
Abstract
Atherosclerosis is a progressive, chronic inflammation in arterial walls. Long noncoding RNAs (lncRNAs) participate in inflammation, but the exact mechanism in atherosclerosis is unclear. Our microarray analyses revealed that the levels of lncRNA-FA2H-2 were significantly decreased by oxidized low-density lipoprotein (OX-LDL). Bioinformatics analyses indicated that mixed lineage kinase domain-like protein (MLKL) might be regulated by lncRNA-FA2H-2. In vitro experiments showed that lncRNA-FA2H-2 interacted with the promoter of the MLKL gene, downregulated MLKL expression, and the binding sites between −750 and 471 were necessary for lncRNA-FA2H-2 responsiveness to MLKL. Silencing lncRNA-FA2H-2 and overexpression of MLKL could activate inflammation and inhibited autophagy flux. Both lncRNA-FA2H-2 knockdown and overexpression of MLKL could significantly aggravate inflammatory responses induced by OX-LDL. We found that the 3-methyladenine (3-MA) and Atg7-shRNA enhanced inflammatory responses induced by knockdown of lncRNA-FA2H-2 and overexpression of MLKL. We demonstrated that the effects of MLKL on autophagy might be associated with a mechanistic target of rapamycin (mTOR)-dependent signaling pathways. In vivo experiments with apoE knockout mice fed a western diet demonstrated that LncRNA-FA2H-2 knockdown decreased microtubule-associated expression of microtubule-associated protein 1 light chain 3 II and lysosome-associated membrane protein 1, but increased expression of sequestosome 1 (p62), MLKL, vascular cell adhesion molecule-1, monocyte chemoattractant protein-1, and interleukin-6 in atherosclerotic lesions. Our findings indicated that the lncRNA-FA2H-2-MLKL pathway is essential for regulation of autophagy and inflammation, and suggested that lncRNA-FA2H-2 and MLKL could act as potential therapeutic targets to ameliorate atherosclerosis-related diseases.
Similar content being viewed by others


Introduction
Atherosclerosis is a complex chronic disease caused by formation of atherosclerotic plaques that respond to accumulation of lipoproteins. This results in upregulation of endothelial dysfunction, leading to the inflammatory response [1,2,3]. Studies have demonstrated that the inflammatory response induced by oxidized low-density lipoprotein (OX-LDL) acts as an independent risk factor of atherogenesis. Hence, reducing the inflammatory response induced by OX-LDL may be favorable for preventing progression of atherosclerosis [4]. Although the exact mechanism in atherosclerosis remains unknown.
Long noncoding RNAs (lncRNAs) are non-protein-coding RNAs that lack an open reading frame [5]. Recently, studies have found that lncRNAs are involved in various cellular functions through different mechanisms such as post-transcriptional, transcriptional [6,7,8]. Zhang et al. revealed that lncRNA LINC00305 promotes inflammation through activation of the aryl hydrocarbon receptor repressor (AHRR)-nuclear factor nuclear factor-kB (NF-κB) pathway [9]. Moreover, OX-LDL also regulates cholesterol metabolism and macrophage-mediated inflammatory responses by inducing expression of lncRNA RP5-833A20.1 and by inhibiting expression of its downstream nuclear factor I-A [10]. However, the underlying mechanism of lncRNAs in atherosclerosis is poorly understood.
Autophagy is a cellular pathway that participates in the degradation of proteins and organelles [5, 11,12,13]. Moderate autophagy can prevent macrophages and smooth muscle cells (SMCs) from forming foam cells, and stabilizing atherosclerotic plaques [14,15,16]. Increased autophagy can protect cells from inflammatory damage [9, 17, 18]. However, overstimulation of autophagy results in destabilization of plaques and inflammatory increases in atherosclerosis [19]. The blockage of autophagic flux in different tissues leads to inflammation [20]. However, the potential involvement of autophagy in atherosclerosis needs further investigation.
For the first time, our results confirmed that lncRNA-FA2H-2 inhibited MLKL expression, which attenuated the OX-LDL-induced inflammatory response through autophagy flux induction. The mechanism of MLKL inhibition of autophagy flux was mainly due to the activation of mTOR-dependent signaling pathways. In the current study, we explored the role of the lncRNA-FA2H-2-MLKL pathway and the mechanisms in mediating autophagy and inflammation in the pathogenesis of atherosclerosis.
Results
Inhibition or impairment of autophagy flux aggravated the inflammatory response induced by OX-LDL in ECs and SMCs
As indicated in Figure S1A, EC, SMC, and THP-1 cells were treated with OX-LDL. The results showed that OX-LDL obviously decreased ECs and SMCs cell survival in a dose-dependent manner and that this decrease was greater than that in THP-1 cells. Next, we mainly investigated the effects of OX-LDL on inflammation in ECs and SMCs. The protein levels of vascular cell adhesion molecule 1 (VCAM-1), monocyte chemoattractant protein 1 (MCP-1), and interleukin (IL)-6 were markedly increased by OX-LDL (Fig. 1a). Furthermore, OX-LDL obviously increased the production of IL-1β, tumor necrosis factor-α (TNF-α), IL-18, IL-8, IL-6, VCAM-1, and MCP-1. Conversely, OX-LDL decreased the production of IL-10, suggesting that OX-LDL promoted the inflammatory response (Table S1-1).

Inhibition or impairment of autophagy flux could aggravate the inflammatory response induced by OX-LDL in ECs and SMCs. a ECs and SMCs were treated with OX-LDL (0, 25, 50, or 100 mg/L) for 24 h and expressions of VCAM-1, MCP-1, and IL-6 were measured by western blotting. bP < 0.01 and cP < 0.001 versus the 0 mg/L group. b ECs and SMCs were incubated with OX-LDL (0, 25, 50, or 100 mg/L) for 24 h, and expressions of LC3II, p62, and LAMP1 were measured by western blotting. aP < 0.05, bP < 0.01, cP < 0.001, and dP < 0.0001 versus the 0 mg/L group. c ECs were transiently transfected with GFP-mRFP-LC3 for 48 h and subsequently treated with OX-LDL (50 mg/L) for 24 h, OX-LDL (50 mg/L) for 24 h plus BafA1 (50 nm) for 3 h, OX-LDL (50 mg/L) for 24 h plus Rap (100 nM) for 12 h, Rap (100 nM) for 12 h, or BafA1 (50 nm) for 3 h alone, and then observed for changes of both green and red fluorescence using a confocal microscope. Scale bar: 20 μm. The numbers of acidified autophagosomes (GFP-RFP+) versus neutral autophagosomes (GFP+RFP+) per cell in each condition were quantified. #P < No significant (NS); *P < 0.05; **P < 0.01. d ECs and SMCs were treated with BafA1 (50 nM) for 3 h before OX-LDL (50 mg/L) for 24 h. The levels of LC3II were measured by western blotting. bP < 0.01 versus treatment with OX-LDL alone. e ECs and SMCs were treated with OX-LDL (0, 25, 50, or 100 mg/L) for 24 h. The protein levels of endogenous CTSD were measured by western blotting. aP < 0.05 and bP < 0.01 versus the 0 mg/L group. f ECs and SMCs were treated with OX-LDL (0, 25, 50, or 100 mg/L) for 24 h, and enzymatic activity was analyzed using fluorogenic kits. *P < 0.05 and **P < 0.01 versus the 0 mg/L group. All values are expressed as the mean ± SD (n = 3). ECs endothelial cells, SMCs smooth muscle cells, OX-LDL oxidized low-density lipoprotein, 3-MA 3-methyladenine, Rapa Rapamycin, CTSD cathepsin D, BafA1 Bafilomycin A1
To investigate the effect of OX-LDL on autophagy, we monitored autophagy by detecting the abundance of microtubule-associated protein 1 light chain 3 II (LC3II) (Fig. 1b). OX-LDL markedly elevated LC3II levels in a dose-dependent manner. Transmission electron microscopy (TEM) also showed that increased autophagosomes were present in OX-LDL-treated ECs (Figure S4A). The ratio of LC3II: LC3I, p62, and lysosomal associated membrane protein 1 (LAMP1) reached a peak at 24 h (Figure S1B). Collectively, these data revealed that OX-LDL led to the accumulation of autophagosomes in ECs and SMCs. To further monitor the process of autophagy maturation, a tandem labeled green fluorescent protein (GFP)-mRFP-LC3 was used. OX-LDL resulted in predominant autolysosomes (red). Bafilomycin A1 (BafA1), which inhibits the merging of autophagosomes and lysosomes [21], led to increased yellow puncta after treatment with OX-LDL, but red puncta were decreased in the merged images. Conversely, rapamycin (Rapa), which induces autophagy by inhibiting mTOR, significantly increased the yellow and red puncta after treatment with OX-LDL (Fig. 1c). Furthermore, BafA1 led to further accumulation of LC3II caused by OX-LDL (Fig. 1d). We also found that the level of p62 was markedly increased by OX-LDL in a dose-dependent manner in ECs and SMCs (Fig. 1b). The above results revealed that the OX-LDL-induced autophagosome accumulation was mainly due to impaired autophagic degradation in ECs and SMCs.
