
Abstract
In response to stroke, astrocytes become reactive astrogliosis and are a major component of a glial scar. This results in the formation of both a physical and chemical (production of chondroitin sulfate proteoglycans) barrier, which prevent neurite regeneration that, in turn, interferes with functional recovery. However, the mechanisms of reactive astrogliosis and glial scar formation are poorly understood. In this work, we hypothesized that repulsive guidance molecule a (RGMa) regulate reactive astrogliosis and glial scar formation. We first found that RGMa was strongly expressed by reactive astrocytes in the glial scar in a rat model of middle cerebral artery occlusion/reperfusion. Genetic or pharmacologic inhibition of RGMa in vivo resulted in a strong reduction of reactive astrogliosis and glial scarring as well as in a pronounced improvement in functional recovery. Furthermore, we showed that transforming growth factor β1 (TGFβ1) stimulated RGMa expression through TGFβ1 receptor activin-like kinase 5 (ALK5) in primary cultured astrocytes. Knockdown of RGMa abrogated key steps of reactive astrogliosis and glial scar formation induced by TGFβ1, including cellular hypertrophy, glial fibrillary acidic protein upregulation, cell migration, and CSPGs secretion. Finally, we demonstrated that RGMa co-immunoprecipitated with ALK5 and Smad2/3. TGFβ1-induced ALK5-Smad2/3 interaction and subsequent phosphorylation of Smad2/3 were impaired by RGMa knockdown. Taken together, we identified that after stroke, RGMa promotes reactive astrogliosis and glial scar formation by forming a complex with ALK5 and Smad2/3 to promote ALK5-Smad2/3 interaction to facilitate TGFβ1/Smad2/3 signaling, thereby inhibiting neurological functional recovery. RGMa may be a new therapeutic target for stroke.
Similar content being viewed by others


Introduction
Ischemic stroke is a common disease that endangers human life and health. After stroke, neurological functional recovery is very difficult, which needs to be improved. Reactive astrogliosis and glial scar formation is one of the main cause of functional recovery difficulty after ischemic stroke, as the presence of which inhibits neurite regeneration [1,2,3]. In response to stroke, astrocytes, the most abundant cell type in central nervous system (CNS), convert to a reactive phenotype (so-called reactive astrogliosis) chiefly characterized by up-regulation of glial fibrillary acidic protein (GFAP) and cellular hypertrophy [4,5,6]. Then the reactive astrocytes migrate to the lesion site and proliferate at the lesion margin to become the major components of the glial scar [1, 6]. Reactive astrocytes of the scar reconstruct into a dense meshwork of entangled filamentous processes that serves as a chief physical barrier of neurite outgrowth. In addition, the reactive astrocytes synthesize and deposit into the extracellular matrix (ECM) abundant amounts of chondroitin sulfate proteoglycans (CSPGs) such as neurocan and phosphacan, an important class of extrinsic growth-inhibitory factors. The high concentrations of CSPGs form a chemical barrier to suppress neurite growth [1, 7]. Thus, the reactive astrocytes of the formed glial scar act as a main constraint of neurite regeneration, thereby inhibiting functional recovery of stroke [1, 2]. Accordingly, a deeper understanding of mechanisms underlying reactive astrogliosis and glial scar formation may therefore help to identify novel therapeutic targets in order to facilitate neurological functional recovery following stroke.
Repulsive guidance molecule a (RGMa) is a glycosylphosphatidylinositol-anchored membrane protein which belongs to repulsive guidance molecule (RGM) family. RGMa is involved in many physiological and pathological processes of the central nervous system [8,9,10]. Our previous study found that RGMa expression was enhanced in the ischemic area in a rat middle cerebral artery occlusion/reperfusion (MCAO/R) model. Knockdown of RGMa promoted neurite regeneration [11, 12]. However, the function of RGMa in reactive astrogliosis and glial scar formation, which plays an important role in neurite regrowth inhibition, has not yet been explored.
It has been reported that transforming growth factor β1 (TGFβ1) could stimulate RGMa expression [13]. TGFβ1, which is rapidly upregulated after CNS injury, is a key regulator initiating reactive astrogliosis and glial scar formation [1, 6, 14, 15]. TGFβ1 promotes astrocyte cellular hypertrophy, GFAP expression, migration, and CSPGs deposition by activating its type I receptor activin receptor-like kinase 5 (ALK5) to phosphorylate its canonical Smad2/3 pathway [15,16,17,18,19]. Based on these findings, we hypothesized that after ischemic stroke, RGMa facilitates reactive astrogliosis and glial scar formation through TGFβ1/Smad2/3 signaling, thereby inhibiting functional recovery.
Here we first employed a rat MCAO/R model and found that RGMa contributes to reactive astrogliosis and glial scarring that, in turn, interferes with functional recovery. Furthermore, by culturing primary astrocytes, we showed that RGMa promotes reactive astrogliosis and glial scar formation through TGFβ1/Smad2/3 signaling. RGMa forms a molecular complex with ALK5 and Smad2/3 to facilitate ALK5-Smad2/3 interaction, thereby facilitating TGFβ1-induced phosphorylation of Smad2/3.
Results
RGMa expression is upregulated in reactive astrocytes of glial scar after MCAO/R
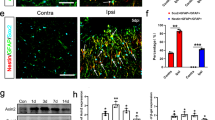
We first examined RGMa expression after MCAO/R. Western blot results showed that RGMa protein expression was increased in ipsilateral hemisphere 7 and 14 days after MCAO/R (Fig. 1a). Immunostaining showed that RGMa was barely detectable in brains of the normal group. After MCAO/R, RGMa was expressed by multiple types of cells in the ischemic areas, including GFAP+ reactive astrocytes, NeuN+ neurons, Nestin+ neural stem/progenitor cells, CC1+ oligodendrocytes, NG2+ oligodendrocyte progenitor cells, Iba1+ microglia/macrophages, CD31+ endothelial cells, and α smooth muscle actin (αSMA)+ vascular smooth muscle cells (Fig. 1c and fig. S1, A to G). In contrast, RGMa was not co-stained with ECM molecules fibronectin and collagen I (fig. S1, H and I). With respect to astrocytes in particular, which we focused on in the present study, RGMa was strongly expressed by the reactive astrocytes that have a strong GFAP immunoreactivity and a hypertrophic morphology in the glial scar of the ischemic areas at 7 and 14 days following MCAO/R. But in the normal group, astrocytes barely expressed RGMa (Fig. 1c). These results suggest that RGMa expression is upregulated in reactive astrocytes of glial scar after MCAO/R.

