
Abstract
Compared to the tidy and immunologically silent death during apoptosis, necrosis seems like a chaotic and unorganized demise. However, we now recognize that there is a method to its madness, as many forms of necrotic cell death are indeed programmed and function beyond lytic cell death to support homeostasis and immunity. Inherently more immunogenic than their apoptotic counterpart, programmed necrosis, such as necroptosis, pyroptosis, ferroptosis, and NETosis, releases inflammatory cytokines and danger-associated molecular patterns (DAMPs), skewing the milieu to a pro-inflammatory state. Moreover, impaired clearance of dead cells often leads to inflammation. Importantly, these pathways have all been implicated in inflammatory and autoimmune diseases, therefore careful understanding of their molecular mechanisms can have long lasting effects on how we interpret their role in disease and how we translate these mechanisms into therapy.
Similar content being viewed by others
Facts
-
Necroinflammation is triggered by the release of inflammatory cytokines and DAMPs from cells undergoing necrotic cell death.
-
Necroptosis, pyroptosis, ferroptosis, and NETosis are forms of programmed necrosis, wherein lytic cell death is mediated by an activatable genetic program.
-
Defects in these programmed necrosis pathways and efferocytosis pathway have been linked to aberrant inflammation and autoimmunity.
Open Questions
-
What crosstalk exists among these types of cell death within a complex organ?
-
How can researchers exploit these pathways as therapeutic targets in vivo?
-
What are the non-canonical roles for some of the key players in these pathways, and how do those roles impact on host defense?
Introduction: Making a Murderer
Inflammation is a complex biological response encompassing clinical signs such as heat, pain, redness, and swelling (calor, dolor, rubor, and tumor in Latin) [1], generally triggered by harmful stimuli including microbial infection and tissue damage [2]. Discovery of the pattern recognition theory by Charles Janeway shed light on the molecular mechanisms by which the innate immune system distinguishes self vs. non-self to mount a response against invasive microbes [3], specifically via innate surveillance for foreign pathological motifs termed pathogen-associated molecular patterns (PAMPs) by classes of pattern recognition receptors (PRRs) [4]. This system, however, is insufficient to explain uncontrolled inflammation, such as autoinflammatory and autoimmune diseases, and spontaneous tumor rejection. Polly Matzinger’s complementary arm to Janeway’s theory, termed the danger theory, describes sensing of endogenous “self” danger signals by the host immune system senses to initiate an appropriate immune response [5]. Accumulating evidence has revealed that various cellular stresses and modes of cell death themselves can trigger inflammation by generating danger-associated molecular patterns (DAMPs), even in the absence of microbe-derived PAMPs [6, 7]. Indeed, many DAMPs signal via PRRs, implicating the integration of signaling pathways in response to both exogenous and endogenous stimuli.
Until recently, apoptosis was considered the sole type of programmed cell death responsible for proper development, homeostasis, and host defense, while necrosis was thought of as cell death in response to physicochemical traumas [8]. We now recognize that many different types of genetically regulated cell death programs, like necroptosis, pyroptosis, ferroptosis, and NETosis, exist and are strongly associated with various inflammatory and autoimmune diseases [9]. Programmed necrosis is defined as a genetically controlled cell death with morphological features such as cellular swelling (oncosis), membrane rupture, and release of cellular contents, in contrast to the organized packaging that occurs during apoptosis. The distinct nature of these cell death processes translates into differing levels of immunogenicity - apoptosis is generally considered immunologically silent, while necrosis is reported to trigger inflammation [2], although impaired clearance of apoptotic cells (a process termed efferocytosis) can result in secondary necrosis, leading to the onset of inflammation [10, 11]. Importantly, cellular contents, normally sequestered safely within the cell to serve innocuous and valuable roles during normal physiology, are released extracellularly during necrotic cell death and can act as DAMPs [12, 13].
In this review, we describe the molecular aspects of distinct programmed cell death pathways, including their execution, their clearance, and their roles as triggers of inflammation. Finally, we explore the association of programmed necrosis and inflammatory and autoimmune diseases, as well as its potential as a therapeutic target [9].
Types of programmed cell death: How to Get Away with Murder
Apoptosis/secondary necrosis: The Walking Dead
Apoptosis (from the Greek for “falling off”) is the quintessential genetically-controlled programmed cell death pathway, designed to prevent unwanted inflammation during development, homeostasis, and infection [8]. The formation of membrane blebs and apoptotic bodies during apoptosis enables safe partitioning and enclosure of cellular contents (potential DAMPs) within the plasma membrane, and the active exposure of distinct cell death extracellular signals ensures effective recognition by phagocytes during subsequent scavenging [8, 10].
Broadly, apoptosis is mediated by a family of cysteine proteases called caspases, which initiate apoptosis via two different pathways [8, 14]. The intrinsic pathway (also known as the mitochondrial pathway) executes apoptosis in response to developmental signals and cellular stresses, such as DNA damage and activates by an imbalance in power between the pro-apoptotic and anti-apoptotic members of the Bcl-2 family. These protein mediate mitochondrial outer membrane permeabilization (MOMP), the release of cytochrome c from mitochondria into the cytosol, and the formation of the apoptosome (with caspase 9 and APAF-1) to activate downstream executioner caspase cascades [15]. During the extrinsic pathway, death receptor engagement by ligands such as tumor necrosis factor (TNF), Fas ligand (CD95L, FasL), and TNF-related apoptosis-inducing ligand (TRAIL) activates caspase 8 to signal to executioner caspases [16]. These caspase signaling cascades not only carry out the actual execution, they also mediate the molecular and cellular changes that contribute to apoptosis’s immunotolerance. Recent studies have demonstrated that the mitochondrial apoptotic pathway triggers potent inflammation in the absence of caspase activity via in concert with cGAS/STING and NF-κB signalings [17,18,19]. Furthermore, caspase signaling also contributes to externalization of “eat-me” signals to facilitate their rapid clearance (discussed below), DNA fragmentation, and membrane blebbing.
When apoptotic cells are not scavenged timely and efficiently, they undergo secondary necrosis (autolysis), characterized by damaged cell membrane and loss of cell integrity. Although secondary necrosis is suggested to be a natural outcome of apoptosis in single cell organisms, it also occurs in multicellular animals [20]. In vivo evidence of secondary necrosis can be observed under both physiological and pathological conditions, wherein apoptotic cells are shed into tissues with reduced phagocyte presence (e.g. airway lumen) or when excessive apoptosis outcompetes the capacity for efferocytosis, respectively [21, 22]. While secondary necrosis results in morphological changes that overlap with programmed necrosis, including cytoplasmic swelling, permeabilization of membranes, and the release of intracellular components, it is generally considered to be less immunogenic than primary necrosis since the DAMPs undergo danger-attenuating modifications during apoptosis [22]. Moreover, secondary necrotic cells can be cleared by micropinocytosis, thus dampening their inflammatory effects [20, 22].