Because the fusion of autophagosomes and lysosomes is mainly dependent on LAMP1 [13], we measured the levels of LAMP1 and showed that it was increased by OX-LDL (Fig. 1b), suggesting that OX-LDL promoted the association of autophagosomes with lysosomes. Moreover, Rapa led to accumulation of LC3II as well as LAMP1, but decreased the levels of p62 regulated by OX-LDL (Figure S1C). Proteases participate in the process of autophagic degradation. Cathepsins are important lysosomal proteases involved in autophagic degradation [22]. We found that OX-LDL markedly decreased expression levels and enzymatic activity of mature-Cathepsin D (CTSD) (Fig. 1e, f), suggesting that this lower enzymatic function decreased lysosomal activity after OX-LDL treatment, which could at least in part account for the observed impairment in autophagic flux.
Recent studies have shown that autophagy is involved in inflammatory reactions. We showed that OX-LDL-induced increases in VCAM-1, MCP-1, and IL-6 levels were further enhanced by 3-methyladenine (3-MA; an autophagy inhibitor), but were markedly decreased by Rapa (Figure S1D). In addition, we used Atg7-silencing to inhibit autophagy (Figure S3A). In addition, 3-MA and Atg7-shRNA markedly increased the production of VCAM-1, MCP-1, IL-6, IL-8, IL-18, IL-1β, and TNF-α induced by OX-LDL, whereas the production of IL-10 was decreased (Table S1, 2 and 1-3). However, 3-MA, Rapa, and BafA1 had no obviously effects on the expression of inflammation factors alone (Figure S1E). Collectively, these results demonstrated that both the impaired autophagy flux or inhibition of autophagy flux might promoted OX-LDL-induced inflammatory responses in ECs and SMCs.
LncRNA-FA2H-2 inhibited the transcription level of MLKL
To observe the effects and mechanisms of OX-LDL on atherosclerosis, we performed microarray analyses in THP-1 macrophages stimulated with 50 mg/L OX-LDL for 24 h, and we found that MLKL was differentially expressed. Previous studies have shown that MLKL plays a crucial role in autophagy and inflammatory disease [23, 24]. These findings suggested that MLKL was associated with OX-LDL-regulated autophagy and inflammation. Next, we investigated the localization of MLKL through use of the UCSC Genome Browser (http://genome.ucsc.edu) and found that lncRNA-FA2H-2 and MLKL were located adjacently on the same chromosome and with opposite directions of transcription (Figure S2). Microarray analyses revealed that lncRNA-FA2H-2 expression was downregulated by OX-LDL (fold change > 3; P < 0.01). Thus, we hypothesized that lncRNA-FA2H-2 might negatively regulate MLKL expression and contribute to OX-LDL-regulated autophagy and inflammation. Our results showed that LncRNA-FA2H-2 mRNA levels were downregulated whereas MLKL mRNA and protein levels were upregulated in vivo and vitro (Fig. 2a–f). Interestingly, lentiviral vector (LV)-siRNA-lncRNA-FA2H-2 (LV-si-lncRNA-FA2H-2) enhanced the protein and mRNA levels of MLKL (Fig. 2g, h). Fluorescence in situ hybridization (FISH) showed that lncRNA-FA2H-2 was mainly localized in the nuclei (Fig. 2i). Next, the enrichment efficiency of the MLKL site1 and MLKL site2 in the lncRNA-FA2H-2 probe group was significantly higher than that of the normal control (NC) group, suggesting that lncRNA-FA2H-2 regulated the transcription of MLKL (Fig. 2j). To verify these results, dual luciferase assays show that luciferase activity obviously increased compared with that of the NC group in the presence of si-lncRNA-FA2H-2 (S121) plus 3713b (−1018 to 387) and si-lncRNA-FA2H-2 (S121) plus 3714b (−750 to 387) (Fig. 2k). However, no significant difference was observed in the si-lncRNA-FA2H-2 and 3715b (−471 to 387) groups, indicating that the binding sites between −750 and 471 were necessary for lncRNA-FA2H-2 responsiveness to MLKL. Together, the above data indicated that lncRNA-FA2H-2 inhibited the transcription levels of MLKL.

LncRNA-FA2H-2 inhibits the transcription levels of MLKL. a ECs and SMCs were treated with OX-LDL (0, 25, 50, or 100 mg/L) for 24 h, and the expression of lncRNA-FA2H-2 was determined by qRT-PCR. aP < 0.05, bP < 0.01, and cP < 0.001 versus the 0 mg/L control group. b ECs and SMCs were treated with OX-LDL (0, 25, 50, and 100 mg/L) for 24 h. MLKL mRNA levels were measured by qRT-PCR. bP < 0.01, cP < 0.001 versus the 0 mg/L control group. c ECs and SMCs were treated with OX-LDL (0, 25, 50, or 100 mg/L) for 24 h. The MLKL protein levels were measured by western blotting. bP < 0.01 and cP < 0.001 versus the 0 mg/L control group. d The lncRNA-FA2H-2 mRNA levels were determined by qRT-PCR in human normal arterial intima (NAI) and advanced atherosclerotic plaque (AAP) (n = 20 for each experimental group). *P < 0.05 versus the NAI group. e MLKL mRNA levels were determined by qRT-PCR in human NAI and AAP (n = 20 for each experimental group). *P < 0.05 versus the NAI group. f MLKL protein levels were measured by western blotting in the NAI and AAP groups (n = 20 for each experimental group). *P < 0.05 versus the NAI group. g ECs and SMCs were transfected with LV-si-lncRNA-FA2H-2 or LV-Mock, and the protein levels of MLKL were measured by western blotting. bP < 0.01 and cP < 0.001 versus the LV-Mock group. h ECs and SMCs were incubated with LV-si-lncRNA-FA2H-2 or LV-Mock, and the mRNA levels of MLKL were measured by qRT-PCR. cP < 0.001 versus the LV-Mock group. i Localization of lncRNA-FA2H-2 was detected by FISH in ECs. Nuclei were stained blue by DAPI. Green fluorescence represents the location of lncRNA-FA2H-2; arrows represent the location of lncRNA-FA2H-2. Scale bar: 20 μm. j The enrichment efficiency of lncRNA-FA2H-2 and MLKL were detected by qRT-PCR. “Odd” and “even” represent the enrichment efficiency of the lncRNA-FA2H-2 probe group; “NC” stands for the negative control probe group; MLKL site 1 and MLKL site 2 represent the binding sites with lncRNA-FA2H-2; **P < 0.01 and ***P < 0.001 versus the NC group. k Luciferase activity was detected by a dual luciferase assay. #P < NS; *P < 0.05; and **P < 0.01 versus the NC group. All values are expressed as the mean ± SD (n = 3). LncRNAs long noncoding RNAs, MLKL mixed lineage kinase domain-like protein, FISH fluorescence in situ hybridization. All other abbreviations are the same as in the Fig. 1 legend
LncRNA-FA2H-2 attenuates inflammatory response by reversing OX-LDL-induced autophagy flux impairment
