
Abstract
Myocardin is a transcriptional co-activator required for cardiovascular development, but also promotes cardiomyocyte survival through an unclear molecular mechanism. Mitochondrial permeability transition is implicated in necrosis, while pore closure is required for mitochondrial maturation during cardiac development. We show that loss of myocardin function leads to subendocardial necrosis at E9.5, concurrent with elevated expression of the death gene Nix. Mechanistically, we demonstrate that myocardin knockdown reduces microRNA-133a levels to allow Nix accumulation, leading to mitochondrial permeability transition, reduced mitochondrial respiration, and necrosis. Myocardin knockdown elicits calcium release from the endo/sarcoplasmic reticulum with mitochondrial calcium accumulation, while restoration of microRNA-133a function, or knockdown of Nix rescues calcium perturbations. We observed reduced myocardin and elevated Nix expression within the infarct border-zone following coronary ligation. These findings identify a myocardin-regulated pathway that maintains calcium homeostasis and mitochondrial function during development, and is attenuated during ischemic heart disease. Given the diverse role of Nix and microRNA-133a, these findings may have broader implications to metabolic disease and cancer.
Similar content being viewed by others


Introduction
The formation of the mammalian heart during embryogenesis is orchestrated by a core set of cardiomyocyte-enriched transcription factors that govern the cellular phenotype by regulating the expression of genes involved in lineage specification, differentiation, patterning, and cell survival [1]. This highly conserved genetic network is reinforced by transcriptional co-activators that modulate cardiac gene expression and dictate target-gene specific activation [2]. Myocardin is a cardiomyocyte- and smooth muscle cell-restricted transcriptional co-activator that physically interacts with several core cardiac transcription factors, including SRF, MEF2C, GATA4, and Tbx5, to regulate gene expression during both cardiovascular development and post-natal cardiovascular remodeling [3,4,5,6]. Cell programming experiments have demonstrated that a transcription factor ‘cocktail’ comprised of GATA4, MEF2C, and Tbx5 (ie. GMT) can directly convert fibroblasts to functional cardiomyocyte-like cells [7]. Interestingly, the addition of myocardin to the GMT and Hand2 cocktail enhances the conversion of human fibroblasts by up to 40% [8]. These observations suggest that myocardin plays a potent co-activator role during cardiomyocyte differentiation. Original gene targeting studies in mice harboring homozygous null alleles for myocardin identified a critical role for this transcriptional co-activator during vascular smooth muscle differentiation and yolk sac vascularization [9]. More recently, myocardin floxed mice were crossed with CMV-Cre and Nkx2.5-Cre transgenic mice, and offspring display exacerbated cardiomyocyte cell death and defects in proliferation, resulting in hypoplastic ventricles, heart failure, and embryonic lethality [10]. Furthermore, the block in cardiomyocyte proliferation was due to a defect in bone morphogenetic protein-10 (BMP10) expression and signaling; however, the direct cause of cell death in the developing ventricles was less clear. Moreover, the direct transcriptional targets of myocardin that regulate this survival phenotype remain unknown. However, electron microscopy identified cardiac nuclear condensation and apoptotic body formation, with the addition of mitochondrial swelling [10]. Conditional cardiac deletion of myocardin in adult mice using a tamoxifen-inducible system led to rapid deterioration in cardiac function, sarcomeric disorganization, and lethal heart failure [11]. Given the rapid deterioration of cardiac function in these animal models, combined with the degree of cell death in the myocardium, we hypothesized that multiple modes of cell death, in addition to apoptosis, were involved in the deterioration of cardiac function when myocardin is genetically inhibited.
Cardiomyocytes rely on mitochondria as a primary source of ATP, and mature cardiomyocytes may contain as much as 35% cellular volume of mitochondria [12]. These observations make cardiomyocytes ideally suited to study mitochondrial-related disease mechanisms. Mitochondrial permeability transition is a term describing the phenomenon where the inner mitochondrial membrane permeabilizes and allows passage of solutes surpassing a kilodalton in size. This results in rapid dissipation of the mitochondrial membrane potential, respiratory uncoupling, and mitochondrial swelling [13]. If prolonged, mitochondrial permeability transition will lead to mitochondrial rupture and cell death resembling a necrotic phenotype. Although originally associated with apoptosis, mitochondrial permeability transition leading to cell death has been recently termed mitochondrial permeability transition-driven regulated necrosis to replace the previous controversial term ‘programmed necrosis’ [14]. The components of the mitochondrial permeability transition pore (MPTP) have been historically elusive; however, recent studies implicate a conformational change in the mitochondrial ATP Synthase as the fundamental pore structure [15], where Bax and Bak serve as outer member modulators of permeability transition [16]. Although these components are ubiquitously expressed in virtually all tissues, recent studies have defined a developmental role for the MPTP in the heart, where pore closure is required for proper cardiac myocyte differentiation, accumulation of mitochondrial content, and mature mitochondrial cristae formation [17].
Accumulating evidence suggests that mitochondrial permeability transition and necrosis are intimately linked with ischemic and mitochondrial-related diseases [14, 18, 19]. An important component of regulated necrosis involves permeability transition triggered by elevations in matrix calcium concentration and reactive oxygen species [14]. Evidence supporting the important nature of regulated necrosis in models of heart disease comes from several studies, by independent laboratories, demonstrating that mice genetically deficient in the mitochondrial matrix protein cyclophilin D, or its pharmacological inhibition by cyclosporine A, are protected against permeability transition and necrosis triggered by cardiac ischemia [20, 21]. Furthermore, mice deficient in the mitochondrial calcium uniporter are protected from ischemia-reperfusion injury [22, 23].
Nix (ie. Bnip3L) is an atypical BH3-only member of the Bcl-2 family that has been demonstrated to regulate apoptosis, necrosis, autophagy, and mitophagy in numerous cell types and several cancers [24]. Nix function has been studied in the heart during pathological remodeling events that lead to heart failure [25, 26]. Since its discovery, it has been noted that Nix, and the homologue Bnip3, could induce cell context-dependent apoptotic or necrotic cell death [27, 28]. Newer findings suggest that Nix can be targeted to either the mitochondria or the endoplasmic/sarcoplasmic reticulum (SR), where mitochondrial-targeted Nix activates apoptotic cell death, and SR-targeted Nix induces necrosis [29, 30]. The mechanism responsible for this alternative cellular targeting remains unknown, as does the exact mode of killing in these different subcellular locations. However, in vivo studies suggest that Nix preferentially localizes to the SR during pressure overload-induced cardiac remodeling [30].
We recently identified microRNA-133a (miR-133a) as an important regulator of mitochondrial function and insulin sensitivity through the repression of Nix expression [31]. Previous reports observed that miR-133a is downregulated in human heart failure and in animal models [32, 33]. These findings prompted us to examine the regulation of miR-133a and Nix expression by myocardin in both the developing heart and in animal models of necrotic cardiac injury, where mitochondrial permeability transition has been demonstrated to play a critical biological role.