Programmed necrosis: Homicide: Life on the Street
Many types of necrosis, both accidental and programmed, share common morphological features, including cellular swelling and plasma membrane permeabilization, resulting in the release of potentially harmful cellular contents and the induction of inflammation [7, 23]. Until recently, necrosis has been considered a solely accidental and non-programmed cell death modality. However, extensive genetic and pharmacological studies have clearly demonstrated that cells can and do undergo programmed necrotic processes, such as necroptosis, pyroptosis, ferroptosis, and NETosis (Fig. 1), that are induced by RIPK3/MLKL, inflammasome activation, iron metabolism, and reactive oxygen species (ROS), respectively [7]. Despite several shared morphological features, each programmed necrotic pathway utilizes distinct molecular process and generates different immunological outcomes. Since programmed necrosis is closely associated with multitude of inflammatory and autoimmune diseases, thorough understanding of these mechanisms is needed to identify effective therapeutic targets for disease [9, 24].

Molecular pathways of cell death and their roles in inflammation. Depending on death stimuli and context, live cells can undergo apoptosis or programmed necrosis. When caspase 3/7-dependent apoptotic cells are timely scavenged by efferocytosis, efferocytes like macrophages release anti-inflammatory cytokines and prevent unwanted inflammation. In the absence of effective clearance, apoptotic cells are proceeded to secondary necrosis and elicit some inflammatory responses. Upon damage signals such as infection or metabolic stress, cells trigger genetically programmed necrosis. Necroptosis and pyroptosis are RIPK3-MLKL- and inflammasome-GSDMD-mediated processes, respectively, displaying typical lytic morphology similar to primary necrosis. In contrast, ferroptosis is triggered by lipid peroxidation, and shows damaged mitochondria and reduced cellular volume. NETosis is a ROS-induced lytic cell death resulting in the extrusion of neutrophil extracellular traps (NETs), consisting of genomic DNA complexed with cellular proteins. Programmed necrotic cells generally release DAMPs and inflammatory cytokines that stimulate innate immune cells and promote necroinflammation
Necroptosis: Murder, She Wrote
Necroptosis was first reported in a seminal study wherein inhibition of caspase 8 sensitized fibroblasts to TNF-α-induced necrotic death [25]. Indeed, engagement of aforementioned death receptor pathways can trigger necroptotic cell death in the absence of caspase 8 activity, via RHIM-containing kinases like receptor-interacting protein kinase 1 (RIPK1) and RIPK3 [26, 27]. Additionally, signals that activate TIR-domain-containing adapter-inducing interferon-β (TRIF) or DNA-dependent activator of interferon-regulatory factors (DAI), both of which encode RHIM domains, by Toll like receptor 3/4 (TLR3/4) agonists including dsRNA, LPS, or viral DNA/RNA, respectively, are known to induce necroptosis [28, 29]. Furthermore, necroptosis can be triggered by other signals including T cell receptor (TCR) engagement, retinoic acid-inducible gene I (RIG-I) stimulation, and type I and type II interferon (IFN) signaling [30,31,32].
Downstream of these triggers are several molecular pathway options, depending on the availability of cellular inhibitor of apoptosis (cIAPs), FLICE-like inhibitory proteins (FLIP), or caspase 8, and the cell’s choice dictates its fate in the form of nuclear factor (NF)-κB-mediated survival, apoptosis, and necroptosis [7, 23]. Death receptor-induced necroptosis is currently the most deeply characterized necrotic pathway. Upon engagement of the TNF receptor (TNFR) by TNF-α, the cytosolic tail of TNFR recruits TNF receptor-associated death domain (TRADD) to promote the assembly of Complex I, in association with RIPK1, cIAP1/2, TNF receptor-associated factor 2/5 (TRAF2/5), and linear ubiquitin chain assembly complex (LUBAC). In this complex, RIPK1 is poly-ubiquitylated by cIAPs, which facilitates its interaction with TGF-β-activated kinase 1 (TAK1) and TAK1-associated binding protein (TAB). Further association with the inhibitor of NF-κB kinase (IKK) complex, promoted by LUBAC-mediated linear ubiquitination, activates canonical NF-κB signaling to promote cell survival, proliferation, and a potent pro-inflammatory response [33]. When RIPK1 is deubiquitinated by the deubiquitinase associated with cylindromatosis (CYLD), the TRADD-dependent Complex IIa is formed and executes apoptosis through caspase 8 activity. Likewise, if cIAPs, TAK1, or NF-κB essential modulator (NEMO) is depleted (by SMAC mimetics), RIPK1-dependent Complex IIb (sometimes referred to as the RIPoptosome) is assembled and dictates an alternative path of caspase 8-dependent apoptosis [33].
In the absence of caspase 8, activation of the necroptotic pathway requires assembly of the RIPK1/RIPK3 complex, called the Necrosome [34, 35]. Auto- and trans-phosphorylation of RIPK1 and RIPK3 enables recruitment and activation of the pseudokinase, mixed lineage kinase-like (MLKL), followed by oligomerization, binding to plasma membrane-associated phosphatidylinositol phosphates, and the disruption of plasma membrane integrity [26, 36, 37]. The RIPK1/RIPK3 complex also facilitates mitochondrially-derived reactive oxygen species (ROS) production, which reinforces the stability of the necrosome [38]. A recent study showed that ESCRT-III regulates the dynamics and kinetics of plasma membrane integrity during necroptosis, thus modulating the level of DAMPs release to facilitate an appropriate subsequent adaptive immune response [39]. As a consequence of plasma membrane rupture, extracellular DAMPs and cytokines affect neighboring cells and trigger inflammation via signaling through their cognate receptors [40]. Indeed, necroptotic cells release many types of DAMPs including HMGB1, nucleic acids, IL-1 family members, ATP, uric acid, and S100 protein, which contribute to the activation of innate immune cells like dendritic cells and macrophages [13, 40], although IL-33 seems to dampen inflammation through modulating regulatory T cells [13, 24]. In addition, coupling RIPK3 with RIPK1-NF-κB axis may play an important role in the subsequent stimulation of T cell response by enhancing inflammatory cytokines like IL-6 and CXCL1 [41].
