
Abstract
TRAF-binding domain (Trabid), one of deubiquitination enzymes, was recently reported to activate Wnt/ β-catenin signaling pathway. However, the role of Trabid in tumors including hepatocellular carcinoma (HCC) and the underlying mechanisms controlling its activity remain poorly understood. Here, we report that Trabid is significantly downregulated in HCC tumor samples and cell lines compared with normal controls and that its expression level is negatively correlated with HCC pathological grading, recurrence, and metastasis. The reintroduction of Trabid expression in tumor cells significantly decreases HCC progression as well as pulmonary metastasis. The effect of Trabid on HCC development occurs at least partially through regulation of Twist1 activity. Mechanistically, Trabid forms a complex with Twist1 and specifically cleaves RNF8-induced K63-linked poly-ubiquitin chains from Twist1, which enhances the association of Twist1 with β-TrCP1 and allows for subsequent K48-linked ubiquitination of Twist1. Knockdown of Trabid increases K63-linked ubiquitination, but abrogates K48-linked ubiquitination and degradation of Twist1, thus enhancing HCC growth and metastasis. Interestingly, Twist1 negatively regulates the promoter activity of Trabid, indicating that a double-negative feedback loop exists. Our findings also identify an essential role for activation of Trabid by AKT-mediated phosphorylation at Ser78/Thr117 in negatively regulating Twist1 signaling, which further provides insights into the mechanisms by which Trabid regulates Twist1 ubiquitination. Our results reveal that Trabid is a previously unrecognized inhibitor of HCC progression and metastasis, which sheds light on new strategies for HCC treatment.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) is the second major cause of cancer-related mortality and fifth most solid malignancy worldwide [1]. Despite the improvements in surgical techniques and diagnosis, long-term survival of patients with HCC after liver transplantation or resection remains low and is less than satisfactory [2]. It has been reported that mainly due to the high recurrence and metastasis rate, the 5-year survival rate after HCC surgical resection is less than 30% [2]. Therefore, to develop novel therapeutic strategies for the treatment of patients with HCC, a better understanding of the molecular mechanisms involved in the initiation and development of HCC is necessary and particularly important. Although many protein-coding genes have been shown to regulate the initiation and progression of HCC over the past decades of years, the molecular mechanisms underlying HCC growth and metastasis remain largely unknown [3].
The deubiquitinase (DUB) Trabid (tumor-necrosis factor receptor-associated factor-binding protein domain), also called “Zranb1”, belongs to the A20 OTU family [4]. Trabid has been reported to “preferentially” cleaves K29-linked, K33-linked, and K63-linked ubiquitin chains [5]. Previous studies indicated that Trabid functions as a positive regulator of Wnt/β-catenin signal pathway, however, this role of Trabid remains controversial [6, 7]. Recently, Trabid has been shown to affect TLR4-mediated cellular immune and inflammation [8]. Given the tight relationship between the Wnt/β-catenin, inflammation, and tumor [10], it is possible that Trabid plays a crucial role in cancer pathobiology. However, so far, very little was known about the involvement of Trabid and underlying molecular mechanisms in tumors including HCC.
Therefore, in this study, we explored the clinical relevance and potential role of Trabid in human HCC. Unexpectedly, our findings present the first evidence that knockdown of Trabid promoted HCC growth and metastasis via regulating Twist1 K63-ubiquitination. Thus, this study provides a proof of principle that targeting Trabid/Twist1 axis might represent new opportunities for effective treatment in this deadly disease.
Results
Trabid is markedly decreased in HCC which predicts poor prognosis
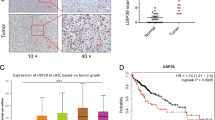
To elucidate the potential role of Trabid in HCC development, we firstly assessed its expression in pairs of human HCC specimens. It was observed that compared with adjacent nontumorous tissues, the levels of Trabid protein and mRNA were drastically reduced in tumor tissues (Fig. 1a, b). Then we examined the levels of Trabid expression in HCC samples with different disease severities based on the AJCC staging system [9]. The results not only verified an almost five-fold downregulation of Trabid in HCC tumor samples compared with healthy control or non-cancerous adjacent tissues (Fig. 1c1), but also indicated that Trabid downregulation is closely correlated with significantly increased recurrence, metastasis, tumor volumes, and disease stage (Fig. 1c2–c5). Additionally, compared with the normal hepatocytes or liver cell line L02, Trabid mRNA and protein expression levels in all of HCC cell lines were substantially decreased (Fig. 1c6).

Trabid is markedly decreased in HCC which predicts poor prognosis. a The levels of Trabid protein in 10 pairs of HCC and matched adjacent nontumorous tissues were measured using Western blot. b The quantitative polymerase chain reaction (qPCR) was used to measure the levels of Trabid mRNA in the above samples. RT-PCR analysis of Trabid expression in healthy control tissues, matched adjacent nontumorous tissues, and HCC tissues (c1). RT-PCR was used to measure Trabid expression in liver tissues from patients with different recurrence statuses (c2), or metastasis statuses (c3), or tumor volumes (c4), or disease stages (c5). c6 qPCR assays and WB of Trabid expression in normal hepatocytes, normal liver cell lines, and HCC cell lines. d Representative images of immunohistochemical (IHC) staining in tissue microarray-based IHC study of Trabid, and the percentage of patients with different expression of Trabid based on TNM stage. e, f Compared with patients with high Trabid levels in two cohorts, patients with low Trabid levels have a higher probability of recurrence and poorer overall survival
To further determine the prognostic value of Trabid in HCCs, an IHC staining on Trabid from a commercial tissue array of 322 HCC patients was performed. To ensure that the system works, we validated Trabid antibody using IHC staining in wild type or Trabid knockout MEF cells (Supplementary Fig. 1A). The IHC assay further indicated that HCC tissues had lower expression of Trabid compared to matched normal tissues and that Trabid expression was negatively correlated with HCC TNM stages (Fig. 1d). Additionally, an analysis of Trabid in 291 HCC tissues from two independent cohorts with comparable clinicopathological features and complete follow-up data (Table S1) was also performed. Notably, low expression of Trabid was found to be significantly associated with recurrence probability (RP) and poor overall survival (OS) in 91 HCC patients of cohort 1 (Fig. 1e). These findings were further confirmed in another larger independent cohort of HCC patients (cohort 2, n = 200) (Fig. 1f). Cox proportional hazards regression analyses identified the expression level of Trabid as an independent prognostic predictor for RP and OS (Table S2), suggesting that Trabid downregulation is significantly associated with bad prognosis of HCCs.