RGMa expression is increased in reactive astrocytes of glial scar after MCAO/R. a Western blot analysis for RGMa expression in the ipsilateral hemisphere at day 7 and 14 after MCAO/R (n = 3). NS, no significance. Data in bar graphs are means ± SEM; one-way ANOVA with Bonferroni post hoc test, **p < 0.01. b Schematic representation of a coronal brain section. The gray area indicates ischemic core. The square fields represent observed regions. c Representative photographs of immunofluorescence staining for RGMa (green) expression in reactive astrocytes (GFAP, red) in the glial scar at day 7 and 14 after reperfusion. Insets show higher magnification images of representative cells. One representative panel per group out of three rats is shown. Scale bar, 50 μm
RGMa mediates reactive astrogliosis and glial scar formation in rats after MCAO/R
To investigate the role of RGMa in reactive astrogliosis and glial scar formation, the RGMa-specific inhibitor six fibronectin type III (6FNIII) or RGMa-specific recombinant adenovirus rAd-shRGMa was injected into the right lateral ventricle of the experimental rats. 6FNIII is a peptide that inhibits RGMa activity by interacting with RGMa [20, 21]. We have previously shown that genetic knockdown of RGMa after MCAO/R increases collapsing response mediator protein-2 (CRMP-2) protein expression and improve the scores of staircase test, a behavior assessment used to assess fine motor function [11, 12]. Here, we found that the medium (0.5 mg/ml) and high (1.0 mg/ml) concentrations of 6FNIII produced similar increases in CRMP-2 expression (fig. S2A) and in the number of retrieved pellets in the staircase test (fig. S2B), and these increases were superior to those observed at the low 6FNIII concentration (0.25 mg/ml). For viral infection, the high titer group (2.5 × 1010 pfu/ml) of rAd-shRGMa was shown to infect the ipsilateral ischemic brain and inhibit RGMa expression most effectively (fig. S2, C and D). Therefore, 0.5 mg/ml of 6FNIII and 2.5 × 1010 pfu/ml of rAd-shRGMa were chosen to be used in the following in vivo study. In addition, phosphate-buffered saline (PBS) or rAd-HK (2.5 × 1010 pfu/ml) was also administrated into the right lateral ventricle as controls.
To examine the role of RGMa in astrogliosis and glial scar formation, we performed a series of experiments with the timeline denoted in Fig. 2a. First, we detected morphology of GFAP+ cells and GFAP expression by immunostaining. Morphologically, GFAP+ cells in 6FNIII or rAd-shRGMa group were less hypertrophic than GFAP+ cells in MCAO/R group. Furthermore, compared with MCAO/R group, rats treated with 6FNIII or rAd-shRGMa showed an obvious reduction in GFAP immunoreactivity at 14 days following MCAO/R (Fig. 2,b, c). Likewise, western blot confirmed that 6FNIII and rAd-shRGMa treatment significantly attenuated GFAP expression at 7 and 14 days post stroke (Fig. 2d). Similar effects were observed with the deposition of CSPGs neurocan and phosphacan. MCAO/R led to an increase in neurocan and phosphacan immunoreactivity at 7 and 14 days, and these increases were inhibited by 6FNIII or rAd-shRGMa treatment (Fig. 2e–h). Together, these results suggest that both 6FNIII and rAd-shRGMa treatment reduce astrocyte activation and glial scarring after MCAO/R, indicating a key role for RGMa in these processes.

RGMa inhibition reduces MCAO/R-induced reactive astrogliosis and glial scar formation in rats and promotes neurological function recovery. a Timeline of experimental design and animal group classification. WB, Western blot. b Representative fluorescence microscope images showing GFAP expression in tissue sections 14 days post reperfusion. Images are representative of three rats per treatment. (i–iii) Composition of low magnification micrographs (× 40). The dotted lines indicate the boundary of glial scar. Scale bar, 1000 μm. (iv–xv) Higher-magnified view of the squared region (R1-R6) in (i–iii) respectively; × 100 (iv–ix), × 200 (x–xv). DAPI (blue) was used to stain cellular nuclei. Scale bar, 100 μm. c Quantification of GFAP expression at R1-R6 (× 200) (n = 3). IR, immunoreactivity. **p < 0.01. d Western blot analysis for GFAP expression in the ipsilateral hemisphere of rats 7 and 14 days after reperfusion (n = 3). **p < 0.01 vs MCAO/R, PBS, and rAd-HK groups. Representative micrographs showing anti-neurocan e and anti-phosphacan g staining in tissue sections 14 days after MCAO/R. Scale bar, 100 μm. Quantification of neurocan f and phosphacan h expression (n = 3). IR, immunoreactivity. **p < 0.01. Staircase test (I) and cylinder test (J) were performed at 2 days before as well as 7, 10, and 14 days after MCAO/R (n = 5). *p < 0.05 vs MCAO/R, PBS, and rAd-HK groups; **p < 0.01 vs MCAO/R, PBS, and rAd-HK groups. Data in bar and line graphs are means ± SEM (c, d, f, h, i, and j); one-way ANOVA with Bonferroni post hoc test
RGMa inhibition promotes neurological function recovery
As glial scar is a main inhibitor of functional recovery [1], we used staircase test and cylinder test (asymmetries in forelimb use for postural support) to evaluate neurobehavioral functions at 2 days before as well as 7, 10, and 14 days following MCAO/R. 6FNIII and rAd-shRGMa treatment significantly attenuated the MCAO/R-induced decrease in pellet retrieval in staircase test and the increase in asymmetry score in cylinder test 7, 10, and 14 days after reperfusion (Fig. 2i, j). Hence, pharmacological- and genetic-induced loss of function of RGMa produced congruent results, promoting recovery of neurological function.
TGFβ1 upregulates RGMa expression in primary astrocytes
We next studied if RGMa expression in astrocytes is modulated by TGFβ1. TGFβ1, which is rapidly upregulated after CNS injury including stroke, is a master regulator of reactive astrogliosis and glial scar formation [1, 14]. The levels of RGMa were increased in primary cultured astrocytes after 1–100 ng/ml TGFβ1 treatment for 3 days (Fig. 3a). And it has been confirmed that the reactive activation of primary cultured astrocytes could be induced by 10 ng/ml TGFβ1 treatment for 3 days [18, 22], so we used this concentration in subsequent experiments. Co-immunostaining analysis confirmed that astrocytes treated with TGFβ1 (10 ng/ml for 3 days) had higher RGMa and GFAP immunoreactivity with a hypertrophic morphology (Fig. 3b). Furthermore, the upregulation of RGMa induced by TGFβ1 was abolished by pretreatment with an antagonist of the ALK5, SB431542, which inhibits the TGFβ1 downstream pathways (Fig. 3c, d). These astrocytes pretreated with SB431542 also displayed reduced GFAP immunoreactivity and were less hypertrophic. These data indicate that RGMa can be upregulated by TGFβ1, concomitant with activation of astrocytes.