In vivo evidence for necroptosis and its pathophysiological role is also rapidly accumulating. Recent studies indicate that necroptosis is very common during damage associated with physical trauma, infection, neurodegeneration, and ischemia. Despite the capacity of necroptosis to induce inflammation, emerging evidence suggests that the main pathological role of necroptosis is host defense. During specific circumstances like infection, cells may adopt this prompt and immunogenic death to eliminate the pathogen’s replicative niche [1, 50]. The phenotypic discrepancies observed between RIPK3- and MLKL-deficient mice support the idea of necroptotic machinery being a critical component of inflammation, beyond the function of necroptosis [42, 43]. RIPK3-deficient mice display enhanced protection against ischemic injury and sterile sepsis models via cell death-independent mechanisms, and RIPK3 deficiency also limits viral pathogenesis of West Nile Virus (WNV) independently of cell death [44, 45]. Moreover, RIPK3 can still promote inflammasome activation, even in the absence of MLKL [46]. Together, these results imply that RIPK3 has a role in regulating inflammation and the immune response independently of MLKL activation and necroptosis.
Pyroptosis: Burn Notice
Pyroptosis (from the Greek, pyro meaning fire), which is another form of programmed cell death mediated by excessive inflammasome activation and displays features common to both apoptosis (caspase dependence, chromatic condensation, and DNA fragmentation) and necrosis (cellular swelling, plasma membrane pore formation, and membrane rupture) [14, 47, 48]. The inflammasome is a multimeric complex responsible for the conversion of inactive pro-forms of inflammatory cytokines into their mature, active forms (IL-1β and IL-18). While the exact composition of the inflammasome can differ depending on the stimuli, it is broadly comprised of caspase 1 and a PRR that senses PAMPs or DAMPs, such as NOD-like receptors NLRP1 (anthrax lethal toxin), NLRP3 (various PAMPs and DAMPs), NLRC4 (flagellin, type III/IV secretion systems), AIM2 (dsDNA), and Pyrin (bacterial toxins) [10, 49, 50]. These PRRs contain either a caspase activation and recruitment domain (CARD) or a pyrin domain (PYD). Receptors containing CARD like NLRP1 and NLRC4 can either directly interact with pro-caspase 1, or indirectly associate with pro-caspase 1 via apoptosis-associated speck-like protein containing a CARD (ASC) to induce caspase 1 activation. PRRs without a CARD domain, such as NLRP3, AIM, and Pyrin require ASC to complex with pro-caspase 110,50. Caspase 11 was also reported to induce IL-1β maturation and pyroptosis in response to gram negative bacterial infections [51]. Recent studies have revealed that inflammatory caspases (human caspase 1/4/5, murine caspase 1/11) can cleave a conserved site at gasdermin D (GSDMD), and activate the pore-forming capacity of GSDMD to elicit the lytic feature of pyroptosis [51,52,53]. While pyroptosis plays a critical role in the containment of bacterial spread, the activation of the inflammatory cytokines, IL-1β and IL-18, by caspase 1 reveals another layer of inflammatory outcomes in addition to various DAMPs release [47, 48].
Ferroptosis: Iron Chef
The term ‘ferroptosis’ was coined to describe a distinct form of cell death observed in RAS-transformed tumor cells induced by inhibition of system XC– (using small molecules, like erastin) or inhibition of the downstream component glutathione peroxidase 4 (GPX4) [54]. A cell’s ability to undergo ferroptosis seems to be linked with its state of amino acid, polyunsaturated fatty acid (PUFA), and iron metabolic activity, as well as biosynthesis of glutathione (GSH) [55]. Ferroptosis exhibits unique morphological changes, like decreased cell volume and damaged mitochondria. Apoptotic features (DNA fragmentation, membrane blebbing) or necrotic features (oncosis, necrotic membrane rupture) are largely absent in ferroptosis [54]. Despite this, ferroptosis can still potently induce inflammation via the release of HMGB1, IL-33, and other unidentified pathways [54, 55]. Mechanistically, erastin treatment inhibits the system XC-Cys/Glu antiporter, which facilitates the exchange of extracellular L-Cys for intracellular L-Glu. This imbalance results in a depletion of intracellular GSH and inhibition of GPX4, the net effect being high intracellular levels of H2O2 and excessive lipid peroxidation, ultimately causing cell death in a manner independent of apoptotic or necroptotic machinery [54,55,56].
Intracellular cysteine availability is a rate-limiting step for GSH synthesis, and inhibiting the 1:1 exchange of cystine for glutamate through the antiporter is critical for the initiation of ferroptosis. Thus, excessive level of extracellular glutamate may trigger ferroptosis under physiological conditions [57]. In some cells, cysteine can be biosynthesized via the trans-sulfuration pathway using methionine, resulting in ferroptotic resistance [58]. Importantly, PUFAs are highly susceptible to lipid peroxidation, and the accumulation of PUFA hydroperoxides proves to be the lethal step in ferroptosis, as lipid peroxides can be broken into lipid radicals that react with proteins and nucleic acids [55].
Iron availability is a critical factor in determining ferroptosis, as lipid peroxidation is mediated by the activity of lipoxygenases via a phosphorylase b kinase gamma catalytic chain, liver/testis isoform (PHKG2)-dependent iron pool [59]. Silencing of iron-responsive element-binding protein 2 (IREB2), a master regulator of iron metabolism, decreases ferroptosis [54], while selective autophagic clearance of ferritin enhances susceptibility of ferroptosis by releasing free iron [60, 61].
NETosis: Deadliest Catch
Neutrophils are a short-lived but critical component at the frontline of host defense. In addition to phagocytosis, neutrophil produce and release of antimicrobiocidal granule contents (degranulation), ROS (oxidative bursts) and neutrophil extracellular traps (NETs) [62]. NETs are web-like structures comprised of decondensed chromatin bound to cytosolic and granular proteins. The NETs are able to trap, immobilize, inactivate, and kill microorganisms, as well as stimulate the immune response [62, 63]. An extracellular release of NETs is usually accompanied by a lytic form of cell death termed NETosis, which can be induced by microbial infections (bacteria, fungi, viruses, and parasites) or endogenous ‘sterile’ stimuli (uric acid crystal, cholesterol crystal, and immune complex) [62, 63].
