
Abstract
Caspases, the cysteine proteases which facilitate the faithful execution of apoptosis, are tightly regulated by a number of mechanisms including phosphorylation. In response to cAMP, PKA phosphorylates caspase-9 at three sites preventing caspase-9 activation, and suppressing apoptosis progression. Phosphorylation of caspase-9 by PKA at the functionally relevant site Ser-183 acts as an upstream block of the apoptotic cascade, directly inactivating caspase-9 by a two-stage mechanism. First, Ser-183 phosphorylation prevents caspase-9 self-processing and directly blocks substrate binding. In addition, Ser-183 phosphorylation breaks the fundamental interactions within the caspase-9 core, promoting disassembly of the large and small subunits. This occurs despite Ser-183 being a surface residue distal from the interface between the large and small subunits. This phosphorylation-induced disassembly promotes the formation of ordered aggregates around 20 nm in diameter. Similar aggregates of caspase-9 have not been previously reported. This two-stage regulatory mechanism for caspase-9 has likewise not been reported previously but may be conserved across the caspases.
Similar content being viewed by others


Introduction
Caspase proteases irreversibly cleave protein substrates causing gain or loss of function, coordinating the faithfully execution of apoptosis. Caspases display exquisite substrate specificity, cleaving after an aspartate, glutamate [1, 2], or phosphoserine [3]. Uncontrolled activation of initiators (casp-2/-8/-9) or executioners (casp-3/-6/-7) is lethal, so caspases are synthesized as inactive zymogens (procaspases) prior to apoptosis. Activation relies on recruitment to an activating scaffold (initiators) or cleavage at the intersubunit linker forming mature caspases (executioners) [4]. Most procaspases are homodimers of a protease core, which is cleaved into two large and two small subunits. While executioners are constitutive dimers, initiators equilibrate between monomers and dimers, with casp-9 predominantly monomeric [5, 6]. Upon activation, the highly dynamic active-site loops undergo conformational rearrangements to bind and cleave substrates, thereby initiating apoptosis. Ultimately, caspase activity is intrinsic to apoptosis, so caspase expression and activation is tightly regulated by a number of mechanisms which exert control at various cellular checkpoints. Improper regulation of caspases is associated with diseases ranging from cancer to neurodegeneration [7,8,9]. Thus, caspases are attractive drug targets for apoptosis-associated diseases.
Unlike other classic signaling pathways, apoptosis does not directly utilize kinases, but the related activity of kinases can determine a cell’s susceptibility to death [10,11,12]. All apoptotic caspases are kinase substrates and caspases usually cleave the very kinases that phosphorylate them. Efforts to map kinase-caspase co-regulation and competition are ongoing [10, 12,13,14] and some caspases have multiple phosphorylation sites involving several kinases. Thus, although cell-based studies are fundamental in identifying cognate kinases and in determining the functional endpoint as either caspase inhibition or activation, biochemical and structural studies are essential in identifying critical residues and elucidating molecular details of diverse regulatory mechanisms arising from phosphorylation.
Among caspases, casp-9 is most prone to phosphorylation, which is mediated by multiple kinases in response to specific cellular signals [15]. For example, in many cell types elevated cAMP levels confer protection from apoptosis [16,17,18,19] due to sustained Protein Kinase A (PKA) activity. PKA phosphorylates casp-9 at three sites—Ser-99, Ser-183, and Ser-195—leading to failure of casp-9 activation and suppression of apoptosis [20]. Intriguingly, phosphorylation of these sites was reported not to directly inhibit casp-9. It was instead suggested that PKA phosphorylates a more upstream substrate such as Apaf-1 to prevent casp-9 activation. This observation evokes silent phosphorylation in casp-9. While non-functional phosphorylation does occur in proteins [21, 22], the idea that PKA phosphorylates all three casp-9 sites without functional consequence differs from the sensitivity of other caspases to phosphorylation. Recent studies from our group [23,24,25] and others [26] revealed that caspase activity is directly affected by phosphorylation both orthosterically and allosterically. In addition, the high conservation of phosphorylation sites across species [15, 27] suggests that phosphorylation yields functional effects, as functional phosphorylation sites evolve more slowly than non-functional sites [22]. Given the complexity of multiple phosphorylation sites, differentiating sites critical to inhibition from non-functional sites is paramount for unraveling the dynamic control of casp-9 by phosphorylation.
Here we identify Ser-183 as the functional site leading to direct casp-9 inhibition upon phosphorylation by PKA. Phosphorylation of the casp-9 zymogen prevents substrate binding, whereas phosphorylation of the mature form destabilizes the casp-9 core leading to disassembly and ordered aggregate formation. Understanding the multi-level mechanisms by which phosphorylation controls casp-9 should provide new avenues to tap casp-9’s therapeutic potential in apoptosis-related disease.
Results
Phosphorylation of casp-9 by PKA directly results in inhibition
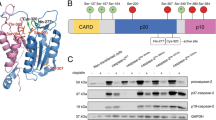
All three reported sites of phosphorylation are in the CARD (caspase activation and recruitment domain) plus large (CARD + Lg) region of casp-9 (Fig. 1a, b). Building on the report that PKA directly phosphorylates casp-9 [20], we tested whether PKA affects procasp-9’s self-processing. Procasp-9 possesses basal activity [5, 28, 29] that allows in trans cleavage, under favorable conditions, to generate mature, active casp-9. In homogeneous preparations of wild-type (WT) procasp-9, the intersubunit linker is rapidly cleaved, generating the CARD + Lg and small (Sm) subunits (Fig. 2a). Addition of PKA with ATP greatly attenuated the rate of procasp-9 self-cleavage (Fig. 2a).

Sites of PKA phosphorylation in casp-9. a Domain architecture of casp-9 showing the caspase activation and recruitment domain (CARD) (yellow green) and the protein core composed of the large (dark green) and small (light green) subunits connected by an intersubunit linker with three cleavage sites indicated by arrows: E306 (minor, self-cleavage), D315 (major, self-cleavage, and by casp-8) and D330 (by casp-3). The sites of PKA phosphorylation are indicated by ℗. S99 is in the CARD while both S183 and S195 are in the large subunit. b Structure of the casp-9 dimer with phosphorylation sites (yellow spheres) noted. There is no structure of full-length casp-9, so this model was built based on the CARD-deleted casp-9 structure (aa 138-416; PDB ID: 1JXQ) and the casp-9 CARD structure (aa 1–95; PDB ID: 3YGS) from a dimeric complex with Apaf-1 CARD. The region containing S99 is not present in either structure and is potentially highly disordered, so it was modeled as a dark gray coil. S99 is in the CARD; both S183 and S195 are located in the α1 helix

Phosphorylation of casp-9 by PKA results in significant inhibition of casp-9. a Active PKA inhibits self-processing of procasp-9. Full-length (FL) WT procasp-9 rapidly undergoes self-cleavage to generate the CARD + Large (Lg) and Small (Sm) subunits as assessed by Coomassie-stained SDS-PAGE analysis. Addition of PKA and ATP to procasp-9 slows the rate of self-cleavage, so that casp-9 remains predominantly in the full-length, uncleaved form even after 4 h incubation. b Addition of active PKA to cleaved casp-9 WT results in a two-fold decrease in caspase activity after 2 h as measured by the hydrolysis of the fluorogenic casp-9 substrate Ac-LEHD-afc, suggesting that PKA treatment leads to inhibition. Percent casp-9 activity is normalized against activity in the absence of PKA. c Casp-9 is phosphorylated by PKA only in the CARD + Lg region, as detected by autoradiography after in vitro phosphorylation using [γ-32P] ATP for 4 h. There is no visible phosphorylation in the Sm subunit. PKA is phosphorylated during overexpression in bacteria [66], so it does not get efficiently labeled by [γ-32P] ATP. Treatment of PKA-phosphorylated casp-9 with λ protein phosphatase (λPP) results in dephosphorylation as manifested by loss of signal in the autoradiogram (labeled here and in the succeeding figures as 32P). d Phosphorylation of casp-9 is reversible. Casp-9 phosphorylated by PKA is inhibited. Treatment of phosphorylated casp-9 with λ protein phosphatase (λPP) relieves the inhibition. Percent inhibition for phosphorylated casp-9 (with both PKA and ATP present) was normalized against activity in the non-phosphorylated form (with PKA but no ATP present). e Unphosphorylatable alanine variants (single, double and triple alanine substitutions at phosphorylated serines) and catalytic site-inactivated variant C287A were subjected to in vitro phosphorylation by PKA for 4 h. Double alanine variants show (observed by Coomassie-stained SDS-PAGE analysis in the first of each pair of panels) decreased levels of phosphorylation as the weaker intensity of the bands in the autoradiogram (second of each pair of panels and Table S1). The triple alanine variant shows only weak, non-specific phosphorylation in the small subunit, indicating that all three sites S99, S183, and S195 are phosphorylated by PKA. f Inhibition by phosphorylation of casp-9 WT and alanine variants. Only when S183 is available to be phosphorylated (WT, S99A, S195A, and S99A/A195A) does casp-9 experience significant inhibition. All S183A variants are insensitive to PKA-mediated inhibition. The catalytic parameters of alanine variants are indicated in Table 1. Percent inhibition for phosphorylated casp-9 was determined as in d. For b, d and f, data shown are means (± SEM) of three independent trials done on three separate days.