In this report, we present evidence of subendocardial necrosis in myocardin knockout embryos, concurrent with elevated Nix expression. Through detailed gain-of-function and loss-of-function approaches, we define a genetic pathway regulated by myocardin that drives mitochondrial function, maintains calcium homeostasis, and reduces cardiomyocyte sensitivity to permeability transition-driven necrosis. Moreover, we provide evidence that this genetic pathway is attenuated within the infarct border-zone following coronary ligation, but not during doxorubicin-induced cardiac necrosis. These findings highlight the specificity of this genetic pathway and suggest that necrosis within the infarct border-zone is uniquely regulated compared to toxicity-induced necrosis in the heart.
Results
Silencing of myocardin increases Nix expression through a miR-133a-dependent mechanism
Consistent with a role for myocardin in the regulation of necrosis, in addition to apoptosis, examination of E9.5 myocardin-null embryos revealed elevated Nix expression in the heart (Fig. 1a). E9.5 is 1-day prior to the reported cardiovascular failure and embryonic lethality of myocardin knock-out mice [10]. Previous studies have implicated Nix in cardiac necrosis [29, 30]; therefore, we stained myocardin-null embryos for an early and specific marker of necrosis, called HMGB1 [34,35,36]. HMGB1 is a chromatin-binding protein that is exported into the cytoplasm during necrosis, and is eventually released into the interstitium where it contributes to the inflammatory response [35]. We observed cytoplasmic HMGB1 in the subendocardial region of hearts from myocardin-null embryos, compared to a nuclear localization in control embryos, suggesting removal of myocardin function during cardiac development leads to myocyte necrosis (Fig. 1b).

Silencing of myocardin increases Nix expression through a miR-133a-dependent mechanism. a, b E9.5 wild-type (WT) and myocardin knock-out (KO) mouse embryos were fixed, mounted and stained for confocal immunofluorescence. Dapi (blue) identifies nuclei. Inserts identify myocardium. c, d Primary ventricular neonatal cardiomyocytes were transduced with an shRNA targeting myocardin (sh-Myocd) lentivirus or scrambled control. Cells were fixed and stained, as indicated, and visualized by fluorescent microscopy. e H9c2 cells were transfected with sh-Myocd or scrambled control. Protein extracts were immunoblotted as indicated. f H9c2 cells were transfected with sh-Myocd, sh-SRF, as indicated, and RNA extracts were analyzed by real-time PCR for microRNA-133a (miR-133a). g H9c2 cells were transfected sh-Myocd or a scrambled vector, and miR-133a. Protein extracts immunoblotted for Nix expression. h H9c2 cardiac myocytes were transfected as in e. Isolated RNA was analyzed by PCR-based array. Data are expressed as mean ± SE. *p < 0.05 compared to control, determined by one-way ANOVA
Recently, we identified miR-133a as an important regulator of Nix expression in cardiac, skeletal, and smooth muscle cells [31]. Thus, we tested the hypothesis that myocardin regulates Nix expression through a miR-133a dependent mechanism. We knocked-down myocardin expression in both primary neonatal rat ventricular cardiomyocytes and in the H9c2 myocyte cell line, using lentiviral and plasmid-based shRNA vectors, respectively. Knock-down of myocardin in these cellular models resulted in simultaneous decreased expression of miR-133a and increased expression of Nix (Fig. 1c–f). Since myocardin does not bind to DNA directly, and regulates gene expression primarily through interaction with SRF, we knocked-down SRF and observed a comparable decrease in miR-133a expression, and no additional decrease in miR-133a expression when both SRF and myocardin were knocked-down (Fig. 1f). Furthermore, reconstitution of miR-133a expression attenuated Nix expression following myocardin knock-down (Fig. 1g), suggesting that miR-133a is sufficient to repress Nix expression independent of myocardin function. In addition, we performed a PCR-based gene expression array in H9c2 cells following myocardin knockdown using a commercially available cell death pathway finder array (Fig. 1h and Supplemental Table 1). Interestingly, few if any survival genes were reduced by 50% or more, and only three death genes were increased by 2-fold using this array. Although not exhaustive, this observation led us to speculate that the genetic pathway connecting myocardin function to Nix expression was biologically relevant in cardiac myocytes.
Myocardin regulates mitochondrial function and permeability transition
In order to understand how inactivation of myocardin leads to cardiomyocyte necrosis, we used RNA interference in primary cardiomyoctes and H9c2 cells and assessed mitochondrial- and cell viability-parameters by fluorescent microscopy. In primary cardiomyocytes, knockdown of myocardin led to loss of mitochondrial membrane potential and mitochondrial permeability transition, determined by the calcein-cobalt chloride staining method (Fig. 2a–c). Similar findings were observed in the H9c2 cell line (Fig. 2d, e). Furthermore, knockdown of myocardin increased the percentage of necrotic cells, determined by ethidium homodimer staining in both primary cardiomyocytes and H9c2 cells (Fig. 2f–h). As a positive control, we treated primary cardiomyocytes with isoproterenol (100 μM for 18 h), as excessive β-adrenergic stimulation has been previously implicated as an important trigger for permeability transition and cardiac necrosis [37]. Shown in Supplemental Fig. 1A-C, isoproterenol affected mitochondrial membrane potential, permeability transition, and ethidium homodimer staining in a comparable manner to myocardin knockdown. We also evaluated respiration in cardiomyocytes using a metabolic flux analyzer (Seahorse, XF24). While myocardin knockdown had no effect on basal respiration in neonatal rat ventricular myocytes. When myocardin knockdown cells were challenged with the mitochondrial uncoupler FCCP, reduced respiration was observed (Fig. 2i, j). In addition, knockdown of myocardin reduced cellular ATP levels, determined by the ATeam1.03 ATP biosensor (Fig. 2k). Finally, we observed that mitochondrial permeability transition, induced by myocardin knockdown, was inhibited by the cyclophilin-D inhibitor (cyclosporine A; 1 μM), the inositol triphosphate receptor (IP3R) inhibitor (2APB; 10 μM), and the mitochondrial calcium uniporter inhibitor (Ru360; 10 μM; Fig. 2m); however, the ryanodine receptor (RyR) antagonist Dantrolene (Dan; 10 μM) had no effect on permeability transition induced by myocardin knockdown (Fig. 2l). These findings suggest the involvement of an IP3R-dependent SR-to-mitochondria calcium transfer in the regulation of permeability transition when myocardin expression is reduced.