TGFβ1 stimulates RGMa protein expression in primary astrocytes. a Western blot analysis for RGMa expression in primary astrocytes treated with or without TGFβ1 (1–100 ng/ml) for 3 days (n = 3). b Representative fluorescence photographs of RGMa (green) and GFAP (red) expression in cultured astrocytes in the absence or presence of TGFβ1 (10 ng/ml for 3 days). Similar results were obtained using two additional cell batches. c Western blot analysis for RGMa expression in astrocyte cultures pretreated with or without SB431542 (30 μM for 1 h) before TGFβ1 (10 ng/ml for 3 days) stimulation (n = 3). d Immunostaining of RGMa (green) and GFAP (red) expression in cultured primary astrocytes pretreated with or without SB431542 (30 μM for 1 h) before TGFβ1 (10 ng/ml for 3 days) stimulation. One representative panel per group out of three independent cell cultures is shown. Scale bar, 100 μm. Data in bar graphs are means ± SEM a and c; one-way ANOVA with Bonferroni post hoc test, **p < 0.01
RGMa regulates key steps of TGFβ1-induced reactive astrogliosis and glial scar formation
To further explore the role of RGMa in TGFβ1-induced astrogliosis and glial scar formation, we infected rAd-shRGMa into cultured astrocytes (fig. S3A), which led to a significant reduction in RGMa levels (fig. S3B). Because the process of reactive astrogliosis and glial scar formation normally involves cellular hypertrophy, upregulation of GFAP, cell migration and proliferation, and secretion of CSPGs, we evaluated the role of RGMa in each of these aspects [1, 4]. Morphologically, TGFβ1-induced astrocyte hypertrophy was attenuated in the presence of rAd-shRGMa infection (Fig. 4a). Moreover, rAd-shRGMa infection also abolished the upregulation of GFAP expression induced by TGFβ1 treatment (Fig. 4a, b). Next, the transwell cell culture chambers were used to investigate the function of RGMa on TGFβ1-induced astrocyte migration. The results showed that TGFβ1 notably stimulated the migration of astrocytes, whereas this effect was significantly inhibited in the presence of rAd-shRGMa (Fig. 4c, d). As it remains unclear whether TGFβ1 affects astrocyte proliferation, we studied the effects of TGFβ1 and rAd-shRGMa on cell proliferation using CCK8 assay. Neither TGFβ1 nor rAd-shRGMa significantly altered astrocyte proliferation (Fig. 4e). Finally, we found that TGFβ1-triggered secretion of neurocan and phosphacan in astrocyte supernatants was attenuated by rAd-shRGMa infection (Fig. 4f, g). Collectively, these results show that RGMa is a critical modulator in TGFβ1-induced reactive astrogliosis and glial scar formation.

Knockdown of RGMa reduces key steps of TGFβ1-triggered reactive astrogliosis and glial scar formation. a Immunofluorescence of GFAP (red) in culture of primary astrocytes infected with rAd-shRGMa or rAd-HK 3 days before TGFβ1 (10 ng/ml for 24 h) treatment. One representative panel per group out of three independent experiments is shown. b Western blot analysis of GFAP expression in primary astrocytes infected with rAd-shRGMa or rAd-HK, followed by TGFβ1 (10 ng/ml for 3 days) treatment (n = 3). c, d The migration capability of astrocytes determined by a transwell chamber assay. The astrocytes were cultured in different conditions (Control, TGFβ1, TGFβ1 + rAd-shRGMa, and TGFβ1 + rAd-HK). c Representative fluorescence microscope images of the lower surface of the filter. The cells were stained with DAPI (blue). d Quantification of the cell number of astrocytes that migrated to the lower side of the filter in each group (n = 3). e The proliferation ability of astrocytes measured by a CCK8 assay (n = 3). The cultured astrocytes were infected with rAd-shRGMa or rAd-HK before exposure to TGFβ1 (10 ng/ml for 3 days). NS, no significance. f, g Western blot analysis of neurocan f and phosphacan g expression in the supernatant of cultured astrocytes infected with rAd-shRGMa or rAd-HK 3 days before TGFβ1 (10 ng/ml for 3 days) treatment (n = 3). Scale bar, 100 μm. Data in bar graphs are means ± SEM (b, d, e, f, g); one-way ANOVA with Bonferroni post hoc test, **p < 0.01
RGMa mediates TGFβ1-induced phosphorylation of Smad2/3 signaling
The canonical Smad2/3 signaling pathway has been confirmed to be responsible for TGFβ1-induced reactive astrogliosis and glial scar formation [1, 18]. To determine the effect of RGMa on Smad2/3 pathway, the phosphorylation of Smad2 and Smad3 in astrocytes was examined by western blot. The infection of rAd-shRGMa in cultured astrocytes markedly inhibited the TGFβ1-induced phosphorylation of Smad2 and Smad3 (Fig. 5a, b), indicating that RGMa regulates TGFβ1-induced reactive astrogliosis and glial scar formation through Smad2/3 signaling pathways.

RGMa inhibition abolishes TGFβ1-induced phosphorylation of Smad2/3. a, b Western blot analysis for phosphorylation and expression of Smad2 a and Smad3 b in primary astrocyte cultures infected with rAd-shRGMa or rAd-HK 3 days before exposure to TGFβ1 (10 ng/ml for 3 days; n = 3). NS, no significance. Data in bar graphs are means ± SEM (a, b); one-way ANOVA with Bonferroni post hoc test, **p < 0.01
RGMa forms a complex with ALK5 and Smad2/3 to facilitate phosphorylation of Smad2/3 by ALK5
We next investigated how RGMa activates Smad2/3 signaling in astrocytes. As Smad2/3 is phosphorylated by TGFβ1 receptor ALK5 directly, Smad2/3 phosphorylation could be regulated by alteration of ALK5 expression or ALK5-Smad2/3 interaction [23, 24]. As shown in Fig. 6a, the expression levels of ALK5 were not affected by the knockdown of RGMa. In contrast, the ALK5 kinase inhibitor SB431542 attenuated the upregulation of RGMa induced by TGFβ1 (Fig. 3c, d), indicating that TGFβ1 regulates RGMa expression through ALK5. The effects of RGMa on the ALK5-Smad2/3 interaction were examined by a co-immunoprecipitation assay. Reducing RGMa expression in astrocytes significantly weakened the ability of ALK5 to form a complex with Smad2/3 in response to TGFβ1 (Fig. 6a). We also detected RGMa in the ALK5 immunoprecipitates. The amount of RGMa that associated with ALK5 was increased upon TGFβ1 treatment and this effect was reduced by rAd-shRGMa infection, which is in line with the alteration of RGMa expression in cell lysates. In view of these results, we speculated that RGMa may form a complex with ALK5 and Smad2/3 to enhance their interactions to modulate Smad2/3 activity. As expected, RGMa is able to associate with Smad2/3 and ALK5 in astrocytes (Fig. 6b). The profiles of the amount of Smad2/3 and ALK5 associating with RGMa and the overall RGMa expression in astrocytes were similar in that RGMa were enhanced after TGFβ1 stimulation and it were alleviated by RGMa knockdown. Taken together, these data suggest that RGMa facilitates ALK5-induced Smad2/3 phosphorylation by forming a complex with both ALK5 and Smad2/3.