To quantify the inhibition, we assayed WT casp-9 activity upon phosphorylation using the substrate Ac-LEHD-afc. Casp-9 activity decreased two-fold with active PKA (Fig. 2b), suggesting that PKA significantly inhibits casp-9 function. In vitro phosphorylation using PKA and [γ-32P]ATP showed CARD + Lg phosphorylation (Fig. 2c), with a corresponding inhibition of casp-9 activity (Fig. 2d). This sensitivity of casp-9 to phosphorylation was reversible. Treatment with lambda protein phosphatase (λPP) removed phosphates from casp-9 (Fig. 2c), and casp-9 activity was relieved from inhibition (Fig. 2d).
Ser-183 is the critical residue leading to casp-9 inactivation upon PKA phosphorylation
To pinpoint the residue(s) responsible for inhibition, the phosphorylation states of unphosphorylatable alanine variants (Fig. 2e) were correlated with casp-9 inhibition (Fig. 2f). All single alanine variants were phosphorylated only in the CARD + Lg, but only S183A was not highly inhibited upon phosphorylation. Among the double alanine variants, only S99A/S195A displayed inhibition similar to phosphorylated WT. Having all three sites unphosphorylatable (S99A/S183A/S195A) resulted in low levels of non-specific phosphorylation, especially with the appearance of a band corresponding to a phosphorylated small subunit (Fig. 2e). This extra, non-specific phosphorylation had no substantial influence in casp-9 activity, since only background levels of inhibition for the triple alanine mutant were observed. These results clearly suggest that Ser-183 is the dominant site responsible for the inhibition due to PKA phosphorylation.
PhosphoS183 casp-9 and phosphomimetic S183E are completely inactive
Due to low levels of non-specific phosphorylation observed in vitro, we prepared versions of casp-9 that are unambiguously phosphorylated at only one site. We used a genomically recoded E. coli method to genetically encode site-specific phosphoserine incorporation [30] during protein production. All phosphocasp-9 variants (phosphoS99, phosphoS183 and phosphoS195) and unphosphorylated WT casp-9 were predominantly in the zymogen form (Fig. 3a). WT and both phosphoS99 and phosphoS195 were able to self-process, while phosphoS183 remained in its full-length/zymogen form (Fig. 3a), suggesting that phosphoS183 limits casp-9 activity. Consistent with self-processing, phosphoS99 and phosphoS195 showed LEHDase activity, whereas phosphoS183 had no measurable activity (Fig. 3b), clearly demonstrating that phosphorylation at Ser-183 inactivates casp-9.

Casp-9 phosphorylated at S183 and phosphomimetic S183E are inactive. a Uncleaved zymogen versions of casp-9 (WT and phosphocaspase, indicated with ℗ preceding the phosphoserine residue) purified from E. coli C321.ΔA which allows site-specific incorporation of phosphoserine, were subjected to self-cleavage for 2 h and assessed by Coomassie-stained SDS-PAGE analysis. Only casp-9 specifically labeled at S183 (phosphoS183) did not self-process, indicating that its activity is inhibited. PhosphoS99 and S195 were able to self-process with kinetics similarly to WT casp-9. b LEHDase activities of phosphocasp-9 variants. Activity was measured after 2 h to allow self-cleavage/activation of phosphocasp-9. PhosphoS183 exhibited no LEHDase activity. Data shown are means (± SEM) of three independent trials on three separate days. c Phosphomimetic S183E was expressed as full-length, uncleaved casp-9, unlike WT and phosphomimetics S99E and S195E which were expressed in a mature, cleaved state following expression, as assessed by Coomassie-stained SDS-PAGE analysis. The constitutively two-chain (CT) version of casp-9 is generated from the independent translation of the CARD + Lg and Sm subunits. CT WT and CT S183E are the cleaved forms of casp-9. d Catalytic parameters for casp-9 phosphomimetic variants using Ac-LEHD-afc as substrate. Only the phosphomimetic S183E has a dramatic effect on casp-9 activity in both full-length, uncleaved and CT versions. Data shown are means (± SEM) of three independent trials on three separate days. e Structure of casp-9 dimer (PDB ID IJXQ) highlighting the substrate-binding groove and critical interactions between S183 (yellow sticks) and the conserved S1 subsite R180 (green sticks). R180 is an important residue that makes contacts with P1 aspartate in the substrate (light gray sticks). f Models for inhibition of casp-9 by S183E and phosphorylation at S183. Substitution at S183 by glutamate or phosphoserine are predicted to result in a steric clash with R180, thus disorienting the active-site loops
Although the phosphocaspase variants are the biologically relevant forms, the yield of phosphoserine-incorporated casp-9 was extremely low. To obtain sufficient quantities for thorough interrogation of the effects of phosphorylation on casp-9 function and structure, we generated glutamate phosphomimetic variants. Following expression, S99E was in the mature, cleaved form and was catalytically active, with a modest three-fold reduction of activity compared to the WT (Fig. 3c, d). S195E was also in the mature, cleaved state and had WT-like activity. In contrast, the S183E substitution adversely affected casp-9 activity. In addition to retaining a zymogen form, it was also fully inactive (Fig. 3c, d). The expression and activity profiles of the phosphomimetics were consistent with what was observed for the phosphocaspase counterparts, indicating that they are robust phosphomimetics.
While cleavage is not an absolute requirement for casp-9 activation, we sought to test whether the loss of activity of S183E is due to its zymogen nature, or due to the intrinsic changes brought about by the phosphomimetic mutation. S183E expressed from a constitutively two-chain (CT) construct, in which the CARD + Large is translated independently from the small subunit, yields the mature/cleaved form (Fig. 3c). Even in this mature version, CT S183E was still catalytically inactive (Fig. 3d), showing at least 1000-fold less efficient kcat/KM than WT casp-9. A significantly higher KM and decreased kcat suggests that both substrate binding and catalytic turnover are impacted. S183A, which retains similar size and is uncharged like a serine residue, has a 10-fold lower catalytic efficiency than WT, primarily due to its decreased kcat (Fig. 3d). The triple alanine mutant S99A/S183A/S195A also shows a 10-fold decrease in kcat/KM (Table 1). This suggests that phosphorylation or glutamate substitution, which results in a dramatic 1000-fold decrease in activity, is the major cause of inhibition, rather than simply generic sensitivity of the Ser-183 site. These data strongly suggest that Ser-183 phosphorylation directly impairs substrate binding.