Trabid significantly inhibits HCC growth and metastasis
Next, we evaluated the functions of Trabid in HCC. To this end, we firstly tested four Trabid-specific shRNAs to knock down the endogenous Trabid expression in Hep3B and SMMC-7721 cells. Given that shTrabid#1 induced the most significant knockdown effect, it was adopted for further study (Fig. 2a). We also constructed a shRNA-resistant Trabid plasmid, TrabidΔ, to eliminate the possibility of off-target effects (Fig. 2b1). We found that exogenous upregulation of Trabid in HCCLM3 and MHCC97H induced significant decreases in the proliferation and foci formation of HCC cells (Fig. 2b1, b2, c1). Knockdown of Trabid enhanced HCC cell proliferation and foci formation while the reintroduction of Trabid expression with shRNA-resistant TrabidΔ vector brought the increased proliferation back to almost normal levels (Fig. 2b3, c2).

Trabid significantly inhibits HCC growth and metastasis. a Western blot results demonstrate transfection of lentiviral vectors containing individual shRNA against Trabid (shRNA 1–4) (LV-sh-Trabid) induced significant downregulation of Trabid in HCC cells. ShTrabid-1# produced the best knock down effect on Trabid. b1 Western blot for Trabid in the indicated HCC cells transfected with Trabid or shTrabid or the shRNA-resistant expression construct, TrabidΔ, or their matched controls. The effects of Trabid overexpression (b2) or knockdown (b3) on HCC cell proliferation and foci formation. The effects of Trabid overexpression (c1) or knockdown (c2) on HCC cell foci formation. Representative images of the whole plate are shown in the right. d Indicated HCC cells were injected subcutaneously into six NOD/SCID mice. The growth of xenograft tumors was monitored at different time points. e The volume of MHCC97H orthotopic hepatic tumors was determined at various time points, and representative bioluminescence images or orthotopic tumors are shown
To verify these in vitro findings, we investigated the effects of Trabid expression on the growth of xenograft and orthotopic hepatic tumors in NOD/SCID mice. As expected, compared with control cells, HCC cells overexpressing Trabid developed smaller tumors, while silencing Trabid expression in SMCC-7721 cells accelerated HCC growth in vivo (Fig. 2d, e).
Collectively, these functional studies demonstrate that Trabid inhibits HCC growth.
Trabid knockdown enhances HCC cell invasion and lung metastasis
We further examined the effects of Trabid on HCC cell migration and invasion. Wound healing and transwell assays indicated that the invasive capability of the Trabid-transfected HCCLM3 and MHCC97H cells was smaller than that of the control cells (Fig. 3a, b, and Supplementary Fig. S1C and S1D). Conversely, Trabid knockdown in SMMC-7721 and Hep3B cells caused a robust increase in the invasion ability, which was reversed by reintroducing TrabidΔ (Fig. 3a, b and Supplementary Fig. S1C and S1D).

Trabid inhibits HCC invasion and lung metastasis by inducing EMT. a The effects of Trabid expression on wound closure in indicated HCC cells. b The effects of Trabid expression on the invasion of HCC cells. Western blotting (c), immunofluorescence (d), and e real-time PCR were used to measure the levels of vimentin, fibronectin, E-cadherin, and ZO-1 expression in the indicated HCC cells. f, g An in vivo lung metastasis analysis. The indicated HCC cell lines were injected into the mice through the tail vein. The metastatic lung foci were counted and detected using bioluminescence imaging. g Representative images of mice lungs and H&E staining (n = 10/group) are shown. h Silencing Trabid prolonged overall survival of xenograft mice models bearing HCCs
Since epithelial-mesenchymal transition (EMT) significantly increased HCC invasion [10], we wondered whether Trabid suppressed the invasiveness of HCC via blocking EMT. Immunofluorescence, immunoblotting, and real-time polymerase chain reaction (PCR) data indicated that Trabid overexpression decreased the expression of the mesenchymal markers (vimentin and fibronectin), but enhanced the levels of epithelial markers ZO-1 and E-cadherin in HCC cells, whereas Trabid knockdown in SMCC-7721 cells showed the opposite phenotype (Fig. 3c–e). All of these results clearly pointed out that Trabid knowdown induced EMT in HCC cells.
To confirm the above in vitro findings, we studied the effects of Trabid expression on lung metastasis of HCC. Compared to those in the control group, MHCC97H cells with Trabid overexpression had less and smaller lung metastases nodules, while the lung metastases in NOD/SCID mice injected with SMCC-7721-shTrabid cells significantly increased, which was further confirmed by histologic analyses (Fig. 3f, g). Similar results were obtained in HCCLM3 and Hep3B cells (Supplementary Fig. S1E). As expected, Trabid overexpression prolonged overall survival of HCC xenograft mice models (Fig. 3h). Our results suggest that Trabid may be critical for the inhibition of migration and invasion of HCC cells.