To explore whether lncRNA-FA2H-2 affected autophagy and inflammation, as shown in Fig. 3a, expressions of VCAM-1, MCP-1, and IL-6 were significantly increased by LV-si-lncRNA-FA2H-2. Moreover, the protein levels of VCAM-1, MCP-1, and IL-6 induced by OX-LDL were further enhanced by LV-si-lncRNA-FA2H-2 (Fig. 3b). Additionally, LV-si-lncRNA-FA2H-2 also significantly increased the production of VCAM-1, IL-6, MCP-1, IL-8, IL-18, IL-1β, and TNF-α induced by OX-LDL, whereas it decreased the production of IL-10 (Table S2-1), suggesting that lncRNA-FA2H-2 alleviated OX-LDL-induced inflammatory responses. Subsequently, to determine whether autophagy was involved in the inflammatory response regulated by lncRNA-FA2H-2. The expressions of LC3II and LAMP1 were significantly decreased, whereas expression of p62 was markedly increased by LV-si-lncRNA-FA2H-2. Interestingly, BafA1 had no obviously effect on the protein levels of LC3II, p62, and LAMP1 after treatment with LV-si-lncRNA-FA2H-2 (Fig. 3c). As expected, the number of autophagosomes was decreased by LV-si-lncRNA-FA2H-2 in ECs (Figure S4B). Our results also showed that yellow and red puncta were significantly decreased by LV-si-lncRNA-FA2H-2 and were increased by LV-lncRNA-FA2H-2 in GFP-mRFP-LC3 adenovirus-infected ECs. BafA1 had no obvious effect on the red and yellow puncta after treatment with LV-si-lncRNA-FA2H-2. In contrast, the red and yellow puncta were increased by Rapa addition after treatment with LV-si-lncRNA-FA2H-2 (Fig. 3d). Together, these results indicated that lncRNA-FA2H-2 induced autophagy flux. Increasing evidence has suggested that p62 is a known Nuclear factor-kB (NF-kB) target gene [25]. The activity of NF-κB was increased by LV-si-lncRNA-FA2H-2 (Figure S3B). Interestingly, lncRNA-FA2H-2 interacted with the promoter of NF-κB, through use of CHIRP-Seq, suggesting the decrease in p62 induced by lncRNA-FA2H-2 was might due to the decrease of NF-κB (Figure S3C). Moreover, we confirmed that LV-si-lncRNA-FA2H-2 decreased the expression of LC3II and LAMP1, and increased the expression of p62 regulated by OX-LDL (Figure S3D). These results indicated that lncRNA-FA2H-2 reversed the impaired autophagy flux induced by OX-LDL. Moreover, treatment with 3-MA further increased the protein levels of VCAM-1, MCP-1, and IL-6 caused by LV-si-lncRNA-FA2H-2 (Fig. 3a). In addition, 3-MA and Atg7-shRNA markedly increased the production of VCAM-1, MCP-1, IL-6, IL-8, IL-18, IL-1β, and TNF-α induced by LV-si-lncRNA-FA2H-2, whereas the production of IL-10 was decreased (Table S2-2 and S2-3). Moreover, Atg7-shRNA had no obvious effect on the expression of inflammatory factor alone (Figure S1E). These results revealed that lncRNA-FA2H-2 alleviated the inflammatory responses induced by OX-LDL though enhancing autophagy flux.

LncRNA-FA2H-2 attenuates inflammatory response by reversing OX-LDL-induced autophagy flux impairment. a ECs and SMCs were incubated with 3-MA (3 mM) for 24 h after LV-si-lncRNA-FA2H-2 or LV-Mock treatments, and the expressions of VCAM-1, MCP-1, and IL-6 were measured by western blotting. aP < 0.05 and bP < 0.01 versus LV-si-lncRNA-FA2H-2 alone. b ECs and SMCs were treated with LV-si-lncRNA-FA2H-2 before OX-LDL (50 mg/L) for 24 h, and the expressions of VCAM-1, MCP-1, and IL-6 were determined by western blotting. aP < 0.05, bP < 0.01, and cP < 0.001 versus OX-LDL alone. c ECs and SMCs were incubated with BafA1 (50 nm) for 3 h before treatment with LV-si-lncRNA-FA2H-2 or LV-Mock, and the expressions of LC3II, p62, and LAMP1 were measured by western blotting. bP < 0.01, cP < 0.001, and #P < NS versus the LV-si-lncRNA-FA2H-2 group. d ECs were transiently transfected with GFP-mRFP-LC3 for 48 h and subsequently treated with LV-Mock, LV-si-lncRNA-FA2H-2, or LV-si-lncRNA-FA2H-2 plus BafA1 (50 nm) for 3 h, LV-si-lncRNA-FA2H-2 plus Rap (100 nM) for 12 h, BafA1 (50 nm) for 3 h or Rap (100 nM) for 12 h alone, and then observed for the change of both green and red fluorescence using confocal microscopy. Scale bar: 20 μm. Lower panel, the numbers of acidified autophagosomes (GFP-RFP+) versus neutral autophagosomes (GFP+RFP+) per cell in each condition were quantified. #P < NS; *P < 0.05; and **P < 0.01. All values are expressed as the mean ± SD (n = 3). All other abbreviations are the same as in the Fig. 1 legend
A detailed mechanism for the necroptotic cell death execution pathway by receptor interacting serine/threonine kinase 1/3 (RIPK1/3) and MLKL has been reported [26]. Thus, we wanted to address whether the LncRNA-FA2H-2-affected inflammatory responses were due to the RIPK1-RIPK3-MLKL-mediated necroptosis pathway. Interestingly, LV-si-lncRNA-FA2H-2 had no apparent effect on the expression levels of total and phosphorylated RIPK1 and RIPK3 (Figure S3E). Taken together, these data suggested that lncRNA-FA2H-2 alleviated the inflammatory responses induced by OX-LDL primarily through reversal of the impaired autophagy flux, therefore, the mechanism might not be due to the RIPK1-RIPK3-MLKL-dependent pathway.
The MLKL-aggravated inflammatory response was due to autophagy flux inhibition
To verify whether MLKL induced inflammatory responses, we found that inflammatory responses were significantly enhanced by pcDNA-MLKL (Fig. 4a and S5A). We also found that OX-LDL induced increases of VCAM-1, MCP-1, and IL-6 protein levels, and these were further upregulated by pcDNA-MLKL (Fig. 4b and S5B). In addition, pcDNA-MLKL significantly aggravated the effect of lncRNA-FA2H-2 knockdown and OX-LDL treatment on VCAM-1, MCP-1, and IL-6 protein levels (Figure S6A). The inflammatory responses caused by necroptotic cell death, in part, contribute to the release of damage-associated molecular pattern molecules (DAMPs), such as high mobility group box 1 (HMGB1) [27]. As shown in Figure S5C, pcDNA-MLKL alone had no obvious effect on HMGB1. Moreover, the production of VCAM-1, MCP-1, IL-6, IL-8, IL-18, and IL-1β induced by OX-LDL were further enhanced by pcDNA-MLKL, whereas the production of IL-10 was decreased (Table S3-1). These results indicated that MLKL aggravated the inflammatory response stimulated by OX-LDL. Subsequently, our results showed that LC3II and LAMP1 levels were significantly decreased by pcDNA-MLKL, whereas p62 levels were increased. In addition, BafA1 had no obvious effects on LC3II, LAMP1, and p62 after treatments with pcDNA-MLKL (Fig. 4c and S5E). Moreover, yellow and red puncta were significantly decreased by pcDNA-MLKL in GFP-mRFP-LC3 adenovirus-infected ECs. BafA1 had no obvious effect on the red and yellow puncta after treatment with pcDNA-MLKL. Furthermore, the red and yellow puncta were increased by Rapa after treatment with pcDNA-MLKL (Fig. 4d). Together, these results revealed that MLKL inhibited autophagy flux. In addition, we used gene silencing to inhibit the expression of MLKL (Figure S5D). As expected, we found that the MLKL-shRNA further increased levels of LC3II and LAMP1 induced by OX-LDL, whereas inhibited levels of p62 (Fig. 4e and S5F). These results indicated that silencing MLKL reversed the impaired autophagy flux induced by OX-LDL. We also found that pcDNA-MLKL significantly alleviated the effect of lncRNA-FA2H-2 knockdown and OX-LDL treatment on LC3II and LAMP1 protein levels, but aggravated p62 protein levels (Figure S6B). Next, we found that 3-MA obviously increased the protein levels of VCAM-1, MCP-1, and IL-6 induced by pcDNA-MLKL (Fig. 4a and Figure S5A). Furthermore, 3-MA and Atg7-shRNA markedly increased the production of VCAM-1, MCP-1, IL-6, IL-8, IL-18, IL-1β, and TNF-α induced by pcDNA-MLKL, and decreased the production of IL-10 in ECs (Table S3-2 and S3-3). Collectively, these results indicated that lncRNA-FA2H-2 suppressed MLKL expression, which alleviated the inflammatory response induced by OX-LDL through induction of autophagy flux.