Once activated, ROS is produced by either the NADPH oxidase (NOX) or mitochondrial pathway, resulting in the translocation of granule-associated enzymes like myeloperoxidase (MPO) and neutrophil elastase (NE) to the nucleus, where NE and MPO synergize to disrupt and decondense chromatin [64]. Following the disintegration of the nuclear envelope, the chromatin decondensation process expands into the cytoplasm of living cells, where DNA and cytoplasmic and granular components co-mingle. Finally, the plasma membrane is permeabilized, and NETs are released into the extracellular space, typically 3-8 h after neutrophil activation [62]. While DNase I is responsible for the clearance of NETs, the mechanism governing this process is unclear, and NET-associated proteins persist after DNA degradation, indicative of possible additional mechanisms [65]. In vitro studies have demonstrated that, in addition to DNase I, macrophages play an important scavenging role in NET clearance [66].
The morphology of NETosis suggests that this type of cell death is pro-inflammatory, owing to its lytic nature and the release of histones, nucleic acids, and cytotoxic granular molecules. Indeed, there is evidence that NETosis can initiate inflammation via the release of DAMPs. Crystal-induced NETs trigger the transcription of IL-6 and IL-1β in macrophages [67]. Moreover, oxidized nucleic acids can stimulate STING-dependent type I IFN production in myeloid cells. cathelicidin-DNA complexes promote the IFN response via endosomal TLRs in plasmacytoid dendritic cells, and cathelicidin can activate NLRP3 inflammasome in LPS-primed macrophages [63]. However, the immunological consequences of NETosis remain controversial as it is also suggested that NETs can help resolve local inflammation at a high concentration [68].
Efferocytosis: Curb your Enthusiasm
While the role of innate immunity to sense and control invasive foreign microbes has long been appreciated, its equally important role of mediating tolerance in the face of the body’s most abundant threat, dying cells, is often overlooked. Billions of cells undergo programmed cell death in an organism daily, and their silent and proficient clearance, a process termed efferocytosis (from the Latin meaning “to take to the grave”), ensures proper development and immunological homeostasis [69, 70]. For decades, efferocytotic research focused on the clearance of apoptotic cells, considered the archetypal programmed cell death. That characterization is not without merit - apoptosis is a critical component of an organism’s proper development and arguably the most abundant programmed cell death in vivo. The apoptotic cell packages itself into immunologically inert pieces and promotes its own removal via the active advertising of its presence. Thus, the efferocytosis of apoptotic cells can be viewed as the gold standard of dying cell clearance, as the mechanisms underlying apoptosis and efferocytosis are rooted in effective immunotolerance [71].
Efferocytosis is a coordinated event, wherein dying cells recruit, prime, and present themselves to both professional phagocytes (macrophages and dendritic cells) and non-professional phagocytes (epithelial cells). The release of “find-me” signals, such as ATP, UTP, sphingosine-1-phosphate (S1P), lysophosphatidylcholine (LPC), and CX3CL1 (fractalkine), by dying cells results in the recruitment of local phagocytes to the sites of cell death, as well as licenses the phagocyte to upregulate molecules required for efferocytosis [72]. The release of these “find-me” signals is often caspase-dependent, though recent studies have demonstrated that caspase-independent deaths result in high local concentrations of these signals, undoubtedly due to their lytic nature [73]. While “find-me” signals are an important first step in efferocytosis, their function, specifically in vivo, lies beyond their chemoattractant ability [74, 75].
Phagocytes distinguish dying cells from living cells by their display of “eat-me” signals, which are actively exposed by dying cells via caspase-dependent mechanisms. While molecules such as ICAM3, oxidized LDL-like molecules, calreticulin (CRT), and C1q-bound serum proteins have been described to act as “eat-me” signals [14, 71, 73], the best characterized “eat-me” signal is phosphatidylserine (PS), a lipid confined to the inner leaflet of the plasma membrane in living cells, but translocated to the outer leaflet by caspase-3-mediated activation of the scramblase Xkr8 [76, 77]. Recent reports demonstrate that exposure of PS on dying cells precedes actual death and morphological features of apoptosis, and PS exposure is sufficient to trigger efferocytosis, though concurrent expression of don’t eat me” signals, such as CD47, can hinder this effect [78]. Thus, PS exposure acts as a first line mechanism to preemptively clear dying cells before secondary necrosis and the release of DAMPs occurs.
Phagocytes are armed with receptors that allow them to sense and migrate to areas of cell death, as well as recognize and internalize cellular corpses for degradation and processing [71]. “Find-me” signals are detected via receptors like P2Y2 (ATP, UTP), S1PRs (S1P), G2A (LPC), and CXCR3 (CX3CL1), and engagement of these receptors mediates chemoattraction as well priming the phagocyte for engulfment [79]. Phagocytes employ a variety of PS-specific receptors (PSRs) to recognize “eat-me” signals on dying cells, and engagement of these receptors mediates engulfment. Examples of these PSRs include membrane-bound receptors, such as T cell immunoglobulin mucin receptor 4 (TIM4), brain-specific angiogenesis inhibitor 1 (BAI1), and stabilin-2, or bridging molecules, such as milk fat globule-EGF factor 8 (MFG-E8) and Gas6, which further link to integrins or Tryo3-Axl-Mer (TAM) receptors [80, 81]. Different tissues preferentially express different PSRs including BAI1 (bone marrow, spleen, brain), TIM4 (kidney), and stabilin-2 (sinusoidal endothelial cells), suggesting tissue-specific PS receptor mechanisms [71, 74, 82]. While downstream molecules used by specific PSRs can differ (and are discussed in greater detail here [14]), engulfment mediated by PSRs activates the Rho family of small GTPases, converging on evolutionarily conserved Rac1 [14].
Once engulfed, phagocytes must degrade, digest, and process their cellular corpse cargo. Rab5 and Rab7, small GTPases, are recruited to the phagosome to mediate fusion to the lysosomal network, which contains acidic proteases and nucleases that digest cellular corpses into their basic factors [14]. A recently described form of non-canonical autophagy, termed LC3-associated phagocytosis (LAP), was found to be crucial for the homeostatic clearance of dying cells, as well as shaping the appropriate immune response during efferocytosis [69, 70]. While LAP can be triggered by pathogen sensing (viaTLRs or FcR) during uptake, LAP is also activated by PSR engagement during efferocytosis, resulting in the recruitment of a distinct subset of autophagic machinery to the cargo-containing, single-membraned vesicle [69, 83]. LAP requires the activity and localization of Rubicon (RUN domain protein as Beclin-1 interacting and cysteine-rich containing), whereas canonical autophagy (induced by starvation or rapamycin treatment) is Rubicon-independent [84]. LAP facilitates the rapid destruction of the engulfed cargo via fusion with the lysosomal pathway and is crucial in mediating the immunotolerant response associated with efferocytosis [69, 83, 84]. Strikingly, Rubicon-/- mice develop systemic lupus erythematosus (SLE)-like disease with age, whereas autophagy-deficient mice do not [70].