Downregulation of Twist1 protein, not mRNA level is required for Trabid-induced suppression of HCC proliferation and EMT
Snai1, Twist1, Zeb1, Slug, and SIP1 are well-known transcription factors (TFs) which decide EMT [10]. Next, we evaluated whether Trabid regulated these TFs. Although the mRNA levels of these TFs failed to be significantly changed in shTrabid-transfected HCC cells compared with control cells, the level of Twist1 protein, not other EMT-related genes, was significantly increased in shTrabid-transfected SMCC7721 cells, while downregulated in MHCC97H-Trabid cells when compared to their counterparts (Fig. 4a, b). Moreover, sh-Twist1 significantly decreased the loss of E-cadherin expression induced by shTrabid in SMCC-7721-HCC cells, whereas overexpression of Twist1 markedly inhibited the increase in E-cadherin expression in MHCC97H-Trabid cells (Fig. 4b). Similar results were also observed in Hep3B and HCCLM3 cells (Supplementary Fig. S1F). Immunofluorescence assays further confirmed the above findings (Fig. 4c). Thus, Twist1 may be required for shTrabid-induced EMT in HCC cells.

Downregulation of Twist1 is required for Trabid-induced inhibition of proliferation and EMT of HCC cells. a The relative mRNA and protein levels of EMT-related genes in indicated HCC cells were determined using qPCR. b Indicated cells were transfected with indicated plasmids for 24 h, and then cell lysates were probed with the indicated antibodies. c E-cadherin and vimentin in HCC were analyzed using IF staining. d The invasion assay in indicated HCC cells. e The luciferase-expressing HCC cell lines were injected by way of the tail vein into the NOD/SCID mice. Tumor metastasis was monitored by bioluminescence imaging. The incidence of lung metastases observed in the different experimental groups of NOD/SCID mice. Representative images showing lung and H&E staining of lung tissues from the different experimental groups. f The BrdU assay of HCC cells treated as indicated was determined at indicated times
Indeed, we observed that overexpression of Twist1 significantly increased the invasive capabilities of MHCC97H-Trabid cells, while shTrabid-induced invasive capabilities were restored by overexpression of shTwist1 (Fig. 4d). In vivo experiments confirmed that the number of lung metastasis was decreased in SMMC7721-shTrabid group after expression of shTwist1, while Twist1 overexpression significantly increased the metastatic capabilities of MHCC97H-Trabid cells (Fig. 4e). Moreover, overexpression of Twist1 was observed to abrogate the Trabid-mediated inhibition of proliferation in MHCC97H-Trabid cells, but specific knockdown of Twist1 apparently blocked the increased cell proliferation caused by Trabid depletion (Fig. 4f). Collectively, our data clearly suggest that Trabid-induced suppression of HCC growth and metastasis is mainly mediated through inhibition of Twist1 expression.
Trabid inactivates Twist1 by cleaving RNF8-mediated Twist1 K63-linked ubiquitination
To elucidate the mechanisms by which Trabid suppresses Twist1, we firstly examined the interaction between Trabid and Twist1. We detected that Flag-Trabid is associated with HA-Twist1 in HEK293T cells (Fig. 5a1) and that endogenous Twist1 and Trabid in HCC cells could be efficiently co-precipitated by each other (Fig. 5a2). Pull-down assays demonstrated that Twist1 was co-precipitated with GST-Trabid but not with GST (Fig. 5a3). We further mapped their interaction regions. It was observed that GST-full-length (FL) Trabid and Trabid (1–354) strongly interacted with Twist1, whereas GST-Trabid (355–708) showed no interaction (Fig. 5b1), indicating that the N-terminal of Trabid should contain a primary interaction domain for Twist1. We also demonstrated that Trabid mainly interacted with the FL and the 169–202 amino acid fragment (Twist1 box domain) of GST-Twist1, but not with other Twist1 fragments and GST alone (Fig. 5b2). Thus, we concluded that Trabid directly interacts with Twist1.

Trabid inhibits Twist1 activity by cleaving RNF8-mediated Twist1 K63-linked ubiquitination. a1 HEK293T cells were co-transfected with indicated plasmids. Co-IP was performed with indicated antibody. a2 co-IP assays for endogenous Twist1 and Trabid from HCC cells. a3 GST pulldown assays. b1 An illustration to depict Trabid full-length (FL) and its deletion constructs. The interaction between Twist1 and Trabid domains is indicated by the number of plus signs (+), and detected via immunoblotting. b2 Schematic diagram of the functional domains of human Twist1 protein. And the Twist1 domain required for the interaction of Trabid was shown. c1 HCC cells were transfected with shRNA targeting Trabid for 72 h, and the lysates were analyzed to determine the K63 ubiquitination on Twist1. HEK293T cells were transfected with HA-tagged K63-only ubiquitin (c2) or K48-only ubiquitin (c3) and other indicated plasmids. Then all samples were immunoprecipitated with anti-Flag antibody, followed by probing. c4 In vitro K63-linked deubiquitination of Twist1 by Trabid. Purified recombinant Trabid or Trabid–C443A was incubated with K63 ubiquitinated Twist1 in vitro, and then probed with indicated antibody. d1 Trabid removes Twist1 K63 ubiquitination mediated by RNF8. HEK293 cells were cotransfected with indicated plasmids. Following Myc-Twist1 IP, K63 ubiquitination was detected. d2 HEK293T cells were transfected with shcontrol or Trabid shRNA and treated with 20 μM MG132. Then lysates were immunoprecipitated using an antibody against RNF8 (upper panel) or Twist1 (lower panel), and then immunoblotting was performed. d3 HEK293T cells were treated as indicated, and then cell lysates were immunoprecipitated and analyzed. e β-TrCP1 knockdown abolished Trabid-induced polyubiquitination of Twist1. HEK293T cells were treated as indicated, and then lysate was used in immunoprecipitation
Since Trabid can deubiquitinate K63 ubiquitin linkage [11], we considered the possibility that Trabid might regulate HCC progression by deubiquitinating K63 ubiquitinated Twist1. Indeed, shRNA of Trabid led to increases in the levels of K63 ubiquitination on endogenous Twist1 (Fig. 5c1). Conversely, overexpression of Trabid significantly reduced the K63 but increased K48, ubiquitination levels of Twist1 (Fig. 5c2, c3). In vitro incubation of purified wild-type Trabid, but not inactive Trabid-C443A [8], with K63 ubiquitinated Twist1 led to its deubiquitination (Fig. 5c4), indicating that Trabid can directly deubiquitinate Twist1 K63 ubiquitination.