The MLKL-aggravated inflammatory response was due to autophagy flux inhibition. a ECs were treated with 3-MA (3 mM) for 24 h after pcDNA-MLKL (overexpressed MLKL) or pcDNA (pcDNA-mock), and the expressions of VCAM-1, MCP-1, and IL-6 were measured by western blotting. bP < 0.01, cP < 0.001, and dP < 0.0001 versus treatment with pcDNA-MLKL alone. b ECs were incubated with pcDNA-MLKL before OX-LDL (50 mg/L) for 24 h, and the expressions of VCAM-1, MCP-1, and IL-6 were determined by western blotting. aP < 0.05, bP < 0.01 versus treatment with OX-LDL alone. c ECs were exposed to pcDNA or pcDNA-MLKL before BafA1 (50 nm) for 3 h, and the expression of LC3II, p62, and LAMP1 were measured by western blotting. bP < 0.01, cP < 0.001, and #P < NS versus treatment with pcDNA-MLKL alone. d ECs were transiently transfected with GFP-mRFP-LC3 for 48 h and subsequently transfected with pcDNA or pcDNA-MLKL, pcDNA-MLKL plus BafA1 (50 nm) for 3 h, pcDNA-MLKL plus Rap (100 nM) for 12 h, BafA1 (50 nm) for 3 h, or Rap (100 nM) for 12 h alone, and then observed for the change of both green and red fluorescence using confocal microscopy. Scale bar: 20 μm. The numbers of acidified autophagosomes (GFP-RFP+) versus neutral autophagosomes (GFP+RFP+) per cell in each condition were quantified. #P < NS; *P < 0.05; and **P < 0.01. e ECs were treated with MLKL-shRNA before OX-LDL (50 mg/L) for 24 h, and protein levels of LC3II, p62, and LAMP1 were measured by western blotting. bP < 0.01 and cP < 0.001 versus treatment with OX-LDL alone. All values are expressed as the mean ± SD (n = 3). The abbreviations are defined in the Figs 1, 2 legends
MLKL-inhibited autophagy was mainly due to activation of the mTOR-dependent signaling pathway
The above mentioned data confirmed that MLKL inhibited autophagy flux, but the mechanism remains unknown. Increasing evidence suggests that the mTOR-dependent signaling pathway plays a key role in autophagy [28]. Our results indicated that the phosphorylation levels of phosphatidylinositol 3-kinase (PI3K), protein kinase B (AKT), and mTOR were increased by pcDNA-MLKL, whereas AMP-activated protein kinase (AMPK) were decreased, with no change in PI3K, AKT, AMPK, and mTOR total protein expression levels (Fig. 5a). BEZ235 treatment (a PI3K inhibitor) resulted in obvious decreases in phosphorylation levels of AKT and mTOR. However, AMPK phosphorylation levels were decreased and mTOR were increased after AMPK inhibitor (Compound C, CC) treatment (Figure S7A). We also found that MLKL-induced AKT and mTOR phosphorylation level increases were suppressed by BEZ235 (Fig. 5b) [29]. In addition, when AMPK was suppressed by CC[17], MLKL-induced mTOR phosphorylation level increases were further enhanced (Fig. 5c). As shown in Fig. 5e, the phosphorylation levels of p70S6K and 4EBP1 were increased by pcDNA-MLKL, with no change in p70S6K and 4EBP1 protein expression levels. Furthermore, the expression levels of LC3II and LAMP1 suppressed by pcDNA-MLKL were obviously enhanced by BEZ235. In contrast, the expression levels of p62 were obviously decreased (Fig. 5d). Thus, these data showed that MLKL-inhibited autophagy mainly contributed to the activation of the mTOR-dependent signaling pathway.

MLKL-inhibited autophagy was mainly due to activation of the mTOR-dependent signaling pathway. a ECs and SMCs were treated with pcDNA-MLKL or pcDNA. Total and phosphorylation levels of PI3K, AKT, mTOR, and AMPK were determined by western blotting. aP < 0.05, bP < 0.01, cP < 0.001, and dP < 0.0001 versus treatment with pcDNA alone. b ECs and SMCs were pretreated with PI3K inhibitor (1 μm BEZ235) for 72 h before pcDNA-MLKL or pcDNA. Total and phosphorylated levels of AKT and mTOR were determined by western blotting. bP < 0.01, cP < 0.001 versus treatment with pcDNA-MLKL alone. c ECs and SMCs were treated with the AMPK inhibitor (5 μm Compound C, CC) for 1 h before pcDNA-MLKL or pcDNA. Total and phosphorylation levels of AMPK and mTOR were measured by western blotting. aP < 0.05 and bP < 0.01 versus treatment with pcDNA-MLKL alone. d ECs and SMCs were treated with 1 μm BEZ235 for 1 h before pcDNA-MLKL or pcDNA. The levels of LC3II, LAMP1 and p62 were measured by western blotting. aP < 0.05, bP < 0.01 versus treatment with pcDNA-MLKL alone. e ECs and SMCs were treated with pcDNA-MLKL or pcDNA. Total and phosphorylation levels of 4EBP1 and p70S6K were determined by western blotting. bP < 0.01 and cP < 0.001 versus treatment with pcDNA alone. All values are expressed as the mean ± SD (n = 3). The abbreviations are defined in the Figs 1, 2 legends
The lncRNA-FA2H-2-MLKL was associated with the mTOR-dependent signaling pathway upon OX-LDL exposure
Subsequent experiments were performed to ascertain the effects of the mTOR-dependent signaling pathway on lncRNA-FA2H-2-MLKL when exposed to OX-LDL. Our results showed that phosphorylation levels of PI3K, AKT, and mTOR were decreased, whereas phosphorylation levels of AMPK were increased in a dose-dependent manner by treatment with OX-LDL (Fig. 6a). The decreased phosphorylation levels of AKT and mTOR were further suppressed by treatments with BEZ235 (Fig. 6b). Furthermore, the increased phosphorylation levels of AMPK regulated by OX-LDL were further suppressed by CC, and the decreased phosphorylation levels of mTOR regulated by OX-LDL were increased by CC (Fig. 6c). Next, LV-si-lncRNA-FA2H-2 reversed the decreased levels of phosphorylation of PI3K, AKT, and mTOR and inhibited the increased levels of phosphorylation of AMPK caused by OX-LDL (Fig. 6d), suggesting that lncRNA-FA2H-2 was associated with the OX-LDL-regulated mTOR-dependent signaling pathways. The pcDNA-MLKL also reversed the decreased levels of phosphorylation of PI3K, AKT, and mTOR and inhibited the increased levels of phosphorylation of AMPK caused by OX-LDL in ECs and SMCs (Fig. 6e). These results suggested that OX-LDL regulated the mTOR-dependent signaling pathway partly through the lncRNA-FA2H-2-MLKL pathway.