Phosphorylation at Ser-183 disorients Arg-180 leading to impaired substrate binding
Ser-183 resides just below Loop 1 (L1) which, together with L3, L4, L2, and L2’, form the active-site loop bundle (Fig. 3e). L1 contains the highly conserved and critical Arg-180. Arg-180 plays two important roles: directly binding substrate P1 residues and orienting Arg-355, another critical residue that makes bidentate interactions with substrate. The proper positioning of Arg-180 is mediated by an H-bond to the side chain of Ser-183, with another set of H-bonds provided by their amide backbones (Fig. 3e). These interactions ideally situate Arg-180 to orient the P1 aspartate in the substrate catalytic cleavage. The inability of S183E and phosphoS183 to bind substrate appears to stem from a steric clash between Glu-183 or phosphoS183 with Arg-180 (Fig. 3f). The additional bulk and charge from these groups should cause Arg-180 to become displaced from the S1 pocket, consequently disorienting the active-site loop bundle, thus making casp-9 incompetent to bind substrate.
Phosphomimetic S183E impacts recognition by casp-8
Casp-9’s canonical function is cleaving executioner caspases, but it is also cleaved and activated by casp-8 (at Asp-315) [31, 32] and casp-3 (at Asp-330) [33,34,35], providing feedback regulation. We assessed changes in the ability of other caspases to cleave casp-9 phosphomimic. Whereas WT casp-9 fully self-processes immediately, full-length S183E was completely devoid of self-processing activity (Fig. 4a) so any processing can be attributed to other caspases. S183E, like the full-length catalytically inactive variant C287A, was efficiently cleaved by casp-3 (Figs. 4b, d) but remained completely inactive even after cleavage (Fig. 4c), consistent with the observation that CT S183E remained inactive in the mature form. It was also striking that S183E remained mostly full-length/uncleaved after incubation with casp-8, suggesting that S183E transforms casp-9 into a non-optimal substrate of casp-8 (Fig. 4d). Phosphorylation has been reported to alter a substrate’s susceptibility to caspase cleavage [10] and is commonly observed when phosphorylation is adjacent to the caspase cleavage site. In this case, however, Ser-183 and the linker cleavage site Asp-315 are distal in sequence (Fig. 1a). Thus, it is possible that the long linker of casp-9 (which is not ordered in any crystal structure) has a conformation bringing it in the vicinity of Ser-183 where phosphorylation alters the ability of casp-8 to cleave and activate casp-9. Alternatively, S183E could negatively influence the interaction between casp-9 and casp-8, perhaps by exploiting an allosteric mechanism or by disrupting a possible exosite on casp-9 required for interaction with casp-8.

S183E impacts recognition by casp-8. a WT and S183E uncleaved zymogens were incubated for 8 h to allow self processing. S183E shows no self-processing activity as assessed by Coomassie-stained SDS-PAGE analysis. b S183E is cleaved by casp-3 at D330. The catalytic-site substituted C287A and S183E phosphomimetic are cleaved in a similar manner by casp-3 at D330 as assessed by Coomassie-stained SDS-PAGE analysis. Since C287A and S183E are both catalytically inactive, the cleavage products (CARD + Lg and Sm) result from the activity of casp-3. In contrast, WT casp-9 can self-process, resulting in a small subunit spanning residues 316–422 (Sm316–422) or can be cleaved by casp-3, resulting in a small subunit spanning residues 331–422 (Sm331-422). c The catalytic-site inactivated variant C287A, the S183E phosphomimetic or WT casp-9 were incubated for 2 h with casp-3 to allow processing and then tested in an LEHDase activity assay. S183E does not gain activity even after cleavage by casp-3. Data shown are means (± SEM) of three independent trials on three separate days. d The catalytic-site inactivated variant C287A or the S183E phosphomimetic were incubated for 6 h with casp-3 or casp-8 to observe processing using Coomassie-stained SDS-PAGE analysis. Whereas C287A is fully processed by both casp-3 and casp-8, S183E is only cleaved by casp-3. S183E remains almost uncleaved after incubation with active casp-8, suggesting that S183E is a poor substrate for casp-8
CT S183E breaks the interaction between the large and small subunits
The catalytic core of a casp-9 monomer is formed from the regions that become the large and small subunits after cleavage [5, 36] (Fig. 1). The strong association between caspase large and small subunits is maintained by critical interactions that form and stabilize the core of both monomeric and dimeric casp-9. Although the subunits are expressed independently of each other in the CT version of WT casp-9, they assemble to form a properly folded protein monomer composed of one small and one large subunit. This tight association between subunits manifests through their co-elution on an ion exchange gradient during protein purification, coupled with co-varying band intensities of the subunits visible in a Coomassie-stained denaturing gel. In CT WT casp-9, the CARD + Lg and Sm subunits co-eluted along a salt gradient and their band intensities on a denaturing gel clearly co-varied (Fig. 5a). This suggests that CT WT is properly folded, as supported by the data that it is as active as the FL WT (Fig. 3d). In contrast, separate elution of subunits in CT S183E was observed (Fig. 5b), implying that S183E breaks the interactions between the large and the small subunits, making the protein unstable. We also observed that traditional caspase purification schemes were not suitable to purify CT S183E, as we always obtained impure protein, thus CT S183E was purified from inclusion bodies and refolded (see Methods). In both approaches, CT S183E displayed no caspase activity, likely due to loss of or weakening of interactions between the large and small subunits of casp-9.

S183E breaks interactions within the core of casp-9. a Anion exchange chromatogram (bottom) and Coomassie-stained gel of peak fractions (top) for CT WT casp-9. CARD + Lg and Sm subunits co-elute and corresponding band intensities co-vary along the salt gradient during an anion exchange column, suggesting tight interaction of the subunits in the casp-9 core. b Anion exchange chromatogram (bottom) and Coomassie-stained gel of peak fractions (top) for CT S183E. The independent elution of the Sm subunit from the CARD + Lg indicates dissociation of core subunits of S183E
Phosphorylation of Ser-183 unfolds and disassembles the casp-9 core
The fact that S183E decreased the interactions between the large and small subunits suggests that phosphorylation of Ser-183 would have a similar effect. We used casp-3 to cleave full-length (FL) versions of casp-9 phosphomimetics (Fig. 6a, f) and assessed the impact of cleavage of a phosphorylated zymogen on the stability of the casp-9 core by native gel electrophoresis and thermal shift. FL C287A and FL S183E showed distinct migration on a native gel (Fig. 6b) with no significant aggregation nor additional bands for each protein, suggesting that the proteins retained their native, properly folded states. Upon cleavage by casp-3, C287A’s mobility only shifted slightly. In striking contrast, cleaved S183E’s mobility was dramatically altered (Fig. 6d), suggesting a shift in conformation away from properly folded, cleaved casp-9.

S183E is highly destabilized upon cleavage. a Full-length (FL) S183E phosphomimetic and C287A catalytic site-inactivated variant are cleaved by casp-3 in the same manner and with similar efficiencies as assessed by denaturing Coomassie-stained SDS-PAGE. b FL C287A, FL S183E, and cleaved WT casp-9 showed similar, compact migration along the native (non-denaturing) Coomassie-stained agarose gels, indicative of properly-folded proteins. c Thermal melting curves generated from differential scanning fluorimetry using SYPRO® orange. Both FL C287A and FL S183E showed typical melting transitions. FL S183E’s stability is less than that of FL C287A and comparable to that of a cleaved WT casp-9 (Table S2). d Casp-3-cleaved S183E exhibits a dramatic shift in mobility on a Coomassie-stained native agarose gel relative to casp-3-cleaved C287A casp-9. e Thermal melting curves of casp-3-cleaved C287A and S183E. Casp-3-cleaved C287A shows a melting transition similar to uncleaved C287A. No melting curve is observed for casp-3-cleaved S183E. f FL phosphomimetic variants and FL S183A were cleaved by casp-3 in the same manner and assessed by denaturing Coomassie-stained SDS-PAGE. g Coomassie-stained native agarose gel of FL phosphomimetic variants and FL S183A. Compact migration on the native gel suggests these casp-9 variants are properly folded. h Thermal melting curves of full-length phosphomimetic variants and S183A. All variants displayed normal melting transitions. i Cleavage by casp-3 of phosphomimetic variants and S183A did not significantly alter mobilities on a Coomassie-stained native agarose gel. Only casp-3-cleaved S183E showed a significant shift in mobility, indicating a substantial change in the folded state of the protein. j Thermal melting curves of casp-3-cleaved phosphomimetic variants and S183A. All variants displayed normal melting transitions, except for casp-3-cleaved S183E. Note: For c, e, h, and j, data shown are means of three independent trials. Fluorescence values were normalized against the lowest and highest values in each data set. For f-j, asterisk indicates that the variant was constructed in the background of C287A to prevent self-processing.