RGMa forms a complex with ALK5 and Smad2/3 to facilitate Smad2/3 phosphorylation by ALK5. a Interaction of ALK5 with Smad2/3 and RGMa in astrocytes analyzed by a co-immunoprecipitation assay. ALK5 was isolated by immunoprecipitation from astrocytes infected with rAd-shRGMa or rAd-HK before exposure to TGFβ1 (10 ng/ml for 3 days), and ALK5-associated proteins were analyzed by Western blot with the indicated antibodies (top panels). Controls of protein levels in whole-cell lysates are displayed on the bottom. Similar results were obtained using two additional cell batches. b Association of RGMa with Smad2/3 and ALK5 in primary cultured astrocytes infected with rAd-shRGMa or rAd-HK, followed by TGFβ1 (10 ng/ml for 3 days) stimulation. Smad2/3 and ALK5 that associated with RGMa were isolated by co-immunoprecipitation with anti-RGMa and detected by western blot with the indicated antibodies on the top panels. Controls of protein expression in whole-cell lysates are shown on the bottom. Similar results were obtained using two additional cell batches
Discussion
In this study, we present evidence for the critical role of RGMa in regulation of reactive astrogliosis and glial scar formation and suggest the following working model depicted in Fig. 7. After stroke, TGFβ1 expression is rapidly increased and activates its cell surface receptor ALK5 in astrocyte, which then enhances RGMa expression. RGMa forms a complex with ALK5 and Smad2/3 to promote ALK5-Smad2/3 interaction, facilitating phosphorylation of Smad2/3. By regulating the TGFβ1/Smad2/3 pathway, RGMa mediates astrocyte reactive astrogliosis and glial scar formation. Thus, suppression of RGMa genetically or pharmacologically in vivo reduces reactive astrogliosis and glial scarring and promotes functional recovery after stroke.

Proposed model of RGMa regulation of TGFβ1/Smad2/3 signaling in astrocytes. By binding to a complex of its cell surface type II receptor (RII) and type I receptor activin-like kinase 5 (ALK5), TGFβ1 activate ALK5, leading to RGMa expression. RGMa forms a complex with ALK5 and Smad2/3 to facilitate ALK5-Smad2/3 interaction, promoting phosphorylation of Smad2/3. By regulating TGFβ1/Smad2/3 pathway, RGMa mediates astrocyte reactive astrogliosis and glial scar formation
Initially identified as an axonal repulsive guidance molecule [25], RGMa now has emerged as a molecule which could be expressed by a wide variety of cells and regulates various functions depending on its cellular and environmental context [8, 26, 27]. However, the role of RGMa in stroke has not been elucidated. Our current study shows that RGMa expression is upregulated in a rat MCAO/R model. Multiple types of cells in the ischemic areas express RGMa, including reactive astrocytes, neurons, neural stem/progenitor cells, oligodendrocytes, oligodendrocyte progenitor cells, microglia/macrophages, endothelial cells, and vascular smooth muscle cells. In contrast, we do not find the presence of RGMa in ECM components fibronectin and collagen I. Based on these results, we speculate that RGMa play multiple roles in stroke, which is associated with the cell types expresses it. Our present study focuses on the role of RGMa in reactive astrogliosis and glial scar formation, which has not been studied previously. We first indicate that RGMa, the expression of which is strongly increased in reactive astrocytes of the glial scar, regulates reactive astrogliosis and glial scarring after MCAO/R. Furthermore, we find that RGMa promotes TGFβ1-induced astrocyte cellular hypertrophy, upregulation of GFAP, migration, and secretion of CSPGs, all of which are the key steps of reactive astrogliosis and glial scar formation [1, 4]. It should be noted that in response to CNS injury, a portion of reactive astrocytes will proliferate modestly, which also contribute to the glial scar formation [1, 6]. However, we find that neither RGMa nor TGFβ1 alter the proliferation of astrocytes. The effect of TGFβ1 on astrocyte proliferation remains debatable. Although our results are consistent with a previous study showing that TGFβ1 do not affect astrocyte proliferation [16], others have reported that TGFβ1 suppresses the proliferation ability of astrocytes [28, 29]. This discrepancy may be attributed to the different experimental conditions or methods used to assess proliferation.
The present study demonstrates that suppression of RGMa promotes motor function recovery in a rat MCAO/R model. These findings are consistent with our previous results demonstrating that RGMa inhibits axon growth and function recovery [11, 12]. In vitro studies have furthermore shown that RGMa inhibits neurite outgrowth and induces growth cone collapse, suggesting that RGMa acts as a growth-inhibitory factor to suppress neurite regeneration and functional recovery [11, 30]. The results of our present study suggest that promotion of reactive astrogliosis and glial scar formation may be another important mechanism by which RGMa inhibits neurite regrowth and functional recovery. The glial scar, mainly formed by reactive astrocytes, is widely regarded as a critical impediment of neurite regeneration and neurological functional recovery after CNS injury [1, 2]. Although the glial scar may have some beneficial effects in early stage of injury, such as prevention of the spread of inflammation, these beneficial effects are counteracted by the inhibition of neurite regeneration during the later stage [31]. When reactive astrogliosis and glial scar formation are experimentally prevented after CNS injury, such as SCI and MCAO/R, the functional recovery and neurite regrowth are enhanced [3, 32,33,34], which is in line with our results. It should be noted that a recent report using photothrombosis to induce focal cortical ischemia shows that axon growth and function recovery are reduced in mice with gene knockout of GFAP and vimentin, the two major astrocytic intermediate filament proteins [35]. Unlike MCAO/R and SCI model, the photothrombosis model may activate some astrocytes into a subpopulation of lacking GFAP and vimentin but still secret CSPGs to inhibit neurite growth. Further study is needed to reveal it.
What induces RGMa expression in reactive astrocytes? A previous report shows that TGFβ1 stimulates RGMa expression in human astrocytes [13]. Similarly, we find that RGMa expression is upregulated by TGFβ1 in rat astrocytes. Furthermore, we show that RGMa upregulation is concomitant with activation of astrocytes. In astrocytes, TGFβ1 mediates its effect by binding to the constitutively active TβRII to cause heterodimerization and phosphorylation of the ALK5 and the subsequent activation of its downstream pathways [23, 24, 36]. Here we show that ALK5 is required for RGMa expression. In addition, it is well known that CNS injury, including stroke, would increase the expression of TGFβ1 [1, 14, 37]. Taken together, we speculate that after stroke, the rapidly elevated expression of TGFβ1 may stimulate RGMa expression in astrocytes through ALK5 activation. It should be noted that this TGFβ1-dependent RGMa expression appears to be cell type-specific, given that it has been reported that RGMa do not mediate TGFβ signaling in HEK 293 cells and LLC-PK1 cells [38].
TGFβ1 acts through a multitude of pathways to regulate a wide array of cellular functions [39]. The canonical Smad2/3 pathway has been widely confirmed to mediate the TGFβ1-induced reactive astrogliosis and glial scar formation [15, 18, 19, 40]. However, a recent study indicates that TGFβ1 induces CSPGs expression through non-Smad-dependent activation of phosphatidylinositol 3-kinase/Akt signaling [41]. These contrary results may arise from the differences of experimental conditions or animal species/strains used to obtain astrocytes. Our present study shows that knockdown of RGMa reduces not only TGFβ1-triggered reactive astrogliosis and glial scarring, but also the phosphorylation of Smad2/3 induced by TGFβ1, indicating that RGMa facilitates TGFβ1-induced reactive astrogliosis and glial scar formation via the canonical Smad2/3 signaling.
