
Abstract
Reorganization of the podosome into the sealing zone is crucial for osteoclasts (OCLs) to resorb bone, but the underlying mechanisms are unclear. Here, we show that tuberous sclerosis complex 1 (TSC1) functions centrally in OCLs to promote podosome organization and bone resorption through mechanistic target of rapamycin complex 1 (mTORC1) and the small GTPases Rac1/Cdc42. During osteoclastogenesis, enhanced expression of TSC1 downregulates mTORC1 activity. TSC1 deletion in OCLs reduced podosome belt formation in vitro and sealing zone formation in vivo, leading to bone resorption deficiency and osteopetrosis. Mechanistically, TSC1 promoted podosome superstructure assembly by releasing mTORC1-dependent negative feedback inhibition of Rac1/Cdc42. Rapamycin and active Rac1/Cdc42 restore podosome organization and bone resorption and alleviate osteopetrotic phenotypes in mutant mice. Our findings reveal an essential role of TSC1 signaling in the regulation of bone resorption. Targeting TSC1 represents a novel strategy to inhibit bone resorption and prevent bone loss-related diseases.
Similar content being viewed by others


Introduction
Skeletal homeostasis depends on the balanced action of bone-resorbing osteoclasts (OCLs) and bone-forming osteoblasts [1,2,3]. OCLs are multinucleated cells which arise from fusion of the monocyte/macrophage lineage. OCLs adhere to mineralized bone matrix and become polarized: a ruffled membrane surrounded by an actin ring, also known as the sealing zone, mediates attachment to the bone surface [1, 4]. To resorb bone, the OCL sealing zone creates an extracellular compartment into which the OCL actively transports H+ and Cl− ions through the ruffled-border apical membrane and secretes proteases, lowering the pH and dissolving the mineral phase of the bone matrix [1, 4]. Understanding how OCLs degrade bone is necessary to understand bone physiology and pathology, and to develop therapeutic strategies for the treatment of OCL dysfunction-related bone diseases [5].
The most prominent cytoskeletal structures, podosomes, is crucial for OCL adhesion, spreading, migration and bone degradation [6,7,8] and podosome patterning is highly dynamic. They are found in a clustered pattern in OCL precursors, but form rings that expand into a stable podosome belt at the periphery of the mature OCL. Attachment to the bone surface causes polarization of OCLs, and the podosomes reorganize to form the sealing zone, a dense array of connected podosome and a hallmark of resorbing OCLs which separates the resorption lacuna beneath the OCL from the surrounding bone microenvironment [6, 9, 10]. The reported regulators of podosome formation and assembly include tyrosine kinases, actin regulators, the microtubule system and Rho family GTPases such as RhoA, Rac1, Cdc42 [11,12,13,14]. Although the intrinsic molecular composition of podosomes is well described, the signaling pathways that govern their collective organization into the superstructures remain ill defined.
The tumor suppressor genes, TSC1 and TSC2, encode hamartin and tuberin, respectively, which are key upstream negative regulators of the mechanistic target of rapamycin complex 1 (mTORC1) [15,16,17]. TSC1/2 complex expresses GTPase activating protein activity towards small GTPase Rheb, thereby inducing conversion of active GTP-bound Rheb to inactive GDP-bound Rheb. Active Rheb promotes mTORC1 activation, controlling protein translation by phosphorylating the ribosomal protein S6 kinase (S6K1) and inhibiting eukaryotic initiation factor 4E–binding protein-1, and controls cap-dependent protein translation. Mutations of TSC1 or TSC2 induce abnormal activation of mTORC1 and development of the genetic disorder tuberous sclerosis complex (TSC) which is characterized by benign hamartomas in multiple organ systems [18, 19]. There is also frequent involvement of TSC in the skeletal system such as sclerotic bone lesions with unknown mechanisms [20]. Although the roles of TSC/mTORC1 in cell metabolism and metabolic diseases have been extensively investigated, their functions and regulatory mechanisms in bone metabolism are less well understood.
Recently, we and other groups revealed an essential role of mTORC1 in the regulation of osteoblastogenesis and bone formation [21, 22]. Several in vitro studies have also demonstrated that mTORC1 is required for OCL progenitor proliferation and OCL differentiation and survival [23,24,25,26], but the functional role of TSC/mTORC1 in bone resorption in vivo remains undefined, and its role in podosome organization and the underlying mechanisms have not been reported.
In this study, we demonstrate that enhanced TSC1 expression, which downregulates mTORC1 activity, is crucial for podosome superstructure assembly during osteoclastogenesis. Deletion of TSC1 in OCLs reduced podosome belt formation in vitro and sealing zone formation in vivo, leading to a deficiency of bone resorption and osteopetrosis in mice. Mechanistically, mTORC1 prevents podosome assembly partially through negative feedback inhibition of Rac1/Cdc42. Our findings uncover a previously unknown but essential role of TSC1/mTORC1 signaling in the regulation of OCL podosome assembly and bone resorption.
Results
TSC1/2 is upregulated during OCL differentiation and podosome superstructure assembly
To examine the relationship between osteoclastogenesis and TSC/mTORC1 signaling under physiological conditions, we analyzed the TSC1/2 expression and mTORC1 activity during osteoclastic differentiation in Raw264.7 cell line and primary bone marrow monocytes (BMMs). High levels of the phosphorylation of mTOR (P-mTOR) and its downstream target, the ribosomal protein S6 (P-S6), a marker of mTORC1 pathway activation, were detected in proliferating Raw264.7 cells. However, P-mTOR and P-S6 began to decline after 1 day induction, and decreased by 80% on day 5 (Fig. 1a). The decrease in mTORC1 signaling was accompanied by a time-dependent increase in TSC1 mRNA (Fig. 1b, d) and in TSC1/2 protein levels of BMMs (Fig. 1c, e), although no significant change in TSC2 mRNA level (Supplementary Fig. 1A, 1B). Immunofluorescence staining confirmed the deactivation of mTORC1 upon OCL differentiation (Fig. 1f, g), which is consistent with a time-dependent increase in OCLs formation and Fusion Index [27] (Supplementary Fig. 1C, D). Interestingly, in multinucleated cells with podosome belts, mTORC1 activity was barely detectable (Fig. 1h). A similar pattern of changes in TSC1/2 expression and mTORC1 activity was observed in primary BMMs (Supplementary Fig. 1E–J) but not in osteoblasts (Supplementary Fig. 1K), which suggested that a low level of mTORC1 activity is required for podosome ring and belt formation in vitro. We further compared the level of P-S6 in monocytes with levels in mature OCLs in vivo and in vitro. Notably, activation of mTORC1 was observed in BMMs but not in multinucleated OCLs in distal femur sections of mice (Fig. 1i) and in primary cultured mature OCLs (Fig. 1j, k). Taken together, our results demonstrate that mTORC1 is inactivated in mature OCLs with assembled podosome superstructures, likely caused by an elevated expression of TSC1/2.