Thermal stability shift assays were performed on casp-9 before and after casp-3 cleavage. Both FL C287A and FL S183E exhibited typical melting curves, indicating that they are properly folded (Fig. 6c), however, FL S183E is less stable than FL C287A (Table S2). After cleavage by casp-3, no drastic change in the melting transition of C287A was observed, however, cleaved S183E was severely destabilized and no melting transition was observed (Fig. 6e). The magnitude of the initial fluorescence was also very high, suggesting that cleaved S183E was already unfolded or in a molten globule state at the start of the thermal denaturation. This destabilization upon cleavage appears to be a direct consequence of the S183E mutation since other phosphomimetics (S99E and S195E), as well as the conservative mutant S183A were not unstable upon cleavage by casp-3. All showed properties of properly-folded proteins as demonstrated by their distinct migration on a native gel (Figs. 6g, i) and by exhibiting typical protein melting curves (Fig. 6h, j Table S2). Moreover, phosphorylation at Ser-183 by PKA recapitulated what was observed for cleaved S183E. Using S183A and S99A/S195A to direct phosphorylation to Ser-99/Ser-195 or Ser-183, respectively, we found that only phosphorylation at Ser-183 resulted in mobility shift similar to cleaved S183E (Fig. 7). This demonstrates that Ser-183 phosphorylation destabilizes the casp-9 core, and subsequent cleavage at the linker causes the core subunits to dissociate, rendering casp-9 non-functional.

Phosphorylation of casp-9 by PKA at S183 results in destabilization similar to S183E. a Mature, cleaved forms of casp-9 unphosphorylatable alanine variants were incubated with PKA and ATP to direct phosphorylation specifically to S99 and S195 or to S183. No impact on mobility was observed upon Coomassie-stained denaturing SDS PAGE. b Mobility shifts of unphosphorylated (no ATP) and phosphorylated (+ ATP) S183A or S99A/S195A casp-9 following non-denaturing native agarose gel electrophoresis after 15 min or 1 h of phosphorylation. After 1 h of phosphorylation, S99A/S195A (conditions promoting S183 phosphorylation) exhibited a drastic shift in migration, similar to what was observed in cleaved S183E, indicating that S183 phosphorylation destabilizes casp-9 leading to a shift in the conformation
Casp-3-cleaved S183E casp-9 forms ordered aggregates
Destabilized and partially unfolded globular proteins are known to have an increased propensity to aggregate in vitro [37]. Thus, the unfolded nature of S183E following cleavage could potentially render casp-9 prone to aggregation. The tendency of cleaved S183E to form aggregates was assessed by Thioflavin T (ThT) binding. A large increase in ThT fluorescence was observed for cleaved S183E (Fig. 8a). In contrast, there was little to no increase in fluorescence observed for FL S183E, and cleaved or FL C287A. Cleaved S183E was found exclusively in the insoluble fraction, while most of cleaved C287A remained soluble (Fig. 8b). This magnitude of increase in ThT fluorescence, as well as the rapid kinetics of aggregate formation, strongly indicate that cleaved S183E forms aggregates which assume a regularity in structure, particularly an assembly or stacking of β-sheets, since amorphous or early aggregates are not known to bind ThT [38, 39].

Cleaved S183E forms ordered aggregates. a In situ ThT fluorescence monitored for 12 h. Only casp-3-cleaved S183E showed significant increase in ThT fluorescence, suggesting that it forms higher order oligomers or aggregates. Very little increase in ThT fluorescence was observed for FL S183E or cleaved C287A. Data shown are means (± SEM) of three independent trials done on three different days. b SDS-PAGE analysis by Coomassie staining of the pellet (P) and supernatant (S) fractions of FL casp-9 C287A or S183E after 12 h of cleavage by casp-3. The casp-9 CARD + Lg and Sm subunits fractionated mostly into the soluble supernatant for cleaved C287A, while for cleaved S183E, the subunits are found exclusively in the insoluble/ pellet fraction. c TEM images of aggregates of cleaved S183E. Individual units (e.g., casp-9 monomers) appear to form ordered aggregates (enlarged images). The scale bar is 40 nm wide and 10 nm for selected enlarged images; all images are magnified ×400,000
The size and morphology of the aggregates were visualized by transmission electron microscopy (TEM). The shape of the aggregates varied from circular to elongated clusters that also ranged in size from 10 to 40 nm (round) and 40 to 80 nm (elongated) (Fig. 8c, Fig. S3). At ×400,000 magnification, individual units were resolved to be ~2−5 nm in diameter, which corresponds to the diameter of casp-9 monomers (~2.5 nm in diameter). Moreover, it appears that the aggregates were assembled from proteins arranged adjacent to one another. Although not sufficient to form fibrils, this arrangement appears to confer enough structural regularity to form ordered aggregates that bind ThT.
Discussion
Phosphorylation is recognized as a global regulator of caspase function, but the molecular basis of how phosphorylation impacts caspase function, especially of casp-9, is vastly understudied. Our results demonstrate that casp-9 phosphorylation at Ser-183 by PKA is sufficient to directly inactivate casp-9 and block the apoptotic cascade. This result agrees well with the report identifying three sites of PKA-mediated phosphorylation [20] however our new data on the kinetic activity of the Ser-183 substitution variants (Table 1) sheds light on the conclusions made in the earlier work. The prior report [20] measured DEVDase activity in cell extracts immunodepleted of casp-9, supplemented with either WT or unphosphorylatable S99A/S183A/S195A casp-9 by in vitro translation and activated to apoptosis with cytochrome c. In those assays, the DEVDase activity in cell extracts supplemented with either WT or S99A/S183A/S195A were sensitive to PKA activity, leading to the conclusion that PKA does not directly act on casp-9[20]. Casp-9 shows strong LEHDase activity, but very weak DEVDase activity [40]. The major DEVDases activated by cytochrome c are casp-3 and casp-7, so the earlier assay [20] measured the downstream activity of casp-3 and casp-7 effectively, but could not directly assess casp-9 activity effectively. The assumption in that experiment was that casp-3 and casp-7 would be activated by the added WT or S99A/S183A/S195A casp-9, however the intrinsic activity of S99A/S183A/S195A casp-9 was not reported at that time. Our work has shown that the intrinsic activity of S99A/S183A/S195A (Table 1, Fig. S2) is only ~10% of WT casp-9 activity. Due to the inherent differences in the catalytic activity of WT and S99A/S183A/S195A casp-9, if casp-9 had been responsible for activating the measured DEVDase activity, then the activity in the cell extracts supplemented with S99A/S183A/S195A should have been only ~10% of the WT levels. However, the S99A/S183A/S195A DEVDase activity in the absence of PKA was ~84% of WT caspase-9. This suggests that the DEVDase activity was not due solely to casp-9, but was probably due to activation of casp-3 and casp-7 by another factor, likely casp-8. Our kinetic data on the intrinsic activity of S99A/S183A/S195A allows an updated interpretation of the prior data, which is consistent with direct inactivation of casp-9 by PKA. However, Martin et al.’s finding that PKA prevents processing of the S99A/S183A/S195A variant [20] also suggests that PKA can influence casp-9 activity when these residues are not phosphorylated. It may be possible that phosphorylation of casp-9 by PKA at a site that we did not observe or phosphorylation of another factor may prevent casp-9 processing. Thus, it appears that PKA could potentially inactivate casp-9 both directly and indirectly. While the ideal experiment is to determine the effect of phosphorylation on casp-9 activity intracellularly, we found that transfection of active casp-9 is toxic (Fig. S5), thus preventing exploration of the function of casp-9 in a whole-cell context. In addition, based on our in vitro experiments, both S183A and S99A/S183A/S195A variants are expected to be inactive intracellularly, hence the use of these unphosphorylatable variants in cells would confound the interpretation of any change in casp-9 activity upon PKA activation. Nevertheless, our in vitro results showing direct inactivation of casp-9 by phosphorylation at Ser-183 suggest that PKA-mediated inhibition of casp-9 will likewise occur in vivo.