Because RNF8 and β-TrCP1 induced K63 and K48 ubiquitination of Twist1 respectively [12, 13], we then sought to determine whether Trabid decides the binding ratio of Twist1 to RNF8 or β-TrCP1. RNF8 overexpression indeed induced Twist1 K63 ubiquitination, while coexpression of Trabid completely prevented the appearance of Twist1 K63 ubiquitination (Fig. 5d1), suggesting that Trabid can cleave K63 ubiquitin chains attached to Twist1. Interestingly, Trabid knockdown in HCC cells increased the interaction between endogenous RNF8 and Twist1, while decreased the association of Twist1 with β-TrCP1 (Fig. 5d2). Furthermore, Trabid-C443A, not WT-Trabid, significantly increased the interaction of endogenous RNF8 with Twist1, while decreased the binding of Twist1 to β-TrCP1 (Fig. 5d3), suggesting that Trabid is critical to for the binding of Twist1 to RNF8 or β-TrCP1.
We further analyzed the importance of Trabid and β-TrCP1 on Twist1 degradation. As expected, Trabid overexpression enhanced polyubiquitination of Twist1. However, the Trabid-mediated ubiquitination of Twist1 was completely abolished by β-TrCP1 downregulation (Fig. 5e), suggesting that both Trabid and β-TrCP1 are essential for Twist1 degradation. Taken together, Trabid inactivates Twist1 by cleaving RNF8-mediated Twist1 K63-linked ubiquitination, facilitating the complex formation of β-TrCP1 with Twist1, whereby enhancing K48-linked ubiquitination of Twist1 by β-TrCP1.
Twist1 directly targets Trabid promoter and inhibits its transcription in HCC
Given that Twist1 was a transcriptional repressor [14], and that sequence analysis of Trabid promoter from position −1845 to +49 revealed four putative Twist1 binding sites (E-box, CANNTG) (Fig. 6a), we hypothesized that Twist1 would inhibit transcription of the Trabid promoter-luciferase reporter. Serial deletion (Fig. 6b1) and site-directed mutagenesis (Fig. 6b2) in the Trabid promoter region demonstrated that four Twist1-binding sites were critical for Trabid expression. Chromatin immunoprecipitation (ChIP) further confirmed that Twist1 directly binds to the Trabid promoter in HCC cells (Fig. 6b3, c) and clinical HCC tissues (Fig. 6b4, c). Consistently, increased Twist1 levels decreased Trabid expression in HCC cells (Fig. 6d). These results strongly define Trabid as a direct target of the Twist1 transcription factor in HCC cells, thus exerting a negative feedback loop.

Twist1 negatively regulated Trabid expression in HCC. a The nucleotide sequences and positions of four putative Twist1 binding site in the Trabid proximal promoter are shown. Luciferase activity assays of the pGL3-Trabid promoter-reporter. b1 Serially truncated and mutation analysis b2 identified twist1-bindinf sites in the Trabid promoter. ChIP assay demonstrated the direct binding of Twist1 to the Trabid promoter in HCC cells (b3) and human HCC tissues (b4). Hepatocytes were separated from liver tissues of healthy controls (HC) and HCC patients. c ChIP assays in clinical HCC tumor samples (case #1 and case #2) and HCC cell lines. d Indicated HCC cells were transduced with or without Ad-Twist1. The levels of Twist1 and Trabid were determined using Western blot
Akt-induced phosphorylation regulates Trabid DUB activity toward Twist1
Since both AKT and Trabid are required for β-TrCP1-mediated Twist1 ubiquitination and degradation [15], we next investigated the possible relationship between Trabid and AKT. Among Trabid orthologs, the amino acid sequences around Ser78 and Thr117 are highly evolutionarily conserved (Fig. 7a), and both of them are predicted to be AKT phosphorylation sites by Scansite. We, therefore, reasoned that Trabid might be a substrate of AKT. We first confirmed the interaction between Trabid and AKT (Supplementary Fig. S1G and S1H). The quantitative phosphoproteomic assay indicated that the phosphorylation levels of Ser78 and Thr117 on Trabid were significantly enhanced in HEK293T cells expressing activated AKT (myristoylation signal [MGSSKSKPK]-attached AKT [Myr-AKT]) (Fig. 7b1). More phosphorylated Trabid was also observed in PTEN knockout mouse embryonic fibroblasts (MEFs) than that of WT MEFs, which was blocked by AKT inhibitors (Fig. 7b2). In vitro kinase assay clearly demonstrated that co-incubation of recombinant Trabid and activated AKT led to Trabid phosphorylation (Fig. 7b3). As compared to WT-Trabid, either S78A or T117A mutants showed partially decreased phosphorylation, while that of double mutant was completely abolished (Fig. 7b4), suggesting both of S78 and T117 as major phosphorylation sites of AKT. An AKT phosphorylation motif (R × × S/T) antibody further confirmed the phosphorylation of Trabid by AKT, while lambda phosphatase eliminated the reactivity of Trabid with this antibody (Fig. 7b5). These results suggest that Trabid is an AKT substrate.