TSC1/2 is upregulated while mTORC1 activity is downregulated during OCL differentiation and podosome superstructure assembly. a Raw264.7 cells were induced to differentiate into OCLs by treatment with 50 ng/mL Rankl. Phosphorylation of S6 (P-S6) and mTOR (P-mTOR) and expression of TSC1 were detected by western blot. b The TSC1 mRNA expression increased during osteoclastogenesis (n = 3). c Both mTORC1 activity and TSC1 expression increased during osteoclastogenesis in BMMs (n = 3) (treated with 25 ng/mL M-CSF and 50 ng/mL RANKL for 2–8 days) d The TSC1 mRNA of BMMs (n = 3). e Western blot analysis of TSC1 and P-S6 in BMMs osteoclastogenic cultures with or without rapamycin. f Immunofluorescence staining confirmed the deactivation of mTORC1 upon OCL differentiation. Scale bar, 100 μm. g The number of P-S6-negative OCLs as a percentage of total OCLs. h mTORC1 activity was barely detectable in OCLs with a podosome belt. Scale bar, 20 μm. i The mTORC1 activity of OCLs and BMMs in the distal femur (8-week-old female C57BL/6 mice, n = 3). The arrowheads indicate mTORC1 activity of monocyte/macrophage. Scale bar, 5 μm. j Immunofluorescence staining showed the mTORC1 activity in BMMs and primary mature OCLs with podosome clusters and belts (n = 3). Scale bar, 10 μm. k P-S6 fluorescence density in BMMs and primary mature OCLs with podosome clusters or belts (n = 3). All experiments were repeated in triplicate
Deletion of TSC1 in OCLs causes osteopetrosis in mice
Upregulation of TSC1 in the late stages of OCL differentiation suggests a potential role of TSC1 in mature OCLs. To drive Cre expression specifically in OCLs, the promoters cathepsin-K (CTSK) and tartrate-resistant acid phosphatase (TRAP) were used. CTSK is a lysosomal cysteine protease that degrades type I collagen and is selectively expressed in OCLs [28, 29] but not in their precursors (such as TRAP), CTSK-Cre mice are better for understanding the podosome and functions of mature OCLs. To characterize the function of OCL TSC1 in bone resorption, we generated mice in which TSC1 was selectively ablated in CTSK-positive OCLs and selected mice with the genotype referred to as Tsc1ctsk−/− for detailed analysis. Tsc1ctskflox/- littermates served as controls (referred to as CTRL). Furthermore, a double-fluorescent Cre reporter mouse (mT/mG), which expresses membrane-targeted tandem dimer Tomato (mT) prior to Cre-mediated excision and membrane-targeted green fluorescent protein (mG) after excision [30], was used to verify the specificity of CTSK-mediated excision. We observed that mG labeling in CTSK+/−mT/mGflox/flox mice was CTSK-dependent, complementary to mT in OCLs showing green fluorescence (Fig. 2a). Tsc1ctsk−/− mice were born at the expected Mendelian frequency, and genotypes of transgenic offspring were determined by PCR (Supplementary Fig. 2A). Western blot analysis of cancellous bone lysates showed a dramatic decrease in TSC1 (~ 90% recombination occurs in homozygous) and increase in P-S6 in Tsc1ctsk−/− mice (Fig. 2b), indicating that mTORC1 was activated by disruption. Immunofluorescence staining of distal femur sections confirmed the activation of mTORC1 in multinucleated OCLs (Fig. 2c, d).

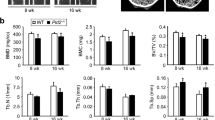
Deletion of TSC1 in OCLs causes osteopetrosis in mice. a Confirmation of the specificity of Cre-mediated excision by CTSK+/−mT/mGflox/flox mice. (white arrows: osteocyte, red arrows: osteoblast) b Western blot analysis of cancellous bone lysates (12-week-old female mice, n = 3, repeat three times). c Immunofluorescence staining of distal femur sections confirmed the activation of mTORC1 in OCLs (12-week-old female mice, n = 3, repeat four times). The arrowheads indicate the sealing zone. Scale bar, 10 μm. d The percentage of P-S6 positive OCLs (n = 5, repeat three times). e A representative image of square skulls and craniofacial abnormalities. The arrowheads indicate maxillofacial heave. Scale bar, 100 μm. f Whole-body X-ray analysis. g The three-dimensional structure of trabecular bone. Scale bar, 1.0 mm. h Quantification of cancellous bone mass in Tsc1ctsk−/− and CTRL mice (n = 6, repeat four times). i Quantification of cancellous bone architecture, including BV/TV, Tb.N, Tb.Th, Tb.Sp. (n = 5) j The three-dimensional structure of the femoral mid-shaft. Scale bar=1.0 mm. k Cortical thickness was increased by 25% in Tsc1ctsk−/− mice. (n = 5, repeat three times) l HE staining of the femoral cortical bones (18-week-old female). Scale bar=100 μm. m The percentage of porous occupation in cortical bone. (n = 5, repeat four times) n SEM showed more holes in the Tsc1ctsk−/− mice (12-week-old female). The arrowheads indicate the pores. Scale bar, 30 μm. o Comparison of pore number in cortical bone between CTRL and Tsc1ctsk−/− mice (n = 5, repeat four times)
At the gross level, 18-week-old Tsc1ctsk−/− mice exhibited square skulls and dwarfism (Fig. 2e and Supplementary Fig. 2B). The body weight and length were both significantly lower in Tsc1ctsk−/− mice (Supplementary Fig. 2C, D). Whole body X-ray analysis showed increased bone mass in the skull, vertebrae and long bones (Fig. 2f). Because of thickened cortical bone, the Tsc1ctsk−/− mice had pale bones (Supplementary Fig. 2E). Micro-CT analysis revealed a progressive osteopetrosis in Tsc1ctsk−/− mice (Fig. 2g, Supplementary Fig. 2F). Eighteen-week-old Tsc1ctsk−/− mice demonstrated a marked increase in cancellous bone mass (Fig. 2h, i). The cortical bone mass also increased in Tsc1ctsk−/− mice (Fig. 2j, k, Supplementary Fig. 2G, H, I). However, HE staining, scanning electron microscopy (SEM) and Micro-CT radiograms showed more porous areas in Tsc1ctsk−/− mice (Fig. 2l–o, Supplementary Fig. 2G), which resulted in a uniform 11% decrease in bone mineral density (BMD) (Supplementary Fig. 2J), a parameter reflecting the mass of mineral per volume of bone and the amount of bone mineral in bone tissue. Interestingly, the tooth eruption of the Tsc1ctsk−/− mice were not disturbed (Supplementary Figure 3).