TGFβ1 activates Smad2/3 signaling through the interaction between ALK5 and Smad2/3. The activated ALK5 phosphorylates Smad2/3, which next complexes with Smad4 and co-translocates into the nuclei to regulate target gene expression [24]. Recruitment of Smad2/3 to ALK5 can be regulated by several proteins, such as Smad anchor for receptor activation [24, 42]. In this investigation, we identify a novel ALK5-Smad2/3 interaction regulation protein that RGMa enhances their interaction by forming a complex with them, thereby modulating TGFβ1/Smad2/3 activity.
Previous studies have shown that RGMa exerts its function through two molecular mechanisms. First, RGMa could bind to the transmembrane protein neogenin, a member of the immunoglobulin superfamily, to regulate multiple downstream signaling, including RhoA, PKC, and focal adhesion kinase [43,44,45]. RGMa-neogenin has been reported in many processes that RGMa mediates, such as neurite growth [43], cell migration [27], and cell death [46]. Secondly, RGMa could function as a bone morphogenetic protein (BMP) co-receptor [47]. BMPs are a large subgroup of the TGFβ superfamily and exhibit a diverse array of biological effects on various cell types, including cell proliferation, differentiation, chemotaxis, and apoptosis. Similar to TGFβs, BMPs transduce their signals by binding to their type I and II serine/threonine kinase receptors to induce Smad1/5/8-dependent and non-Smad dependent signalings [23, 48]. Though the biological function of BMP signaling has not been clearly linked to RGMa, RGMa has been confirmed to bind to BMP2, BMP4, and BMP type I receptor ALK6, leading to enhancement of BMP signaling [38, 48]. Our present study reveals a new molecular mechanism of RGMa that RGMa regulates reactive astrogliosis and glial scarring through forming a complex with ALK5 and Smad2/3 to facilitate TGFβ1-induced Smad2/3 phosphorylation. It is possible that RGMa mediates an orchestrated cross-talk among TGFβ, neogenin, and BMP pathways. Further work will be required to clarify the relationships and interactions among RGMa, neogenin, BMPs, and TGFβs.
In summary, our findings indicate that RGMa may have a critical role in reactive astrogliosis and glial scar formation after stroke. TGFβ1, which is rapidly upregulated after stroke, induces RGMa expression in reactive astrocytes through ALK5. RGMa promotes reactive astrogliosis and glial scar formation by forming a molecular complex with ALK5 and Smad2/3 to facilitate ALK5-Smad2/3 interaction, thereby facilitating TGFβ1-induced phosphorylation of Smad2/3. Inhibition of RGMa suppresses reactive astrogliosis and glial scar formation, promoting functional recovery after stroke. RGMa may be a novel target for stroke treatment.
Materials and methods
Experimental animals
Adult male Sprague–Dawley (SD) rats (male, 200–250 g) and neonatal (1–3 day old) SD rats were supplied by Laboratory Animal Center of Chongqing Medical University (Chongqing, China). All animals were maintained on a 12 h light and dark cycle with free access to water and food. Adequate measures were taken to minimize pain or discomfort during surgeries. All animal procedures were approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University.
In vivo 6FNIII and adenovirus administration
The rats were anesthetized with 3.5% chloral hydrate (350 mg/kg) and placed in a stereotaxic apparatus. 6FNIII is an inhibitor of RGMa (provided by Professor Philippe P. Monnier, Krembil Research Institute, University Health Network, Toronto, Canada). It was injected into the right lateral ventricular (anterior–posterior −1.1 mm, medial–lateral −1.5 mm, dorsal–ventral −4.0 mm from the bregma; 5 μl/injection) 30 min before MCAO/R and 7 days after the first injection. For the initial dose–response experiment of 6FNIII, measurements were made at 7 days after the first injection without the second injection at 7 days. For rats undergoing adenovirus treatment, 5 μl of RGMa-specific recombinant adenovirus rAd-shRGMa or empty carrier recombinant adenovirus rAd-HK (2.5 × 1010 pfu/ml, Wuhan Cell Marker Biotechnology Co., Ltd, Wuhan, China) were injected into the right lateral ventricle (at the same coordinates used for 6FNIII injection). Two days later, MCAO/R was performed in these animals. The efficiency of RGMa knockdown was evaluated with enhanced green fluorescent protein (EGFP) in brain sections and western blot analysis in ipsilateral ischemic tissues. The most appropriate concentration of adenovirus and 6FNIII was selected from testing three doses and was then selected for use in the subsequent study. PBS, which was used to dilute adenovirus and 6FNIII, was also injected into the right lateral ventricle as a control.
Middle cerebral artery occlusion/reperfusion model
MCAO/R was performed using the intraluminal filament method as described previously [49]. In brief, the rat’s right middle cerebral artery was occluded though the coagulated external carotid artery stump with a nylon filament suture rounded by paraffin wax at the head end. After 90 min of occlusion, the filament was withdrawn to allow blood reperfusion. Sham-operated rats underwent the identical surgery except for inserting the suture to the artery. Regional cerebral blood flow (rCBF) was monitored with laser-Doppler flowmetry. Other physiological parameters also were monitored, and body temperature was maintained at 37.0 ± 0.5 °C with a heating pad during the whole process. Rats without at least a 75% reduction of rCBF during MCAO or neurological deficits after reperfusion, evaluated by Longa method [50], were excluded from the study. Rats were killed at the indicated time.
Behavioral assessment
The staircase test and the cylinder test were performed at 2 days before as well as 7, 10, and 14 days after MCAO/R to evaluate neurobehavioral function. And both tests were carried out by an investigator blinded to the experimental conditions.
Staircase Test. The staircase test was used to evaluate fine motor function [51, 52]. Rats were given 1 week of daily training before the MCAO/R surgery. The rats were placed into a staircase apparatus consisting of a chamber with a central platform for the rat to climb onto and a set of seven steps located on either side. Three chow pellets (45 mg) were placed in the wells of each stair. The number of pellets grasped by their affected forelimb was recorded during each 15 min test.
Cylinder Test. The cylinder test was used to assess asymmetries in forelimb use for postural support [52, 53]. Rats were placed in a transparent cylinder (20 cm diameter and 30 cm height). Vertical movements of rats along the cylinder wall were recorded: (a) independent use of the right or left forelimb for contacting the wall, (b) simultaneous use of both the left and right forelimb for contacting the wall, and (c) subsequent use of the other forelimb against the wall following unilateral forelimb placement was scored as ‘both’ as well. A total of 20 movements were recorded during a 10-minute trial. The final score was calculated as 100 × (ipsilateral forelimb use + 1/2 bilateral forelimb use)/total forelimb use.