Trabid is an AKT substrate and this phosphorylation activates the DUB activity of Trabid. a Putative Akt phosphorylation sites predicted by Scansite within Trabid orthologs. b1 Electrospray ionization mass spectrometry (ESI-MS) analysis reveals the levels of S78 and Thr117 phosphorylation in Trabid from HEK293T cells. b2 Endogenous Trabid in Pten knockout mouse embryonic fibroblasts (MEFs) cells treated with or without AKT inhibitor were immunoprecipitated and probed with phospho-Ser/Thr antibody. b3 The phosphorylation of Trabid was found using a phospho-Ser/Thr antibody. b4 HEK293T cells were transfected with wild-type or mutant Trabid constructs, then phosphorylation of Trabid was detected using the phospho-Ser/Thr antibody. b5 The level of phosphorylated Trabid from HEK293T cells treated with or without λ-phosphatase. c1 Recombinant Trabid protein (1 μg) was mixed with or without active AKT (1 μg) in vitro, and Ub-AMC assay was performed. Relative fluorescence units (RFU). c2, c3 AKT activates Trabid in cells. Trabid was immunoprecipitated from HEK293T cells coexpressed with activated AKT (c2) or treated with 10 μM MK2206 (c3), and followed by Ub-AMC hydrolysis assay. c4 S78A/T117A mutation blocks Trabid activation by AKT. d Trabid−/− MEF cells were virally transfected with indicated plasmids, and the K63-Ubiquitination of Twist1 was analyzed. e Trabid−/− MEF cells were virally transfected with indicated plasmids with or without Myr-Akt, and the K63-Ubiquitination of Twist1 was analyzed. f Trabid−/− HCC cells were virally transfected with indicated plasmids with or without 1 µM MK2206, and the K63-Ubiquitination of Twist1 was analyzed
To examine whether AKT-induced phosphorylation might affect the DUB activity of Trabid, we performed an Ub-AMC (ubiquitin-7-amido-4-methylcoumarin, a fluorogenic substrate) hydrolysis assay [16]. As shown in Fig. 7c1, either purified Trabid or AKT alone showed trace hydrolyzing activity towards Ub-AMC, while Trabid co-incubated with AKT indicated high activity (Fig. 7c1). In vivo, Trabid from HEK293T cells co-expressing Myr-AKT showed higher activity than that expressed alone (Fig. 7c2), whereas Trabid activity in the Ub-AMC assay was reduced by MK2206, an AKT inhibitor (Fig. 7c3). Notably, the stimulating effect of AKT on the activity of Trabid was substantially inhibited by S78A/T117A mutation (Fig. 7c4), suggesting that the phosphorylation of Trabid at S78 and T117 is an important regulatory mechanism for its activity.
Then we studied whether phosphorylation of Trabid by AKT was required for its DUB activity toward K63 ubiquitination of Twist1. As shown in Fig. 7d, either WT-Trabid or the Trabid-S78D/T117D mutant, but not the Trabid-S78A/T117A mutant, was able to inhibit the K63 ubiquitination of Twist1. However, even in the presence of activated AKT, Trabid-S78A/T117A mutant failed to inhibit ubiquitination of Twist1 (Fig. 7e). Upon inhibition of AKT, WT-Trabid lost its deubiquitination activity toward Twist1, while the Trabid-S78D/T117D mutant was still able to reduce ubiquitination levels of Twist1 (Fig. 7f), indicating that the phosphorylation of Trabid induced by AKT promotes its K63 deubiquitinating activity toward Twist1.
Inverse correlation between Twist1 and Trabid expression in HCC
To validate the above findings, we examined Trabid, RNF8, and Twist1 expression in human HCC specimens. Firstly, we confirmed the antibodies using IHC staining in HCCLM3 cells with RNF8 or Twist1 knockdown (Supplementary Fig. 1B). As shown in Fig. 8a, b, Trabid expression negatively correlated with both RNF8 and Twist1 level. The Kaplan–Meier analysis indicated that both Trabid low level and Twist1 or RNF8 high expression in HCC correlated with bad OS. It suggests that Trabid-Twist1 inverse regulation in HCC was associated with reduced survival, and that Trabid downregulation in combination with RNF8 or Twist1 overexpression is a stronger predictor of poor survival than Trabid alone (Fig. 8c).

Clinical correlation analysis between Trabid, RNF8, and Twist1 in HCC patients. a Immunohistochemistry pictures of Trabid, RNF8, and Twist1 expressions in 20 representative HCC patients. Scale bar, 200 μm. b The percentage of patients with different expression of Trabid, RNF8, and Twist1 according to different clinical parameters. c The Kaplan–Meier analysis of OS of HCC patients as indicated. d Schematic model of the mechanism underlying Trabid mediated HCC growth and metastasis inhibition
Discussion
For the first time, the present study identifies the critical role of Trabid in HCC and the underlying mechanisms. We demonstrate the following: (1) Trabid expression is significantly downregulated in HCC cell lines and two independent cohorts of HCC patients’ tissues; (2) Trabid expression is negatively correlated with pathological grading, recurrence, and poor survival; (3) Reintroduction of Trabid in HCC cells significantly decreases HCC growth and pulmonary metastasis; (4) The inhibitory effect of Trabid on HCC development occurs at least partially through regulation of Twist1 activity; (5) Trabid forms a complex with Twist1 and specifically cleaves RNF8-induced K63-linked poly-ubiquitin chains from Twist1, which enhances the association of Twist1 with β-TrCP1 and allows for subsequent K48-linked ubiquitination of Twist1; (6) AKT-mediated phosphorylation at Ser78/Thr117 is essential for the role of Trabid in negatively regulating Twist1 signaling; and (7) Twist1 negatively regulates the promoter activity of Trabid, indicating a double-negative feedback loop. These findings shed new light on the pathogenesis of HCC and encourage further study of its potential therapeutic implications.
