
Abstract
p53 is one of the most important tumour suppressor proteins currently known. It is activated in response to DNA damage and this activation leads to proliferation arrest and cell death. The abundance and activity of p53 are tightly controlled and reductions in p53’s activity can contribute to the development of cancer. Here, we show that Fam83F increases p53 protein levels by protein stabilisation. Fam83F interacts with p53 and decreases its ubiquitination and degradation. Fam83F is induced in response to DNA damage and its overexpression also increases p53 activity in cell culture experiments and in zebrafish embryos. Downregulation of Fam83F decreases transcription of p53 target genes in response to DNA damage and increases cell proliferation, identifying Fam83F as an important regulator of the DNA damage response. Overexpression of Fam83F also enhances migration of cells harbouring mutant p53 demonstrating that it can also activate mutant forms of p53.
Similar content being viewed by others


Introduction
p53 is an important tumour suppressor gene. It is mutated in about 50% of human cancer and tumours that retain wild-type p53 frequently show inactivation of genes that impinge on p53 function. Cells normally express low levels of p53 and its expression is strongly induced after genotoxic stress [1, 2]. The increase in p53 abundance together with post-translational modifications of p53, leads to enhanced transcriptional activity of p53 and to p53-induced cell cycle arrest or apoptosis [3].
p53 is mostly regulated at the protein level and the increase in its abundance after cell stress is usually caused by an increase in its protein half-life. p53 is a short-lived protein that is rapidly degraded by cellular 26S proteasomes in an Mdm2-dependent manner [4, 5]. While Mdm2 targets p53 for degradation, it is itself a target gene for p53, meaning p53 and Mdm2 are connected by a negative feedback-loop [6]. This feedback-loop ensures that p53 levels are kept low when p53 activity is not needed and cells are able to proliferate. Several proteins can target this feedback-loop either by binding to p53 or to Mdm2 or by post-translationally modifying one or both proteins [3].
Fam83F is a member of a family of genes with sequence similarity 83 (Fam83). This family comprises eight genes (Fam83A–H) that are all characterised by the presence of a DUF- (domain of unknown function) 1669 domain in the N-terminus. Several members of this family function as intermediates in the EGFR/MAPK and PI3K/AKT pathways. They have oncogenic potential and are frequently upregulated in human tumours [7, 8]. Fam83F has 500 amino acids and a molecular weight of 55 kDa. It is highly expressed in platelets, pancreas, the gastrointestinal tract, and testis (www.genecards.org; www.proteinatlas.org).
We show that Fam83F stabilises p53, leading to an increase in p53 levels. Mechanistically, it interacts with the C-terminus of p53 and reduces Mdm2-mediated ubiquitination of p53. Fam83F also enhances p53 transcriptional activity. We also show that Fam83F is induced by DNA damaging agents and downregulation of Fam83F reduces p53 activity after DNA damage.
Results
Fam83F stabilises the p53 tumour suppressor protein
We previously identified Fam83F in a cell culture-based expression screen for novel regulators of p53 where it was shown to regulate p53 levels in an Mdm2-dependent manner [9]. We performed a more detailed Fam83F overexpression titration, both in the presence and absence of Mdm2, and looked at the effect on p53. In the presence of co-expressed Mdm2, Fam83F overexpression increased p53 abundance in a dose-dependent manner without altering Mdm2 levels (Fig. 1a, lanes 1–3). In the absence of co-expressed Mdm2 the Fam83F-dependent increase of p53, although still detectable, is significantly reduced (Fig. 1a, lanes 4–6, weak exposure). Fam83F was present at very low levels in a variety of different cell lines tested and immunoprecipitation from cell lysates was frequently required for detection. Indeed, downregulation of endogenous Fam83F in RKO cells by shRNA lentiviral infection resulted in only a modest reduction of p53 levels (Fig. 1b).

Fam83F is inducing p53 abundance. a H1299 cells were transfected with plasmids encoding p53 and Mdm2 together with increasing amounts of a plasmid encoding Fam83F or with vector DNA. 24 h after transfection, cells were harvested and analysed for abundance of p53, Mdm2, and Fam83F by Western Blotting. Immunodetection of PCNA was performed for loading control. b RKO cells were infected with lentiviruses carrying a shRNA targeted against Fam83F (Fam83F#I; Fam83F#II) or a control sequence (ctrl). WB: The cells were lysed and monitored for p53 abundance by Western blotting. Immunodetection of PCNA was performed for loading control. IP: Fam83F was immunoprecipitated and abundance of Fam83F was monitored by Western blotting. IgG levels are shown for control
Several regulators of p53 are also p53 target genes that function with p53 in feedback loops. Examples are Mdm2, Pirh2 and Cop-1 [10,11,12,13,14]. To test whether this is also the case for Fam83F, we overexpressed p53 in the p53-negative cell line H1299 and monitored the levels of fam83F by qRT-PCR. Unlike the p53 target genes p21 and mdm2 the expression of fam83F was not altered in a p53-dependent manner (Supplementary figure 1).
p53 is mainly regulated at the protein level and increases in p53 abundance are often accomplished by inhibiting the proteasomal degradation of p53 [3]. To test if Fam83F can alter p53 degradation, we co-transfected H1299 cells with p53, Mdm2, and Fam83F, either alone or in combination, in the presence or absence of the proteasome inhibitor MG132. Fam83F was not able to increase p53 levels in the presence of MG132, indicating that it does inhibit proteasomal degradation of p53 (Fig. 2a). We next blocked protein synthesis using cycloheximide in the presence of Mdm2 with or without Fam83F overexpression. Under these conditions, we observed a strong increase in p53 half-life after overexpression of Fam83F, showing that Fam83F stabilises p53 (Fig. 2b). This Fam83F-dependent increase in p53 protein abundance was not due to an increase of p53 RNA levels and indeed a reduction of p53 mRNA was actually observed (Fig. 2c).