The hierarchical nature of casp-9 activation allows phosphorylation to exert multiple levels of regulation during the casp-9 life cycle. It is conceivable that Ser-183-phosphorylated casp-9 would still be recruited to the apoptosome since CARD:CARD interactions are not likely disrupted, however this phosphorylated casp-9 is inherently non-activatable. It appears that Ser-183 phosphorylation should not favor a conformation in either the zymogen or cleaved form that would allow casp-9 to bind substrate, even in the apoptosome, the ultimate casp-9 activating platform. Thus, phosphorylation at Ser-183 acts as an upstream block in the caspase activation cascade. Ser-183 resides near the active site. Other caspases (casp-6/-7) are similarly phosphorylated at sites neighboring the active-site loops, which prevents these loops from assuming an active conformation [23, 24, 26]. This mechanism likewise appears pertinent to casp-9 Ser-183 phosphorylation, thus orthosteric inhibition emerges as a common theme among phosphorylated caspases.
It is evident that Ser-99 and Ser-195 are phosphorylated by PKA, yet phosphorylation and phosphomimicry showed little or no influence in any of the interrogated casp-9 activities. Perhaps these are “functionally neutral” sites that have persisted evolutionarily as they confer no phosphorylation-associated disadvantages [21, 22]. Alternatively, their phosphorylation may influence a different aspect of casp-9 function. Given that Ser-99 resides within the CARD, phosphorylation may impact casp-9 recruitment or apoptosomal activation. Ser-195 is highly surface exposed so phosphorylation might mediate interactions with casp-9 substrates. These putative mechanisms warrant further study.
Crosstalk between caspases and cognate kinases [10] is essential for the balance between cell death and survival. Typically, caspases cleave cognate kinases, either freeing regulatory elements and relieving inhibition, or rendering the kinase inactive [10]. Our in vitro phosphorylation assays showed no casp-9 cleavage of PKA. A casp-9 recognition site within PKA and homotypic binding motifs in both enzymes are requisite for co-regulation, however, no casp-9 cleavage sites are predicted in PKA, potentially suggesting a heavy weighting towards casp-9 inhibition. Moreover, the phosphomimetic S183E greatly attenuates its cleavage by casp-8 (Fig. 4d) suggesting that once phosphorylated, casp-9 could no longer be alternatively activated by the extrinsic pathway. Thus PKA phosphorylation would prevail over several modes of casp-9 activation. PKA is overexpressed in many cancers [41,42,43] and phosphorylation of casp-9 to prevent its full-scale activation could be one mechanism by which unregulated PKA promotes tumorigenesis, proliferation and transformation.
Ser-183 appears to be a hotspot for phosphorylation-mediated inactivation, utilizing divergent mechanisms to limit casp-9 activity. In addition to directly blocking substrate binding, Ser-183 phosphorylation breaks critical interactions within the casp-9 core causing conformational transformations. This dramatic transition is fascinating. Since Ser-183 is not situated at the large- small subunit interface, this dissociation must stem from conformational strain translated across the casp-9 core. We believe this is the first report of such an inactivation mechanism. This phosphorylation-induced destabilization appears to affect the mature (cleaved) form of casp-9 more severely than the zymogen, likely due to covalently-induced proximity.
On one hand, in the context of casp-9 activation on the apoptosome, a Ser-183-phosphorylated casp-9 would remain non-activatable and would likely disengage from the apoptosome because it is no longer structurally and functionally intact after maturation. On the other hand, it is possible that altered-conformation casp-9 could enter a different pathway with novel functions beyond activating executioner caspases. Unfolding itself can be a functional signal [44, 45]. Here we observed that unfolded, phosphorylated casp-9 serves as a precursor to forming ordered aggregates.
Accumulation of unfolded protein is a hallmark of protein aggregation diseases - Alzheimer’s, Huntington’s and amyotrophic lateral sclerosis (ALS) [46, 47]. Phosphorylation is a common feature in toxic aggregation [48] in Tau, amyloid-β and α-synuclein [49,50,51]. While that is reminiscent of phosphorylation-induced unfolding of casp-9, to date no disease has been attributed to casp-9 aggregation. Whereas amyloid-like aggregates and fibrils are usually associated with disease, functional aggregates have also been exploited in many species [37, 52] to play a number of valuable roles. Cell death and inflammation pathways utilize functional aggregates or higher-order structures to amplify and propagate signals, increase local concentration, facilitate recruitment or even direct subcellular localization [53,54,55,56,57,58,59]. Other cell death-related proteins like casp-1 [60], ASC [61], Bax and Bak [62] assemble into higher-order structures in vivo. Thus the idea of functional casp-9 aggregates is not unprecedented. Nevertheless, whether these aggregates actually form in vivo and their functional roles are in the cell remain to be explored. One could speculate that for Ser-183-phosphorylated casp-9, aggregates may prevent apoptosis, yielding either protective or, more likely, proliferative effects. Regardless, this is the first report of casp-9 forming higher-order structures in the absence of any other protein or recruitment platform. PKA recognition sites are present in the sequences of other caspases including casp-3/-6/-7/-8/-10, although PKA-mediated regulation of these caspases has not been reported. Given the high structural homology among caspases, it is likely that other caspases may also form similar ordered aggregates in response to phosphorylation at the homologous site.
Materials and Methods
DNA constructs and E. coli strains
The casp-9 (C9) full-length wild-type (C9FL) expression construct consists of the full-length, C-terminal His6x-tagged human casp-9 gene (amino acids 1-416 plus the six terminal Histidines) in pET23b [29] (gift of Guy Salvesen). The casp-9 constitutively two-chain (C9 CT) expression construct consists of a synthetic gene (Genscript, Piscataway NJ) in pET21b that encodes E. coli codon-optimized human casp-9 that is built for separate expression of the casp-9 large subunit (amino acids 1- 315) independently from the small subunit (amino acids 316-416 + His6x) which was under the control of a second ribosome binding site. Casp-9 variants encoding amino acid substitutions were generated using the QuikChange site-directed mutagenesis method (Stratagene/Agilent, Santa Clara CA). The casp-3 wild type expression construct consists of the full-length, C-terminal His6x-tagged human casp-3 gene (amino acids 1-279 plus six terminal His) in pET23b [63] (gift of Guy Salvesen). The gene for the catalytic subunit of PKA in pET15b [64] was expressed from a construct supplied by Susan Taylor (Addgene plasmid # 14921). The SepOTSλ (phosphoserine orthogonal translation system) plasmid, pBAD-GST-AmpR vector, and the fully recoded E. coli strain C321.ΔA [30] were gifts from Jesse Rinehart (Yale University).
Expression and purification of proteins
Purification of soluble casp-9 proteins
Casp-9 variants (except C9 CT S99A/S183A/S195A) were transformed into BL21(DE3) strain of E. coli. The cultures were grown in 2xYT media with 100 µg/mL of ampicillin at 37 °C with vigorous shaking until they reached an optical density (OD600) between 1 to 1.2. The temperature was lowered to 15 °C and protein expression was induced by adding 1 mM of IPTG (Anatrace, Maumee OH). Protein expression was allowed to proceed for 3 h (except for C9 FL WT zymogen which was expressed for only 30 min, C9FL S183A, C9FL S99A/S183A, and C9FL S195A/S183A which were expressed for 16 h) and cells were harvested by centrifugation at 4700×g for 10 min at 4 °C. Cell pellets were stored at −80 °C, freeze-thawed and lysed in a microfluidizer (Microfluidics, Inc., Westwood MA) in lysis buffer (50 mM sodium phosphate pH 7.0, 300 mM NaCl, and 2 mM imidazole). Cell lysates were centrifuged at 30,600×g for 50 min at 4 °C to remove cellular debris. The supernatant was filtered through 0.45 µm PVDF (EMD Millipore, Billerica MA) filter and loaded unto a 5-mL HiTrap Ni-affinity column (GE Healthcare, Pittsburgh PA). Proteins were eluted using a linear gradient of 2–100 mM imidazole in lysis buffer. Protein fractions were analyzed by SDS PAGE and fractions containing casp-9 were pooled and diluted 8× with a buffer containing 20 mM Tris pH 8.5 and 5 mM DTT (buffer A) and loaded onto a HiTrap Q-column (GE Healthcare). Proteins were eluted by a linear gradient from 0–275 mM of NaCl in buffer A. Casp-9 eluted in buffer A at 180 mM NaCl. Peak fractions were analyzed by SDS PAGE for purity and were stored in −80 °C until use.