The lncRNA-FA2H-2-MLKL was associated with the mTOR-dependent signaling pathway upon OX-LDL exposure. a ECs and SMCs were treated with OX-LDL (0, 25, 50, and 100 mg/L) for 24 h. Total and phosphorylation levels of PI3K, AKT, mTOR, and AMPK were determined by western blotting. aP < 0.05, bP < 0.01, cP < 0.001, and dP < 0.0001 versus the 0 mg/L group. b ECs and SMCs were pretreated with PI3K inhibitor (1 μm BEZ235) for 72 h before OX-LDL (50 mg/L) for 24 h. Total and phosphorylated levels of AKT and mTOR were measured by western blotting. aP < 0.05, bP < 0.01 versus treatment with OX-LDL alone. c ECs and SMCs were pretreated with AMPK inhibitor (5 μm Compound C) for 1 h before OX-LDL (50 mg/L) for 24 h. Total and phosphorylation levels of AMPK and mTOR were measured by western blotting. bP < 0.01, cP < 0.001 versus treatment with OX-LDL alone. d ECs and SMCs were pretreated with LV-si-lncRNA-FA2H-2 before OX-LDL (50 mg/L) for 24 h. Total and phosphorylated levels of PI3K, AKT, AMPK, and mTOR were detected by western blotting. aP < 0.05, bP < 0.01, and cP < 0.001 versus treatment with OX-LDL alone. e ECs and SMCs were treated with pcDNA-MLKL before OX-LDL (50 mg/L) for 24 h. Total and phosphorylation levels of PI3K, AKT, mTOR, and AMPK were determined by western blotting. aP < 0.05, bP < 0.01, cP < 0.001, and dP < 0.0001 versus treatment with OX-LDL alone. All values are expressed as mean ± SD (n = 3). The abbreviations are defined in the Figs. 1, 2 legends
The lncRNA-FA2H-2 activated autophagy and restrained inflammation in vivo
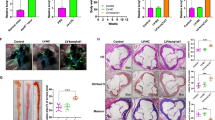
We explored the effects of lncRNA-FA2H-2 on atherosclerotic lesions by hematoxylin and eosin (HE) staining in the aortic root and aortic valve of ApoE−/− mice fed a high-fat diet. The lesion areas in the aortic root and aortic valve were obviously increased (67.34% and 74.72%, respectively; P < 0.05) by LV-si-lncRNA-FA2H-2 treatment. The intima of the aortic root and aortic valve were proliferative and disarranged, and had a large number of lipid-laden foam cells and cholesterol crystals in the LV-si-lncRNA-FA2H-2 group (Fig. 7a, b). As expected, LV-si-lncRNA-FA2H-2 induced obvious cell death in ApoE−/− mice (Figure S7C). The expression of LC3II and LAMP1 in the LV-si-lncRNA-FA2H-2-treated group was decreased, whereas p62 expression was increased (Fig 7c, d). This suggested that lncRNA-FA2H-2 activated autophagy in ApoE−/− mice fed a high-fat diet. Consistent with the results in vitro, we also found that the expression of MLKL was increased by LV-si-lncRNA-FA2H-2. The expressions of VCAM-1, MCP-1, and IL-6 in the aortic root and aortic valve were also increased in the LV-si-lncRNA-FA2H-2. Interestingly, there were no significant increases in HMGB1, RIPK1, and RIPK3 in aortic valves of ApoE−/− mice infected with LV-si-lncRNA-FA2H-2 (Figure S7B). The above findings suggested that the levels of inflammatory factors were significantly increased in atherosclerosis lesions, which was mainly due to autophagy inhibition induced by LV-si-lncRNA-FA2H-2 in ApoE−/− mice. Taken together, these results showed that lncRNA-FA2H-2 alleviated the development of atherosclerosis.

The lncRNA-FA2H-2 activated autophagy and restrained inflammation in vivo. a Representative staining of an aorta root with HE in the LV-si-lncRNA-FA2H-2 or LV-Mock mice group. b Representative staining of an aortic valve with HE staining in the LV-si-lncRNA-FA2H-2 or LV-Mock mice group. c Aorta roots from apoE−/− mice were stained by immunohistochemistry for LC3II, LAMP1, p62, MLKL, VCAM-1, MCP-1, and IL-6 in the LV-si-lncRNA-FA2H-2 or LV-Mock mice group. d Aorta valves from apoE−/− mice were stained by immunohistochemistry for LC3II, LAMP1, p62, MLKL, VCAM-1, MCP-1, and IL-6 in the LV-si-lncRNA-FA2H-2 or LV-Mock mice group (n = 10 mice/group). Magnification: 400 × . *P < 0.05 versus the LV-Mock group. HE hematoxylin and eosin. All other abbreviations are defined in the Figs 1, 2 legends
Discussion
Inflammation is present throughout the course of atherosclerosis [17]. Autophagy is a homeostatic mechanism that participates in the clearance of denatured proteins, invading pathogens by a lysosomal degradation pathway [30]. Studies have shown that in advanced atherosclerosis, autophagy prevents plaque necrosis [31, 32]. Some studies have revealed that the induction of autophagy alleviated inflammatory injury [6, 33]. However, in the advanced stages of atherosclerosis, damaged autophagy flux contributes to the activation of inflammation [34]. In this study, we found that the inflammatory response was stimulated by OX-LDL, and OX-LDL induced impaired autophagy flux. Interestingly, we found that the impaired autophagy induced by OX-LDL was mainly due to dysfunctional proteases. Our data suggested that the inhibition of autophagy flux or impaired autophagy flux further aggravated the inflammatory responses when cells are exposed to OX-LDL. The underlying molecular mechanism of lysosomal proteases in impaired autophagy flux activated by OX-LDL requires further exploration.
Numerous studies have shown that lncRNAs play important roles in endothelial cell dysfunction, vascular inflammation [35]. Yan et al. [36] have shown that lncRNA-MIAT acts as a regulator of EC function by forming a feedback loop with vascular endothelial growth factor and miR150-5p. Our results showed that lncRNA-FA2H-2 expression was decreased and MLKL was increased in vivo and vitro. In most cases, lncRNAs exert their function by regulating expressions of targeted mRNAs at the transcriptional or post-transcriptional levels [37]. In the present study, MLKL expression was obviously suppressed by lncRNA-FA2H-2 at the transcriptional level. We found that lncRNA-FA2H-2-alleviated inflammatory response was mainly due to reverse impaired autophagy flux. Furthermore, the activity of NF-kB was decreased by lncRNA-FA2H-2 and lncRNA-FA2H-2 interacted with the promoter of NF-kB. Thus, the NF-kB pathway might be involved in the autophagy and inflammation regulated by lncRNA-FA2H-2. However, further investigations are needed to verify these results. Accumulating data indicate that the RIPK1-RIPK3-MLKL pathway plays a crucial role in inflammatory responses and necroptosis [38]. In this study, we found that lncRNA-FA2H-2 had no obvious effect on the levels of RIPK1 and RIPK3 in vivo and in vitro. The inflammatory response was enhanced and autophagy was inhibited in ApoE−/− mice by knockdown of lncRNA-FA2H-2. Our data showed that the lncRNA-FA2H-2-alleviated inflammatory response was mainly due to reverse impaired autophagy flux, and the mechanism was not due to RIPK1-RIPK3-MLKL-dependent pathway effects.
Necroptosis, a newly discovered process of programmed cell death, participates in inflammatory diseases. MLKL is essential for necroptosis, which can release intracellular molecules, resulting in the occurrence of an inflammation response. Recent studies have shown that expression of MLKL is increased in humans with unstable carotid atherosclerosis, and phosphorylation of MLKL, which plays an important role in necroptosis, can be detected in advanced atheromas [39]. Further study of the biological functions of MLKL found that it was closely related to autophagy and inflammation. TNF/z-VAD-fmk (zVAD) activates necroptosis through suppressing autophagic flux [40]. Heat shock protein 70 downregulates cardiomyocyte necroptosis through autophagy during ischemic reperfusion [41]. MLKL activation triggers potassium efflux, assembly of the NLRP3 inflammasome, caspase-1 activation, and secretion of IL-1β [42]. Here, we found that MLKL aggravated inflammatory response caused by OX-LDL. Moreover, lncRNA-FA2H-2 suppressed MLKL expression, which alleviated the inflammatory response induced by OX-LDL through induction of autophagic flux. These results provided evidence for the underlying mechanisms of proinflammatory effects of MLKL, in which autophagy may play a key role. Previous studies have revealed that the Janus kinase pathway is activated by overexpression of MLKL [43]. MLKL has also been shown to contribute to inflammasome activation through signaling pathways [44]. In addition, MLKL can activate p38 MAPK, resulting in the activation of the PI3K pathway [45]. In our study, we confirmed that MLKL inhibited autophagy was mainly due to the activation of the mTOR-dependent signaling pathway. We also demonstrated that lncRNA-FA2H-2-MLKL was associated with mTOR-dependent signaling pathways regulated by OX-LDL, which verified the involvement of these pathways in autophagy. Detailed mechanisms of MLKL in autophagy will be further explored in future studies.