Myocardin regulates mitochondrial function and permeability transition. a Primary ventricular neonatal cardiomyocytes (PVNM) were infected with sh-Myocd or a scrambled control. Cells were stained with TMRM or with calcein-AM and cobalt chloride (CoCl2, 5 µM) to determine mitochondria permeability transition. b Quantification of TMRM; fluorescence signal normalized to cell area (relative fluorescence) quantified in 10 random fields. c H9c2 cells transfected with sh-Myocd or scrambled vector; stained with TMRM and quantified. d PVNM treated as in a; stained with calcein-AM and CoCl2, and quantified. e H9c2 cells treated as in c; stained with calcein-AM and CoCl2, and quantified. f PVNM treated as in a, stained with calcein-AM and ethidium homodimer-1 (Eth HD-1) to identify living (green) and necrotic (red) cells, respectively. g Quantification of fluorescent images in f by calculating the percentage of necrotic cells (Eth HD-1 Positive) in 10 random fields and over 200 cells per condition. h H9c2 cells treated as in c, Eth HD-1 Positive cells quantified. (i) PVNM treated as in a; to assess oxygen consumption rate (OCR). PVNMs were injected with oligomycin (1 µM) [A], FCCP (1uM) [B], and antimycin A (1 µM) and rotenone (1uM) [C]. OCR was corrected by total cell number following analysis (n = 9). j Calculated respiration rates from i. k H9c2 cells were transfected as in c, along with ATeam 1.03. Cells were analyzed by excitation of CFP and taking the emission ratio of YFP to CFP (FRET Ratio) in 15 random fields and 35 cells per condition. l PVNM treated as in a, following treatment with Dantrolene (DAN, 10uM; 18 h); DMSO used as a control vehicle. Cells stained with calcein-AM and CoCl2 and quantified. (M) PVNM treated as in a, following treatment with cyclosporine A (CsA, 1uM), 2-aminoethoxydiphenyl borate (2APB, 2uM), or Ruthenium360 (Ru360, 10uM; 18 h). DMSO used as a control vehicle. Cells stained with calcein-AM and CoCl2 and quantified. Data are expressed as mean ± SE. *p < 0.05 compared to control, **p < 0.05 compared to treatment, determined by one-way ANOVA
In order to perform the reciprocal gain of function experiments, we used H9c2 cells transfected with myocardin and evaluated the sensitivity of these cells to agents known to induce mitochondrial permeability transition and necrosis. Ionomycin is a calcium ionophore, previously demonstrated to induce MPTP-dependent necrosis (Supplemental Fig. 1E–F) [16]. Following myocardin transfection, or empty vector control, we treated cells with ionomycin (2μM). Myocardin expression substantially reduced the number of ethidium homodimer-positive cells and reduced mitochondrial permeability transition (Fig. 3a–d). As a control, we treated cells with cyclosporine A (1μM), and we observed a near complete reversal of permeability transition following ionomycin treatment (Fig. 3e). We also treated myocardin expressing H9c2 cells with staurosporine (2μM), previously shown to induce mitochondrial outer membrane permeability (MOMP) and caspase-dependent apoptosis (Supplemental Fig. 1G-H) [16]. As determined by MTT assay, myocardin expression had no effect on cell viability when H9c2 cells were exposed to staurosporine (Fig. 3f).

Myocardin opposes necrosis and mitochondrial dysfunction. a H9c2 cells were transfected with myocardin (Myocd) or an empty vector control, and treated with ionomycin (Iono, 2 µM, 18 h); DMSO was used as a control vehicle. Cells were stained with calcein-AM and Eth HD-1 to identify living (green) and necrotic (red) cells, respectively; b stained with calcein-AM and CoCl2. CMV-dsRed identifies positive cells. c Quantification of a by calculating the percentage of necrotic cells (Eth HD-1 Positive) and over 200 cells per condition and d by calculating the percentage of transfected cells with mitochondrial puncta, in 10 random fields. e H9c2 cardiac myocytes were pre-treated with CsA (1 µM, 2 h) followed by ionomycin treatment (Iono, 2 µM, 18 h). Cells were stained with calcein-AM and CoCl2 and quantified. (f) H9c2 cardiac myocytes transfected with Myocd or an empty vector control, treated with staurosporine (STS, 2uM, 18 h); cell viability was assessed by MTT analysis (n = 4). Data are expressed as mean ± SE. *p < 0.05 compared to control, **p < 0.05 compared to treatment, determined by 1-way ANOVA
Myocardin regulates myocyte calcium homeostasis
To more fully investigate the role of cellular calcium in myocardin-regulated permeability transition, we used organelle-targeted and genetically encoded calcium biosensors, called GECOs (Genetically Encoded Ca2+ indicators for Optical imaging) [38]. We used the mitochondrial matrix-targeted red GECO, known as Carmine (mito-carmine), and ER-targeted LAR-GECO (ER-LAR-GECO) [39, 40]. We rationalized that sustained activation of protein kinase-A (PKA) would trigger an intracellular calcium release from the SR that would be buffered by the mitochondrial. As a proof-of-concept experiment, we transfected these GECO constructs into H9c2 cells and treated cells with a combination of the adenylate cyclase activator forskolin (FSK; 10μM) and the pan-phosphodiesterase inhibitor IBMX (500 μM), termed FSK-I. Shown in Fig. 4a–c, FSK-I treatment reduced SR calcium and increased mitochondrial calcium, while the IP3R inhibitor 2APB blocked the effects of FSK-I. Interestingly, the RyR blocker dantrolene had no effect on FSK-I induced calcium release (Supplemental Fig. 2A). Furthermore, transfection of cells with myocardin prevented both the SR calcium release and the mitochondrial calcium accumulation triggered by FSK-I treatment (Fig. 4d, e). Consistent with our hypothesis that an SR-dependent calcium release leads to mitochondrial permeability transition, we observed that FSK-I treatment reduced mitochondrial membrane potential, reduced calcein-cobalt chloride staining, increased mitochondrial superoxide production, and the percentage of ethidium homodimer positive cells (Fig. 4f–i). Moreover, expression of myocardin rescued permeability transition and necrosis induced by FSK-I treatment (Fig. 4f–i). In addition, FSK-I induced mitochondrial membrane depolarization and permeability transition were blocked by 2APB treatment, while isoproterenol increased mitochondrial calcium in primary cardiomyocytes (Supplemental Fig. 2B–D). Myocardin expression also reduced the expression of Nix, without affecting other mitochondrial regulators, such as the mitochondrial biogenesis inducer PGC-1α or mitofusin-2 (Fig. 4j). Finally, myocardin knockdown increased mitochondrial calcium accumulation, determined by the mito-carmine biosensor (Fig. 4k, l). Collectively, these findings suggest that myocardin can alter the sensitivity of the SR-dependent calcium release, which serves as a proximal event to mitochondrial calcium uptake and MPTP opening.