Purification of casp-9 from inclusion bodies
Casp-9 variants C9 CT S99A/S183A/S195A and C9 CT S183E were transformed into BL21(DE3) strain of E. coli. The cultures were grown in 2xYT media with 100 µg/mL of ampicillin at 37 °C with vigorous shaking until they reached OD600 = 0.8. The temperature was lowered to 30 °C and protein expression was induced by addition of 1 mM IPTG. Protein expression was allowed to proceed for 3 h and cells were harvested by centrifugation at 4700×g for 10 min at 4 °C. Cells were freeze-thawed and lysed in a microfluidizer in a buffer containing 10 mM Tris pH 8.0 and 1 mM EDTA. The lysate was centrifuged at 27,000×g for 1 h at 4 °C. Inclusion bodies were purified from the pellet by was washing 3× with buffer containing 100 mM Tris pH 8.0, 1 mM EDTA, 500 mM NaCl, 2% Triton-X and 1 M urea, centrifuging at 17,000×g for 15 min between washes. The pellet was then washed 3× with 100 mM Tris pH 8.0 and 1 mM EDTA to remove urea and performing the same centrifugation between washes. The pellet was then resuspended in minimal volume of 6 M guanidine chloride with 20 mM β-mercaptoethanol and placed in a rotating platform for 1 h. The mixture was centrifuged at 20,000×g for 20 min at 4 °C. Supernatant containing denatured casp-9 was added dropwise to a refolding buffer containing 100 mM Tris pH 8.0, 10% sucrose, 0.1% CHAPS, 150 mM NaCl, and 10 mM β-mercaptoethanol. Refolding was allowed to proceed by dialyzing the solution against 10 mM Tris pH 8.0, 0.1 mM EDTA, and 10 mM β-mercaptoethanol. The dialysate was centrifuged at 20,000×g for 10 min at 4 °C to remove aggregates and the supernatant was filtered through a 0.45 µm PVDF membrane and loaded onto a 5-mL HiTrap Q-column. The column was developed by a linear gradient from 0–250 mM NaCl. Refolded casp-9 eluted in a buffer containing 20 mM Tris pH 8.0, 200 mM NaCl and 5 mM β-mercaptoethanol. Fractions were analyzed by SDS PAGE for purity and were stored in −80 °C until use.
Purification of casp-3 protein
The gene for full-length wild-type Casp-3 in pET23b was transformed into BL21(DE3) E. coli. Cultures were grown in 2xYT media with 100 µg/mL Ampicillin at 37 °C with shaking until OD600 = 0.8. Protein expression was induced by addition of 1 mM IPTG at 30 °C for 3 h and cells were harvested by centrifugation at 4700×g for 10 min at 4 °C. Cells were freeze-thawed, lysed in a microfluidizer in a lysis buffer containing 50 mM sodium phosphate pH 8.0, 300 mM NaCl and 2 mM imidazole, and centrifuged at 30,600×g for 50 min at 4 °C to remove cellular debris. The supernatant was loaded onto a 5-mL HiTrap Ni-affinity column, washed with 50 mM imidazole and proteins were eluted with 250 mM imidazole in lysis buffer. The eluted protein fraction was diluted six-fold with 20 mM Tris pH 8.0 with 2 mM DTT (buffer A) and loaded onto a HiTrap Q-column. Proteins were eluted by a linear gradient from 0–500 mM of NaCl in buffer A. Casp-3 eluted in buffer A with 250 mM NaCl. Peak fractions were analyzed by SDS PAGE for purity and were stored in −80 °C until use.
Purification of PKA catalytic subunit
PKA in pET15b was transformed into the BL21(DE3) strain of E. coli. Cultures were grown in 2xYT media with 100 µg/mL of ampicillin at 37 °C with vigorous shaking until OD600 = 0.6. Protein expression was induced by addition of 0.5 mM IPTG at 16 °C for 12 h. Cells were harvested by centrifugation at 4700×g for 10 min at 4 °C. PKA was purified as reported [65] with modifications. Cells were resuspended in lysis buffer (50 mM KH2PO4 pH 8.0 and 20 mM Tris-HCl), lysed by microfluidizer and centrifuged at 30600×g for 45 min at 4 °C to remove cellular debris. The supernatant was loaded onto a 5-mL HiTrap Ni-affinity column and the column was washed with 50 mM imidazole in lysis buffer. Proteins were eluted with 500 mM imidazole in lysis buffer, diluted 6× with a buffer containing 50 mM KH2PO4 pH 7.15, 20 mM KCl and 1 mM DTT and loaded onto a 5-mL HiTrap Q-column. The column was developed by a linear gradient from 20–250 mM KCl. PKA eluted in a buffer of 50 mM KH2PO4 pH 7.15, 150 mM KCl and 1 mM DTT. Fractions were analyzed by SDS-PAGE to determine purity (Fig. S4) and were stored in −80 °C until further use.
In vitro phosphorylation and dephosphorylation of casp-9
Autophosphorylation of PKA
PKA was incubated in kinase buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 0.1 mM EDTA and 0.01% Brij 35) with 250 µM ATP at 30 °C for 30 min.
Phosphorylation of casp-9
Casp-9 was added to the autophosphorylated PKA reaction and supplemented with 250 µM [γ-32P]ATP (10 µCi/µL, Perkin Elmer) and incubated for 30 min to 4 h at 32 °C. Reactions were stopped by adding SDS-PAGE loading dye and boiling for 10 min.
Dephosphorylation of casp-9
Lambda protein phosphatase (NEB) was used to dephosphorylate casp-9. 100 U of phosphatase were used for every 10 µM of phosphate attached to casp-9. The reaction was allowed to proceed for 1 h at 30 °C. The reaction was stopped by addition of SDS-PAGE loading dye and boiling for 10 min. Removal of phosphates was confirmed by loss of band intensity in the phosphorimage. Phosphorylation levels were quantified from an ATP standard curve on the same phosphorimage (Fig. S1). Bands were imaged using Typhoon FLA 7000 (GE Healthcare) and quantified using ImageQuant TL software (GE Healthcare).
Assay for casp-9 activity
Casp-9 was diluted in casp-9 activity assay buffer (100 mM MES pH 6.5, 10% PEG 8000, 5 mM DTT) to a final concentration of 800 nM. A substrate titration was performed in the range of 0–3 mM fluorogenic substrate Ac-LEHD-afc (Ex 365/Em 495) (Enzo Life Sciences). Enzyme concentrations were determined by labeling the active-site using a quantitative inhibitor z-VAD-fmk. The rate of LEHD cleavage was measured with a fluorescence plate reader (SpectraMax M5, Molecular Devices, Sunnyvale CA).