Altogether, these data suggest that OCL-specific TSC1 disruption induces activation of mTORC1 in OCLs and causes osteopetrosis in mice.
TSC1 deficiency disturbs OCL podosome assembly and suppresses bone resorption in vitro and in vivo
We next analyzed the cellular basis for the increased bone mass in Tsc1ctsk−/− mice. Surprisingly, the number of OCLs increase and the number of osteoblasts decrease in Tsc1ctsk−/− mice compared with littermate controls (Fig. 3a, b and Supplementary Fig. 4A, B). Consistently, the levels of osteoclast differentiation marker CTSK were enhanced in Tsc1ctsk−/− mice (Supplementary Fig. 4C, D). However, levels of both procollagen 1 N-terminal propeptide (P1NP), a serum bone formation marker, and C-terminal telopeptides of type 1 collagen (CTX1), a serum bone resorption marker, were markedly reduced in Tsc1ctsk−/− mice (Fig. 3c, d). In addition, the Eroded surface (ES/BS) (bone resorption), mineral apposition rate (MAR) (bone formation) and bone formation rate (BFR) all declined significantly in Tsc1ctsk−/− mice (Fig. 3e–g). These data suggest that osteopetrosis in Tsc1ctsk−/− mice may result from reduced bone resorption but not increased bone formation, and that TSC1 deficiency in OCLs inhibits the OCLs’ function of resorbing bone but not their differentiation or secretion of proteases. Increased numbers of OCLs may result from feedback regulation of osteoclastogenesis to compensate for the impaired bone resorption, as increased proliferation of OCL progenitors (Fig. 3h, i) was observed. In addition, OCL apoptosis was inhibited in Tsc1ctsk−/− mice (Supplementary Fig. 4E, F).

TSC1 deficiency disturbs OCL podosome assembly and suppresses bone resorption in vitro and in vivo. a Representative images of TRAP staining of femurs from female mice. Scale bar=100 μm. b Number of TRAP positive OCLs (n = 5). c Serum CTX1 (12-week-old females, n = 5). d Serum P1NP (12-week-old females, n = 6). e The ES/BS decreased markedly in Tsc1ctsk−/− mice (n = 5). f Representative image of double-labeling fluorescent analysis. g The MAR and BFR, as bone formation parameter, decreased in the Tsc1ctsk−/−mice (n = 5). h Ki67 staining of BMMs-derived OCLs and BMMs. Scale bar, 20 μm. i Quantification of Ki67+ OCL progenitors (n = 5). j The immunostaining of F-actin and Tubulin in primary mature OCLs from CTRL and Tsc1ctsk−/− mice, showing abnormity in the width of podosome belt and podosome location in TSC1-null OCLs, but no significant change in tubulin structure. The arrowheads indicate the podosome pattern. Scale bar, 10 μm. k–n For OCL differentiation, BMMs from Tsc1ctsk−/− and CTRL mice were cultured in α-MEM containing M-CSF (25 ng/mL) and RANKL (50 ng/mL) for 8–10 days. k TSC1-null OCLs were unable to organize their podosomes into rings and belts, although the number increased markedly. Scale bar=100, 20 μm. l Fluorescence density of F-actin (n = 3). m The smaller and scattered lacunae in TSC1-null OCLs. Scale bar=200 μm. n Tsc1ctsk−/− BMMs-derived OCLs reduced resorbing capacity (n = 4). o TEM analysis indicated absence of the sealing zone formation in 6-week-old female Tsc1ctsk−/− mice (n = 5). SZ, sealing zone; BM, bone mineral; RB ruffle border, NU nucleus. Scale bar=4 μm. p Abnormal RB formation was observed in 6-week-old female Tsc1ctsk−/− mice (n = 5). Scale bar=600 nm. q SEM analysis revealed superficial lacunae in the femurs of 12-week-old female Tsc1ctsk−/− mice (n = 5). The arrowheads indicate absorption lacunae. Scale bar=30 μm. r TRAP staining showed that OCLs of 18-week-old female Tsc1ctsk−/− were round and fat, and inadequately spread (n = 5). The arrowheads indicate the cealing zone. Scale bar=100 μm. All experiments were repeated in triplicate
To determine the mechanism underlying the impaired bone resorption in transgenic bone, we cultured primary mature OCLs from Tsc1ctsk-/- and control mice. Interestingly, we observed most OCLs from Tsc1ctsk−/− mice were unable to form podosome ring and belt in vitro (Fig. 3j) and these TSC1 deficient cells demonstrated hyper-activation of mTORC1 (Supplementary Fig. 4I). Furthermore, although the number increased, OCLs differentiated from BMMs of Tsc1ctsk−/− mice had reduced capacity in organizing their podosomes into rings and belts (Fig. 3k, l) and resorption of bone in vitro (Fig. 3m, n). The ablation of TSC1 and enhanced activation of mTORC1 (P-S6) in OCLs but not in BMMs were confirmed by immunostaining (Fig. 3k and Supplementary Fig. 4G, H). These results indicate that TSC1 is crucial for OCL podosome organization and bone resorption in vitro.
We further assessed the role of TSC1 in podosome assembly and bone resorption in mice. Analysis by Transmission electron microscope (TEM) indicated that formation of the sealing zone and ruffled-border was abrogated in Tsc1ctsk−/− mice (Fig. 3o, p). Scanning electron microscope (SEM) observation further revealed more superficial resorption lacunaes in Tsc1ctsk−/− mice (Fig. 3q and Supplementary Fig. 4J). Interestingly, Immunofluorescence, TRAP staining, and SEM showed that these TSC1-deficient OCLs were morphologically round and fat, and lacked the capacity to spread on the bone surface (Figs. 2c and 3r).