Myocardin regulates mitochondrial calcium homeostasis. a H9c2 cells were transfected with a SR-targeted (above) or mitochondrial (mito)-targeted (below) calcium biosensor. Cells were pre-treated with 2APB (2 µM, 2 h) followed by co-treatment with Forskolin (FSK, 10 µM) and 3-isobutyl-1-methylxanthine (IBMX, 500uM; FSK-I, 18 h), or DMSO as a control vehicle. b Quantification of SR-calcium and c mito-calcium from a. Fluorescence signal normalized to cell area (relative fluorescence) was quantified in 10 random fields. d H9c2 cardiac myocytes were transfected with Myocd or an empty vector, and a SR-targeted calcium biosensor or (e) a mito-targeted calcium biosensor; treated with FSK-I, and quantified. f H9c2 cardiac myocytes transfected with Myocd or an empty vector and treated with FSK-I; stained with TMRM, or (g) calcein-AM and CoCl2, or (h) Mito-Sox to evaluate mitochondrial super oxide, or (i) calcein-AM and Eth HD-1; 10 random fields were quantified for each end-point. j H9c2 cells were transfected with Myocd or empty vector. Protein extracts analyzed as indicated. k H9c2 cells were transfected with sh-Myocd or scrambled control, with the mito-calcium biosensor and stained with Hoechst to visualize nuclei. l Quantification of k. Data are expressed as mean ± SE. *p < 0.05 compared to control, **p < 0.05 compared to treatment, determined by one-way ANOVA
The myocardin-dependent microRNA, miR-133a, regulates mitochondrial function and permeability transition
To evaluate the role of miR-133a in mitochondrial permeability transition and necrosis, we transfected H9c2 cells with miR-133a and assessed cell sensitivity to FSK-I treatment. Shown in Fig. 5a, b, cells expressing miR-133a displayed improved mitochondrial membrane potential when exposed to FSK-I, and did not undergo permeability transition to the same extent as control cells. Furthermore, expression of miR-133a reduced the number of necrotic cells following FSK-I treatment (Fig. 5c). Next, we expressed the SR- and mitochondrial-targeted calcium biosensors to assess if miR-133a expression could impact calcium homeostasis. Cells expressing miR-133a were desensitized to FSK-I induced SR calcium release, compared to control cells (Fig. 5d). Furthermore, miR-133a expression prevented mitochondrial calcium accumulation triggered by FSK-I treatment (Fig. 5e). We also evaluated respiration in H9c2 cells treated with FSK-I. Shown in Fig. 5f, g, 18-h treatment with FSK-I significantly reduced basal and maximal respiration in these cells; however, miR-133a attenuated the effects of FSK-I on mitochondrial respiration. Next, we performed a rescue experiment, where we determined whether miR-133a expression could overcome mitochondrial calcium accumulation when myocardin was knocked-down. Shown in Fig. 5h, myocardin knockdown increased mitochondrial calcium levels; however, when cells were co-transfected with miR-133a, mitochondrial calcium levels were restored to control levels. Finally, we reconstituted Nix function by expressing a Nix construct, which lacks the miR-133a 3’-UTR binding site, and demonstrate that FSK-I induced elevations in mitochondrial calcium were maintained, even in the presence of ectopic miR-133a expression (Fig. 5i).

Myocardin-dependent micro-RNA, miR-133a, regulates mitochondrial function and permeability transition. a–c H9c2 cells were transfected with miR-133a or an empty vector, and treated with FSK-I (18 h), or DMSO as a control vehicle; stained with TMRM (a) or (b) with calcein-AM and CoCl2, or (c) with calcein-AM and Eth HD-1. d H9c2 cells were transfected with a SR-targeted calcium biosensor, treated with FSK-I, and quantified. e H9c2 were transfected with miR-133a or an empty vector, a mito-targeted calcium biosensor, treated with FSK-I, and quantified. f H9c2 cells were transfected with a miR-133a mimic, and treated with FSK-I or vehicle control. Cells were injected with oligomycin (1 µM) (a), FCCP (1 µM) (b), and antimycin A (1 µM) and rotenone (1 µM) (c). OCR was corrected by total cell number following analysis (n = 4). g Calculated respiration rates from f. h H9c2 cells were transfected with sh-Myocd or scrambled control, miR-133a, a mito-targeted calcium biosensor, and quantified. i H9c2 cells were transfected with miR-133a or an empty vector control, Nix and a mito-targeted calcium biosensor. Cells were treated with FSK-I (18 h), or DMSO as a control vehicle and quantified. Data are expressed as mean ± SE. *p < 0.05 compared to control, **p < 0.05 compared to treatment, determined by one-way ANOVA
Myocardin regulates mitochondrial permeability transition and calcium homeostasis through Nix
To further describe the role of Nix as a down-stream effector of myocardin-regulated calcium homeostasis, we performed a double-knockdown experiment and evaluated mitochondrial calcium accumulation. Shown in Fig. 6a, b, knockdown of myocardin increased mitochondrial calcium and induced permeability transition; however, simultaneous knockdown of Nix returned mitochondrial calcium to control levels and prevented permeability transition. Furthermore, expression of Nix in H9c2 cells induced mitochondrial permeability transition, which was inhibited by CsA treatment (Fig. 6c). In addition, expression of Nix reduced SR calcium and increased mitochondrial calcium levels (Fig. 6d, e), and knockdown of Nix prevented FSK-I induced permeability transition and necrosis (Fig. 6f and Supplement Fig. 2E). Next, we performed cell fractionation studies following myocardin knock-down. Although Nix expression is greater in the mitochondria fraction than in the ER/SR fraction, the primary induction of Nix following myocardin knock-down occurred in the ER/SR fraction. The mitochondrial fraction remained relatively unchanged (Fig. 6g). Taken together, our data suggests that suppression of Nix expression at the ER/SR is an important component of myocardin-regulated calcium homeostasis and survival phenotype.