In this manuscript, we used different cohorts of HCC patients to evaluate the relationship between Trabid expression level, HCC patient clinicopathologic parameters, and outcome. Our data proved that Trabid is a significant prognosis biomarker. Further, both univariate and multivariate cox harzard regression analysis indicated that Trabid expression is an independent progonostic predictor. However, unlike the other digestive cancers which were referred to a routine post-surgical chemotherapy, most of the patients included in this study didn’t receive a chemotherpay post-surgically. Only two patients in stage IV received sorafenib after palliative hepatectomy. Particularly, based on our follow-up, we found that HCC patients in China prefer to use various drugs, such as Chinese herb, as an adjuvant therapy for HCC after surgery. Namely, these patients didn’t receive a unified treatment after surgery. Thus we were not able to take the post-surgical treatment as an independent factor for further multivariate cox analysis. Therefore, more standardized management of post-surgical HCC patients is necessary for a more accurate prognosis predictor analysis.
One interesting novel finding in this paper is that we identified Trabid as a DUB responsible for cleaving RNF8-mediated K63-linked poly-ubiquitin chains of Twist1 and promoting the K63-K48 ubiquitination transition. Twist1 is a well-known EMT-related TF, and persistent activation of Twist1 in HCC correlates closely with a poorer survival [10]. However, the regulatory mechanisms of Twist1 activation remain less understood. Previous studies reported that β-TrCP1 and FBXL14 could inactivate Twist1 via K48-linked ubiquitination [13]. Most recently, RNF8-mediated K63 ubiquitination in breast cancer has been found to be critical for Twist1 activation and subsequent EMT [12]. However, it is entirely unknown about how these different regulators of Twist1 are reconciled in HCC? Here, we provide substantial evidence to demonstrate that Trabid acts as a new and key switcher of Twist1 activity. When the Trabid expression is up, Twist1 binds to β-TrCP1, leading to Twist1 K48-linked ubiquitination. Conversely, Trabid knockdown promotes the association of Twist1 with RNF8 and subsequent Twist1 K63 ubiquitination, which is reminiscent of A20, another deubiquitinase [17]. A20 and Trabid belong to the same DUB family, but their structures and functions are different [11]. Interestingly, A20 was also shown to inhibit Twist1 activity [18]. However, unlike Trabid, A20 controls Twist1 expression through affecting the transcriptional activity of NF-κB on which Trabid was shown to have a very weak effect [18]. Additionally, unlike A20, Trabid also can cleave K29 and K33 ubiquitination chains [5]. Whether K29-linked or 33-linked polyubiquitin chain affects Twist1 activation definitely needs further investigation.
Another novel component of the current study is the identification of the crosstalk between Trabid and AKT during HCC development. The highly conserved AKT kinase family are well-known to enhance cancer development and considered excellent anti-cancer target [19]. However, our finding that AKT-mediated phosphorylation at Ser78/Thr117 is essential for Trabid in negatively regulating Twist1 signaling seems counterintuitive. Notably, contrary to our views, the previous study reported that AKT-mediated Twist1 phosphorylation promoted metastatic tumor development by regulating TGF-β2 and activating PI3K/AKT pathway [20]. Therefore, our data unexpectedly describe a novel signaling event linking AKT inactivation in HCC cells to the regulation of Twist1 hyperactivation. How to coordinate these opposite views? In fact, it has already been reported that as a key sensor of multiple intracellular signaling pathways, AKT regulates both anabolic and catabolic mechanisms [21] and that the oncogenic activities of the AKT isoforms are far more complex [22]. For example, AKT1 knockdown in MCF 10A cells promotes stem cell-like phenotype and EMT by decreasing miR-200 abundance [22]. Similarly, cellular and genetic studies also suggest that AKT1, not AKT2 or AKT3, is a negative regulator of EMT in breast cancer, although the underlying mechanism by which AKT1 inhibits metastasis remains largely unknown [22]. Here, our observation that phosphorylation of Trabid by AKT is required for β-TrCP1-mediated Twist1 ubiquitination and degradation, maybe provide an explanation about how AKT inhibits EMT. Therefore, it seems that AKT can control Twist1 activity via direct and indirect manners. Together, these findings uncover new insights into the role of crosstalk between Trabid and AKT in HCC progression.
Our data firmly demonstrated that Trabid is an important inhibitor of HCC growth and metastasis, which is in contrast to previously published data that Trabid was shown to activate Wnt/β-catenin pathway and hence promoted CRC cell proliferation [6]. Thus, it would be necessary to reconcile the conflicting data. A natural explanation for such an observation is that Trabid can function as either a tumor suppressor or an oncogene in a context-dependent manner. Another potential reason is that the oncogenic role of Trabid in CRC cells might not be real or at least remains controversial. Several lines of evidence support this notion. Firstly, although Trabid indeed inactivated the APC tumor suppressor protein [6], the previous study did not provide in vivo data to confirm this alleged oncogenic role of Trabid in CRC. Secondly, the authors did not use cancer cell lines other than CRC to confirm their preliminary data. Thirdly, despite using the same materials, the other group did not observe Trabid having any effect on Wnt/β-catenin pathway [7]. Fourthly, the data from fly also suggested that Trabid might not be a core component of Wnt/β-catenin signaling [6]. Conversely, our comprehensive studies not only explicitly provide the molecular basis for the inhibitory action of Trabid in HCC development, but also support the notion that Trabid could be an attractive candidate marker to stratify HCC patients into distinct risk subgroup and guide individualized therapy. However, whether the potential role of Trabid as a tumor suppressor is tissue type-specific definitely needs further study.