Fam83 increases the half-life of p53. a H1299 cells were transfected with plasmids encoding p53 and Mdm2 together with a plasmid encoding Fam83F or with vector DNA. 24 h after transfection, MG132 (20 µM f.c.) was added for 16 h where indicated. Cells were harvested and analysed for the abundance of p53, Mdm2, and Fam83F by Western blotting. Immunodetection of PCNA was performed for loading control. b H1299 cells were transfected with plasmids encoding p53 and Mdm2 together with a plasmid encoding Fam83F or with vector DNA. 24 h after transfection, cycloheximide (CHX; 60 µg/ml f.c.) was added. Cells were harvested at the indicated time points and analysed for abundance of p53, Mdm2, and Fam83F by Western blotting. Immunodetection of PCNA was performed for loading control. The signals for p53 and PCNA were quantified and the relative amount of p53 was calculated. The relative amount of p53 at the time of CHX addition was set to 100%. The graph shows mean values and standard deviations of three independent experiments. c H1299 cells were transfected with plasmids encoding p53, Mdm2, and Fam83F or with vector DNA, for control, in the indicated combinations. 24 h after transfection, cells were harvested. The cells were divided into two aliquots. One of the aliquots was used to monitor the abundance of p53, Mdm2, and Fam83F by Western blotting. From the second aliquot RNA was prepared and analysed for the presence of p53 RNA and of the housekeeping gene β-actin. The relative amount of p53 RNA was calculated by the 2ΔCT equation. The graph shows mean values and standard deviations of three independent experiments. Relative amounts of p53 in the absence of transfected Mdm2 and Fam83F were set to 1. d H1299 cells were transfected with plasmids encoding His-tagged ubiquitin, p53, Mdm2, and Fam83F or with vector DNA, for control, in the indicated combinations. 24 h after transfection, cells were harvested and divided into two aliquots. One of the aliquots was used to monitor the abundance of p53, Mdm2, and Fam83F in the total cell lysate by Western blotting. From the second aliquot, ubiquitinated proteins were purified by adsorption to Ni2+ agarose. Ubiquitinated p53 was monitored by Western blotting
Since Fam83F can regulate the stability of p53 in the presence of Mdm2, we tested whether this was due to an effect on the ubiquitination of p53. Indeed, when we overexpressed Fam83F, Mdm2-induced p53 ubiquitination was significantly reduced (Fig. 2d).
Since Fam83F reduced p53 ubiquitination, we reasoned that Fam83F might interact with p53 and/or Mdm2. To test for direct protein–protein interactions, we performed in vitro pulldown experiments with purified proteins. Bacterially expressed, GST-tagged Fam83F indeed associated specifically with p53 (Fig. 3a.i) and, to a lesser extent, with Mdm2 (Fig. 3a.ii). We next investigated whether Fam83F associates with p53 and Mdm2 in cells. To increase the abundance of Fam83F, we treated cells with the DNA damage-inducing agent hydroxyurea. In contrast to the result using purified proteins, in cell lysates Mdm2 did not co-precipitate with Fam83F, however p53 did (Fig. 3b). Using a series of Fam83F and p53 C-terminal deletion constructs, we next determined their respective interaction sites. Fam83F interacted with full-length p53 but not with deletions removing 93 or more residues from the C-terminal region, indicating that Fam83F associates with the C-terminus of p53 (Fig. 3c). In contrast, when either the C-terminal region or the C-terminal region together with the central region of Fam83F were deleted, Fam83F was still able to interact with p53 (Fig. 3d), indicating that the N-Terminus of Fam83F is required for p53 interaction. In line with the finding that the N-terminus of Fam83F interacts with p53, the N-terminus of Fam83F appears sufficient to increase p53 levels (Fig. 3e).

The N-terminus of Fam83F associates with the C-terminus of p53. a.i His-tagged p53 and GST-tagged Fam83F or GST, all separately expressed in bacteria and purified were mixed. 10% of the mixture was taken for input control and analysed by Western blotting for the presence of p53, Fam83F, and GST. Glutathione-sepharose was added to the rest and the mixture was incubated for 24 h. The sepharose was collected by centrifugation and washed. Bound proteins were eluted and p53, Fam83F, and GST were monitored by Western blotting. a.ii Flag-tagged Mdm2 expressed in insect cells and purified was mixed with GST-tagged Fam83F or with GST expressed in bacteria and purified. 10% of the mixture was taken for input control and analysed by Western blotting for the presence of Mdm2, Fam83F, and GST. Glutathione-sepharose was added to the remaining mixture and incubated for 4 h. The sepharose was collected by centrifugation and washed. Bound proteins were eluted and Mdm2, Fam83F, and GST were monitored by Western blotting. b RKO cells were treated with hydroxyurea (HU) for 24 h. IP: Fam83F was precipitated and associated p53 and Mdm2 were monitored by Western blotting. Immunoprecipitation with IgG was performed for control. Input: An aliquot of the lysate was used to monitor p53, Mdm2, and Fam83F levels in the total cell lysate. c H1299 cells were transfected with plasmids encoding V5-tagged wild-type p53 or the indicated deletion mutants (see c.i for a schematic drawing) together with a plasmid encoding Flag-tagged Fam83F or with vector DNA. The Flag-tagged Fam83F was precipitated and associated p53 was monitored by Western blotting. Input: An aliquot of the lysate was used to monitor p53 and Fam83F in the total cell lysate. d H1299 cells were transfected with plasmids encoding Flag-tagged wild type Fam83F or the indicated deletion mutants (see d.i for a schematic drawing) together with a plasmid encoding p53 or with vector DNA. Fam83F was precipitated and associated p53 was monitored by Western blotting. Input: An aliquot of the lysate was used to monitor p53 and Fam83F in the total cell lysate. e H1299 cells were transfected with plasmids encoding Flag-tagged wild type Fam83F or the indicated deletion mutants together with plasmids encoding p53 and Mdm2. Abundance of p53, Mdm2, and Fam83F was monitored by Western blotting. Immunodetection of PCNA was performed for loading control. f H1299 cells were transfected with plasmids encoding Fam83F, p53, and Mdm2 in the indicated combinations. Mdm2 was precipitated and associated p53 and Fam83F were monitored by Western blotting. Input: An aliquot of the lysate was used to monitor p53 and Fam83F in the total cell lysate
The interaction of p53 and Fam83F implies that both proteins reside in the same subcellular compartment and cell fractionation studies indeed showed Fam83F, like p53, to be mainly nuclear (Supplementary figure 2).