In summary, as shown in the schematic diagram (Fig. 8), our results indicated that lncRNA-FA2H-2 suppressed MLKL expression, which alleviated the inflammatory response induced by OX-LDL through induction of autophagy flux. Moreover, the autophagy suppressed by MLKL was associated with mTOR-dependent signaling pathway activation. These novel findings are relevant to enhancing our understanding of the pathogenesis of atherosclerosis and indicate that lncRNA-FA2H-2 and MLKL could act as novel potential therapeutic targets in atherosclerosis.

Schematic diagram of the main findings of this study. Our results indicated that lncRNA-FA2H-2 suppressed MLKL expression, which alleviated the inflammatory response induced by OX-LDL through induction of autophagy flux. Moreover, the autophagy suppressed by MLKL was associated with mTOR-dependent signaling pathway activation. (Purple arrowheads represent activation, and red arrowheads represent inhibition)
Materials and Methods
Tissues
In this study, 20 patients with atherosclerotic plaque and normal arterial tissues were all from atherosclerotic patients who underwent carotid endarterectomy at Southern Hospital of Southern Medical University from 2013 to 2015. Arteries without macroscopic evidence of atherosclerosis were collected from individuals who died from a traffic accident or cerebral edema. After washing the specimens with saline, the outer fat tissues and surrounding normal tissues of the plaque were separated. The plaque tissue was separated into appropriate sizes, and immediately placed in liquid nitrogen. The collected tissue was pathologically tested and diagnosed as primary atherosclerosis. The exclusion criteria were patients with diabetes, cancer, congestive heart failure, valvular heart disease, hematological system diseases, autoimmune disease, and/or infections. The basic information of the patients included name, age, and sex. The study was approved by the Committee for Ethical Review of Research Involving Human Subjects, Nanfang Hospital, Southern Medical University, Guangzhou, China. Informed consent was obtained from the participants or relatives of deceased individuals.
Reagents
Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum were purchased from Gibco (Grand Island Biological Company, New York, NY, USA). Antibodies against rabbit MLKL (A5579), AKT (A10605), LAMP1 (A2582), LC3II (A7198), p62 (A0682), phospho-AKT1-S473 (AP0140), phospho-AMPKα-T172 (AP0116), phospho-mTOR-S2448 (AP0409), mTOR (A5520), and PI3K (A0265) were obtained from Abclonal (Woburn, MA, USA). Antibodies against human MLKL, HMGB1 (ab228624), RIPK1 (ab202985), RIPK3 (ab152130), phospho-4EBP1-T37 (ab75767), and phospho-S6K1-T89 (ab2571) were purchased from Abcam (Cambridge, MA, USA). Antibodies against rabbit AMPKα1 (YT0215), and phosphp-PI3K-p85α (YP0765) were purchased from Immunoway (Plano, TX, USA). Antibodies against rabbit IL-6, VCAM-1, and MCP-1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). CC (P5499), Rapa (S1039), and BEZ235 (S1009) were purchased from Selleck Chemicals (Shanghai, China). BafA1 (B1793) and 3-MA (M9281) were purchased from Sigma-Aldrich (St. Louis, MO, USA). ACTB (RM2001) was purchased from Abcam. The (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) cell proliferation assay (Beyotime Biotechnology, Haimen, China; C0038) was performed according to the manufacturer’s instructions.
OX-LDL preparation
The OX-LDLs were derived from human low-density lipoprotein (LDL). At 37 °C, 2 mM CuSO4 was added overnight to oxidize (OX) the LDL, and oxidation was stopped with an excess of sodium EDTA. Agarose gel electrophoresis was used to analyze the migration of LDL. The colorimetric determination of thiobarbituric acid reactive species used malondialdehyde (MDA) as a standard, at 12.0 nM MDA per mg protein for the OX-LDL.
Cell culture
Human monocytic cells (THP-1; ATCC TIB-202; ATCC, Manassas, VA, USA), human umbilical vein endothelial cells (ECs; ATCC CRL-1730), and human aorta vascular smooth muscle cells (SMCs; ATCC CRL-1999) were obtained from American Type Culture Collection. THP-1 cells were cultured in RPMI 1640 medium with 10% fetal bovine serum. ECs and SMCs were cultured in DMEM with 10% fetal bovine serum. Cells were incubated at 37 °C in an atmosphere of 5% CO2. Cells were seeded in 6- or 12-well plates or 60-mm dishes and grown to 70–80% confluence before use.
ELISA
The production of TNF-α, IL-8, IL-18, IL-1β, VCAM-1, IL-6, MCP-1, IL-10, and HMGB1 in cell culture supernatants were measured via ELISA assays. The kits were purchased from Abcam.
Immunoblot analyses
The total protein was measured by the bicinchoninic acid protein assay kit (P0010-1; Beyotime, China). The cleavage products of each sample were separated by 12.5% SDS-PAGE. The western blots were incubated for 8 h with 1:1000-diluted primary antibodies, and then incubated 2 h at room temperature with horseradish-peroxidase-conjugated secondary antibodies. Visualization of proteins used chemiluminescence (ECL Plus Western Blot Detection System; Amersham Biosciences, Foster City, CA, USA).
TEM
ECs were fixed in 4% glutaraldehyde followed by 1% osmium tetroxide at 4 °C. Samples were washed and dehydrated in a graded series of alcohol. Ultrathin sections of 50 nm were obtained using a Leica microtome. The sections were stained with uranyl acetate and lead citrate. Autophagosomes was detected using a Hitachi H-7500 using TEM.
GFP-mRFP-LC3 adenoviral transfection
ECs and SMCs plated on glass-bottomed cell culture dishes were infected with adenoviral vectors containing GFP-mRFP-LC3 (HanBio Technology, Shanghai, China). The culture medium was replaced with fresh medium and the cells were incubated for 24 h and observed with a confocal laser scanning microscope (LSM 880 with Airyscan; Zeiss, Dublin, CA, USA) to detect autophagy flux and estimate the number of yellow and green dots.
qRT-PCR
Total RNA from cultured cells and tissues was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and reverse transcribed. Real-time PCR was performed on the lightCycler 480 II (Roche, Pleasanton, CA, USA) with SYBR Green Dye detection (TaKaRa Bio, Mountain View, CA, USA). All samples were assayed in triplicate. The data were analyzed using the ΔΔCt method, with U6 RNA as a reference in the lncRNA analysis and GAPDH as a reference in the mRNA analysis. The primer sequences are listed in Supplementary Table 6.
Lentivirus construction
LncRNA-FA2H-2 cDNA was amplified by PCR and then cloned into the pLOV-EF1a-PuroR-CMV-EGFP-2A-3FLAG vector, and in this construct the correct sequence of the LncRNA-FA2H-2 gene was verified by sequencing.
Lentivirus infection
ECs and SMCs were cultured. Packed empty LV vectors (LV-Mock) (TTCTCCGAACGTGTCACGT) and LV-mediated si-lncRNA-FA2H-2 vector (CCGGATCATAGCGTAAATT) were generated. ECs and SMCs were transfected at a multiplicity of infection of 20 for ECs and SMCs transfecting units per cell in the presence of 5 mg/mL of polybrene. The cells were washed with fresh complete medium after 24 h. The efficiency of lncRNA-FA2H-2 knockdown was verified by qRT-PCR.