Casp-9 cleavage assays
Caspase-3 (30 nM) or caspase-8 (300 nM) was diluted in their respective activity buffers (caspase-3: 20 mM HEPES pH 7.5, 150 NaCl 5 mM CaCl2, 10% PEG 400 and 2 mM DTT; caspase-8: 10 mM PIPES pH 7.2, 100 mM NaCl, 1 mM EDTA, 0.05% CHAPS, 10% sucrose, and 2 mM DTT). Casp-9 (3 µM) (catalytic site-inactive variant C287A or FL S183E) was added and the cleavage reactions were incubated at 37 °C for times indicated. For self-cleavage of casp-9, 3 µM of casp-9 zymogen (FL uncleaved WT casp-9) was diluted in casp-9 minimal activity buffer (100 mM MES, 20% PEG 400 and 5 mM DTT) and incubated at 37 °C at each time point indicated. For cleavage of FL casp-9 C287A phosphomimetics and FL S183E by caspase-3 for native gel electrophoresis and thermal shift assays, 500 nM of casp-3 was prepared in 50 mM Tris-Cl pH 7.5, 150 mM NaCl, and 2 mM DTT (cleavage buffer). Casp-9 was then added to a final concentration of 50 µM and cleavage was allowed to proceed for 15 min at 37 °C. Reactions were stopped by adding SDS-PAGE loading dye and boiling for 10 min. Cleavage of the full-length casp-9 band was analyzed by SDS-PAGE and densitometry using ImageLabTM (BioRad). For cleavage of FL casp-9 as parallel samples for the ThT fluorescence assay, 75 µM of casp-9 FL C287A or FL S183E was incubated with casp-3 (500 nM) in casp-3 cleavage buffer at 37 °C for 15 min. The cleavage reactions were transferred to a 96-well black plate and further incubated at 30 °C for 12 h, after which the samples were centrifuged at 18,000×g to pellet the aggregates. The pellet was dissolved in 2% SDS and both the pellet and supernatant fractions were analyzed by SDS PAGE.
Construction, expression, and purification of site-specific phosphocasp-9
C9FL + His6x (1–422) was subcloned into NdeI and HindIII sites of pBAD-GST-AmpR vector to generate the plasmid C9F-pBAD. Codons that code for S99 (TCG), S183 (TCC) and S195 (TCC) were replaced by TAG using a QuikChange mutagenesis approach to generate phosphocasp-9 expression plasmids (C9pBAD-SepC9). Site-specific phosphoincorporation was performed as reported by Pirman, et al. [30]. Briefly, recoded E. coli (rEcoli) C321.ΔA were co-transformed with the C9pBAD-SepC9 and SepOTSλ plasmids. Cultures were grown in LB media supplemented with 2 mM O-phospho-L-serine (Sigma) pH 7.5, 100 µg/mL Ampicillin and 25 µg/mL Kanamycin at 30 °C with shaking until an OD600 = 0.8. The temperature was lowered to 18 °C and protein expression was induced by 0.4% L-arabinose (Acros Organics, NJ) and 1 mM IPTG for 20 h. Cells were harvested by centrifugation at 4700×g for 10 min at 4 °C. Cells were lysed and casp-9 proteins were purified as described above, except for the addition of glycerol (5%), phosphatase inhibitors NaF (20 mM) and β-glycerophosphate (2 mM) in all purification buffers, as well as the use of a MonoQ 5/50 GL column (GE Healthcare) for the final ion exchange purification using a gradient of 0–300 mM NaCl. Yields of the phosphoserine-incorporated casp-9 were extraordinarily low: ~0.05–0.10 mg from 12 L of expression.
Native agarose gel electrophoresis
Casp-9 (full-length and casp-3-cleaved versions of phosphomimetics and alanine variants) (20 µg) was mixed with 2x native gel sample buffer and loaded onto a 0.8% agarose gel (prepared using 25 mM Tris-Cl pH 8.5 and 192 mM glycine). The gel was run for 90 min at 60 V. Protein bands were stained with Coomassie dye, imaged and analyzed using ImageLabTM (BioRad).
Thermal shift assay by differential scanning fluorimetry
The thermal stability of casp-9 variants in 10 µM in 50 mM Tris-Cl pH 7.5, 150 mM NaCl and 2 mM DTT was analyzed in the presence of SYPRO® Orange (ThermoFisher) (0.5× final concentration) using a CFX Connect Real-Time PCR detection system (BioRad). Measurements were performed in a 96-well plate in 50 µL reactions. The fluorescence intensity was monitored by increasing the temperature in 0.5 °C increments from 25 to 95 °C. Thermal melting points (Tm) were determined by curve fitting analysis using Prism (GraphPad) software.
In situ ThT fluorescence assay
Casp-9 samples (FL and casp-3-cleaved C287A and S183E versions of casp-9) (75 µM) were mixed with Thioflavin T (ThT, Sigma) to a final concentration of 10 µM and incubated at 30 °C in black 96-well plates that were sealed to prevent evaporation. The total reaction volume was 100 µL. The ThT fluorescence (Ex 450/Em 485) intensity of each sample was recorded every 10 min using a SpectraMax M5 plate reader over the course of 12 h.
Transmission electron microscopy
A 50 µL sample of casp-3-cleaved S183E (75 µM) was incubated at 30 °C for 12 h in parallel with the ThT fluorescence samples. After 12 h, the sample was centrifuged for 10 min at 18,000×g and the pellet was washed twice and resuspended in 25 µL nanopure water. A 3 µL sample of a three-fold diluted suspension was embedded on an ultra thin carbon film supported by a lacey carbon film on a 400-mesh copper grid (Ted Pella, Inc.) for 5 min. The grid was blotted to remove excess sample and was washed twice with nanopure water. The grid was then incubated upside-down on a drop of 2% uranyl acetate for 30 s, washed with water to remove excess stain and dried overnight for EM analysis. A JEOL JEM-2200 FS EFTEM (Energy Filtered Transmission Electron Microscope) operating at 200 kV was used to obtain micrographs at ×200,000 to ×400,000 magnification (UMass Amherst Electron Microscopy Center). In order to enhance contrast all images were zero-loss filtered using an energy slit width of 20 eV.
References
Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–6.
Chęcińska A, Giaccone G, Rodriguez JA, Kruyt FAE, Jimenez CR. Comparative proteomics analysis of caspase-9-protein complexes in untreated and cytochrome c/dATP stimulated lysates of NSCLC cells. J Proteom. 2009;72:575–85.
Seaman JE, Julien O, Lee PS, Rettenmaier TJ, Thomsen ND, Wells JA. Cacidases: caspases can cleave after aspartate, glutamate and phosphoserine residues. Cell Death Differ. 2016. https://doi.org/10.1038/cdd.2016.62.
Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. 2009;284:21777–81.
Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS. Dimer formation drives the activation of the cell death protease caspase-9. Proc Natl Acad Sci USA. 2001;98:14250–5.
Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, et al. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell. 2003;11:519–27.
McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5:1–27.
Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N Engl J Med. 2003;348:1365–75.
Howley B, Fearnhead HO. Caspases as therapeutic targets. J Cell Mol Med. 2008;12:1502–16.
Kurokawa M, Kornbluth S. Caspases and kinases in a death grip. Cell. 2009;138:838–54.
Franklin RA, McCubrey JA. Kinases: positive and negative regulators of apoptosis. Leukemia. 2000;14:2019–34.
López-Otín C, Hunter T. The regulatory crosstalk between kinases and proteases in cancer. Nat Rev Cancer. 2010;10:278–92.
Parrish AB, Freel CD, Kornbluth S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol. 2013;5:1–24.
Dix MM, Simon GM, Wang C, Okerberg E, Patricelli MP, Cravatt BF. Functional interplay between caspase cleavage and phosphorylation sculpts the apoptotic proteome. Cell. 2012;150:426–40.
Allan LA, Clarke PR. Apoptosis and autophagy: regulation of caspase-9 by phosphorylation. FEBS J. 2009;276:6063–73.
Rossi AG, Cousin JM, Dransfield I, Lawson MF, Chilvers ER, Haslett C. Agents that elevate camp inhibit human neutrophil apoptosis. Biochem Biophys Res Commun. 1995;217:892–9.
Martin MC, Dransfield I, Haslett C, Rossi AG. Cyclic AMP regulation of neutrophil apoptosis occurs via a novel protein kinase A-independent signaling pathway. J Biol Chem. 2001;276:45041–50.
Orlov SN, Thorin-Trescases N, Dulin NO, Dam T-V, Fortuno MA, Tremblay J, et al. Activation of cAMP signaling transiently inhibits apoptosis in vascular smooth muscle cells in a site upstream of caspase-3. Cell Death Differ. 1999;6:661–72.
Insel PA, Zhang L, Murray F, Yokouchi H, Zambon AC. Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol. 2012;204:277–87.
Martin MC, Allan LA, Lickrish M, Sampson C, Morrice N, Clarke PR. Protein kinase A regulates caspase-9 activation by Apaf-1 downstream of cytochrome c. J Biol Chem. 2005;280:15449–55.