Culture of primary astrocytes
Primary astrocytes were isolated from the cerebral cortex of neonatal (1–3 days old) SD rats as described previously with slight modifications [16]. In brief, brain cortices were mechanically dissociated, digested with 0.25% trypsin (SH30042.01 Hyclone, Logan, WV, USA), centrifuged, and resuspended into single-cell suspension. The dissociated cells were plated into 25 cm2 flasks and grown in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12; C11330500BT Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; P30-2602 PAN-Biotech, Adenbach, Germany) and 1% Penicillin/Streptomycin (C0222 Beyotime, Shanghai, China) at 37 °C and 5% CO2 atmosphere. The culture media were replaced every 3 days. When confluent, astrocytes were purified by shaking overnight (260 rpm, 37 °C) to remove microglia and oligodendrocytes. After rinsing with PBS, the adherent cells were trypsinized and re-seeded in the culture media. After reaching confluency, the astrocytes were used for experiments. Culture purity was considered satisfactory when more than 95% of the cells were positive for GFAP.
In vitro drug treatment and adenovirus infection
Astrocytes were starved in serum-free media for 24 h prior to drug treatments. To detect RGMa expression, astrocytes were treated with different concentrations of TGFβ1 (240-B R&D Systems, Minneapolis, MN, USA) for 3 days without media change. SB431542 (30 μΜ; S4317 Sigma-Aldrich, Schnelldorf, Germany), an ALK5 kinase inhibitor, or dimethyl sulfoxide, the solvent for SB431542, were added to the media 1 h before TGFβ1 treatments. For knockdown of RGMa expression, astrocytes were treated with media containing rAd-shRGMa (MOI = 4) or rAd-HK (MOI = 8) for 2 h, and then replaced back to the regular media and cultured for 2 days. The efficiency of RGMa knockdown was assessed by EGFP fluorescence and western blot. To determine the involvement of RGMa in reactive astrogliosis, after infecting with adenovirus and serum starvation, astrocytes were incubated with TGFβ1 (10 ng/ml) for 3 days.
Sample preparation and western blot
Sample preparation. To extract protein of ipsilateral hemisphere brain tissues and cultured astrocytes, tissues and cells were lysed in radio immuno precipitation buffer (P0013B Beyotime) with protease and phosphatase inhibitor cocktail (78441 Thermo Fisher, Waltham, MA, USA) and cleared of debris by centrifugation at 14000 g for 15 min at 4 °C. After measuring protein concentrations by bicinchoninic acid (BCA) protein assay reagent (P0010S Beyotime), the lysates were boiled with SDS for 10 min. For detection of astrocyte-secreted neurocan and phosphacan, astrocyte conditioned media was obtained and concentrated by Amicon Ultra-15 Centrifugal Filter Devices (UFC905096 Millipore, Darmstadt, Germany) according to the manufacturer’s instructions. BCA protein assay reagent was used to measure protein content. The samples were treated with 0.01 U/ml chondroitinase ABC (C3667 Sigma-Aldrich) for 3 h at 37 °C to remove chondroitin sulfate sidechains and then heat denatured with SDS at 100 °C for 10 min. All protein samples were stored at −80 °C until needed.
Western blot. Equal amounts of protein were loaded into SDS-PAGE gels. The gels were electrophoresed and transferred to a 0.45 μm polyvinylidene fluoride membrane (IPVH00010 Millipore). The membranes were blocked with 5% nonfat milk or bovine serum albumin, according to the instructions provided by manufactures of primary antibodies, in tris-buffered saline containing 0.1% Tween 20 for 2 h. The membranes were then incubated overnight at 4 °C with the following primary antibodies: anti-RGMa (1:10000, ab169761 Abcam, Cambridge, UK), anti-GFAP (1:1000, 12389 Cell Signaling Technology, Leiden, The Netherland), anti-neurocan (1:5000, N0913 Sigma-Aldrich), anti-phosphacan (1:5000, P8874 Sigma-Aldrich), anti-Phospho-Smad2 (1:1000, 3108 Cell Signaling Technology), anti-Smad2 (1:1000, 5339 Cell Signaling Technology), anti-Phospho-Smad3 (1:1000, 9520 Cell Signaling Technology), anti-Smad3 (1:1000, 9523 Cell Signaling Technology), anti-ALK5 antibody (1:750, ab31013 Abcam), anti-Smad2/3 (1:1000, 8685 Cell Signaling Technology), anti-Phospho-Smad2/3 (1:1000, 8828 Cell Signaling Technology), anti-GAPDH (1:1000, 2118 Cell Signaling Technology), and anti-β-Actin (1:1000, 4970 Cell Signaling Technology). After being washed, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:1000, A0208 goat anti-rabbit IgG or A0216 goat anti-mouse IgG, Beyotime) for 1 h at room temperature. Images were captured and quantified by Fusion FX5 image analysis system (Vilber Lourmat, F-77601 Marne-la-Vallée cedex 3, France).
Co-immunoprecipitation
Co-immunoprecipitation was performed using a cross-link immunoprecipitation kit (26147 Thermo Fisher) according to the manufacturer’s instructions, which could eliminate antibody contamination. First, the Protein A/G Plus Agarose was incubated with anti-RGMa (ab169761 Abcam) or anti-ALK5 antibody (ab31013 Abcam) on a rotator for 1 h at 4 °C. Then, to cross-link the bound antibody, the disuccinimidyl suberate was added to the Protein A/G on the resin and incubated for 1 h at room temperature. After washing and centrifuging, the lysates of astrocytes pre-cleared by the Control Agarose Resin were added into the antibody-crosslinked resin and incubated overnight at 4 °C. After being washed, the antigen was eluted by the Elution Buffer. After adding the Lane Marker Sample Buffer and heating for 5 min, the samples were used for Western blot performed as described above.
Cell migration
Astrocyte migration was assessed by a transwell chamber assay (8 μm pore size; MCEP24H48 Millipore). Astrocytes were cultured in different conditions: (1) DMEM/F-12 (Control); (2) TGFβ1 (10 ng/ml); (3) TGFβ1 (10 ng/ml) + rAd-shRGMa (4) TGFβ1 (10 ng/ml) + rAd-HK. The cells then were trypsinized and added into the upper chamber without FBS (1×105 cells/chamber). To induce migration, the lower chambers were filled with 600 μl serum-free DMEM/F-12 with 10 ng/ml TGFβ1. After 24 h of incubation, free cells and debris were removed from the upper surface of the filters. Then the filters were fixed with ice-cold methanol and subjected to 4',6-diamidino-2-phenylindole (DAPI) (C1005 Beyotime) staining. Finally, the average number of migrated cells on the lower surface of each filter was evaluated by counting four randomly selected microscopic fields at × 100 magnification with a fluorescence microscope (IX71, Olympus, Tokyo, Japan).