Altogether, these in vitro and in vivo data collectively establish the essential role of TSC1 in controlling OCL podosome assembly and bone resorption.
TSC1 mediated OCL podosome organization and bone resorption are mTORC1 dependent
To further clarify the role of mTORC1 in OCLs, rapamycin as the specific mTORC1 inhibitor was used during osteoclastogenesis. Rapamycin treatment inhibited mTORC1, prevented the proliferation of Raw264.7 cells (Fig. 4a,b) and BMMs (Supplementary Fig. 5A, B) and reduced the number of differentiated OCLs (Fig. 4c, d and Supplementary Fig. 5C, D). However, podosome ring and belt formation were accelerated by low doses of rapamycin (Fig. 4e, f and Supplementary Fig. 5E, F). As expected, the deactivation of mTORC1 in bone marrow cells was observed after rapamycin (2 mg/kg/d) treatment. Rapamycin-treated mice exhibited a slight decrease in the number of OCLs but no change in osteoblasts (Fig. 4g–k), although this did not affect bone mass in these mice (Fig. 4l, m). The decreased OCLs in rapamycin-treated mice may have resulted from reduced OCL progenitor proliferation. These results demonstrate that mTORC1 promotes OCL progenitor proliferation but inhibits podosome assembly, indicating a stage-dependent regulation of osteoclastogenesis by mTORC1.

Podosome ring and belt formation was accelerated by low doses of Rapamycin. a Raw264.7 cells were induced to differentiate into OCLs by treating with 20 nM rapamycin. mTORC1 activity was analyzed by Western blot. b CCK8 assay (n = 3). c Representative image of TRAP staining (after 6 days of induction). Scale bar=100 μm. d Number of TRAP-positive cells (n = 3). e The formation of podosome rings and belts in Raw264.7-derived OCLs was accelerated by rapamycin after 5 days of induction. Scale bar=100 μm. f The percentage of OCLs with podosome belts (n = 3). g Eight-week-old female C57BL/6 mice were administered rapamycin (2 mg/kg/d) for 12 weeks, control mice were treated with PBS (n = 5). Analysis of mTORC1 activity in bone marrow cells by Western blot. h Representative image of TRAP staining. Scale bar, 100 μm. i Quantification of OCL number (n = 6). j Representative image of Osterix immunostaining. Scale bar=100 μm. k Quantification of osteoblast number (n = 6). l The three-dimensional structure of trabecular bone. Scale bar=1.0 mm. m No significant effect was observed on BMC of normal mice after treatment with rapamycin (n = 6). All experiments were repeated two times
TSC1/2 has been shown to regulate various cellular processes through mTORC1-dependent or -independent mechanisms [18, 21, 31]. To verify the role of mTORC1 in TSC-controlled OCL podosome organization and function, we examined whether rapamycin was able to rescue the phenotype of Tsc1ctsk−/− mice. As expected, rapamycin significantly ameliorated the osteopetrotic phenotypes of Tsc1ctsk−/− mice, as manifested by marked reductions in bone mass and cortical bone thickness after 3 months of rapamycin treatment (2 mg/kg/d) (Fig. 5a, d and Supplementary Fig. 6A, B). Accordingly, OCL number was reduced (Fig. 5e, f), while ES/BS and serum CTX1 and P1NP levels were enhanced by rapamycin (Fig. 5g–i). Importantly, rapamycin promoted formation of the podosome ring/belt and resorption of bone matrix in vitro by cultured OCLs differentiated from BMMs of Tsc1ctsk−/− mice (Fig. 5j–m) or mature TSC1-deficient OCLs (Supplementary Fig. 6C), as well as sealing zone formation by Tsc1ctsk−/− mice in vivo (Fig. 5n). In addition, the number and shape of OCL resorption lacunae were recovered (Fig. 5o, p, Supplementary Fig. 6D). These results indicate that TSC1 regulates OCL podosome organization and bone resorption through inhibition of mTORC1.

TSC1-mediated OCL Podosome Organization and Bone Resorption Are mTORC1 Dependent. a Six-week-old male CTRL and Tsc1ctsk−/− mice were treated with rapamycin (2 mg/kg/day) for 12 weeks. Rapamycin almost abrogated the osteopetrotic phenotypes of Tsc1ctsk−/− mice. The three-dimensional structure of the distal femur. Scale bar=1 mm. b Quantification of cancellous bone mass (n = 6). c Micro-CT radiograms of femoral mid-shaft. Scale bar=1 mm. d Quantification of cortical thickness (n = 6). e Representative image of TRAP staining. Scale bar, 100 μm. f Number of TRAP-positive OCL (n = 5). g The ES/BS was restored by rapamycin (n = 5). h Serum CTX1 (n = 5). i Serum P1NP (n = 5). j BMMs from CTRL and Tsc1ctsk-/- mice were induced to form OCLs for 8 days (n = 3). Rapamycin (20 nM) administered after 3 days induction improved the podosome assembly and podosome belt formation in TSC1-null OCLs. Scale bar, 100 μm. k Quantification of F-actin fluorescence density (n = 3). l Pit resorption assay. Scale bar=100 μm. m Quantification of pit resorption, showing that rapamycin partially restored bone resorbing capacity of Tsc1ctsk−/− OCLs in vitro (n = 4). n TEM results suggested the rapamycin partially reversed sealing zone formation of Tsc1ctsk-/- mice in vivo. Scale bar, 1 mm. o SEM revealed that OCL morphology after treatment with rapamycin. Scale bar=2 μm. p Rapamycin markedly restored the shape of the OCLs in Tsc1ctsk−/− mice (n = 5). All experiments were repeated two times
TSC/mTORC1 regulates Rac1/Cdc42 activity in OCLs
We next investigated the mechanism by which TSC/mTORC1 regulates podosome organization and bone resorption in OCLs. Rac1 and Cdc42 are members of the GTPase family that play essential roles in the regulation of podosome formation and assembly in OCLs [24, 32, 33]. Previous studies have shown that their activities could be modulated by TSC1/2 and mTOR in an Akt-dependent mechanism in fibroblasts and renal tumor cells [34, 35]. To determine if TSC1 regulates Rac1/Cdc42 activity in OCLs, active Rac1/Cdc42 were isolated with Rac1/cdc42 Activation Magnetic Beads Pulldown Assay Kit. This assay uses the downstream effector of Rac1/Cdc42, p21-activated protein kinase (PAK1), to isolate the active GTP-bound form of Rac1/Cdc42 from the lysates. The p21-binding domain of PAK1 was expressed as a GST-fusion protein and coupled to magnetic beads. After magnetic rack separation, an immunoblot was performed and the activated Rac1/Cdc42 was detected with specific monoclonal antibodies [36,37,38]. Analysis of trabecular bone lysates from Tsc1ctsk−/− mice revealed a remarkable reduction in GTP binding of Rac1/Cdc42 without affecting protein levels (Fig. 6a). Interestingly, correlated with the upregulation of TSC1, Rac1/Cdc42 activity increased during OCL differentiation (Fig. 6b, c). Similarly, OCLs derived from Tsc1ctsk−/− mice had much lower levels of Rac1/Cdc42 activity compared with those of littermate controls (Fig. 6d), indicating that TSC1 is essential for Rac1/Cdc42 activation in OCLs. Furthermore, rapamycin treatment restored Rac1/Cdc42 activity in TSC1-deficient OCLs (Fig. 6e), suggesting that mTORC1 is an upstream negative regulator of Rac1/Cdc42.