FISH
At room temperature, cells were fixed with 4% paraformaldehyde for 20 min, washed 5 min/twice with 0.1% diethlypyrocarbonate water, subjected to protease K digestion for 20 min at 37 °C, and washed 5 min/twice with PBS, then fixed again in 1% paraformaldehyde at room temperature for 10 min, and washed 5 min/twice with PBS. The samples were frozen at −20 °C, and dehydrated in 70, 85, and 100% alcohol for 5 min. FITC fluorescent dye probe and probe diluent were mixed into a probe hybridization mixture, and denatured at 73 °C for 8 min on ice. The solution was dropped onto the slice and incubated overnight at 42 °C, then washed for three times for 5 min with preheated (42 °C) 50% formamide. The nuclei were stained with DAPI and washed again three times for 5 min with PBS and then observed with fluorescence microscopy.
Luciferase activity assay
The cells were inoculated and incubated by Lipofectamine 3000 transfection reagent (Invitrogen; 11668019) for 24 h and then transfected with firefly and Renilla luciferase plasmids at a confluence of 80–90%. After washing with PBS, PLB buffer was added and the cells were incubated at room temperature for 15 min, and cell lysates were centrifuged at 10,000 rpm for 5 min at 4 °C. The supernatant was discarded, and 20 μL of sample was transferred to a 96-well plate and mixed with 100 μL Dual-Glo Luciferase Assay System (Promega, Madison, WI, USA). The cell lysis buffer alone was used to treat the control group. Finally, 100 μL Stop & Glo reagent was added and then we measured the relative light units.
ChIRP qRT-PCR
Using singlemoleculefish.com, the probes of lncRNA-FA2H-2 were designed. Oligonucleotides were biotinylated at the 3′ end with an 18-carbon spacer arm. ECs were collected and subjected to ChIRP as previously described [46]. The probes or RNA sequences are listed in Supplemental Tables 4, 5.
Recombinant plasmid construction
A plasmid containing the full-length MLKL cDNA was purchased from OriGene (Rockville, MD, USA). MLKL cDNA was amplified by PCR and subcloned into the pcDNA3.1( + ) vector. The correct sequence of the MLKL cDNA in the recombinant plasmid was verified by sequencing and named pcDNA-MLKL. The plasmid was used to transfect ECs and SMCs using Lipofectamine 3000 transfection reagent.
siRNA assay
Control siRNA (negative control), siRNA-MLKL, and siRNA-ATG7 were purchased from Ribo Targets (Guangzhou, China). ECs and SMCs were transfected with 50 nM siRNA. Control samples for all experimental procedures were processed with a non-targeting control mimic sequence of equal concentration. Western blotting was performed to observe the efficiency of siRNA protein knockdown. The sequence of siRNA-ATG7, siRNA-MLKL, and control siRNA are shown in Supplementary Table 7.
Animals
All investigations conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and were approved by the Animal Experimental Committee of Nanfang Hospital. Male C57BL/6 mice (6 weeks of age, 20 g) and apoE−/− mice with a C57BL/6 background were obtained from the Laboratory Animal Center of Peking University (Beijing, China). To ascertain the effect of lncRNA-FA2H-2 on atherosclerosis, the male (6 weeks of age) ApoE−/− mice were randomized into two groups of 10 mice (LV-Mock and LV-si-lncRNA-FA2H-2-treated groups). The high-fat diet mice were injected with control lentivirus (LV-Mock) or lentivirus si-RNA-lncRNA-FA2H-2 (LV-si-lncRNA-FA2H-2) through the tail vein. Mice (6 weeks of age) were fed a high-fat diet (21% protein, 24% carbohydrate, and 55% fat) for 16 weeks. At week 16, mice were anesthetized with inhalation of 2% sodium valproate, and 1.5 mL of blood was removed by cardiac puncture. The mice were euthanized by cervical dislocation, and tissues were collected for further analyses.
Immunohistochemistry (IHC)
The sections of aortic root and aortic valve were stained with different antibodies using rabbit polyclonal antibodies to LC3II (1:100 dilution; Abclonal, Woburn, MA, USA), p62 (1:200 dilution; Abclonal), MLKL (1:150 dilution; Abcam), LAMP1 (1:100 dilution; Abclonal), IL-6 (1:100 dilution; Affinity, Ancaster, ON, Canada), MCP-1 (1:100; Abclonal), VCAM-1 (1:100; Abcam), RIPK1(1:100; ProteinTech, Rosemont, IL, USA), RIPK3 (1:100; ProteinTech), and HMGB1(1:100; Abcam), using an IHC detection kit (Abcam). The immunohistochemical evaluation for each protein was performed by a semi-quantitative method.
Microarray analysis
Briefly, total RNA were isolated from THP-1 macrophages (n = 3) and THP-1 macrophage-derived foam cells (n = 3) and were quantified using the NanoDrop ND-1000 spectrophotometer (ThermoFisher, Scotts Valley, CA, USA). The Agilent Array platform (Agilent, Santa Clara, CA, USA) was employed for microarray analyses. The microarray analyses were performed as described previously [10].
CHIRP-seq
ChIRP-Sequencing library preparation was performed according to Illumina’s protocol Preparing Samples for ChIRP Sequencing of DNA. Ten nanograms of DNA of each sample was converted to phosphorylated blunt-ended; an “A” base was added to the 3′ end of the blunt phosphorylated DNA fragments; Illumina’s genomic adapters were ligated to the A tailed DNA fragments; and, PCR amplification was performed to enrich ligated fragments. The enriched product of ~200–1500 bp was size-selected using AMPure XP beads. The library was denatured with 0.1 M NaOH to generate single-stranded DNA molecules, and loaded onto channels of the flow cell at 8 pM concentration, amplified in situ using the HiSeq 3000/4000 PE Cluster Kit (#PE-410-1001; Illumina). Sequencing was carried out by running 2 × 150 cycles on an Illumina HiSeq 4000 according to the manufacturer’s instructions. After the sequencing platform generated the sequencing images, the stages of image analyses and base calling were performed using Off-Line Basecaller software (OLB V1.8). After passing the Solexa CHASTITY quality filter, the clean reads were aligned to Human reference genome HG19 using BOWTIE (V2.1.0). Aligned reads were used for peak calling of the ChIRP regions using MACS V1.4.2. Statistically significant ChIRP-enriched regions (peaks) were identified by comparison of two samples, using a p-value threshold of 10−5. Then, the peaks were annotated by the nearest gene using the newest UCSC RefSeq database.
Statistical analysis
Data were analyzed using SPSS version 13.0 (SPSS, Chicago, IL, USA) software. Data are presented as the mean ± SD or median (interquartile range) unless otherwise indicated. The results were analyzed by one-way analysis of variance or unpaired Student’s t-tests when continuous variables were normally distributed. A two-tailed P value < 0.05 was considered statistically significant.
References
Zhang H, Zhao Z, Pang X, Yang J, Yu H, Zhang Y, et al. Genistein detection protects against Ox-LDL-induced inflammation through microRNA-155/SOCS1-mediated repression of NF-kB signaling pathway in HUVECs. Inflammation. 2017;40:1450–9.
Yu X, Xing C, Pan Y, Ma H, Zhang J, Li W. IGF-1 alleviates ox-LDL-induced inflammation via reducing HMGB1 release in HAECs. Acta Biochim Biophys Sin. 2012;44:746–51.
Li J, Chen S, Cai X, Wang H, Wang X, Wang W. TLR2 expression doesn’t change in ox-LDL mediated inflammation in Human umbilical vein endothelial cells under high glucose culture. Int J Clin Exp Med. 2015;8:22004–10.
Xue F, Nie X, Shi J, Liu Q, Wang Z, Li X, et al. Quercetin inhibits LPS-induced inflammation and ox-LDL-induced lipid deposition. Front Pharmacol. 2017;8:40.
Chen S, Liang H, Yang H, Zhou K, Xu L, Liu J, et al. LincRNa-p21: function and mechanism in cancer. Med Oncol. 2017;34:98.
Yi H, Peng R, Zhang LY, Sun Y, Peng HM, Liu HD, et al. LincRNA-Gm4419 knockdown ameliorates NF-kappaB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis. 2017;8:e2583.
Yu F, Zhou G, Huang K, Fan X, Li G, Chen B, et al. Serum lincRNA-p21 as a potential biomarker of liver fibrosis in chronic hepatitis B patients. J Viral Hepat. 2017;24:580–8.