In summary, this study uncovers a previously unrecognized tumor-inhibiting function of Trabid and elucidate a new molecular mechanism of Trabid in HCC growth and metastasis. Such knowledge may provide new therapeutic targets for the treatment of patients with HCC.
Materials and methods
In vitro deubiquitination assay
The in vitro deubiquitnation assay was adjusted from a previously described study [23]. Ubiquitinated Twist1 and recombinant myc-Trabid were purified from the cell extracts using an anti-FLAG affinity column. The proteins were eluted with FLAG peptides (Sigma) after washing with lysis buffer. Ubiquitinated Twist1 protein was mixed with recombinant myc-Trabid in a 14.5 μL reaction mixture containing (10 mM DTT, 50 mM Tris-HCl [pH 8.0], 50 mM NaCl, 1 mM EDTA, and 5% glycerol) at 37 °C for 1 h and it was immunoprecipitated with anti-FLAG antibody. Then the beads were washed three times with washing buffer (50 mM Tris-Cl [pH 8.0], 150 mM NaCl, 0.4% NP-40) and resuspended in 2 × SDS sample buffer for immunoblot analysis. With the exclusion of the enzyme, a control reaction mixture was set up under identical conditions. The reaction mixture was stopped by addition of 5 × SDS-PAGE sample loading buffer, and then proteins were separated by SDS-PAGE.
Co-immunoprecipitation
All tissues or cells were lysed in RIPA buffer containing protease inhibitors at 4 °C followed by vortex and centrifugation at 14,000 rpm at 4 °C for 10 min. Total proteins (500 μg/sample) were pre-cleaned with 40 μl A–G beads (Santa Cruz) before immunoprecipitation with 3 μg control IgG (Santa Cruz Biotechnology), indicated primary antibodies overnight. After incubation with 40 μl A–G beads at 4 °C for 6 h, the immunoprecipitates were washed with PBS containing 0.2% NP-40 for five times. The beads were taken up in 40 μl of SDS Laemmli buffer and heated for 5 min at 95 °C. The immunoprecipitated protein complexes were then analyzed by Western blot.
Tissue microarray and immunohistochemistry
The HCC tissue sections and tissue microarray (TMA) were deparaffinized in xylene and rehydrated with ethanol and then preincubated with 10% normal goat serum in PBS (pH 7.5) followed with incubation with the following primary antibodies: anti-RNF8, 1:200; anti-Twist1, 1:300; anti-Trabid, 1:100 overnight at 4 °C. For cellular immunohistochemistry, The trypsin-digested cells were fixed in 4% paraformaldehyde at room temprature for 10 min. After gentle centrifugation, the cell blocks were subjected to paraffin embedding and section preparation. Cellular antigen retrieval procedure was the same as tissues. Afterwards, the sections were incubated with the above primary antibodies under the same condition. Anti-rabbit or anti-mouse biotinylated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were applied or 1 h at room temperature, followed by the Vectastain Elite ABC reagent (Vector lab) for 30 min. The peroxidase reaction was developed with diaminobenzidine (DAB kit; Vector lab) and the slides were counterstained with hematoxylin (Sigma).The percentage of staining positive cells and intensity were used to get scores for the slides. And protein staining was subsequently quantified using a scoring system based on the Allred score for immunohistochemistry as previously described [24]. The proportion score was defined as followed: 0 for 0% IEC (% positive IEC); 1 for 1–10% IEC; 2 for 10–30% IEC; 3 for 30–50% IEC; 4 for 50–80% IEC; and 5 for 80–100% IEC. The intensity was defined as followed: negative (0 for very low or no staining), weak (1+ for intensity just above background level), intermediate (2+ for clearly visible at low magnification), and strong (3+ for striking at low magnification). Finally, it leads to an Allred score between 0 and 8. And at least two tissues cores were available for each patient on the TMA. According to the indicated protein expression, all patients were stratified into two groups based on Allred score >6 (high) and <6 (low). This cut-off was arbitrarily used to produce two comparable groups.
Statistical analysis
All statistical analyses were performed using the SPSS 16.0 statistical software package (Abbott Laboratories, USA) and Graphpad Prism 5.0 software package for Windows. Statisticians in the Department of Health Statistics, Jilin University, confirmed all results. Quantitative values of all experiments are expressed as the mean ± SD (standard deviations). The statistical correlation between the clinical parameters of HCC patients and different Trabid expression group in cohort 1 and cohort 2 was analyzed by Chi-square test or Fisher’s Exact Chi-square test. The Adobe Photoshop CS6 was used to compose images. Kaplan–Meier method and log-rank test assays were used to plot survival curves. Student’s t-test or one-way ANOVA was applied to determine the significance between groups. Statistical analyses between different treatments, in different cell cohorts or at different time points were performed using two-way ANOVA with the Bonferroni’s correction. The correlation between the expression levels of two proteins was determined using Spearman’s correlation analysis. P < 0.05 was accepted to be statistically significant in all samples. **P < 0.01 and ***P < 0.001.
A detailed description of the materials and methods used in this study can be found in the Supplementary Supporting Materials.
References
Hong X, Song R, Song H, Zheng T, Wang J, Liang Y, et al. PTEN antagonises Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to hepatocarcinogenesis. Gut. 2014;63:1635–47.
Portolani N, Coniglio A, Ghidoni S, Giovanelli M, Benetti A, Tiberio GA, et al. Early and late recurrence after liver resection for hepatocellular carcinoma: prognostic and therapeutic implications. Ann Surg. 2006;243:229–35.
Song R, Song H, Liang Y, Yin D, Zhang H, Zheng T, et al. Reciprocal activation between ATPase inhibitory factor 1 and NF-κB drives hepatocellular carcinoma angiogenesis and metastasis. Hepatology. 2014;60:1659–73.