Myocardin regulates mitochondrial permeability transition and calcium homeostasis through Nix. a H9c2 cells were transfected with sh-Myocd, sh-Nix, as indicated, a mito-targeted calcium biosensor and quantified (b). H9c2 cells were transfected with sh-Myocd, sh-Nix; as in (A) and stained with calcein-AM and CoCl2 and quantified. c H9c2 cells were transfected with Nix or empty vector control, treated with cyclosporine A (CsA, 1 µM, 18 h); stained with calcein-AM and CoCl2 and quantified. d, e H9c2 cells were transfected with Nix or empty vector control and (d) the SR-targeted calcium biosensor or (e) the mito-targeted calcium biosensor, and quantified. f H9c2 cells were transfected with sh-Nix or a scrambled control, treated with FSK-I (18 h), or DMSO as a control vehicle; stained with calcein-AM and CoCl2 and quantified. g H9c2 cells were transfected with sh-Myocd or scrambled control. Mitochondrial and ER fractions were immunoblotted as indicated. h H9c2 cells were tansfected with Nix, mitochondrial-targeted Nix (Nix-Mito), or SR-targeted Nix (Nix-SR), and the mito-targeted calcium biosensor, and quantified. i H9c2 cells were transfected with Nix, wild-type Bcl2, mitochondrial-targeted Bcl-2 (Bcl2-M), or SR-targeted Bcl-2 (Bcl2-SR), or empty vector control, stained with calcein-AM and CoCl2 j or transfected with mito-targeted calcium biosensor, and quantified
In order to further understand how Nix regulates mitochondrial permeability transition, we engineered SR- and mitochondrial-targeted Nix constructs (Supplemental Fig. 2G) and evaluated mitochondrial calcium accumulation. Expression of wild-type Nix increased mitochondrial calcium levels (Fig. 6h). Interestingly, mitochondrial-targeted Nix had no effect on mitochondrial calcium levels, while SR-targeted Nix increased mitochondrial calcium (Fig. 6h). These data implicate an SR-dependent calcium release as an important aspect of Nix-induced permeability transition. Previous studies have implicated Bcl-2 as a regulator of IP3-dependent calcium release, and Nix has been demonstrated to functionally interact with Bcl-2 [27, 41, 42]. Thus, we utilized organelle-targeted Bcl-2 constructs to further interrogate mitochondrial calcium accumulation [43]. Shown in Fig. 6i, j, expression of wild-type Bcl-2 attenuated Nix-induced calcium accumulation and permeability transition. Consistent with our organelle-targeted Nix constructs, only SR (ie. ER)-targeted Bcl-2 could prevent Nix-induced mitochondrial calcium accumulation and permeability transition, while mitochondrial-targeted Bcl-2 had no effect on these end-points (Fig. 6i, j).
Myocardin expression is reduced in the border-zone following myocardial infarction
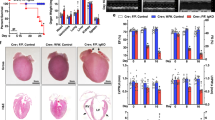
To evaluate whether this pathway connecting myocardin and Nix through miR-133a operates in vivo, we utilized two animal models of cardiac necrosis. First, we used a rat coronary ligation model, and dissected the infarct border-zone from the scar tissue, 4-weeks post-ligation [44]. The border-zone is the area at risk within the left anterior descending territory, that does not infarct within the first 24-hours following coronary ligation. By 4-weeks post-ligation, this tissue is located immediately adjacent to the infarct scar tissue. Left ventricle tissue from sham-operated animals was used as control. We observed a 40% reduction in myocardin mRNA within the infarct border-zone, and a concurrent 45% reduction in miR-133a expression (Fig. 7a, b). Consistent with our hypothesis, Nix expression was also increased within the border zone (Fig. 7c, d). However, these gene expression changes were returned to control levels by 8-weeks post-infarction (not shown). Second, we used a doxorubicin (Dox)-induced cardio-toxicity model [45]. C57BL6 mice received weekly intraperitoneal injections of Dox (8 mg/kg body weight) for 4 weeks. In contrast to the infarction model, we observed a modest increase in miR-133a expression and no change in myocardin expression in Dox-treated animals (Fig. 7e, f). Furthermore, we observed a trend towards decreased Nix expression, but this was not statistically significant in Dox-treated hearts (Fig. 7g, h). These findings suggest that cardiac necrosis in the infarct border-zone and Dox models have unique features and the myocardin-Nix pathway is specifically and temporally regulated within the infarct border-zone.

In vivo analysis of the myocardin-miR133a-Nix pathway. a–d Sprague Dawley rats were subjected to left coronary artery ligation, or sham operation as a control. Following 4-weeks of recovery, the viable infarct border-zone was harvested from the left ventricle. Extracts were analyzed by real-time PCR for Myocardin (a), or miR-133a (b) expression (n = 4). Protein extracts were immunoblotted for Nix (c) expression and subjected to densitometry (d, n = 3). (E-H) C57BL6 mice received weekly intraperitoneal injections of doxorubicin (Dox; 8 mg/kg), or vehicle control, for 4 weeks. Extracts were analyzed by real-time PCR for Myocardin (e), or miR-133a (f) expression (n = 4). Protein extracts were immunoblotted for Nix (g) expression (n = 4) and subjected to densitometry (h). Data are expressed as mean ± SE. *p < 0.05 compared to control, determined by Student's T-test
Discussion
Mitochondrial permeability transition in cardiomyocytes has been implicated in both developmental processes and in regulated necrosis occurring during cardiac pathologies; however, the transcriptional regulation of these events remains poorly defined. Here, we describe a myocardin-regulated pathway that opposes mitochondrial permeability transition through a calcium-dependent mechanism. We also demonstrate that this pathway is operational during embryonic cardiac development and within the infarct border-zone during the recovery from coronary ligation. To our knowledge, this is the first evidence of a direct myocardin-target gene regulating cardiomyocyte survival.
Detailed evaluation of myocardin expression during mouse embryogenesis previously documented that myocardin transcripts are detectable as early as E9.5 in the embryonic heart, and are robustly expressed throughout the embryonic ventricles by E14.5 [46]. This is temporally correlated with developmental closure of the MPTP [17]. Collectively, these findings are consistent with our data demonstrating myocardin is a regulator of mitochondrial permeability transition. In addition, conditional myocardin knockout mouse models have demonstrated that there are developmental and post-natal ‘windows’ where genetic inactivation of myocardin produces lethal cardiac defects [10]. For example, genetic removal of myocardin using the Nkx2.5-Cre line results in lethality at E13.5 due to cardiac chamber maturation defects [10]. Furthermore, using MerCreMer line for the tamoxifen-inducible removal of myocardin in the mature adult mouse heart, which is highly dependent on mitochondrial oxidative metabolism for ATP production, results in rapid deterioration to heart failure [11]. Conversely, l knockouts generated with the αMHC-Cre line, which removes myocardin function after chamber maturation and MPTP closure occurs, results in a delay in mortality by up to 10 months, suggesting that loss of myocardin function at this developmental stage can be partially compensated for, at least transiently [11].
Previous studies have implicated myocardin as a regulator of pathological hypertrophy elicited by aortic banding [47]. Myocardin expression is increased following aortic banding and produces hypertrophy when delivered to cultured cardiomyocytes [47]. Our findings that myocardin expression is reduced following coronary artery ligation may at first seem contradictory to this study. However, the infarct border-zone following coronary ligation is a unique pathological environment, where myocytes are exposed to metabolic stress induced by hypoxia/ischemia, adrenergic stimulation, and mechanical stretch, which is undoubtedly different than the afterload-induced and/or neurohumoral-induced stress imposed by aortic banding.