Pan Y, Li C, Chen J, Zhang K, Chu X, Wang R, et al. The emerging roles of long noncoding RNA ROR (lincRNA-ROR) and its possible mechanisms in human cancers. Cell Physiol Biochem. 2016;40:219–29.
Zhang C, Syed TW, Liu R, Yu J. Role of endoplasmic reticulum stress, autophagy, and inflammation in cardiovascular disease. Front Cardiovasc Med. 2017;4:29.
Hu YW, Zhao JY, Li SF, Huang JL, Qiu YR, Ma X, et al. RP5-833A20.1/miR-382-5p/NFIA-dependent signal transduction pathway contributes to the regulation of cholesterol homeostasis and inflammatory reaction. Arterioscler Thromb Vasc Biol. 2015;35:87–101.
Raft C, Martens S. Mechanisms and regulation of autophagosome formation. Curr Opin Cell Biol. 2012;24:496–501.
Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy Fights disease through cellular self-digestion. Nature. 2008;451:1069–75.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42.
Li BH, Yin YW, Liu Y, Pi Y, Guo L, Cao XJ, et al. TRPV1 activation impedes foam cell formation by inducing autophagy in oxLDL-treated vascular smooth muscle cells. Cell Death Dis. 2014;5:e1182.
Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–53.
Grootaert MO, da Costa Martins PA, Bitsch N, Pintelon I, De Meyer GR, Martinet W, et al. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neo intima formation and atherogenesis. Autophagy. 2015;11:2014–32.
Chen ML, Yi L, Jin X, Liang XY, Zhou Y, Zhang T, et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy. 2013;9:2033–45.
Zhang Y, Hu X, Gao G, Wang Y, Chen P, Ye Y. Autophagy protects against oxidized low density lipoprotein-mediated inflammation associated with preeclampsia. Placenta. 2016;48:136–43.
Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy. 2016;12:245–60.
Lavandero S, Chiong M, Rothermel B, A Hill JA. Autophagy in cardiovascular biology. J Clin Invest. 2015;125:55–64.
Yang J, Yu J, Li D, Yu S, Ke J, Wang L, et al. Store-operated calcium entry-activated autophagy protects EPC proliferation via the CAMKK2-MTOR pathway in ox-LDL exposure. Autophagy. 2017;13:82–98.
Appelqvist H, Waster P, Kagedal K, Ollinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol. 2013;5:214–26.
Fitzwalter BE, Thorburn A. Recent insights into cell death and autophagy. FEBS J. 2015;282:4279–88.
Basit F, Cristofanon S, Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013;20:1161–73.
Ling J, Kang Y, Zhao R, Xia Q, Lee D,F, Chang Z, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–20.
He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11.
Liu x, Cao H, Li J, Wang B, Zhang P, Dong Zhang X, et al. Autophagy induced by DAMPs facilitates the inflammation response in lungs undergoing ischemia-reperfusion injury through promoting TRAF6 ubiquitination. Cell Death Differ. 2017;24:683–93.
Wang S, Livingston MJ, Su Y, Dong Z. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy. 2015;11:607–16.
Aziz SA, Jilaveanu LB, Zito C, Camp RL, Rimm DL, Conrad P. et al. Vertical targeting of the phosphatidylinositol-3 kinase pathway as a strategy for treating melanoma. Clin Cancer Res. 2010;16:6029–39.
Schrijvers DM, De Meyer GR, Martinet W. Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler Thromb Vasc Biol. 2011;31:2787–91.
Fan X, Wang J, Hou J, Lin C, Bensoussan A, Chang D, et al. Berberine alleviates ox-LDL induced inflammatory factors by up-regulation of autophagy via AMPK/mTOR signaling pathway. J Transl Med. 2015;13:92.
Dai S, Wang B, Li W, Wang L, Song X, Guo C, et al. Systemic application of 3-methyladenine markedly inhibited atherosclerotic lesion in ApoE-/- mice by modulating autophagy, foam cell formation and immune-negative molecules. Cell Death Dis. 2016;7:e2498.
Jin X, Chen M, Yi L, Chang H, Zhang T, Wang L, et al. Delphinidin-3-glucoside protects human umbilical vein endothelial cells against oxidized low-density lipoprotein-induced injury by autophagy upregulation via the AMPK/SIRT1 signaling pathway. Mol Nutr Food Res. 2014;58:1941–51.
Hu YW, Wu SG, Zhao JJ, Ma X, Lu JB, Xiu JC, et al. VNN1 promotes atherosclerosis progression in apoE-/- mice fed a high-fat/high-cholesterol diet. J Lipid Res. 2016;57:1398–411.
Aryal B, Rotllan N, Fernandez-Hernando C. Noncoding RNAs and atherosclerosis. Curr Atheroscler Rep. 2014;16:407.
Yan B, Yao J, Liu JY, Li XM, Wang XQ, Li YJ, et al. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015;116:1143–56.
Nan A, Chen L, Zhang N, Liu Z, Yang T, Wang Z, et al. A novel regulatory network among LncRpa, CircRar1, MiR-671 and apoptotic genes promotes lead-induced neuronal cell apoptosis. Arch Toxicol. 2017;91:1671–84.
Humphries F, Yang S, Wang B, Moynagh PN. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ. 2015;22:225–36.
Dalton RG, Anderson NF, Sanders JH. Observations on repeated courses of antilymphocytic globulin on skin allograft survival in rats and the formation of antibody to antilymphocytic globulin. Transplantation. 1972;14:65–69.
Ogasawara M, Yano T, Tanno M, Abe K, Ishikawa S, Miki T, et al. Suppression of autophagic flux contributes to cardiomyocyte death by activation of necroptotic pathways. J Mol Cell Cardiol. 2017;108:203–13.
Liu X, Zhang C, Zhang C, Li J, Guo W, Yan D, et al. Heat shock protein 70 inhibits cardiomyocyte necroptosis through repressing autophagy in myocardial ischemia/reperfusion injury. In Vitro Cell Dev Biol Anim. 2016;52:690–8.
Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SW, et al. MLKL activation triggers NLRP3-mediated processing and release of IL-1beta independently of gasdermin-D. J Immunol. 2017;198:2156–64.
Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA. 2012;109:5322–7.
Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282.
Wang X, He Z, Liu H, Yousefi S, Simon HU. Neutrophil necroptosis is triggered by ligation of adhesion molecules following GM-CSF priming. J Immunol. 2016;197:4090–4100.
Chu C, Quinn J, Chang HY. Chromatin isolation by RNA purification (ChIRP). J Vis Exp, 2012; 61:3912.
Acknowledgements
This work was supported by the National Natural Sciences Foundation of China (grant numbers 81772244, 81572051, and 81472009), the Science and Technology Program of Guangzhou (Grant numbers 201604020015, 201704020213), the Natural Science Fund of Guangdong (grant numbers 2017A030313532, 2017A030313535, and 2015A030313245), the Science and Technology Program of Guangzhou (grant numbers 201510010091, 201604020015, 201607010267, 201512385434869, and 201707010034), and the Outstanding Youths Development Scheme of Nanfang Hospital, Southern Medical University (JQ201402).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Guo, FX., Wu, Q., Li, P. et al. The role of the LncRNA-FA2H-2-MLKL pathway in atherosclerosis by regulation of autophagy flux and inflammation through mTOR-dependent signaling. Cell Death Differ 26, 1670–1687 (2019). https://doi.org/10.1038/s41418-018-0235-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0235-z
This article is cited by
-
Nrf2-mediated redox balance alleviates LPS-induced vascular endothelial cell inflammation by inhibiting endothelial cell ferroptosis
Scientific Reports (2024)
-
Mediators of necroptosis: from cell death to metabolic regulation
EMBO Molecular Medicine (2024)
-
Acute lung injury: a view from the perspective of necroptosis
Inflammation Research (2024)
-
Long noncoding RNA signatures in intrauterine infection/inflammation-induced lung injury: an integrative bioinformatics study
BMC Pulmonary Medicine (2023)
-
Klotho inhibits renal ox-LDL deposition via IGF-1R/RAC1/OLR1 signaling to ameliorate podocyte injury in diabetic kidney disease
Cardiovascular Diabetology (2023)