Fernando MD, Kounatidis I, Ligoxygakis P. Loss of trabid, a new negative regulator of the drosophila immune-deficiency pathway at the level of TAK1, reduces life span. PLoS Genet. 2014;10:e1004117.
Virdee S, Ye Y, Nguyen DP, Komander D, Chin JW. Engineered diubiquitin synthesis reveals Lys29-isopeptide specificity of an OTU deubiquitinase. Nat Chem Biol. 2010;6:750–7.
Tran H, Hamada F, Schwarz-Romond T, Bienz M. Trabid, a new positive regulator of Wnt-induced transcription with preference for binding and cleaving K63-linked ubiquitin chains. Genes Dev. 2008;22:528–42.
Shi T, Bao J, Wang NX, Zheng J, Wu D. Identification of small molecule TRABID deubiquitinase inhibitors by computation-based virtual screen. BMC Chem Biol. 2012;12:4.
Jin J, Xie X, Xiao Y, Hu H, Zou Q, Cheng X, et al. Epigenetic regulation of the expression of Il12 and Il23 and autoimmune inflammation by the deubiquitinase trabid. Nat Immunol. 2016;17:259–68.
Qu C, He D, Lu X, Dong L, Zhu Y, Zhao Q, et al. Salt-inducible kinase (SIK1) regulates HCC progression and WNT/β-catenin activation. J Hepatol. 2016;64:1076–89.
Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471–4.
Licchesi JD, Mieszczanek J, Mevissen TE, Rutherford TJ, Akutsu M, Virdee S, et al. An ankyrin-repeat ubiquitin-binding domain determines TRABID’s specificity for atypical ubiquitin chains. Nat Struct Mol Biol. 2011;19:62–71.
Lee HJ, Li CF, Ruan D, Powers S, Thompson PA, Frohman MA, et al. The DNA damage transducer RNF8 facilitates cancer chemoresistance and progression through twist activation. Mol Cell. 2016;63:1021–33.
Zhong J, Ogura K, Wang Z, Inuzuka H. Degradation of the transcription factor twist, an oncoprotein that promotes cancer metastasis. Discov Med. 2013;15:7–15.
Vesuna F, van Diest P, Chen JH, Raman V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem Biophys Res Commun. 2008;367:235–41.
Li CW, Xia W, Lim SO, Hsu JL, Huo L, Wu Y, et al. AKT1 inhibits epithelial-to-mesenchymal transition in breast cancer through phosphorylation-dependent twist1 degradation. Cancer Res. 2016;76:1451–62.
Lee JI, Woo SK, Kim KI, Park KC, Baek SH, Yoo YJ, et al. A method for assaying deubiquitinating enzymes. Biol Proced Online. 1998;1:92–9.
Komander D, Barford D. Structure of the A20 OTU domain and mechanistic insights into deubiquitination. Biochem J. 2008;409:77–85.
Chen H, Hu L, Luo Z, Zhang J, Zhang C, Qiu B, et al. A20 suppresses hepatocellular carcinoma proliferation and metastasis through inhibition of Twist1 expression. Mol Cancer. 2015;14:186.
Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene. 2005;24:7482–92.
Xue G, Restuccia DF, Lan Q, Hynx D, Dirnhofer S, Hess D, et al. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer Discov. 2012;2:248–59.
McCarthy JJ, Esser KA. Anabolic and catabolic pathways regulating skeletal muscle mass. Curr Opin Clin Nutr Metab Care. 2010;13:230–5.
Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, et al. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal. 2009;2:ra62.
Dar A, Shibata E, Dutta A. Deubiquitination of Tip60 by USP7 determines the activity of the p53-dependent apoptotic pathway. Mol Cell Biol. 2013;33:3309–20.
Mohsin SK, Weiss H, Havighurst T, Clark GM, Berardo M, Roanh LD, et al. Progesterone receptor by immunohistochemistry and clinical outcome in breast cancer: a validation study. Mod Pathol. 2004;17:1545–54.
Acknowledgements
This study was supported by the University Grant of Zunyi Medical University. National Natural Science Foundation of China (No. 81360309, No.81572738, No.81602149). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
The first four authors contribute equally to this paper. ZZY, ZYK, QC and HXH: study design, acquisition of data, analysis and interpretation; LMH, LYM, LFQ, XXL, XXQ, WQ, PDX, YT, HJ, LYF and XYW: acquisition of data, technical support. ZZY, HQ, JFJ and YCY: drafting of the manuscript and material support. ZZY: study design, drafting and critical revision of the manuscript and obtaining funding.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
These authors contributed equally: Yuekun Zhu, Chao Qu, Xuehui Hong, Yanyan Jia.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, Y., Qu, C., Hong, X. et al. Trabid inhibits hepatocellular carcinoma growth and metastasis by cleaving RNF8-induced K63 ubiquitination of Twist1. Cell Death Differ 26, 306–320 (2019). https://doi.org/10.1038/s41418-018-0119-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0119-2
This article is cited by
-
Emerging roles of deubiquitinating enzymes in actin cytoskeleton and tumor metastasis
Cellular Oncology (2024)
-
Exploration of Diagnostic Deubiquitinating Enzymes in Endometriosis and Its Immune Infiltration
Biochemical Genetics (2024)
-
Reciprocal regulation of TWIST1 and OGT determines the decitabine efficacy in MDS/AML
Cell Communication and Signaling (2023)
-
SOCS2-enhanced ubiquitination of SLC7A11 promotes ferroptosis and radiosensitization in hepatocellular carcinoma
Cell Death & Differentiation (2023)
-
Deubiquitinases in cancer
Nature Reviews Cancer (2023)

{kind=link}