Since Fam83F interacts with and stabilises p53 by reducing its ubiquitination, we tested whether Fam83F interferes with binding of p53 to Mdm2, which is the major ubiquitin ligase for p53 [3]. We transfected H1299 cells with p53 and Mdm2 with or without Fam83F, then immunoprecipitated Mdm2 and monitored for p53 association by Western Blotting. Fam83F co-expression resulted in a slight increase in the amount of p53 associated with Mdm2, possibly due to the effect Fam83F has on enhancing p53 levels (Fig. 3f). Fam83F is therefore apparently unable to displace p53 from its interaction with Mdm2. Vice versa, Mdm2 was not able to displace p53 from its interaction with Fam83F (Supplementary figure 3).
Since Fam83F promotes p53 levels, we wondered whether this might lead to increased p53 activity. To test this, we performed cell culture reporter gene assays using the PG13 reporter, which carries 13 repeats of the consensus p53-binding site upstream of a luciferase reporter gene. As shown in Fig. 4a, transfection of p53 and Mdm2 increased PG13 reporter activity and this activity was further enhanced by co-transfection of Fam83F (Fig. 4a.i). No effect was seen with the control reporter MG15 that harbours mutated p53-binding sites, confirming specificity with respect to p53 transcriptional activity (Fig. 4a.i). Similarly, when the p53-dependent reporters p21-Luc, Mdm2-Luc, or Pig3-Luc were co-transfected with p53 and Mdm2, an induction of the reporter activities was observed and additional co-transfection of Fam83F further enhanced this (Fig. 4a.ii+iii+v). This was however not the case with a Bax-reporter gene (Fig. 4a.iv). We also performed reporter gene assays with MCF7 cells that express endogenous levels of p53 and Mdm2 to rule out artifacts associated with their overexpression. Again, we observed a significant increase in the activity of the p53-dependent reporter PG13 by Fam83F (Fig. 4b). Although the activity of the Bax reporter in cells expressing p53 and Mdm2 was not further induced by overexpression of Fam83F, it was significantly reduced upon downregulation of Fam83F (Fig. 4c). Also the amount of the p53 target Mdm2 was decreased when Fam83F was downregulated (Fig. 5c.i).

Fam83F is controlling p53 activity. a H1299 cells were transfected with plasmids encoding different p53-dependent firefly luciferase reporter genes together with a plasmid encoding a renilla luciferase reporter gene and with plasmids encoding p53, Mdm2, and Fam83F as indicated. 30 h after transfection, cells were harvested and firefly and renilla luciferase activity was determined. The relative reporter activities were calculated by dividing the readings for firefly luciferase by the readings for renilla luciferase. The graphs show mean values and standard deviations from 3 (PG-13-Luc, Mdm2-luc) to 4 (MG15-Luc, p21-Luc, Bax-Luc, Pig3-Luc) independent experiments. Reporter activity in the absence of p53 and Mdm2 was set to 100% (*p < 0.05; **p < 0.01; n.s.: not significant). b MCF7 cells were transfected with plasmids encoding Fam83F, the p53-dependent firefly luciferase reporter PG13 and renilla luciferase. 24 h after transfection, cells were harvested and analysed for firefly and renilla luciferase activity as described in the legend to part a. Relative firefly activity in the absence of Fam83F was set to 1 (*p < 0.05). c RKO cells that had been infected with lentiviruses carrying a control shRNA (C), or two different shRNAs targeted against Fam83F (I: shRNA I; II: shRNA II) were transfected with plasmids encoding the p53-dependent Bax- luciferase reporter gene and renilla luciferase. 48 h after transfection, cells were harvested and analysed for firefly and renilla luciferase activity as described in the legend to part a. Relative firefly activity in the absence of Fam83F was set to 1. The graph shows mean values and standard deviations of five independent experiments (**p < 0.01; n.s.: not significant)

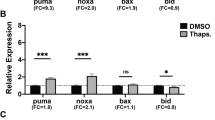
Fam83F enhances p53’s activity in response to DNA damage. a GM38 primary fibroblasts were exposed to UVC light (40 J/m2), etoposide (50 µM f.c.), and MMS (5 mM f.c.). RKO cells were treated with hydroxyurea (1.5 mM f.c.). After the indicated times cells were harvested and analysed by Western blotting for expression of Fam83F. Immunodetection of PCNA was performed for loading control. b GM38 primary fibroblasts were incubated with etoposide (50 µM f.c.) for the indicated times and analysed for the abundance of Fam83F, p53, and Mdm2 by Western blotting. Immunodetection of PCNA was performed for loading control. c RKO cells that had been infected with lentiviruses carrying two different shRNAs targeted against Fam83F (shRNA I; shRNA II) or a control shRNA (ctrl) were treated with etoposide (50 µM f.c.). 18 h after treatment, cells were analysed by Western blotting for the abundance of Mdm2, Fam83F, and p21. Immunodetection of PCNA was performed for loading control. c.ii The signals for Mdm2, p21, and PCNA were quantified and the relative amounts of Mdm2 and p21 were calculated. The graphs show mean values and standard deviations of five independent experiments (*p > 0.05; **p > 0.01; ***p > 0.005). d RKO cells that had been infected with lentiviruses carrying two different shRNAs targeted against Fam83F (shRNA I; or shRNA II) or a control shRNA (ctrl) were treated with etoposide (50 µM f.c.) or exposed to UV light (40 J/m2) as indicated in the figure. Cells were harvested at 4 and 24 h after treatment. RNA was prepared and analysed for the abundance of mdm2 and puma RNA. Abundance of ß-actin RNA was determined for internal control. The graphs show mean values of the obtained 2ΔCT numbers of qRT-PCR signals from three independent experiments. Values of non-treated RKO cells that had been infected with a lentivirus carrying a control shRNA were set to 1. e RKO cells that had been infected with lentiviruses carrying two different shRNAs targeted against Fam83F (shRNA I; or shRNA II) or a control shRNA (ctrl shRNA) were treated for 1 h with the indicated doses of etoposide, hydroxyurea, or MMS 24 h after plating. MTT assays were performed 3 days after treatment. The graphs show mean values and standard deviations of 4–5 independent experiments. Values of untreated cells were set to 100% (*p > 0.05; **p > 0.01; ***p > 0.005)
Fam83F activates p53 during the DNA damage response
In cells, both the expression level and activity of p53 is generally low but it becomes active and accumulates to high levels in response to DNA damage. Since we observed that Fam83F stabilises p53, we speculated that Fam83F might contribute to the activation of p53 in the presence of DNA damage. If this hypothesis is correct, then Fam83F levels should be induced by DNA damage. Indeed, the level of endogenous Fam83F was induced in cells exposed to DNA damaging agents, such as UVC-light, etoposide, MMS, and hydroxyurea (Fig. 5a). Intriguingly, no increase in fam83F RNA levels were detected after DNA damage, suggesting this induction of Fam83F occurs at the protein level (Supplementary figure 4A). The induction of Fam83F after DNA damage coincided with stabilisation of p53 (Fig. 5b). Moreover, when Fam83F was downregulated, DNA-damage-mediated induction of the p53 target p21, was significantly reduced (Fig. 5c; Supplementary figure 4B). Similarly, we observed lower amounts of puma and mdm2 RNA as well as of Mdm2 protein after DNA damage when Fam83F was downregulated (Fig. 5c, d). The decrease in the level of expression of the p53 target genes correlated with an increase in cell numbers and colony-forming abilities (Fig. 5e, Supplementary figure 4C). Altogether, these data suggest that Fam83F contributes to the p53-mediated DNA damage response and may be necessary for the efficient removal of cells with damaged DNA.