TSC/mTORC1 regulate Rac1/Cdc42 activity in OCLs. a GTP-binding of Rac1/Cdc42 was reduced markedly in trabecular bone lysates from Tsc1ctsk−/− mice. b GTP-binding of Rac1/Cdc42 in Raw264.7 cells increased in a time-dependent manner during osteoclastogenesis. c GTP-binding of Rac1/Cdc42 in BMMs-derived OCLs increased significantly. d OCLs differentiated from BMMs of Tsc1ctsk-/- mice had much lower levels of Rac1/Cdc42 activity compared with those of littermate controls. e The downregulation of Rac1/Cdc42 activity in Tsc1-null OCLs was partially restored by rapamycin (20 nM, added after 3 days of OCL induction). f Phosphorylation of the PI3K downstream target Akt (T308) and mTORC2 substrate Akt (S473) were downregulated while phosphorylation of Rictor (T1135) was increased in Tsc1-null OCLs. Notably, rapamycin rescued these changes. g Analysis of the expression and location of Tiam1 in primary mature OCLs by immunostaining. Scale bar=10 μm. h Quantification of the Tiam1 fluorescence density (n = 3). i The expression and location of RacGAP1 in primary OCLs. Scale bar=10 μm. j Quantification of the RacGAP1 fluorescence density (n = 3). k The expression and location of Rich1 in primary OCLs. Scale bar=10 μm. l Quantification of the Rich1 fluorescence density (n = 3). m Analysis of the expression of GEFs and GAPs in BMMs-derived OCLs from CTRL and Tsc1ctsk−/− mice by Western blot. All experiments were repeated in triplicate
We next sought to answer how TSC1/mTORC1 regulates Rac1/Cdc42 in OCLs. It has been shown that Rac1/Cdc42 could be modulated by mTORC1/S6 kinase 1 (S6K1) via negative feedback regulation of two pathways that are essential for Akt activation. S6K1 inhibits PI3K/Akt via phosphorylation and degradation of insulin receptor substrate-1, while inhibits mTORC2/Akt via phosphorylation of mTORC2 essential component Rictor at Thr1135 [34, 35, 39, 40]. Interestingly, phosphorylation of the PI3K downstream target Akt (T308) and the mTORC2 substrate Akt (S473) are downregulated, while phosphorylation of Rictor (T1135) is increased, in TSC1-null OCLs (Fig. 6f). Notably, rapamycin rescues these changes in TSC1-deficient OCLs (Fig. 6f), suggesting that both negative feedback regulations may contribute to reduced Rac1/Cdc42 activation caused by TSC1 deficiency and mTORC1 hyper-activation. These data demonstrate that TSC1/2 may activate Rac1/Cdc42 via blocking of mTORC1-dependent negative feedback mechanisms in OCLs.
Rac1/Cdc42 activities are controlled by guanine nucleotide–exchange factors (GEFs) and GTPase-activating proteins (GAPs). Tiam1 and RacGAP1 are well established as the GEF and GAP for Rac1, respectively [41,42,43,44], and DNMBP and Rich1 are the GEF and GAP for Cdc42, respectively [41, 45, 46]. Interestingly, the expression and activity (Rac1-GTP/GDP) of Tiam1 was also decreased significantly (Fig. 6h, m). In addition, mild increase in RacGap1 and Rich1 (Fig. 6i–m), but not DNMBP was detected in Tsc1-null OCSLs as compared with that of control OCLs (Supplementary Fig. 7A, B). Taking together, these findings implicate that negative feedback regulation of Akt and Tiam1 might contribute to the inhibition of OCLs podosome assembly by mTORC1.
TSC1/mTORC1 regulate OCL podosome assembly and bone resorption partially through Rac1/Cdc42
To further clarify whether Rac1/Cdc42 mediates TSC/mTORC1 dependent podosome organization and bone resorption, we constructed active Rac1/Cdc42 (Rac1-GTP and Cdc42-GTP) lentiviruses separately. We transfected Raw264.7 with these two letiviruses and confirmed their efficiency by Western blot (Supplementary Fig. 8A). Raw264.7-derived, bone marrow-derived and primary mature OCLs, were each transfected with constitutively active Rac1/Cdc42 viruses, and no obvious abnormality of podosome formation or assembly was observed in these cells (Supplementary Fig. 8B, C). However, active Rac1 and Cdc42 increased podosome ring and belt formation in TSC1-null OCLs (Fig. 7a–c), and importantly, partially restored their ability to resorb bone in vitro (Fig. 7d, e). To verify the action of Rac1/Cdc42 on podosome superstructure formation and bone resorption in vivo, we injected these lentivirus vectors expressing active Rac1/Cdc42 into the femur bone marrow cavity of Tsc1ctsk−/− mice (Supplementary Figure 8D). We found that sealing zone formation (Fig. 7f) and the shape of OCLs were partially restored (Fig. 7g, h, Supplementary Fig. 8E). ES/BS and serum CTX1 level were both increased after exogenous expression of active Rac1/Cdc42, but there was no change in P1NP (Fig. 7i–k). TRAP staining indicated no significant change in OCL number but normal morphology of TSC1-null OCLs expressing active Rac1/Cdc42 (Fig. 7g, l, m). Accordingly, Rac1/Cdc42 significantly alleviated the osteopetrotic phenotypes of Tsc1ctsk−/− mice, as manifested by reduced cancellous bone mass and cortical bone thickness 10 weeks after virus injection (Fig. 7n–p and Supplementary Fig. 8F, G). Taken together, these data suggest that TSC1/mTORC1 regulate OCL podosome assembly and bone resorption partially through Rac1/Cdc42.