A dead cell is not like other foreign particles, however, and its engulfment essentially doubles the phagocyte’s levels of cellular components, including fats, sterols, peptides, and nucleotides. To handle this metabolic stress, efferocytosis leads to the activation of peroxisome proliferator-activated receptor γ/δ (PPARγ/δ) and liver x receptor (LXR) families, both critical modulators of cellular lipid homeostasis and immunity [85, 86]. Activation of these factors mediates the upregulation of other phagocytic machinery, such as the TAM family, and basal cholesterol efflux machinery, such as ABCA1 (ATP-binding cassette subfamily A, member 1), to accommodate this increase in cholesterol levels [87]. Indeed, cholesterol homeostasis is important for immunotolerance during the clearance of apoptotic cells, as it is closely linked to the production of anti-inflammatory cytokines, like TGF-β and IL-10, and active suppression of pro-inflammatory cytokines, such as TNF-α, IL-1, and IL-12 [88, 89]. Despite the multi-step process by which efferocytosis occurs, and the functional redundancy of some of the key players, defects in even a single component of this pathway can have deleterious effects, often resulting in autoinflammation or autoimmunity [11], highlighting the importance of this pathway in homeostasis.
DAMPs and inflammation: Jeopardy!
The efficiency with which efferocytosis operates underscores the danger of exposure to DAMPs [90, 91]. DAMPs run the gamut of cellular molecules, including nuclear or cytosolic proteins (high mobility group box 1 [HMGB1], S100 family of calcium-binding proteins, interleukin 1 [IL-1] family members, transcription factor A mitochondrial [TFAM], and histones) or non-protein molecules (ATP, uric acid, DNA, RNA, and mitochondrial DNA), which are both immunologically inert and biologically beneficially within the confines of a cell [92]. Phagocytes employ the same PRRs used to detect PAMPs to sense DAMPs, thus despite their “self” source, DAMPs are viewed as threatening as invading microbes (Table 1) [93].
The immunotolerant beauty of apoptosis lies in its sequestration of cellular contents into membrane-bound blebs which isolates DAMPs from their cognate PRRs. The developmentally and functionally homeostatic process of apoptosis is therefore akin to the nuclear compartmentalization of nucleic acids, permitting the evolution of viral nucleic acid sensors, which are critical for host defense [94]. Programmed necrosis, however, unleashes intracellular and immunostimulatory DAMPs on to phagocyte PRRs, eliciting inflammation and if left unchecked, autoimmune and autoinflammatory disorders (Fig. 2) [82]. The lytic nature of programmed necrosis means that its debris is inadvertently PS-positive, and necroptotic and pyroptotic cells can and do engage PSRs [10]. Studies have demonstrated that both necrotic and necroptotic cells engages TIM4 on phagocytes to mediate LAP, and engagement of PSRs is sufficient to engage the PPAR/LXR immunotolerant pathway [10]. Despite providing the excess cholesterol and fatty acids that tend to suppress inflammation by apoptotic cells, efferocytosis of necrotic cells does not mediate the same immunotolerant response [69]. The key to this quandary lies in the release of DAMPs – the immunostimulatory effects of DAMPs outweighs the tolerance induced by PSR engagement, and the net effect is inflammation [69,95].

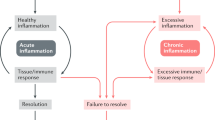
Systemic spread of necroinflammation. Programmed necrosis or defective efferocytosis of dying cells can initiate local inflammation. DAMPs and proinflammatory cytokines derived from necrotic cells and subsequently activated immune cells provide feed-forward signals reinforcing programmed necrosis in more cells. Continuing the vicious death cycle allows the damage of barrier function, the spread of necroinflammation to systemic level, which may ultimately cause devastating multi-organ failure
Programmed necrosis, however, is not solely a losing game. Programmed necrosis can be triggered by an immunologically calamitous event, such as infection. In situations that require immune attention, these programmed necrotic pathways have likely evolved to modulate the immune response. Necrotic cell death achieves immunoregulatory effects beyond mere death – it can destroy the pathogen’s replicative niche, it can result in the increased release of “find-me” signals (which can double as DAMPs) [73], it can engage downstream immune signaling pathways [96], and the increased DAMPs release can heighten the inflammatory cascade. During pyroptosis, pore-induced intracellular traps (PITs) can trap intracellular bacteria within structures and complement and scavenger receptor stimulation can promote the PIT clearance by neutrophils. Therefore, pyroptosis and necroptosis may represent two unique biological pathways in which the host counteracts pathogen evasion mechanisms [97].
Autoimmune and inflammatory diseases: House, MD
Programmed necrosis has a crucial role in defending against invading microorganisms and regulating innate immunity. However, its activity may be a double-edged sword in human pathophysiology, as it is involved in the development of a variety of chronic inflammatory and autoimmune diseases via different mechanisms (Table 2).
Necroptosis is triggered by a vast number of pathogens that express TLR3/4 ligands, resulting in the rupture of infected cells and the suppression of microbial growth [98]. While RIPK1 global knockout mice are neonatally lethal, RIPK3- and MLKL-deficient mice undergo normal development and can be utilized to examine the role of necroptosis in vivo. Mice lacking RIPK3 are highly susceptible to bacterial (Yersinia pestis [99]) and viral pathogens [100], often accompanied by defective pathogen-induced tissue necrosis [35]. Interestingly, although RIPK3-deficient animals were more susceptible to influenza A virus and WNV infections, MLKL-deficient animals were not [45, 101]. These results suggest that control of these viruses in vivo may depend on RIPK3-directed transcriptional responses instead of necroptosis. RIPK1/3 have also been shown to contribute to the progression of ischemic and degenerative diseases. Treatment with the RIPK1 kinase inhibitor, Necrostatin-1, or genetic ablation of RIPK3 effectively protects against brain and kidney ischemia [102, 103], renal allograft inflammation [104], and myocardial infarction [105, 106]. Moreover, RIPK1 deficiency ameliorates retinal degeneration (retinitis pigmentosa) [107] and neuronal degeneration (Huntington’s disease, multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD)) [108,109,110,111] in animal disease models.