Fam83F controls p53 activity and cancer cell behaviour in zebrafish xenografts
Since all previous experiments were performed in cultured cells, we next tested if Fam83F controls p53 activity under more physiologically relevant conditions. Fish models have proven to be an excellent system for investigating p53 activity [15]. We injected zebrafish fam83F mRNA into fertilised zebrafish (Danio rerio) eggs and determined the effect on p53 activity by monitoring the abundance of the p53 target genes mdm2, p21, bax, and puma. To control for any adverse effects of the injection or of the excessive amounts of RNA in the egg and early embryo, we also injected the same amount of gfp mRNA. As shown in Fig. 6a, injection of fam83F mRNA led to a specific and strong increase of all three p53 target genes (Fig. 6a).

Fam83f controls proliferation and invasion in a zebrafish xenograft model. a 200 ng/µl of zebrafish fam83F or of gfp mRNA, for control, were injected into zebrafish eggs. Embryos were harvested 7 h post injection and RNA was extracted. The RNA was transcribed into cDNA and abundance of mdm2, p21, bax, and puma RNA was determined by qRT-PCR. Abundance of β-actin RNA was determined for internal control. The graphs show mean values of the obtained 2ΔCT numbers of qRT-PCR signals of five independent experiments. Abundance of the respective RNA of un-injected embryos was set to 1. b RKO cells expressing mCherry and one of two different shRNAs directed against Fam83F or a control shRNA were implanted into the brain ventricles of 2-day-old zebrafish larvae. Images were taken at 1.5 and 3 days post implantation (dpi). The area occupied by the cells on a dorsal view of a confocal Z projection of the whole cell cluster was determined. The graph shows mean values and standard deviations of seven animals per group (*p < 0.05; **p < 0.01)
Since Fam83F is a strong activator of p53, we tested whether Fam83F affects tumour behaviour in an in vivo transplantation assay. RKO cancer cells transduced with lentiviruses expressing shRNAs targeting either fam83F or a non-targeting control were transplanted into the brain ventricles of 2-day-old zebrafish larvae. For tracing, cells were additionally transduced with a lentivirus encoding mCherry fused to a fragment of the membrane protein GAP43 and the fluorescently labelled cells were monitored by in vivo focal microscopy. As shown in Fig. 6b, transplanted RKO cells expressing fam83F shRNA occupied a significantly larger area of the host brain and showed increased migration into neighbouring tissues (Fig. 6b.i). This was quantified by measuring the area that was occupied by the transplanted tumour cells from a dorsal view, and a statistically significant increase in this area was observed when Fam83F was downregulated (Fig. 6b.ii). These results indicate that Fam83F functions to augment the tumour suppression function of p53.
Fam83F is frequently downregulated in tumours with wild-type p53
p53 suppresses cell proliferation under potentially mutagenic conditions, such as after DNA damage or after activation of oncogenes [16]. The importance of this activity is demonstrated by the fact that p53 is frequently mutated in tumours [17]. We show that Fam83F is an activator of p53 and enhances p53’s activity in response to DNA damage. We hypothesise that Fam83F in the presence of wild-type p53 should be a disadvantage for tumorigenesis. We furthermore hypothesise that in tumours with wild type p53, Fam83F may be downregulated. To investigate this hypothesis, we analysed tumour samples, as well as neighbouring normal tissue from cancer patients. We measured both p53 expression by Western Blotting and fam83F mRNA levels by qRT-PCR. More than 50% of the tumours in our analysis showed elevated expression of p53 (Fig. 7). We sequenced the p53 gene of these tumours and found that 10 out of 17 tumours had mutated p53 (Fig. 7, Supplementary figure 5, Supplementary table 1)

fam83F RNA is decreased in tumours with wild-type p53. Patient samples of tumours (T) and surrounding normal tissue (N) were divided into three aliquots. One of the aliquots was used to monitor abundance of p53 by Western blotting. Ink staining of the membranes is shown for loading control. The second aliquot was used to prepare RNA. The RNA was transcribed into cDNA and the amounts of fam83F and ß-actin mRNAs were determined by qRT-PCR. The graphs show the obtained 2ΔCT numbers of the qRT-PCR signals. The third aliquot was used for histological analysis and sequencing of p53 (exon 4–8). The p53 status and the different mutations are shown. fs: frameshift
Most interestingly, fam83F expression was reduced in over 70% of the tumours with wild-type p53 in comparison to unaffected neighbouring tissue, supporting our hypothesis. In contrast, 50% of tumours with mutated p53 showed upregulation of fam83F RNA (Fig. 7 and Table 1).