TSC1/mTORC1 regulate OCL podosome assembly and bone resorption partially through Rac1/Cdc42. a Primary mature OCLs were transfected with constitutively active Rac1/Cdc42 and NC letiviruses for 12 h. Rac1/Cdc42 lentiviruses ameliorated podosome organization and belt formation in Tsc1-null OCLs (n = 5). The arrowheads indicate the podosome pattern. Scale bar=100 μm b BMMs from Tsc1ctsk−/− and CTRL mice were transfected with lentiviruses after 3 days of OCL induction. Rac1/Cdc42 letiviruses partially improved the podosome belt formation in Tsc1-null OCLs (n = 3). Scale bar, 100 μm. c Quantification of the percentage of OCLs with podosome belts (n = 3). d Pit resorption assay (n = 3). Scale bar, 100 μm. e Quantification of pit resorption, showing that Rac1/Cdc42 lentiviruses partly restored the ability of Tsc1-null OCLs to resorb bone in vitro (n = 4). f For rescue experiments in vivo, Rac1/Cdc42 and NC lentiviruses (5 × 108 TU/10 μL) were injected into the bilateral femurs of 8-week-old female Tsc1ctsk−/− mice for 10 weeks (n = 6–7). TEM measurement showed that SZ and RB formation was partially restored. Scale bar=2 μm. g SEM measurement. h The shape of the partial TSC1-null OCLs were recovered (n = 5). i The ES/BS was restored (n = 5). j Serum CTX1 level (n = 5–6). k Serum P1NP level (n = 5–6). l Representative image of TRAP staining. Scale bar=100 μm. m Quantification of OCL number (n = 5). n The three-dimensional structure of the femur. Scale bar=100 μm. o Cancellous bone mass was reduced compared with Tsc1ctsk−/− mice (n = 6). p Cortical bone thickness reduced slightly (n = 6). All experiments were repeated two times
Discussion
We have identified TSC1/mTORC1 as a critical yet previously unrecognized regulator of OCL podosome organization and bone resorption, partially mediated via the small GTPases Rac1 and Cdc42. Increased TSC1 expression, and downregulation of mTORC1 activity during osteoclastogenesis, is required for OCL Rac1/Cdc42 activation and podosome superstructure assembly, which is partially by releasing mTORC1-dependent negative feedback regulation of Akt and GEF/GAP (Fig. 8). Deletion of TSC1 in OCLs inactivates Rac1/Cdc42 via stimulation of mTORC1, reduces podosome belt formation in vitro and sealing zone formation in vivo, leading to impaired bone resorption and osteopetrosis in mice. Targeting TSC1 represents a novel strategy to inhibit bone resorption and prevent bone loss-related diseases.

Proposed role for Tsc1 and its signaling in osteoclasts. Enhanced TSC1 expression, leading to downregulation of mTORC1 activity during osteoclastogenesis, is required for OCL Rac1/Cdc42 activation and podosome superstructure assembly, by releasing mTORC1-dependent negative feedback regulation
Tuberous sclerosis is an autosomal dominant genetic disorder caused by a mutation in TSC1/2 and characterized by benign hamartomas in multiple organ systems [18, 19]. There is frequent involvement of the skeletal system in TSC patients including sclerotic bone lesions such as increased skull density and thickened calvaria, but it is not well understood or characterized [20, 47]. Mice with either TSC2 disrupted in mature osteoblasts (osteocalcin-cre) or TSC1 disrupted in preosteoblasts (osterix-cre) exhibited high bone mass but impaired differentiation of osteoblasts and mineralized nodule formation [21, 22]. Interestingly, deletion of TSC1 in OCLs in this study also produced a high bone mass phenotype. Although these three models all shared elevated mTORC1 activity, and exhibited uniform high bone mass, the underlying mechanisms were different. We have previously shown that TSC1 deficiency promotes preosteoblast proliferation but prevents osteoblast differentiation and maturation through activation of the Notch pathway [21]. Here we show that TSC1 ablation inactivates Rac1/Cdc42 to prevent podosome organization and bone resorption in OCLs. Loss-of-function mutations in OCLs always causes dental defects in osteoclast-poor osteopetrosis [48,49,50]. However, the tooth eruption of Tsc1ctsk−/− mice was not disturbed, which were different from other osteopetrosis models. The possible explanation is that systemic functional defects of OCLs occurred later than the tooth eruption, as the osteopetrotic phenotype of the mutant mice developed progressively and detectable osteopetrosis occurred at 12 weeks old. Of note, many of the phenotypes (such as growth defects) described for these TSC mutant mice do not appear in TSC patients. In addition, TSC mutation develops focal sclerotic bone lesions in patients while develops systemic sclerosis in mice. Nevertheless, these mouse models may have potential implications for the cellular and molecular basis of the formation of sclerotic bone lesions in tuberous sclerosis.
Several studies have shown that mTOR is implicated in osteoclastogenesis. Macrophage colony-stimulating factor (M-CSF)- and RANKL-dependent mTORC1/S6K1 activity is required for the survival of OCLs and their precursors, as rapamycin reduced their survival in vitro [23, 26]. Rapamycin has been demonstrated to either enhance RANKL-induced OCL differentiation [51] or inhibit OCL formation and bone resorption in vitro through an effect on the CCAAT/enhancer binding protein β (C/EBPβ) isoform ratio and on musculoaponeurotic fibrosarcoma oncogene homolog B (MafB) [25, 52]. Treatment with rapamycin prevents the bone loss induced by ovariectomy in Wistar rats [53], as well as structural damage to the joints of arthritic animals [52]. Interestingly, we found that low mTORC1 activity (not completely inhibited) is essential for Rac1/Cdc42 activation and podosome assembly (podosome belt and sealing zone formation) during OCL maturation. Based on findings from this study, we propose dynamic regulation and stage dependent function of mTORC1 is critical for OCL formation and bone resorption. Dynamic changes in TSC1 expression may contribute to the elegant regulation of mTORC1 activity during osteoclastogenesis. Generation of mice with conditional disruption of mTORC1 or TSC1 in different stages of osteoclastogenesis using Lyz-cre, TRAP-cre and CTSK-cre mice will elucidate the stage-dependent function of mTORC1 in OCL formation and bone resorption.