Pyroptosis acts as a protective host mechanism against microbial infections, via destruction of its replication niche. Inflammasome-defective mice are more susceptible to bacterial, viral, or fungal challenge [97]. Recent studies indicate that parasites can also trigger pyroptosis via inflammasome activation [112]. While inflammasome complexes are essential for pathogen clearance, their uncontrolled and excessive activation is detrimental and pathogenic. Over-activation of pyroptosis by pathogens can lead to widespread cell death, acute organ failure, sepsis, and septic shock [113]. Furthermore, patients carrying a gain-of-function mutation in inflammasome genes (NLRP1, NLRP3, and NLRC4) present with a wide spectrum of multi-organ inflammatory symptoms due to spontaneously increased inflammasome activation and IL-1β production [114,115,116]. Finally, inflammasome and caspase 1 activation have been implicated in myriad chronic inflammatory or autoimmune diseases, including atherosclerosis [117], MS [118, 119], diabetes [120], obesity [121], AD [122], and Parkinson’s disease [123], suggesting that either pharmacological or genetic inhibition of inflammasomes could significantly alleviate symptoms and reduce disease severity in experimental animals [117,118,119,120,121,122,123] or in patients [124].
Ablation of the key enzyme, GPX4, in the ferroptosis pathway results in embryonic lethality in mice, implying an important role of ferroptosis in development [125]. In addition, conditional deletion of GPX4 in murine neuronal cells caused rapid motor neuron degeneration, and inducible GPX4 ablation in adult mice led to neuronal loss and inflammation in the hippocampus [126, 127]. Increased levels of hepcidin, a regulator of iron release, was involved in ischemic brain injury, and its silencing ameliorated damage [128]. Importantly, impaired cysteine biosynthesis resulted in necrotic cell death in a mouse model, and substantial depletion of cystathione γ-lyse (an enzyme for cysteine biosynthesis) was observed in Huntington’s disease patients [129]. Furthermore, renal tubular cells are particularly sensitive to ferroptosis in GPX4-deficient mice [130]. Consistently, highly reactive catalytic iron is considered as a risk factor for acute kidney injury (AKI), and the treatment with iron chelators, like desferrioxamine, displayed some protective effect despite inconsistency [13]. In addition, ferritin, hepcidin, and heme oxygenase 1 are all involved in the pathogenesis of AKI [24].
Human patients deficient for MPO, a key neutrophilic enzyme, have an increased risk for Candida albicans infection [131]. Similarly, MPO deficient mice also failed to clear C. albicans and other large pathogens, but not small bacteria [132]. Despite its role in controlling infection, NETs are now considered a source of self-antigen and a potential initiator in many autoimmune diseases, including SLE [133], rheumatoid arthritis (RA) [134], anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) [135] and type 1 diabetes (T1D). Accordingly, SLE patients have a unique low-density granulocyte (LDG) population with an increased NET formation capacity [133], and have elevated serum anti-dsDNA and anti-ribonucleoprotein proteins that are directed against the NETs components [135]. Likewise, neutrophils from RA and T1D patients produce more NETs in vitro and display increased ROS production [134, 136]. Furthermore, NETs are implicated in vascular diseases, as evidenced by NE-/- RP3-/- mice which are protected against the development of atherosclerosis [67].
Conclusion: Life Goes On
Paradoxical as it may seem, life requires death – for the proper development of an organism, for the regeneration of tissue and cells over time, and for the appropriate immune response during challenge. We now recognize that apoptosis is not the only show in town and that other programmed cell death pathways, such as necroptosis, pyroptosis, ferroptosis, and NETosis, have their own role to play in an organism’s homeostasis and defense. These newly characterized pathways represent possible targets for therapies against inflammatory diseases and infections that have often been refractory to current treatments. Future studies on how these pathways intersect with other key biological processes and their tissue specificities are needed to fully harness their potential.
References
Kearney CJ, Martin SJ. An Inflammatory Perspective on Necroptosis. Mol Cell. 2017;65:965–73.
Wallach D, Kang T-B, Kovalenko A. Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat Rev Immunol. 2013;14:51–59.
Janeway CA. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):1–13.
Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–97.
Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045.
Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell. 2014;54:281–8.
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147.
Wallach D, Kang T-B, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016;352:aaf2154.
Conrad M, Angeli JPF, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348–66.
Kolb JP, Oguin TH, Oberst A, Martinez J. Programmed Cell Death and Inflammation: Winter Is Coming. Trends Immunol. 2017;38:705–18.
Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ. 2016;23:915–26.
Krysko DV, Krysko DV, Garg AD, Garg AD, Kaczmarek A, Kaczmarek A, et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–75.
Sarhan M, Land WG, Tonnus W, Hugo CP, Linkermann A. Origin and Consequences of Necroinflammation. Physiol Rev. 2018;98:727–80.
Martinez J. Prix Fixe: Efferocytosis as a Four-Course Meal. Curr Top Microbiol Immunol. 2017;403:1–36.
Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–32.
Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–90.
Giampazolias E, Zunino B, Dhayade S, Bock F, Cloix C, Cao K, et al. Mitochondrial permeabilization engages NF-κB-dependent anti-tumour activity under caspase deficiency. Nat Cell Biol. 2017;19:1116–29.
Rongvaux A, Jackson R, Harman CCD, Li T, West AP, de Zoete MR, et al. Apoptotic Caspases Prevent the Induction of Type I Interferons by Mitochondrial DNA. Cell. 2014;159:1563–77.
White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–62.
Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 2010;584:4491–99.
Mahajan A, Herrmann M, Muñoz LE. Clearance Deficiency and Cell Death Pathways: A Model for the Pathogenesis of SLE. Front Immunol. 2016;7:35.
Sachet M, Liang YY, Oehler R. The immune response to secondary necrotic cells. Apoptosis. 2017;22:1189–204.
Pasparakis M, Pasparakis M, Vandenabeele P, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–20.
Tonnus W, Linkermann A. The in vivo evidence for regulated necrosis. Immunol Rev. 2017;277:128–49.
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477–85.
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–67.
Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–72.
Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–79.
Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–97.
Ch’en IL, Beisner DR, Degterev A, Lynch C, Yuan J, Hoffmann A, et al. Antigen-mediated T cell expansion regulated by parallel pathways of death. Proc Natl Acad Sci USA. 2008;105:17463–68.
Zou J, Kawai T, Tsuchida T, Kozaki T, Tanaka H, Shin K-S, et al. Poly IC triggers a cathepsin D- and IPS-1-dependent pathway to enhance cytokine production and mediate dendritic cell necroptosis. Immunity. 2013;38:717–28.