Cell proliferation
The proliferation of astrocytes was measured by Cell Counting KIT-8 (CCK8, CK04 Dojindo Laboratories Inc., and Ku mamoto, Kumamoto, Japan) according to the manufacturer’s instructions. After infecting with rAd-shRGMa or rAd-HK, astrocytes were seeded into a 96-well plate and starved in serum-free DMEM/F-12 for 24 h. Then, the cells were incubated with or without 10 ng/ml TGFβ1 for 3 days, after which 10 μl CCK8 solution was added to each well and incubated with cells at 37 °C for 2 h. The absorbance was measured using a microplate reader (MB-530, Heales, Shenzhen, China) at 450 nm. The cell number was correlated with the optical density.
Immunofluorescence
For brain tissue staining, rats were transcardially perfused with cold saline followed by 4% paraformaldehyde under anesthesia. Brain samples were removed, post-fixed, and cut into 10 μm sections. After antigen retrieval using sodium citrate buffer, sections were permeabilized using 0.1% Triton X-100 and blocked with 10% normal donkey serum. Then the sections were incubated overnight at 4 °C with specific primary antibodies as follows: anti-GFAP (1:100, BM0055 Boster, Wuhan, China), anti-RGMa (1:50, ab26287 Abcam), anti-NeuN (1:50, MAB377 Millipore), anti-Nestin (1:100, ab11306 Abcam), anti-CC1 (1:50, ab16794 Abcam), anti-NG2 (1:100, ab50009 Abcam), anti-Iba1 (1:50, NB100-1028 Novus, Littleton, CO, USA), anti-CD31 (1:50, ab64543 Abcam), anti-αSMA (1:50, ab21027 Abcam), anti-fibronectin (1:50, 610077 BD Biosciences, Franklin Lakes, NJ, USA), anti-collagen I (1:100, ab90395 Abcam), anti-neurocan (1:100, N0913 Sigma-Aldrich), and anti-phosphacan (1:100, P8874 Sigma-Aldrich). On the following day, the sections were washed using PBS and incubated with appropriate secondary antibodies conjugated with Alexa Fluor 488 (1:200, A-21206 Thermo Fisher) or 555 (1:200, A0460 Beyotime; or 1:300, A-21432 Thermo Fisher) for 1 h at 37 °C. For cultured astrocyte staining, cells were fixed with 100% methanol for 10 min. After washing with PBS, cells were blocked by 10% normal donkey serum for 1 h at 37 °C and incubated overnight at 4 °C with primary antibodies including anti-RGMa (1:50, ab169761 Abcam) and anti-GFAP (1:100, BM0055 Boster). Cells were washed and for 1 h at 37 °C incubation with secondary antibodies conjugated with Alexa Fluor 488 (1:200, A-21206 Thermo Fisher) or 555 (1:200, A0460 Beyotime). Sections or cells were stained for DAPI (C1005 Beyotime) to visualize nuclei. Images were captured using confocal laser scanning microscope (A1+R, Nikon, Tokyo, Japan) and analyzed by Image J software (National Institutes of Health).
Statistical analyses
All data presented represent results from at least three independent experiments. Quantitative data were expressed as mean ± standard error of the mean (SEM). SPSS 17.0 for windows was used to perform statistical analyses. Statistical differences among groups were compared by using one-way analysis of variance with Bonferroni post hoc test. A difference was considered statistically significant when p < 0.05.
References
Cregg JM, DePaul MA, Filous AR, Lang BT, Tran A, Silver J. Functional regeneration beyond the glial scar. Exp Neurol. 2014;253:197–207.
Liu Z, Chopp M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol. 2016;144:103–20.
Cheon SY, Cho KJ, Song J, Kim GW. Knockdown of apoptosis signal-regulating kinase 1 affects ischaemia-induced astrocyte activation and glial scar formation. Eur J Neurosci. 2016;43:912–22.
Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev. 2014;94:1077–98.
Rossi D. Astrocyte physiopathology: at the crossroads of intercellular networking, inflammation and cell death. Prog Neurobiol. 2015;130:86–120.
Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–47.
Sharma K, Selzer ME, Li S. Scar-mediated inhibition and CSPG receptors in the CNS. Exp Neurol. 2012;237:370–8.
Siebold C, Yamashita T, Monnier PP, Mueller BK, Pasterkamp RJ. RGMs: structural insights, molecular regulation, and downstream signaling. Trends Cell Biol. 2017;27:365–78.
Muramatsu R, Kubo T, Mori M, Nakamura Y, Fujita Y, Akutsu T, et al. RGMa modulates T cell responses and is involved in autoimmune encephalomyelitis. Nat Med. 2011;17:488–94.
Tanabe S, Yamashita T. Repulsive guidance molecule-a is involved in Th17-cell-induced neurodegeneration in autoimmune encephalomyelitis. Cell Rep. 2014;9:1459–70.
Wang T, Wu X, Yin C, Klebe D, Zhang JH, Qin X. CRMP-2 is involved in axon growth inhibition induced by RGMa in vitro and in vivo. Mol Neurobiol. 2013;47:903–13.
Feng J, Wang T, Li Q, Wu X, Qin X. RNA interference against repulsive guidance molecule A improves axon sprout and neural function recovery of rats after MCAO/reperfusion. Exp Neurol. 2012;238:235–42.
Satoh J, Tabunoki H, Ishida T, Saito Y, Arima K. Accumulation of a repulsive axonal guidance molecule RGMa in amyloid plaques: a possible hallmark of regenerative failure in Alzheimer’s disease brains. Neuropathol Appl Neurobiol. 2013;39:109–20.
Doyle KP, Cekanaviciute E, Mamer LE, Buckwalter MS. TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflamm. 2010;7:62.
Yu Z, Yu P, Chen H, Geller HM. Targeted inhibition of KCa3.1 attenuates TGF-beta-induced reactive astrogliosis through the Smad2/3 signaling pathway. J Neurochem. 2014;130:41–49.
Huang XQ, Zhang XY, Wang XR, Yu SY, Fang SH, Lu YB, et al. Transforming growth factor beta1-induced astrocyte migration is mediated in part by activating 5-lipoxygenase and cysteinyl leukotriene receptor 1. J Neuroinflamm. 2012;9:145.
Hsieh HL, Wang HH, Wu WB, Chu PJ, Yang CM. Transforming growth factor-beta1 induces matrix metalloproteinase-9 and cell migration in astrocytes: roles of ROS-dependent ERK- and JNK-NF-kappaB pathways. J Neuroinflamm. 2010;7:88.
Susarla BT, Laing ED, Yu P, Katagiri Y, Geller HM, Symes AJ. Smad proteins differentially regulate transforming growth factor-beta-mediated induction of chondroitin sulfate proteoglycans. J Neurochem. 2011;119:868–78.
Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, et al. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J Neurosci. 2010;30:5843–54.
Tassew NG, Mothe AJ, Shabanzadeh AP, Banerjee P, Koeberle PD, Bremner R, et al. Modifying lipid rafts promotes regeneration and functional recovery. Cell Rep. 2014;8:1146–59.
Tassew NG, Charish J, Seidah NG, Monnier PP. SKI-1 and Furin generate multiple RGMa fragments that regulate axonal growth. Dev Cell. 2012;22:391–402.