Rac1 and Cdc42 are members of the Rho GTPase family that act as essential regulators of actin cytoskeleton and cell migration. Altered Rho family GTPase function have relevance to the clinical manifestations of Tuberous sclerosis such as neural disorder [54]. In skeletal system, these small GTPases also play crucial roles in the differentiation, function, and survival of OCLs [12, 32, 33, 55,56,57,58]. Cdc42 modulates OCL proliferation, survival and podosome formation [12]. Cdc42 loss-of-function mice were osteopetrotic while OCLs generated from Cdc42-KO mice displayed reduced actin ring formation and reduced bone resorption in vitro [12]. Unlike Cdc42 deficiency, the combined absence of Rac1/2 does not impact upon differentiation but promotes severe osteopetrosis by dysregulating the OCL cytoskeleton, as evidenced by reduced motility of the cells and their inability to spread or generate podosome belts, sealing zones and ruffled borders [59]. Understanding the regulation of individual small GTPases in OCL biology may lead to more targeted therapies for the treatment of low bone mass. The present study revealed that Rac1/Cdc42 were both activated by TSC1/2 and suppressed by mTORC1 during the formation of OCLs. Our data suggest that TSC1/2 may activate Rac1/Cdc42 via blocking of mTORC1-dependent S6K1-PI3K and S6K1-Rictor/mTORC2 negative feedback mechanisms in OCLs. It is not surprising that long-term rapamycin treatment inhibits Rac1/Cdc42, although short term rapamycin treatment (5 h) could not induce any significant change in Rac1/Cdc42 activity [34]. A long-term rapamycin treatment may prevent Cdc42 and Rac1 protein synthesis [60], or block assembly of rictor/mTORC2 [61], an upstream activator of Rac1/Cdc42, to regulate actin assembly and organization [62].
It has been shown that PI3K/AKT activates RAC by relocaliaztion of GEFs to the plasma membrane [63] and by accumulation of the Rac1-GEF Tiam1 [64]. Our data demonstrate that negative feedback regulation of Akt and Tiam1 partially contributes to the inhibition of OCLs podosome assembly by mTORC1, although other unknown mechanisms may also be involved in this process.
Nitrogen-containing bisphosphonates which are currently the first-line treatment for osteolytic diseases such as osteoporosis, reduce bone resorption by inhibiting an enzyme required for the post-translational modification of small GTPases in OCLs [65]. However these drugs may inhibit osteoblast activity and bone formation directly or indirectly by inducing OCL apoptosis and disturbing osteoclast–osteoblast coupling [65]. An alternative approach would be to target the small GTPase signaling pathway, choosing proteins specifically involved in the control of sealing zone formation without affecting OCL differentiation. In this context, we identified TSC1 as an activator of Rac1/Cdc42 activation in OCLs. TSC1 deficiency impairs bone resorption without being toxic to OCLs. Thus, the modulation of TSC1 expression and mTORC1 activity shows promising results for future treatment of osteolytic diseases.
Materials and methods
Mice
Eight-week-old female C57BL/6 mice were purchased from Southern Medical University Laboratory Animal Center, and administered with sirolimus for 12 weeks. TSC1flox/flox (Jackson Laboratory, 129S6/SvEvTac, backcrossed to C57BL/6 mice), mT/mG [30] (Jackson Laboratory, C57BL/6 background, stock number 007676) and CTSK-Cre [66] (Generous gift from Professor Shigeaki Kato) transgenic mice were maintained on a pure C57BL/6 background, backcrossing to C57BL/6 mice for 6 generations. To generate OCL-specific TSC1 KO mice, TSC1flox/flox mice were bred with CTSK-Cre mice to obtain Tsc1ctskflox/- mice and then Tsc1ctskflox/- mice were bred with TSC1flox/flox mice to obtain Tsc1ctsk−/− mice and littermate controls (Tsc1ctskflox/− termed CTRL). CTSK+/-mT/mGflox/flox mice were obtained by breeding CTSK-Cre and mT/mG mice in the way as mentioned above. Six-week-old male Tsc1ctsk−/− mice were forced-fed Sirolimus (rapamycin, 2 mg/kg/day) or 4-phenylbutyricacid (PBA, sigma, St Louis, MO, USA; inhibitor of ER stress, 500 mg/kg/day) for 12 weeks. Genomic DNA extracts were prepared from tip of mouse tails and performed to determine genotypes by PCR. Primers used for PCR: TSC1, GTCACGACCGTAGGAGAAGC, GAATCAACCCCACAGAGCAT; CTSK, CCTGGAAAATGCTTCTGTCCGTTTGCC, GAGTTGATAGCTGGCTGGTGGCAGATG. All the animals were housed under conditions of constant temperature and humidity on a 12-h light, 12-h dark cycle. Food and water were available adlibitum. All animal experiments were carried out with the approval of the Southern Medical University Animal Care and Use Committee in accordance with the guidelines for the ethical treatment of animals.
Bone parameter analyses
The narcotized mice were analyzed by X-ray radiography. Bone parameters were quantified using microCT-80 scanner (Scanco Medical AG, Bassersdorf, Switzerland). The three-dimensional structure was constructed and the parameters analyzed were: BMD, BV/TV, Tb.N, Tb.Th, and Tb.Sp. Serum CTSK, CTX-1 and P1NP were measured using an ELISA kit (Cloud-Clone Corp., Houston, TX, USA). After the mice were killed by cervical dislocation to ameliorate suffering, bone samples were resected and fixed in 10% formalin for 48 h, decalcified in 10% EDTA (pH 7.3) for 14 days at 4 ℃ and embedded in paraffin (Leica Microsystems Ltd, Wetzlar, Germany). Five-micrometer-thick sections were processed for hematoxylin and eosin staining and immunostaining using a standard protocol. Eight-micrometer-thick sections were processed for TRAP staining following a standard protocol (Sigma). For dynamic bone histomorphometric analysis, two-month-old mice were subcutaneously injected with calcein (Sigma, 15 mg/kg body weight) in 2% sodium bicarbonate solution 10 days and 3 days before death, and 9 μm-thick sections were prepared for double-labeling fluorescent analysis. A commercial kit (Roche, Basel, Switzerland) that links digoxigenin-nucleotides to DNA by TdT was used for TUNEL staining.