Our findings are consistent with several important developmental and post-natal cardiac remodeling studies involving miR-133a and Nix, and help consolidate these findings into a genetic pathway that operates during cardiac ontogeny and during ischemic heart disease, where myocardin serves as a proximal regulator of this pathway. In this regard, miR-133a-1 and miR-133a-2 double knockout mice display evidence of abnormal mitochondrial cristae formation in the heart and aberrant smooth muscle gene expression [48]. Furthermore, miR-133a repression has been implicated in both rodent and human cardiac hypertrophy [32] and diabetes-related cardiac remodeling [31], while transgenic mice expressing miR-133a under control of the αMHC promoter display reduced levels of fibrosis during pathological cardiac remodelling [33]. Previously, we demonstrated that miR-133a was a direct transcriptional target of MEF2C and SRF in cardiac, skeletal, and smooth muscle cells [31]. However, we also observed that the gene expression pattern of miR-133a was different in the heart than in skeletal muscle [31]. Based on the observations in the present study, we contend that the presence of myocardin creates an addition layer of regulation over miR-133a expression that is present in the heart, but not in myotubes or mature skeletal muscle. Nix expression is induced following pressure overload and in genetic models of heart failure, such as the transgenic Gαq mouse, while Nix knockout mice are protected from heart failure in these models [49, 50]. However, highly purified mitochondria isolated from Nix knockout animals are identical to wild-type mitochondria in terms of permeability transition triggered by exogenous calcium [30]. In addition, Nix preferentially accumulates in the SR following aortic banding resulting in enhanced caffeine-induced calcium release [30]. These findings are consistent with our data, and we provide addition insight into the function of Nix at the SR/ER, and demonstrate that Nix depletes the SR calcium content, which is buffered by mitochondria calcium uptake at the expense of MPTP opening.
Recent progress has been made regarding the mitochondrial components and regulators of the permeability transition pore, and several studies have defined the importance of MPTP-dependent necrosis in the pathophysiology of ischemic cardiac and cerebrovascular diseases [14]. However, little is known regarding the transcriptional regulation of permeability transition, especially the tissue-specific regulation during developmental or during pathological conditions. The data presented here describe a genetic pathway that regulates MPTP function during cardiac development and within a discrete temporal period in the infarct border-zone following coronary ligation. Our data also suggests that the transcriptional regulation over permeability transition occurs through calcium homeostasis, where Nix expression dictates the sensitivity of the ER/SR to IP3R-activating stimuli. Furthermore, previous literature suggests that an ER-to-mitochondrial calcium transfer, occurring at the mitochondrial-associated membrane of the ER, is an important regulatory component of mitochondrial function and cell death [51, 52]. Additional work is needed to determine if this pathway, or a similar pathway, is conserved in multiple tissues, and to determine how MPTP and necrosis is regulated in other pathologies. Given the diverse roles of miR-133a and Nix in regulating mitochondrial function, proliferation, and autophagy, dysregulation of this genetic pathway may have broad implications involving cardiometabolic disease, insulin resistance, and cancer biology.
Materials and Methods
Myocardin-null embryos and Confocal Immunofluorescence
Myocardin-null mice were generated using the homozygous myocardin conditional mutant mice (MyocdF/F), which were intercrossed with CMV-Cre (BALB/c-Tg(CMV-cre)1Cgn/J) transgenic mice, as described previously [10]. Embryos at day 9.5 (E9.5) were dissected, fixed, and mounted as described previously [10]. Sections were prepared for immunofluorescence using antibodies for Nix (Bnip3L, CST #12396), or HMGB1 (CST #3935), as per manufacturer’s protocols, and nuclei were stained with Dapi. Confocal microscopy was performed on a Zeiss Laser Scanning LSM 700.
Plasmids and microRNA mimic
The myocardin-935 plasmid was provided by E. Olson, and described previously [53, 54]. The shRNA targeting myocardin was based on the targeting sequence previously described by Yoshida et al (5′-GTTCCGATCAGTCTTACAG-3′) [55]. Sense and antisense oligonucleotides containing the target sequence were purchased from Sigma, annealed, and ligated into pSilencer 3.0 H1 (Ambion; Addgene #100768) and pLKO.1-puro (Addgene #100769). The Nix (Bnip3L pcDNA3.1) and shNix plasmids were a gift from Wafik El-Deiry (Addgene #17467, 17469) [56]. Myc-tagged Nix was generated using Bnip3L pcDNA3.1 as a PCR template and ligating the subsequent amplicon into a pcDNA3 backbone containing an N-terminal Myc tag (EcoRI-XhoI; Addgene #100795). Flag-Bcl-2, Flag-Cb5, an Flag-MaoB plasmids were gifted from Clark Distelhorst (Addgene #18003, 18004, and 18005) [43]. ER-targeted and mitochondrial-targeted Nix constructs were generating by using Myc-Nix, the ER-targeting domain of cytochrome B5 (from Bcl-2-Cb5), or the outer mitochondrial membrane-targeting domain of monoamine oxidase B (from Bcl-2-MaoB) in an overlapping PCR reaction, respectively. Primers were designed to replace the transmembrane domain of Nix (aa 186 to the stop codon) with the ER-targeting domain or the outer mitochondrial membrane-targeting domain (EcoRI-XhoI; Addgene #100756, #100757). pcDNA3.2/V5 mmu-mir-133a-1 was a gift from David Bartel (Addgene plasmid # 26326) [57]. ATeam1.03-nD/nA/pcDNA3 was a gift from Takeharu Nagai (Addgene plasmid #51958) [58]. CMV-mito-CAR-GECO1 and CMV-ER-LAR-GECO1 were a gift from Robert Campbell (Addgene plasmid #46022, 61244) [39, 40]. Lentiviral mito-CAR-GECO1 was generated by inserting EcoRI and XhoI restriction sites into a PCR amplicon and ligated into pLenti-puro (Addgene #100765). The miR-133a LNA mimic and control oligonucleotide were purchased from Exiqon, described previously [31].
Cell culture, transductions, and transfections
All cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone), containing penicillin, streptomycin, and 10% fetal bovine serum (Hyclone) at 37 degrees Celsius and 5% CO2. The H9c2 cell line was transfected using Qiagen’s Polyfect reagent or JetPrime reagent, as per the manufacturer’s instructions. Rat neonatal ventricular myocytes were isolated with the Pierce Primary Cardiomyocyte Isolation Kit (#88281), as per manufacturer’s protocol. Knockdown of myocardin in primary cardiomyocytes was performed with rat myocardin shRNA lentiviral particles (Santa Cruz; sc-72228-V), or a lentivirus generated in pLKO.1-puro (above), where control shRNA lentiviral particles-A (sc-108080) were used to control for transduction.