Wang H, Katagiri Y, McCann TE, Unsworth E, Goldsmith P, Yu ZX, et al. Chondroitin-4-sulfation negatively regulates axonal guidance and growth. J Cell Sci. 2008;121:3083–91.
Santibanez JF, Quintanilla M, Bernabeu C. TGF-beta/TGF-beta receptor system and its role in physiological and pathological conditions. Clin Sci (Lond). 2011;121:233–51.
Hata A, Chen YG. TGF-beta signaling from receptors to Smads. Cold Spring Harb Perspect Biol. 2016;8:a022061.
Monnier PP, Sierra A, Macchi P, Deitinghoff L, Andersen JS, Mann M, et al. RGM is a repulsive guidance molecule for retinal axons. Nature. 2002;419:392–5.
Schwab JM, Monnier PP, Schluesener HJ, Conrad S, Beschorner R, Chen L, et al. Central nervous system injury-induced repulsive guidance molecule expression in the adult human brain. Arch Neurol. 2005;62:1561–8.
Lah GJ, Key B. Novel roles of the chemorepellent axon guidance molecule RGMa in cell migration and adhesion. Mol Cell Biol. 2012;32:968–80.
Klaver CL, Caplan MR. Bioactive surface for neural electrodes: decreasing astrocyte proliferation via transforming growth factor-beta1. J Biomed Mater Res A. 2007;81:1011–6.
Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-beta 1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol. 1992;117:395–400.
Kitayama M, Ueno M, Itakura T, Yamashita T. Activated microglia inhibit axonal growth through RGMa. PLoS ONE. 2011;6:e25234.
Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–48.
Menet V, Prieto M, Privat A, Gimenez Y, Ribotta M. Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin genes. Proc Natl Acad Sci USA. 2003;100:8999–9004.
Wang Y, Gao Z, Zhang Y, Feng SQ, Liu Y, Shields LB, et al. Attenuated reactive gliosis and enhanced functional recovery following spinal cord injury in null mutant mice of platelet-activating factor receptor. Mol Neurobiol. 2016;53:3448–61.
Li Y, Xu XL, Zhao D, Pan LN, Huang CW, Guo LJ, et al. TLR3 ligand Poly IC attenuates reactive astrogliosis and improves recovery of rats after focal cerebral ischemia. CNS Neurosci Ther. 2015;21:905–13.
Liu Z, Li Y, Cui Y, Roberts C, Lu M, Wilhelmsson U, et al. Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia. 2014;62:2022–33.
Hamby ME, Hewett JA, Hewett SJ. Smad3-dependent signaling underlies the TGF-beta1-mediated enhancement in astrocytic iNOS expression. Glia. 2010;58:1282–91.
O’Brien MF, Lenke LG, Lou J, Bridwell KH, Joyce ME. Astrocyte response and transforming growth factor-beta localization in acute spinal cord injury. Spine (Phila Pa 1976). 1994;19:2321–30.
Babitt JL, Zhang Y, Samad TA, Xia Y, Tang J, Campagna JA, et al. Repulsive guidance molecule (RGMa), a DRAGON homologue, is a bone morphogenetic protein co-receptor. J Biol Chem. 2005;280:29820–7.
Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–39.
Schachtrup C, Ryu JK, Mammadzada K, Khan AS, Carlton PM, Perez A, et al. Nuclear pore complex remodeling byp75(NTR) cleavage controls TGF-beta signaling and astrocyte functions. Nat Neurosci. 2015;18:1077–80.
Jahan N, Hannila SS. Transforming growth factor beta-induced expression of chondroitin sulfate proteoglycans is mediated through non-Smad signaling pathways. Exp Neurol. 2015;263:372–84.
Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95:779–91.
Metzger M, Conrad S, Skutella T, Just L. RGMa inhibits neurite outgrowth of neuronal progenitors from murine enteric nervous system via the neogenin receptor in vitro. J Neurochem. 2007;103:2665–78.
Endo M, Yamashita T. Inactivation of Ras by p120GAP via focal adhesion kinase dephosphorylation mediates RGMa-induced growth cone collapse. J Neurosci. 2009;29:6649–62.
Shabanzadeh AP, Tassew NG, Szydlowska K, Tymianski M, Banerjee P, Vigouroux RJ, et al. Uncoupling Neogenin association with lipid rafts promotes neuronal survival and functional recovery after stroke. Cell Death Dis. 2015;6:e1744.
Koeberle PD, Tura A, Tassew NG, Schlichter LC, Monnier PP. The repulsive guidance molecule, RGMa, promotes retinal ganglion cell survival in vitro and in vivo. Neuroscience. 2010;169:495–504.
Tian C, Liu J. Repulsive guidance molecules (RGMs) and neogenin in bone morphogenetic protein (BMP) signaling. Mol Reprod Dev. 2013;80:700–17.
Xia Y, Yu PB, Sidis Y, Beppu H, Bloch KD, Schneyer AL, et al. Repulsive guidance molecule RGMa alters utilization of bone morphogenetic protein (BMP) type II receptors by BMP2 and BMP4. J Biol Chem. 2007;282:18129–40.
Guo J, Cheng C, Chen CS, Xing X, Xu G, Feng J, et al. Overexpression of fibulin-5 attenuates ischemia/reperfusion injury after middle cerebral artery occlusion in rats. Mol Neurobiol. 2016;53:3154–67.
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91.
Montoya CP, Campbell-Hope LJ, Pemberton KD, Dunnett SB. The “staircase test”: a measure of independent forelimb reaching and grasping abilities in rats. J Neurosci Methods. 1991;36:219–28.
Soleman S, Yip PK, Duricki DA, Moon LD. Delayed treatment with chondroitinase ABC promotes sensorimotor recovery and plasticity after stroke in aged rats. Brain. 2012;135:1210–23.
Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–87.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81271307) and National Key Clinical Specialties Construction Program of China. We greatly thank Professor Raymond C. Koehler (Johns Hopkins University, USA) for manuscript editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, R., Wu, Y., Xie, F. et al. RGMa mediates reactive astrogliosis and glial scar formation through TGFβ1/Smad2/3 signaling after stroke. Cell Death Differ 25, 1503–1516 (2018). https://doi.org/10.1038/s41418-018-0058-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0058-y
This article is cited by
-
Homer1 ameliorates ischemic stroke by inhibiting necroptosis-induced neuronal damage and neuroinflammation
Inflammation Research (2024)
-
Neuroimmune mechanisms and therapies mediating post-ischaemic brain injury and repair
Nature Reviews Neuroscience (2023)
-
Neutralizing RGMa with Elezanumab Promotes Cerebroprotection and Recovery in Rabbit Middle Cerebral Artery Occlusion
Translational Stroke Research (2023)
-
Transcriptomic characterization of the molecular mechanisms induced by RGMa during skeletal muscle nuclei accretion and hypertrophy
BMC Genomics (2022)
-
The neuroprotective mechanism of sevoflurane in rats with traumatic brain injury via FGF2
Journal of Neuroinflammation (2022)