Thapa RJ, Nogusa S, Chen PR, Maki JL, Lerro A, Andrake M, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. P Natl Acad Sci USA. 2013;110:E3109–118.
Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–55.
Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao Y-S, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–50.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27.
Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–81.
Fulda S. Regulation of necroptosis signaling and cell death by reactive oxygen species. Biol Chem. 2016;397:657–60.
Gong Y-N, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell. 2017;169:286–300.e16.
Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity. 2013;38:209–23.
Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science. 2015;350:328–34.
Kang T-B, Yang S-H, Toth B, Kovalenko A, Wallach D. Caspase-8 Blocks Kinase RIPK3-Mediated Activation of the NLRP3 Inflammasome. Immunity. 2013;38:27–40.
Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun. 2015;6:7515.
Qu Y, Shi J, Tang Y, Zhao F, Li S, Meng J, et al. MLKL inhibition attenuates hypoxia-ischemia induced neuronal damage in developing brain. Exp Neurol. 2016;279:223–31.
Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH, Tait SWG, et al. RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell. 2017;169:301–13.e11.
Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 2015; 6. https://doi.org/10.1038/ncomms7282.
Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–61.
Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109.
Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42:245–54.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20.
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–65.
Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–16.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273–85.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell. 2015;59:298–308.
Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23:270–78.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966–75.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–28.
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–9.
Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134–47.
Gupta S, Kaplan MJ. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol. 2016;12:402–13.
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–91.
Kolaczkowska E, Jenne CN, Surewaard BGJ, Thanabalasuriar A, Lee W-Y, Sanz M-J, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673.
Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol. 2013;191:2647–56.
Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–20.
Muñoz LE, Bilyy R, Biermann MHC, Kienhöfer D, Maueröder C, Hahn J, et al. Nanoparticles size-dependently initiate self-limiting NETosis-driven inflammation. Proc Natl Acad Sci USA. 2016;113:E5856–65.
Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci USA. 2011;108:17396–401.
Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533:115–19.
Poon IKH, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14:166–80.
Luo B, Gan W, Liu Z, Shen Z, Wang J, Shi R, et al. Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity. 2016;44:287–302.
Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207:1807–17.
Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol. 2013;5:a008748.
Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–67.
Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–27.
Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341:403–6.
Liu X, Liu X, Pu Y, Pu Y, Cron K, Cron K, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015;21:1209–15.
Elliott MR, Elliott MR, Chekeni FB, Chekeni FB, Trampont PC, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–86.
Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol. 2012;189:3508–20.
Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–50.
Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–630.
Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37:986–97.
Martinez J, Malireddi RKS, Lu Q, Cunha LD, Pelletier S, Gingras S, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17:893–906.
Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. 2009;15:1266–72.
Roszer T, Menéndez-Gutiérrez MP, Lefterova MI, Alameda D, Núñez V, Lazar MA, et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol. 2011;186:621–31.
Han CZ, Ravichandran KS. Metabolic connections during apoptotic cell engulfment. Cell. 2011;147:1442–45.
A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–58.
Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21:643–53.
Medzhitov R. Approaching the asymptote: 20 years later. Immunity. 2009;30:766–75.
Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–5.
Zhang Q, Kang R, Zeh HJ, Lotze MT, Tang D. DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy. 2013;9:451–58.
Tang DL, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–75.
Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38:870–80.
Ferguson TA, Choi J, Green DR. Armed response: how dying cells influence T-cell functions. Immunol Rev. 2011;241:77–88.
Brault M, Oberst A. Controlled detonation: evolution of necroptosis in pathogen defense. Immunol Cell Biol. 2017;95:131–36.
Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265:130–42.
Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18:127–36.
Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci USA. 2014;111:7391–96.
Wang X, Li Y, Liu S, Yu X, Li L, Shi C, et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci USA. 2014;111:15438–43.
Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH, Ingram JP, et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe. 2016;20:13–24.
Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller J-O, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2013;110:12024–29.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–19.
Lau A, Wang S, Jiang J, Haig A, Pavlosky A, Linkermann A, et al. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am J Transplant. 2013;13:2805–18.
Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res. 2014;103:206–16.
Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23:1565–76.
Murakami Y, Matsumoto H, Roh M, Suzuki J, Hisatomi T, Ikeda Y, et al. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc Natl Acad Sci USA. 2012;109:14598–603.
Zhu S, Zhang Y, Bai G, Li H. Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington’s disease. Cell Death Dis. 2011;2:e115.
Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015;10:1836–49.
Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603–8.
Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, et al. Necroptosis activation in Alzheimer’s disease. Nat Neurosci. 2017;20:1236–46.
Zamboni DS, Lima-Junior DS. Inflammasomes in host response to protozoan parasites. Immunol Rev. 2015;265:156–71.
Aglietti RA, Dueber EC. Recent Insights into the Molecular Mechanisms Underlying Pyroptosis and Gasdermin Family Functions. Trends Immunol. 2017;38:261–71.
Zhong FL, Mamaï O, Sborgi L, Boussofara L, Hopkins R, Robinson K, et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell. 2016;167:187–202.e17.
Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med. 2014;211:2385–96.
Nakanishi H, Kawashima Y, Kurima K, Chae JJ, Ross AM, Pinto-Patarroyo G, et al. NLRP3mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc Natl Acad Sci USA. 2017;114:E7766–75.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61.
Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, et al. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol. 1999;163:2403–9.
Matsuki T, Nakae S, Sudo K, Horai R, Iwakura Y. Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int Immunol. 2006;18:399–407.
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT-H, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–15.
Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. P Natl Acad Sci USA. 2011;108:15324–29.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674.
Ferrari CC, Pott Godoy MC, Tarelli R, Chertoff M, Depino AM, Pitossi FJ. Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1beta in the substantia nigra. Neurobiol Dis. 2006;24:183–93.
Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. New Engl J Med. 2007;356:1517–26.
Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502.
Chen L, Hambright WS, Na R, Ran Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J Biol Chem. 2015;290:28097–106.
Yoo S-E, Chen L, Na R, Liu Y, Rios C, Van Remmen H, et al. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic Biol Med. 2012;52:1820–27.
Ding H, Yan C-Z, Shi H, Zhao Y-S, Chang S-Y, Yu P, et al. Hepcidin is involved in iron regulation in the ischemic brain. PLoS ONE. 2011;6:e25324.
Paul BD, Sbodio JI, Xu R, Vandiver MS, Cha JY, Snowman AM, et al. Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature. 2014;509:96–100.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91.