Fluorescent staining
TMRM, Calcein-AM, ethidium homodimer-1, MitoView Green, and Hoechst 33342 were purchased from Biotium. MitoSox was purchased from Life Technologies. MPTP imaging was performed by quenching the cytosolic Calcein-AM signal with 5 µM cobalt chloride during the incubation period. All imaging, including ER and mitochondrial calcium imaging, was done on an Olympus IX70 inverted microscope with QImaging Retiga SRV Fast 1394 camera using NIS Elements AR 3.0 software. Quantification, scale bars, and processing including background subtraction, was done on ImageJ software. ER-LAR-GECO and mito-carmine imaging was performed 48-hours following transfection in H9c2 cells and 18-hours after FSK-I stimulation (additional details are included in figure legends). Calcium imaging in ventricular myocytes was done 48-hours after viral transduction. ATeam 1.03 was imaged with a CFP and FRET (CFP-YFP) cube-set on an Olympus IX70 inverted microscope with QImaging. Images were analyzed and quantified using ImageJ software.
Immunoblotting
Protein extractions were achieved using a RIPA lysis buffer containing proteases inhibitors and phosphatase inhibitors (Santa Cruz). From tissue, protein was extracted in RIPA buffer by homogenization. Protein concentrations were determined using a Bio-Rad Protein assay kit. Fractionation studies utilized the Qiagen QProteome mitochondrial isolation kit. Extracts were resolved using SDS-PAGE and transferred to a PVDF membrane. Immunoblotting was carried out using appropriate primary antibody in 5% powdered milk or BSA in TBST. Appropriate horseradish peroxidase-conjugated secondary antibody (Jackson; 1:4000) was used in combination with chemiluminescence to visualize bands. The following antibodies were used: Myocardin (Santa Cruz sc-33766), Nix (CST #12396), PGC-1 (Santa Cruz sc-13067), and Actin (Santa Crux sc-1616).
Mitochondrial respiration
Mitochondrial respiration was determined on a Seahorse XF-24 Extracellular Flux Analyzer, as described previously [31]. Calculated respiration rates were determined as per manufacturer’s instructions (Mito Stress Kit; Seahorse).
Quantitative PCR
Total RNA was extracted from pulverized frozen tissue or from cultured cells by TRIzol method. For microRNA analysis all primers were purchased from IDT. cDNA was generated using QScript MicroRNA cDNA Synthesis kit (IDT) and q-RT-PCR performed using PerfeCTa SYBR green super mix on a ABI 7500 Real-Time PCR Instrument. microRNA was normalized to RNU6 expression (Quanta). For mRNA analysis, following column purification using Qiagen RNeasy kit and DNase treatment, cDNA was generated with QScript cDNA super mix (Quanta BioSciences) and analyzed as described above, and normalized to β-actin expression. The primers used were: Myocardin FWD 5′-GTGTGGAGTCCTCAGGTCAAAC-3′; REV 5′-TGATGTGTTGCGGGCTCTT-3′; Beta Actin FWD 5′-CTGTGTGGATTGGTGGCTCTA-3′; REV: 5′-AAAACGCAGCTCAGTAACAGTCC-3′. The SA Biosciences Cell Death Pathway Finder Array was purchased from Qiagen and used manufacturer’s protocol.
Coronary Ligation and doxorubicin-induced cardiotoxicity
All procedures in this study were approved by the Animal Welfare Committee of the University of Manitoba, which adheres to the principles for biomedical research involving animals developed by the Council for International Organizations of Medical Sciences. In the in vivo rodent model of myocardial infarction, the left coronary artery of Sprague Dawley rats was ligated approximately 2 mm from its origin, while sham operated rats serve as control [44, 59]. Following recovery for 4 or 8 weeks, animals are anesthetized, the heart excised, and the left anterior descending territory dissected for scar tissue and viable border-zone myocardium. For Dox-induced cardiotoxicity, C57BL6 mice received weekly intraperitoneal injections of Dox (8 mg/kg body weight) for 4 weeks [45]. Mice were anesthetized and the heart excised for protein and RNA analysis.
Change history
22 October 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–7.
Gordon JW. Regulation of cardiac myocyte cell death and differentiation by myocardin. Mol Cell Biochem. 2018;437:119–131. https://doi.org/10.1007/s11010-017-3100-3.
Creemers EE, Sutherland LB, Oh J, Barbosa AC, Olson EN. Coactivation of MEF2 by the SAP domain proteins myocardin and MASTR. Mol Cell. 2006;23:83–96.
Oh J, Wang Z, Wang D-Z, Lien C-L, Xing W, Olson EN. Target gene-specific modulation of myocardin activity by GATA transcription factors. Mol Cell Biol. 2004;24:8519–28.
Wang C, Cao D, Wang Q, Wang D-Z. Synergistic activation of cardiac genes by myocardin and Tbx5. PLoS ONE 2011;6:e24242.
Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, et al. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–62.
Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–86.
Nam Y-J, Song K, Luo X, Daniel E, Lambeth K, West K, et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc Natl Acad Sci USA. 2013;110:5588–93.
Li S, Wang D-Z, Wang Z, Richardson JA, Olson EN. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc Natl Acad Sci USA. 2003;100:9366–70.
Huang J, Elicker J, Bowens N, Liu X, Cheng L, Cappola TP, et al. Myocardin regulates BMP10 expression and is required for heart development. J Clin Invest. 2012;122:3678–91.
Huang J, Lu MM, Cheng L, Yuan L-J, Zhu X, Stout AL, et al. Myocardin is required for cardiomyocyte survival and maintenance of heart function. Proc Natl Acad Sci USA. 2009;106:18734–9.
Barth E, Stämmler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol. 1992;24:669–81.
Kwong JQ, Molkentin JD. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015;21:206–14.
Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, Galluzzi L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016;26:655–67.
Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA. 2013;110:5887–92.
Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife. 2013;2:e00772.
Hom JR, Quintanilla RA, Hoffman DL, de Mesy Bentley KL, Molkentin JD, Sheu S-S, et al. The Permeability Transition Pore Controls Cardiac Mitochondrial Maturation and Myocyte Differentiation. Dev Cell. 2011;21:469–78.
Pérez MJ, Quintanilla RA. Development or Disease: Duality of the Mitochondrial Permeability Transition Pore. Dev Biol 2017. https://doi.org/10.1016/j.ydbio.2017.04.018.
Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44.
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62.
Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8.
Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12:15–22.
Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12:23–34.
Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–46.
Dorn GW II. Nix Nought Nothing: Fairy tale or real deal. J Mol Cell Cardiol. 2011;51:497–500.
Dorn GW, Kirshenbaum LA. Cardiac reanimation: targeting cardiomyocyte death by BNIP3 and NIX/BNIP3L. Oncogene. 2008;27:S158–67.
Chen G, Cizeau J, Vande Velde C, Park JH, Bozek G, Bolton J, et al. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J Biol Chem. 1999;274:7–10.
Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, et al. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–68.
Chen Y, Lewis W, Diwan A, Cheng EH-Y, Matkovich SJ, Dorn GW. Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci USA. 2010;107:9035–42.
Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, et al. Endoplasmic reticulum–mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest. 2009;119:203–12.