Fam83F also activates mutant p53
More recently, Fam83F was reported to be overexpressed in tumours of the oesophagus and to act as an oncogene [18]. Yet, as described above, we show that Fam83F acts like a tumour suppressor protein, via promotion of p53 function. An explanation for this could be that p53 was mutated to its oncogenic form [19] in those oesophageal tumours and that Fam83F increases also the activity of oncogenic p53 mutants. In fact, we frequently found overexpression of Fam83F when p53 was mutated (Fig. 7, Table 1) and that Fam83F also interacts with mutant p53 (Fig. 8a). In contrast to wild type p53 however, we could not see an increase in the abundance of mutant p53 (R175H) when we co-expressed Fam83F in the presence of Mdm2 (Fig. 8b). Since Fam83F increases p53 levels by prolonging its otherwise short half-life, this result may be explained by the longer half-life already possessed by mutant p53 [20]. In order to investigate whether Fam83F can also increase the activity of mutant p53, we turned to cell migration, which is known to be enhanced specifically by oncogenic p53 mutants [21]. We monitored the effect of Fam83F on the migration of p53-negative H1299 cells expressing either wild type or mutant p53. When mutant p53 was co-expressed with Fam83F, we observed a clear increase in migration (Fig. 8c). Thus, Fam83F can activate both wild type and mutant p53 and may behave as a tumour suppressor protein or as an oncogene depending on the status of p53.

Fam83F also activates mutant p53. a H1299 cells were transfected with plasmids encoding Flag-tagged Fam83F and wild type (wt) or mutant (R175H) p53. 24 h after transfection, cells were lysed, Flag-Fam83F was precipitated and associated p53 was monitored by Western blotting. An aliquot of the lysed cells was used to monitor p53 and Flag-Fam83F abundance in the cell lysate. Immunodetection with an antibody targeted against PCNA was performed for loading control. b H1299 cells were transfected with plasmids encoding wild type or mutant p53 (R175H), Mdm2, and Fam83F in the indicated combinations. 24 h after transfection, cells were lysed and the amounts of p53, Mdm2, and Fam83F were determined by Western blotting. Immunodetection with an antibody targeted against PCNA was performed for loading control. c H1299 cells were transfected with plasmids encoding Fam83F and wild type (wt) or mutant (mu; R175H) p53 in the indicated combinations or with vector DNA for control. An aliquot of the transfected cells was pipetted into the wells of a two-well culture insert. After 24 h, the culture insert was removed and the status of the cells was documented by microscopy (0 h). The cells were cultured for further 48 h and the migration was monitored by microscopy (48 h). The area in the gap was quantified and relative migration was calculated. The graph shows mean values and standard deviation of five independent experiments. ***p > 0.0005. The rest of the transfected cells were lysed 24 h after transfection. p53 and Fam83F levels were monitored by Western blotting. Immunodetection with an antibody targeted against PCNA was performed for loading control
Discussion
The abundance and activity of p53 are mainly controlled post-transcriptionally by stabilisation of the protein, leading to an increase in its abundance, and by modifications that enhance its transcriptional activity [3]. The stability of p53 is largely controlled by Mdm2, which binds to p53 and promotes its polyubiquitination and subsequent degradation by the 26S proteasomes. Other p53-regulatory proteins have been identified that modulate ubiquitination by binding to p53 or Mdm2 [3]. As reported here, Fam83F is an additional, novel regulator of p53 that reduces p53 ubiquitination and enhances its activity.
Fam83F-mediated stabilisation of p53 may be a consequence of its ability to reduce the ubiquitination state of p53. This would most likely be mediated through interaction of Fam83F with the C-terminus of p53, thereby masking the ubiquitination sites in the C-terminus of p53 [22]. Although we saw association of Fam83F with Mdm2 in in vitro pulldown experiments employing bacterially expressed Fam83F, we could not confirm this interaction in cell lysates. This suggests that amino acid residues, which form an interaction interface between Fam83F and Mdm2 in vitro may be modified in cells by post-translational modifications.
Our finding that Fam83F enhances p53 activity is in contrast to a previous study that reported a decrease in cell proliferation upon downregulation of Fam83F [18]. Moreover, Yang et al., observed that microRNA miR-455-3p, which targets Fam83F, inhibits cell proliferation and invasion although they did not report whether this is specifically due to the regulation of Fam83F by miR-455-3p [23]. The reasons for the differences in the published literature and our work is not clear. One possible explanation is that p53 was mutated to oncogenic forms in the cells analysed by Mao and colleagues and by Yang and colleagues. About half of human tumours have mutated p53 and oesophageal cancer is one of the cancer types where p53 is frequently mutated (https://p53.fr). Mutant p53 frequently gains oncogenic properties, resulting in the stimulation of cell proliferation and migration of cancer cells [24]. Indeed, we found that Fam83F interacts with and enhances the migration of cells harbouring an oncogenic form of p53. Moreover, we also found overexpression of Fam83F in 50% of tumours with mutated p53. This result would therefore be in line with the observations made by Mao and co-workers and Yang and co-workers, assuming an oncogenic form of p53 was present in the cells that they analysed. In the case of wild type p53, we found that Fam83F enhanced p53 levels and activity and was required for full activity of p53 after DNA damage. Although the size of our cohort of cancer patients was relatively small, in line with this notion, we observed that fam83F mRNA expression was frequently reduced in tumours with wild-type p53. These observations suggest that Fam83F may function either as an oncogene or as a tumour suppressor protein, depending on the status of p53.
Material and methods
Cell lines and their treatment
H1299 cells, MCF7 cells, and RKO cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% foetal bovine serum and 1% penicillin/streptomycin. GM38 cells were cultured in Dulbecco’s modified Eagle’s medium containing 15% foetal bovine serum and 1% penicillin/streptomycin. All cells were maintained at 37 °C and 5% CO2 in a humidified atmosphere. All cell lines were mycoplasma-free and checked against the register for misidentified cell lines (http://iclac.org/databases/cross-contaminations/). H1299 cells were transiently transfected by calcium-phosphate DNA co-precipitation as described [25], or with PromoFectin (#PK-CT-2000-100, Promokine, Heidelberg, Germany) according to the manufacturer’s recommendation. MCF7 cells were transfected with ScreenFect®-A (#S-3001, Screenfect GmbH, Karlsruhe, Germany) according to the manufacturer’s recommendation.