Lehrer RI, Cline MJ. Leukocyte myeloperoxidase deficiency and disseminated candidiasis: the role of myeloperoxidase in resistance to Candida infection. J Clin Invest. 1969;48:1478–88.
Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. 2014;15:1017–25.
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22:146–53.
Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;5:178ra40.
Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23:279–287.
Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21:815–19.
Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25:707–25.
Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet. 2009;41:71–76.
Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti T-D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–91.
Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–U149.
Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH et al. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. MBio 2014; 5. https://doi.org/10.1128/mBio.01117-13.
Rathinam VAK, Jiang ZZ, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–403.
Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–U94.
Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600.
Tomalka J, Ganesan S, Azodi E, Patel K, Majmudar P, Hall BA, et al. A novel role for the NLRC4 inflammasome in mucosal defenses against the fungal pathogen Candida albicans. PLoS Pathog. 2011;7:e1002379.
Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Tengchuan J, et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci USA. 2013;110:E4571–80.
Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–41.
McKeithen DN, Omosun YO, Ryans K, Mu J, Xie Z, Simoneaux T, et al. The emerging role of ASC in dendritic cell metabolism during Chlamydia infection. PLoS ONE. 2017;12:e0188643.
Pedra JH, Sutterwala FS, Sukumaran B, Ogura Y, Qian F, Montgomery RR, et al. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-gamma axis during Anaplasma phagocytophilum infection. J Immunol. 2007;179:4783–91.
Kumar M, Roe K, Orillo B, Muruve DA, Nerurkar VR, Gale M, et al. Inflammasome adaptor protein Apoptosis-associated speck-like protein containing CARD (ASC) is critical for the immune response and survival in west Nile virus encephalitis. J Virol. 2013;87:3655–67.
Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17:151–64.
Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science. 2013;339:975–78.
Hatcher HC, Tesfay L, Torti SV, Torti FM. Cytoprotective Effect of Ferritin H in Renal Ischemia Reperfusion Injury. PLoS ONE. 2015;10:e0138505.
Walker VJ, Agarwal A. Targeting Iron Homeostasis in Acute Kidney Injury. Semin Nephrol. 2016;36:62–70.
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 2010;107:9813–18.
Wang Y, Xiao Y, Zhong L, Ye D, Zhang J, Tu Y, et al. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with β-cell autoimmunity in patients with type 1 diabetes. Diabetes. 2014;63:4239–48.
Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, et al. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med. 1998;4:615–18.
Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193:1539–43.
You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28:1564–73.
Lin J, Li H, Yang M, Ren J, Huang Z, Han F, et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013;3:200–10.
Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57:1773–83.
Pierdomenico M, Negroni A, Stronati L, Vitali R, Prete E, Bertin J, et al. Necroptosis Is Active in Children With Inflammatory Bowel Disease and Contributes to Heighten Intestinal Inflammation. Am J Gastroenterol. 2014;109:279–87.
Liu WJ, Chen BB, Wang Y, Meng CL, Huang HH, Huang XR, et al. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL- dependent mechanism. P Natl Acad Sci USA. 2018;115:E1475–84.
Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994–1006.
Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12:593–605.
Shi FD, Takeda K, Akira S, Sarvetnick N, Ljunggren HG. IL-18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN-gamma by NK cells. J Immunol. 2000;165:3099–104.
Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, Akira S, et al. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc Res. 2003;59:234–40.
Yin Z, Deng T, Peterson LE, Yu R, Lin J, Hamilton DJ, et al. Transcriptome analysis of human adipocytes implicates the NOD-like receptor pathway in obesity-induced adipose inflammation. Mol Cell Endocrinol. 2014;394:80–87.
Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol. 2010;185:974–81.
Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, et al. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci. 2010;30:15811–20.
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65.
Yan YQ, Jiang W, Liu L, Wang XQ, Ding C, Tian ZG, et al. Dopamine Controls Systemic Inflammation through Inhibition of NLRP3 Inflammasome. Cell. 2015;160:62–73.
Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204.
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–5.
Romberg N, Moussawi AlK, Nelson-Williams C, Stiegler AL, Loring E, Choi M, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet. 2014;46:1135–39.
Grandemange S, Sanchez E, Louis-Plence P, Tran Mau-Them F, Bessis D, Coubes C, et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann Rheum Dis. 2017;76:1191–98.
Dumas A, Amiable N, de Rivero Vaccari JP, Chae JJ, Keane RW, Lacroix S, et al. The inflammasome pyrin contributes to pertussis toxin-induced IL-1beta synthesis, neutrophil intravascular crawling and autoimmune encephalomyelitis. PLoS Pathog. 2014;10:e1004150.
French FMFC. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31.
Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med. 2013;19:1132–40.
Bolisetty S, Traylor A, Joseph R, Zarjou A, Agarwal A. Proximal tubule-targeted heme oxygenase-1 in cisplatin-induced acute kidney injury. Am J Physiol Renal Physiol. 2016;310:F385–94.
Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther. 2014;16:R122.
Wang W, Jian Z, Guo J, Ning X. Increased levels of serum myeloperoxidase in patients with active rheumatoid arthritis. Life Sci. 2014;117:19–23.
Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–25.
Willis VC, Gizinski AM, Banda NK, Causey CP, Knuckley B, Cordova KN, et al. N-alpha-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J Immunol. 2011;186:4396–404.
Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis. 2015;74:2199–206.
Seri Y, Shoda H, Suzuki A, Matsumoto I, Sumida T, Fujio K, et al. Peptidylarginine deiminase type 4 deficiency reduced arthritis severity in a glucose-6-phosphate isomerase-induced arthritis model. Sci Rep. 2015;5:13041.
Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118:1952–61.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Kim, E.H., Wong, SW. & Martinez, J. Programmed Necrosis and Disease:We interrupt your regular programming to bring you necroinflammation. Cell Death Differ 26, 25–40 (2019). https://doi.org/10.1038/s41418-018-0179-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0179-3
This article is cited by
-
Major adverse cardiovascular events are associated with necroptosis during severe COVID-19
Critical Care (2023)
-
Update of cellular responses to the efferocytosis of necroptosis and pyroptosis
Cell Division (2023)
-
Necroptosis executioner MLKL plays pivotal roles in agonist-induced platelet prothrombotic responses and lytic cell death in a temporal order
Cell Death & Differentiation (2023)
-
Next-generation RNA sequencing elucidates transcriptomic signatures of pathophysiologic nerve regeneration
Scientific Reports (2023)
-
Caspofungin Suppresses Brain Cell Necroptosis in Ischemic Stroke Rats via Up-Regulation of Pellino3
Cardiovascular Drugs and Therapy (2023)