Mughal W, Nguyen L, Pustylnik S, da Silva Rosa SC, Piotrowski S, Chapman D, et al. A conserved MADS-box phosphorylation motif regulates differentiation and mitochondrial function in skeletal, cardiac, and smooth muscle cells. Cell Death Dis. 2015;6:e1944.
Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–8.
Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, et al. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. 2010;106:166–75.
Berghe TV, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–47.
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5.
Zong W-X, Thompson CB. Necrotic death as a cell fate. Genes Dev. 2006;20:1–15.
Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, et al. Ca2 + - and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–44.
Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, et al. An expanded palette of genetically encoded Ca2 + indicators. Science. 2011;333:1888–91.
Wu J, Prole DL, Shen Y, Lin Z, Gnanasekaran A, Liu Y, et al. Red fluorescent genetically encoded Ca 2 + indicators for use in mitochondria and endoplasmic reticulum. Biochem J. 2014;464:13–22.
Wu J, Liu L, Matsuda T, Zhao Y, Rebane A, Drobizhev M, et al. Improved orange and red Ca 2 + indicators and photophysical considerations for optogenetic applications. ACS Chem Neurosci. 2013;4:963–72.
Rong Y-P, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell. 2008;31:255–65.
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166:193–203.
Wang NS. Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against bax-induced cell Death. J Biol Chem. 2001;276:44117–28.
Ghavami S, Cunnington RH, Gupta S, Yeganeh B, Filomeno KL, Freed DH, et al. Autophagy is a regulator of TGF-β1-induced fibrogenesis in primary human atrial myofibroblasts. Cell Death Dis. 2015;6:e1696.
Cheung KG, Cole LK, Xiang B, Chen K, Ma X, Myal Y, et al. Sirtuin-3 (SIRT3) protein attenuates doxorubicin-induced oxidative stress and improves mitochondrial respiration in H9c2 cardiomyocytes. J Biol Chem. 2015;290:10981–93.
Du KL, Ip HS, Li J, Chen M, Dandre F, Yu W, et al. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol. 2003;23:2425–37.
Xing W, Zhang T-C, Cao D, Wang Z, Antos CL, Li S, et al. Myocardin induces cardiomyocyte hypertrophy. Circ Res. 2006;98:1089–97.
Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, et al. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–54.
Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW. Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation. 2008;117:396–404.
Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC et al. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med 2002;8:725–30.
Murgia M, Giorgi C, Pinton P, Rizzuto R. Controlling metabolism and cell death: at the heart of mitochondrial calcium signalling. J Mol Cell Cardiol. 2009;46:781–8.
Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–18.
Gordon JW, Pagiatakis C, Salma J, Du M, Andreucci JJ, Zhao J, et al. Protein kinase A-regulated assembly of a MEF2middle dotHDAC4 repressor complex controls c-Jun expression in vascular smooth muscle cells. J Biol Chem. 2009;284:19027–42.
Pagiatakis C, Gordon JW, Ehyai S, Mcdermott JC. A novel RhoA/ROCK-CPI-17-MEF2C signaling pathway regulates vascular smooth muscle cell gene expression. J Biol Chem. 2012;287:8361–70.
Yoshida T, Sinha S, Dandré F, Wamhoff BR, Hoofnagle MH, Kremer BE, et al. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ Res. 2003;92:856–64.
Fei P, Wang W, Kim S, Wang S, Burns TF, Sax JK, et al. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell. 2004;6:597–609.
Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009.
Kotera I, Iwasaki T, Imamura H, Noji H, Nagai T. Reversible dimerization of Aequorea victoria fluorescent proteins increases the dynamic range of FRET-based indicators. ACS Chem Biol. 2010;5:215–22.
Dixon IM, Lee SL, Dhalla NS. Nitrendipine binding in congestive heart failure due to myocardial infarction. Circ Res. 1990;66:782–8.
Acknowledgements
This work was support by the Natural Science and Engineering Research Council (NSERC) Canada, through a Discovery Grant to JWG. Seed funding was provided by the Thorlakson Foundation Fund and the Children’s Hospital Foundation of Manitoba. MSP is support by NIH R01-HL102968 and R01-HL094520. I.M.D., V.W.D., and G.M.H. are supported by the Heart and Stroke Foundation of Canada. R.K. is a Thorlakson Chair in Surgical Research, and G.M.H. is a Canada Research Chair in Molecular Cardiolipin Metabolism. V.W.D. is the Ken Hughes Young Investigator and the Allen Rouse Fellow of the Manitoba Medical Services Foundation. J.W.G., V.W.D., G.M.H., and R.K. are members of the DEVOTION Research Cluster. W.M. is supported by a scholarship from the Children’s Hospital Foundation of Manitoba and Research Manitoba, J.F. is supported by a studentship from NSERC Canada, and L.K.C. is supported by a CIHR Fellowship. We thank Fuqin Zhu for assistance with the confocal microscopy.
Author Contributions:
Conceptualization, W.M., A.R.W., V.W.D., I.M.D., and J.W.G.; Methodology, Y.H., L.K.C., S.R., K.G.C., V.W.D., A.R.W., G.M.H., R.K., and I.M.D.; Investigation, W.M., J.F., M.M., D.C., J.H., S.R., S.K., K.G.C., A.R.W., J.W.G.; Writing – Original Draft, W.M. and J.W.G.; Writing – Review & Editing, W.M., A.R.W., G.M.H., W.D-.J., V.W.D., I.M.D., M.S.P., J.W.G.; Funding Acquisition, G.M.H., W.D-.J., V.W.D., I.M.D., M.S.P., and J.W.G.; Resources, Y.H., L.K.C., A.R.W., G.M.H., R.K., V.W.D., I.M.D., M.S.P., and J.W.G.; Supervision, Y.H., L.K.C., W.D-.J., V.W.D., I.M.D., M.S.P., and J.W.G.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Mughal, W., Martens, M., Field, J. et al. Myocardin regulates mitochondrial calcium homeostasis and prevents permeability transition. Cell Death Differ 25, 1732–1748 (2018). https://doi.org/10.1038/s41418-018-0073-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0073-z
This article is cited by
-
Mitochondria bridge HIF signaling and ferroptosis blockage in acute kidney injury
Cell Death & Disease (2022)
-
BNIP3L/NIX-mediated mitophagy: molecular mechanisms and implications for human disease
Cell Death & Disease (2021)
-
Misoprostol treatment prevents hypoxia-induced cardiac dysfunction through a 14-3-3 and PKA regulatory motif on Bnip3
Cell Death & Disease (2021)
-
In vivo tracking on longer retention of transplanted myocardin gene-modified adipose-derived stem cells to improve erectile dysfunction in diabetic rats
Stem Cell Research & Therapy (2019)
-
Autophagy modulates temozolomide-induced cell death in alveolar Rhabdomyosarcoma cells
Cell Death Discovery (2018)