MG132 (#S2619, Selleckchem.com, Houston, TX, USA) was used at 20 µm (f.c.), cycloheximide (#C-1988, Sigma Aldrich, Taufkirchen, Germany) at a final concentration of 60 µg/ml, etoposide (#E-1383, Sigma Aldrich, Taufkirchen, Germany) at 50 µM (f.c.), MMS (#64294, Fluka, now Sigma Aldrich, Taufkirchen, Germany) at 5 mM final concentration and hydroxyurea (#H-8627, Sigma Aldrich, Taufkirchen, Germany) at 1.5 mM final concentration. For UV irradiation, cells were washed once with PBS, irradiated with UVC light (40 J/m2) and cultured in the original culture medium.
Infection of RKO cells with mission shRNA lentivirus particles (Sigma-Aldrich, Taufkirchen, Germany) was performed according to the manufacturer’s recommendation. Infection of RKO cells with lentiviruses containing the mCherry/GAP43 construct was performed with filtered supernatant from HEK-293T cells that had been transfected with the lentiviral vector together with third generation lentiviral packaging vectors. The resulting transduced RKO cells were selected with blasticidin (15 μg/ml f.c.; Invivogen, San Diego, CA, USA) for 7 days.
Fish strains, housing, RNA preparation, injection, and sampling
Wildtype zebrafish (Danio rerio) from the AB inbred line were raised and maintained under standard conditions [26]. Embryos were raised at 28.5 °C in E3 medium (4.92 mM NaCl, 0.17 mM KCl, 0.295 mM CaCl2 × 2H2O, 0.333 mM MgSO4 × 7H2O). 24 h after fertilization 0.003% 1-phenyl-2-thiourea (PTU; Sigma Aldrich, Taufkirchen, Germany) was added to prevent pigmentation. Zebrafish larvae at 2 days post fertilization were anaesthetised with Tricaine mesylate (0.01%, Sigma-Aldrich, Taufkirchen, Germany) and 200–300 RKO cells, suspended in Dulbecco’s modified Eagle’s medium containing 10% foetal bovine serum, were injected into the mid-brain ventricle. After cell injection, larvae were kept in E3-PTU medium at 33 °C. For confocal live imaging (Zeiss LSM 800, Germany) 1.5-day post implantation (dpi) and 3 dpi larvae were anaesthetised and embedded into 1% low-melting-agarose (PEQLAB Biotechnologie, Erlangen, Germany), diluted in E3 medium, on microscope slides and imaged. After imaging, the larvae were removed from embedding and kept under the same conditions for a follow-up at 3 dpi. Z-stacks of confocal images from a dorsal view of the cell implants were overlayed into Max-projections and the area of the red fluorescent cells was measured using ImageJ (NIH, USA).
For mRNA injection, zebrafish fam83F and gfp mRNA were transcribed using the mMESSAGE mMACHINE SP6 Kit (#AM-1340, Ambion, Foster City, CA, USA) according to the manufacturer’s protocol and injected at a concentration of 200 ng/µl into fertilised eggs at the one-cell stage.
Patient samples
Samples from 17 different carcinoma tissues and their matched adjacent normal tissue were obtained from the University hospital of the Saarland (Homburg, Germany). None of the patients had received preoperative treatment. All tissues were frozen immediately after surgery and stored at −80 °C until used. Samples were collected with informed consent from the patients and approved by the ethics committee of the University hospital of Saarland.
Plasmids
The plasmids encoding wild type p53, Mdm2, and His-ubiquitin have been described earlier [25]. The V5-tagged version of p53 and the deletion mutants pcDNA3-V5-p53 aa1-150 and pcDNA3-V5-p53 aa1-300 were generated by PCR amplification using pcDNA3-p53 as a template and primers encoding the V5 tag. The obtained PCR products were cloned into pcDNA3. The plasmid for mutant p53 (R175H) was a gift from David Lane. Fam83F-p3xFLAG-CMV-pBluescriptR was purchased from Open Biosystems. The Flag-tagged deletion mutants of Fam83F were obtained by PCR amplification of the respective parts of Fam83F using Fam83F-p3xFLAG-CMV-pBluescriptR as a template. The plasmid without the Flag-tag was obtained by PCR-amplification of the Fam83F sequence using the p3xFLAG-CMV-pBluescriptR as a template. The obtained PCR product was cloned into the NotI and XbaI sites of pcDNA3. GFP-tagged Fam83F was obtained by amplifying fam83F by PCR and cloning into pGEX-4T-2. For His-tagged p53, p53 was amplified by PCR and cloned into the pQE-vector. All DNAs were confirmed by sequencing. Sequences of primers are available on request. The luciferase reporter for Bax was a gift from Moshe Oren, Rehovot, Israel. The luciferase reporters p21-Luc, PG13-Luc, MG15-Luc, Mdm2-Luc, p21-Luc were given to us by M. Fritsche, Freiburg, Germany. The luciferase reporter for Pig3 was provided by Jean-Christoph Bourdon, Dundee, Scotland.
Antibodies
The p53 (DO-1, sc-126), PCNA (anti-proliferating cell nuclear antigen; PC10, sc-56), Mdm2 (SMP14, sc-965), and Fam83F (K18, sc-102516) antibodies were purchased from Santa Cruz, (Dallas, TX, USA). The p21 antibody (#556430) was obtained from BD Biosciences (Franklin Lakes, NJ, USA) and the Flag-antibody from Cell Signaling (#2368, Danvers, MA, USA). The Mdm2 antibody 2A10 (#D29080) was from Calbiochem (now Merck, Darmstadt, Germany) and the Mdm2 antibody D1V2Z (#86934) from Cell Signaling Technology (Danvers, MA, USA).
For secondary antibodies, we used HRP-coupled anti-mouse and rabbit antibodies (#P0447; #P0448) from Dako (Darmstadt, Germany) and the Clean blot™ IP Detection Reagent (HRP; #21230) from Thermo Fisher (Waltham, MA, USA).