Lienhard GE. Non-functional phosphorylations? Trends Biochem Sci. 2008;33:351–2.
Landry CR, Levy ED, Michnick SW. Weak functional constraints on phosphoproteomes. Trends Genet. 2009;25:193–7.
Velázquez-Delgado EM, Hardy JA. Phosphorylation regulates assembly of the caspase-6 substrate-binding groove. Structure. 2012;20:742–51.
Eron SJ, Raghupathi K, Hardy JA. Dual site phosphorylation of caspase-7 by PAK2 blocks apoptotic activity by two distinct mechanisms. Structure. 2017;25:27–39.
Serrano BP, Szydlo HS, Alfandari DR, Hardy JA. Active-site adjacent phosphorylation at Tyr-397 by c-Abl kinase inactivates caspase-9. J Biol Chem. 2017;292:21352–21365: jbc.M117.811976.
Cao Q, Wang X-J, Liu C-W, Liu D-F, Li L-F, Gao Y-Q, et al. Inhibitory mechanism of caspase-6 phosphorylation revealed by crystal structures, molecular dynamics simulations, and biochemical assays. J Biol Chem. 2012;287:15371–9.
Zamaraev AV, Kopeina GS, Prokhorova EA, Zhivotovsky B, Lavrik IN. Post-translational modification of caspases: the other side of apoptosis regulation. Trends Cell Biol. 2017;27:322–39.
Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, et al. A unified model for apical caspase activation. Mol Cell. 2003;11:529–41.
Stennicke HR, Deveraux QL, Humke EW, Reed JC, Dixit VM, Salvesen GS. Caspase-9 can be activated without proteolytic processing. J Biol Chem. 1999;9:8359–62.
Pirman NL, Barber KW, Aerni HR, Ma NJ, Haimovich AD, Rogulina S, et al. A flexible codon in genomically recoded Escherichia coli permits programmable protein phosphorylation. Nat Commun. 2015;6:8130.
McDonnell MA, Wang D, Khan SM, Vander Heiden MG, Kelekar A. Caspase-9 is activated in a cytochrome c-independent manner early during TNFα-induced apoptosis in murine cells. Cell Death Differ. 2003;10:1005–15.
Gyrd-Hansen M, Farkas T, Fehrenbacher N, Bastholm L, Hoyer-Hansen M, Elling F, et al. Apoptosome-independent activation of the lysosomal cell death pathway by caspase-9. Mol Cell Biol. 2006;26:7880–91.
Fujita E, Egashira J, Urase K, Kuida K, Momoi T. Caspase-9 processing by caspase-3 via a feedback amplification loop in vivo. Cell Death Differ. 2001;8:335–44.
Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, et al. Ordering the cytochrome c-initiated caspase cascade: Hierarchical activation of caspases-2,-3,-6,-7,-8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–92.
Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–57.
Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J. 2004;384:201–32.
Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–66.
Biancalana M, Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta. 2010;1804:1405–12.
Groenning M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status. J Chem Biol. 2010;3:1–18.
McStay GP, Salvesen GS, Green DR. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 2008;15:322–31.
Caretta A, Mucignat-Caretta C. Protein kinase a in cancer. Cancers. 2011;3:913–26.
Sapio L, Di Maiolo F, Illiano M, Esposito A, Chiosi E, Spina A, et al. Targeting protein kinase A in cancer therapy: an update. EXCLI J. 2014;13:843–55.
Nesterova MV, Cho-Chung YS. Significance of protein kinase A in cancer. In: Apoptosis, cell signaling, and human diseases. Rakesh Srivastava, Totowa, NJ: Humana Press; 2006. pp 3–30.
Mitrea DM, Kriwacki RW. Regulated unfolding of proteins in signaling. FEBS Lett. 2013;587:1081–8.
Schultz JE, Natarajan J. Regulated unfolding: a basic principle of intraprotein signaling in modular proteins. Trends Biochem Sci. 2013;38:538–45.
Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10:S10–7.
Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237–48.
Tenreiro S, Eckermann K, Outeiro TF. Protein phosphorylation in neurodegeneration: friend or foe? Front Mol Neurosci. 2014;7:42.
Kumar S, Walter J. Phosphorylation of amyloid beta peptides-A trigger for formation of toxic aggregates in Alzheimer’s disease. Aging. 2011;3:803–12.
Sato H, Kato T, Arawaka S. The role of Ser129 phosphorylation of α-synuclein in neurodegeneration of Parkinson’s disease: a review of in vivo models. Rev Neurosci. 2013;24:115–23.
Samuel F, Flavin WP, Iqbal S, Pacelli C, Sri Renganathan SD, Trudeau L-E, et al. Effects of serine 129 phosphorylation on α-synuclein aggregation, membrane association, and internalization. J Biol Chem. 2016;291:4374–85.
Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid–from bacteria to humans. Trends Biochem Sci. 2007;35:217–224. https://doi.org/10.1016/j.tibs.2007.03.003.
Lin S-C, Lo Y-C, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–90.
Qiao Q, Yang C, Zheng C, Fontá L, David L, Yu X, et al. Structural architecture of the CARMA1/Bcl10/MALT1 signalosome: nucleation-induced filamentous assembly. Mol Cell. 2013;51:766–79.
Siegel RM, Martin DA, Zheng L, Ng SY, Bertin J, Cohen J, et al. Death-effector filaments: novel cytoplasmic structures that recruit caspases and trigger apoptosis. J Cell Biol. 1998;141:1243–53.
Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao Y-S, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–50.
Hou F, Sun L, Zheng H, Skaug B, Jiang Q-X, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–61.
Berson JF, Harper DC, Tenza D, Raposo G, Marks MS. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol Biol Cell. 2001;12:3451–64.
Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006;4:e6. https://doi.org/10.1371/journal.pbio.0040006.
Lu A, Li Y, Schmidt FI, Yin Q, Chen S, Fu T-M, et al. Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat Struct Mol Biol. 2016;23:1–12.
Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, et al. ASC, a Novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 Cells. J Biol Chem. 1999;274:33835–8.
Westphal D, Kluck RM, Dewson G. Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014;21:196–205.
Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS. Target protease specificity of the viral serpin CrmA: analysis of five caspases. J Biol Chem. 1997;272:7797–800.
Narayana N, Cox S, Shaltiel S, Taylor SS, Xuong N. Crystal structure of a polyhistidine-tagged recombinant catalytic subunit of cAMP-dependent protein kinase complexed with the peptide inhibitor PKI(5-24) and adenosine. Biochemistry. 1997;36:4438–48.
Slice LW, Taylor SS. Expression of the catalytic subunit of CAMP-dependent protein kinase. J Biol Chem. 1989;264:20940–6.
Yonemoto W, McGlone ML, Grant B, Taylor SS. Autophosphorylation of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli. Protein Eng. 1997;10:915–25.
Acknowledgements
This work was supported by the National Institutes of Health (GM 080532) to JH. BS was supported in part by the UMass Chemistry-Biology Interface Training Program (National Research Service Award T32 GM 08515 from the National Institutes of Health). We thank Jesse Rinehart (Yale University) for generously providing the E. coli strain C321.ΔA, the pBAD-GST-AmpR and SepOTSλ plasmids and for advice and helpful discussions about phosphoprotein synthesis. We thank Alex Ribbe, Director of the UMass Electron Microscopy facility for the collection of EM images. We thank Tyler Marcinko for his assistance with staining of protein samples and ThT assay. We also thank Scott Eron for providing casp-8 proteins as samples for caspase cleavage assays.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Edited by P. Salomoni
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Serrano, B.P., Hardy, J.A. Phosphorylation by protein kinase A disassembles the caspase-9 core. Cell Death Differ 25, 1025–1039 (2018). https://doi.org/10.1038/s41418-017-0052-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-017-0052-9
This article is cited by
-
How does caspases regulation play role in cell decisions? apoptosis and beyond
Molecular and Cellular Biochemistry (2023)
-
New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury
Angiogenesis (2020)
-
Identification of Allosteric Inhibitors against Active Caspase-6
Scientific Reports (2019)
