, MTZ, JHZ, and JJW performed the data analysis. QJW, MTZ, BY, QRL, MDY, and QJH contributed to writing the manuscript. QJW, MDY, and QJH contributed to the materials. All the authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by H.-U. Simon
Supplementary information
Rights and permissions
About this article

Cite this article
Weng, Q., Zhao, M., Zheng, J. et al. STAT3 dictates β-cell apoptosis by modulating PTEN in streptozocin-induced hyperglycemia. Cell Death Differ 27, 130–145 (2020). https://doi.org/10.1038/s41418-019-0344-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-019-0344-3
This article is cited by
-
Talin-1 inhibits Smurf1-mediated Stat3 degradation to modulate β-cell proliferation and mass in mice
Cell Death & Disease (2023)
-
Syngeneically transplanted insulin producing cells differentiated from adipose derived stem cells undergo delayed damage by autoimmune responses in NOD mice
Scientific Reports (2022)