Lentiviruses
MISSION® pLKO.1-puro non-target control transduction particles and MISSION® lentiviral transduction particles carrying an shRNA against TRIM25 (clone ID TRCN0000438014 (shRNA II) and clone ID TRCN000434615 (shRNA I)) were purchased from Sigma-Aldrich, St. Louis, MD, USA)
For generating lentiviruses carrying the mcherry-gap43 construct, HEK-293T cells were transfected with plasmids encoding mcherry-gap43, gag-pol (pDMLg/pRRE), rev (pRSV-Rev), and vsv-g (pCMV-VSV-G) using Lipofectamine 2000 (#11668019, Invitrogen, Carlsbad, CA, USA). 6 h after transfection, the medium was changed. 24 h after the medium change, the supernatant was collected, filtered, and transferred to RKO cells. Fresh medium was added to the HEK-293T cells and incubated for further 24 h. The supernatant was again filtered and transferred to the RKO cells that had already received the supernatant 24 h before. 24 h later, the medium of the RKO cells was replaced with fresh culture medium. 24 h later blasticidin (15 μg/ml f.c.; #ant-bl-05; Invivogen, San Diego, CA, USA) was added to select the transduced cells and replaced every 2 days for 1 week.
SDS–PAGE and Western blotting
SDS–PAGE and Western blotting was performed as described [25].
Ubiquitination assay
Ubiquitination assays were performed as described [25].
GST-pull-down
Ten micrograms of purified GST-tagged Fam83F were mixed with 10 µg of purified His-tagged p53 (that has been expressed in and purified from bacteria) or Flag-tagged Hdm2 (that has been expressed in and purified from insect cells) in 400 µl buffer P (10 mM HEPES pH 7.9, 50 mM NaCl, 0.5 M sucrose, 0.5% Triton-X-100). 10% of the mixture was used for input control. To the rest, 10 µl glutathione sepharose (#17527901, GE-healthcare, Chalfont ST. Gilles, GB) were added and incubated over night at 4 °C with end-over-end rotation. The next day, the beads were washed three times with buffer P, mixed with 2x sample buffer, vortexed, heated for 10 min to 95 °C and loaded onto a SDS–PAGE gel.
Immunoprecipitation
For co-immunoprecipitating endogenous proteins, cells were lysed in buffer P for 15 min on ice. The lysate was cleared by centrifugation for 10 min at 13,000 rpm and the protein concentration was determined. Two milligrams of protein were mixed with 1.5 µl of the anti-Fam83F antibody K18 or the same amount of IgG in 750 µl of buffer P and incubated overnight at 4 °C with end-over-end rotation. The next morning, 40 µl of a 1:1 slurry of protein A and G agarose (#20334 and #20399, Pierce, Rockford, IL, USA) pre-incubated with BSA were added and incubated for 1 h. The agarose was pelleted by centrifugation, washed six times with buffer P, mixed with 2x protein sample buffer, vortexed, heated for 10 min to 95 °C and loaded onto an 8% SDS–PAGE gel.
Quantitative reverse transcription PCR
Quantitative reverse transcription PCR was performed as described [27]. Sequences of primers are available on request.
Luciferase assay
H1299, were transfected with plasmids encoding Fam83F and firefly and renilla luciferase reporter genes together with plasmids encoding p53 and Mdm2 or vector DNA. MCF7 cells were transfected with the PG13-firefly reporter and renilla luciferase and RKO cells that had been infected with lentivirus carrying a shRNA targeted against Fam83F or a control shRNA were transfected with the Bax-firefly reporter gene and renilla luciferase. Twenty-four hours after transfection, cells were lysed in passive lysis buffer (#E194A, Promega, Madison, WI, USA) and incubated for 4 h at −80 °C. Activities of firefly and renilla luciferases were determined with the luminometer Victor Light 1420 (Perkin Elmer, Waltham, MA, USA). Reporter activity was calculated by dividing the readings for the firefly luciferase by the readings for renilla luciferase.
Migration assay
H1299 cells were transfected using PromoFectin. Seventy microliters of transfected cells were pipetted into a two-well culture insert (ibidi; Planegg, Germany) that had been inserted into a well of a six-well plate. Twenty-four hours after transfection, 2.5 ml DMEM medium (w/o FCS or Pen/strep) were added to the six-well plate with the two-well culture insert. The two-well culture insert was removed and the cells were photographed for the first time (0 h). Forty-eight hours later, a second image was taken.
MTT assay
RKO cells that had been infected with lentivirus carrying a shRNA targeted against Fam83F or a control shRNA were plated into six-well plates. Twenty-four hours after plating, etoposide, hydroxyurea, or methyl methanesulfonate (MMS) were added at different concentrations and incubated for 1 h. The medium was aspirated, the cells were washed with medium, fresh culture medium was added and cells were incubated for 72 h. (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT) was added at a concentration of 40 µg/ml (f.c.) for 5 h. The medium was aspirated and cells and formazan salt were solubilized in isopropanol. The absorbance was measured at λ = 550 nm.
Sequencing of tumour samples
Tumour samples were divided into two halves. One half was embedded into tissue freezing medium (Leica, Wetzlar, Germany). Tissue sections were made and stained with eosin and hematoxylin according to standard protocols. The tumour sections were analysed by a pathologist and the relative percentage of tumour cells was determined. From the other half of the tumour sample, DNA was prepared using the DNeasy Blood and tissue Kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s recommendation. Analysis of the p53 gene has been performed using single-molecule DNA sequencing (SMRT) as described previously [28]. Briefly, a 2.8 kb amplicon that encompasses exons 4–8 have been amplified using advantage 2 PCR buffer (Advantage HF 2 PCR Kit, #639124, Clontech Laboratories, Mountain View, CA, USA) and sequenced on the PacBio RS II instrument using C4 chemistry, P6 polymerase and a 240-min movie time. p53 variants were analysed using Seshat, a specific web service developed for annotating p53 information derived from sequencing data [29].
Statistics
Statistical analysis was performed with a two-sided t-test.
The mean areas occupied by Fam83f knock-down RKO cells versus control RKO cells in brain ventricles of zebrafish larvae (n = 7 larvae per group) was compared using the one-sided t-test, after Shapiro–Wilk test for normality, with α = 0.05.
References
Levine AJ. p53, the cellular gatekeeper for growth and division. Cell . 1997;88:323–31.
Kubbutat MH, Ludwig RL, Ashcroft M, Vousden KH. Regulation of Mdm2-directed degradation by the C terminus of p53. Mol Cell Biol. 1998;18:5690–8.
Boehme KA, Blattner C. Regulation of p53—insights into a complex process. Crit Rev Biochem Mol Biol. 2009;44:367–92.
Maki CG, Huibregtse JM, Howley PM. In vivo ubiquitination and proteasome-mediated degradation of p53. Cancer Res. 1996;56:2649–54.
Blattner C, Tobiasch E, Litfen M, Rahmsdorf HJ, Herrlich P. DNA damage induced p53 stabilization: no indication for an involvement of p53 phosphorylation. Oncogene. 1999;18:1723–32.
Picksley SM, Lane DP. The p53-mdm2 autoregulatory feedback loop: a paradigm for the regulation of growth control byp53? Bioessays. 1993;15:689–90.
Cipriano R, Miskimen KL, Bryson BL, Foy CR, Bartel CA, Jackson MW. Conserved oncogenic behavior of the FAM83 family regulates MAPK signaling in human cancer. Mol Cancer Res. 2014;12:1156–65.
Bartel CA, Parameswaran N, Cipriano R, Jackson MW. FAM83 proteins: fostering new interactions to drive oncogenic signaling and therapeutic resistance. Oncotarget. 2016;7:52597–612.
Zhang P, Kratz AS, Salama M, Elabd S, Heinrich T, Wittbrodt J, et al. Expression screening using a Medaka cDNA library identifies evolutionarily conserved regulators of the p53/Mdm2 pathway. BMC Biotechnol. 2015;15:92.
Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–91.
Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–08.
Varshavsky A. The ubiquitin system. Trends Biochem Sci. 1997;22:383–7.
Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27.
Kastan MB, Canman CE, Leonard CJ. P53, cell cycle control and apoptosis: implications for cancer. Cancer Metastas-Rev. 1995;14:3–15.
Zhang P, Elabd S, Hammer S, Solozobova V, Yan H, Bartel F, et al. TRIM25 has a dual function in the p53/Mdm2 circuit. Oncogene. 2015;34:5729–38.
Pluquet O, Hainaut P. Genotoxic and non-genotoxic pathways of p53 induction. Cancer Lett. 2001;174:1–15.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9.
Mao Y, Liu J, Zhang D, Li B. miR-143 inhibits tumor progression by targeting FAM83F in esophageal squamous cell carcinoma. Tumour Biol. 2016;37:9009–22.
Sabapathy K. The contrived mutant p53 oncogene—beyond loss of functions. Front Oncol. 2015;5:276.
Buschmann T, Minamoto T, Wagle N, Fuchs SY, Adler V, Mai M, et al. Analysis of JNK, Mdm2 andp14(ARF) contribution to the regulation of mutant p53 stability. J Mol Biol. 2000;295:1009–21.
Yeudall WA, Wrighton KH, Deb S. Mutant p53 in cell adhesion and motility. Methods Mol Biol. 2013;962:135–46.
Lohrum MA, Woods DB, Ludwig RL, Balint E, Vousden KH. C-terminal ubiquitination of p53 contributes to nuclear export. Mol Cell Biol. 2001;21:8521–32.
Yang H, Wei YN, Zhou J, Hao TT, Liu XL. MiR-455-3p acts as a prognostic marker and inhibits the proliferation and invasion of esophageal squamous cell carcinoma by targeting FAM83F. Eur Rev Med Pharmacol Sci. 2017;21:3200–6.
Blandino G, Di Agostino S. New therapeutic strategies to treat human cancers expressing mutant p53. J Exp Clin Cancer Res. 2018;37:30.
Kulikov R, Letienne J, Kaur M, Grossman SR, Arts J, Blattner C. Mdm2 facilitates the association of p53 with the proteasome. Proc Natl Acad Sci USA. 2010;107:10038–43.
Westerfield M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio). Eugene: University of Oregon Press; 2000.
Solozobova V, Rolletschek A, Blattner C. Nuclear accumulation and activation of p53 in embryonic stem cells after DNA damage. BMC Cell Biol. 2009;10:46.
Lodé L, Ameur A, Coste T, Ménard A, Richebourg S, Gaillard JB, et al. Single-molecule DNA sequencing of acute myeloid leukemia and myelodysplastic syndromes with multiple TP53 alterations. Haematologica. 2018;103:e13–e16.
Tikkanen T, Leroy B, Fournier JL, Risques RA, Malcikova J, Soussi T, et al. A Web service for accurate annotation, validation, and analysis of TP53 variants generated by conventional and next-generation sequencing. Hum Mutat. 2018;39:925–33.
Acknowledgements
We thank Christina Bauer and Nicole Zettler for technical assistance, the KIT for support and the DAAD for providing fellowships for Mohammed Salama and Diego Benitez-Riquelme. We are particularly grateful to our colleagues for plasmids and discussions and to Andy Cato for help with the manuscript. We also like to acknowledge support of the National Genomics Infrastructure (NGI)/Uppsala Genome Center and UPPMAX for providing assistance in massive parallel sequencing and computational infrastructure. Work performed at NGI/Uppsala Genome Center has been funded by RFI/VR and Science for Life Laboratory, Sweden,
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Oren
Supplementary information
Rights and permissions
About this article

Cite this article
Salama, M., Benitez-Riquelme, D., Elabd, S. et al. Fam83F induces p53 stabilisation and promotes its activity. Cell Death Differ 26, 2125–2138 (2019). https://doi.org/10.1038/s41418-019-0281-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-019-0281-1
This article is cited by
-
P53: A Key Target in the Development of Osteoarthritis
Molecular Biotechnology (2024)
-
Mutant p53 gains oncogenic functions through a chromosomal instability-induced cytosolic DNA response
Nature Communications (2024)
-
Upregulation of FAM83F by c-Myc promotes cervical cancer growth and aerobic glycolysis via Wnt/β-catenin signaling activation
Cell Death & Disease (2023)
-
Comprehensive analysis of the expression, prognostic significance, and function of FAM83 family members in breast cancer
World Journal of Surgical Oncology (2022)
-
The p53 family member p73 in the regulation of cell stress response
Biology Direct (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}