Scanning and transmission electron microscopy measurement
The surface of the MMA embedded femurs were polished and acid-etched with 37% phosphoric acid for 2–10 s. After washing for 5 min with 5% sodium hypochlorite they were coated with gold and palladium before examining with SEM (S-3700N, Hitachi, Japan). The samples of the fimur were fixed for 16 h at 4 °C in 2.5% glutaraldehyde, and then post-fixed for 2 h at 4 °C in 2% OsO4. After fixations, they were dehydrated through an ascending ethanol series, and then passed through propylene oxide, and finally embedded in resin. Transmission electron microscope was carried out to determine sealing zone morphology, and selected area electron diffraction was recorded by high-resolution transmission electron microscopy (HRTEM, Hitachi).
Cell
The RAW264.7 cell line (TIB-71) (ATCC, Manassas, VA, USA) was cultured in DMEM. Cell proliferation was evaluated using Cell Counting Kit-8 (CCK8, Dojindo, Japan), which allows very convenient assays by utilizing highly water-soluble tetrazolium salt for the determination of the number of viable cells.
The primary osteoblastic cells were prepared from the calvaria of neonatal mice. Cells were maintained in α-MEM at 37℃ with 5% CO2.
Ex vivo OCL culture and differentiation
Mature OCLs and BMMs were isolated from CTRL and Tsc1ctsk−/− neonatal murine long bones by mechanical disaggregation and replated onto glass coverslips, then left untreated or treated with rapamycin (20 nM for 10 h) at 37°C and fixed, stained, and imaged 6-12 h later. For OCL differentiation, BMMs were obtained from 6 to 8-week-old tibia/femur and cultured in α-MEM. OCL-like cells were obtained by differentiation of Raw264.7 in the presence of RANKL (50 ng/mL) for 1–7 days or differentiation of BMMs in α-MEM containing M-CSF (25 ng/mL) and RANKL (50 ng/mL) for 6-8 days. For rapamycin treatment, the cells were incubated with 20 nM rapamycin for 5–10 days. The cells were fixed with 3.7% formaldehyde in PBS for 10 min and immunofluorescence stained with P-S6, Ki67 (Cell Signaling Technologies), TRITC-Actin (Sigma), FITC-Actin (Sigma) and Tubulin-Trakcer Red (Sigma). DAPI (Life Technologies) was used to detect the nuclei. The coverslips were imaged using confocal microscope (Olympus). To analyze pit resorption, BMMs of CTRL and Tsc1ctsk−/− mice were seeded in 96-well plates (BD BioCoat™ Osteologic™ Bone Cell Culture System) and allowed to differentiate into OCLs in the presence of M-CSF and RANKL for 10 days, with a change of medium after 2 days. In some assays, multinucleated OCLs ( > 3 nuclei) were identified as TRAP-positive by TRAP staining kit (sigma). The fusion index was calculated as the percentage of nuclei in OCLs over the total number of nuclei.
Real-time PCR and protein analyses
Total RNA was isolated from cells using TRIZOL Reagent (Invitrogen), reverse transcribed into cDNA using the PrimeScriptTM RT reagent kit (Takara Bio Inc., Shiga, Japan). To quantify gene expression, real-time PCR was performed on an ABI 7300 Real-Time PCR System (Applied Biosystems) using SYBR green (Takara). Real-Time PCR data was analyzed according to the literature [67]. The primers included TSC1 (ACCACTGTTGGCTCACTTCC; CACGCTGTCCTCATCACACT) and TSC2 (GGGTGAAGAGAGCCGTATCA; ATGGAGCGGAATCAGAGATG). For protein analysis, cells were lysed in RIPA buffer containing 25 mM Tris, pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, protease inhibitor and phosphatase inhibitor cocktail (Roche). Protein samples were subjected to SDS–PAGE on NuPAGE gel and visualized by peroxidase solution and Luminol Enhancer solution. Extracts were loaded on different blots to determine the phosphorylated and total levels of respected proteins. The Western blot data was analyzed by BandScan. The following primary antibodies were used: phospho-S6 ribosomal protein(Ser235/236) (#2211, 1:3000), phospho-Akt(Thr308) (#9275, 1:1500), phospho-Akt (Thr473) (#9271,1:1000), phosphor-Rictor(Thr1135) (#3806, 1:1000), phospho-p70 S6K1(T389) (#9206, 1:1000) and Rictor (#9476, 1:1000) were from Cell Signaling Technologies. Hamatin (ab32936) was from Abcam. S6 ribosomal protein (sc-74459, 1:3000), Osterix (sc-22538, 1:200), and Akt (sc-8312, 1:1000) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); Tuberin (BS1604) (1:1000) was from Bioworld Technology (St Louis Park, MN, USA).
Pulldown assay for Cdc42 and Rac1 activity
The active Rac1/Cdc42 (GTP-bound form) in Tsc1ctsk−/− OCLs were isolate following the protocol provided by “Rac1/cdc42 Activation Magnetic Beads Pulldown Assay Kit” from Millipore (stock number: 17-10394). For rapamycin treatment, the cells were incubated in medium containing 20 nm rapamycin for 12 h. The amount of total and active Cdc42 and Rac1 was detected using Cdc42 and Rac1 antibodies (Millipore #05-389, 1:2000; #05-542, 1:500).
Constructed Rac1-GTPase and Cdc42-GTPase lentivirus and lentivirus transfected
The lentiviral expression plasmid constitutively active mutant of Rac1-GTPase (Plenti6/v5-CA-Rac1) and the constitutively active mutant of Cdc42-GTPase (Plenti6/v5-CA-Cdc42) were gifted. The two plasmids were packaged using the ViraPowerTM lentiviral expression system (Invitrogen) to produce constitutively active Rac1 and constitutive active Cdc42 lentiviruses. Mature OCLs of Tsc1ctsk−/− mice were transfected with lentiviruses for 12 h. BMMs from Tsc1ctsk−/− mice were transfected with lentiviruses at a multiplicity of infection of 5 for 72 h after 3 days of OCL induction. The NC lentiviruses were used as control. For rescue experiments in vivo, Rac1/ Cdc42 and NC lentiviruses (5 × 108 TU/10 μL) were injected into the bilateral femur of 8-week-old female Tsc1ctsk−/− mice for 10 weeks, after which femurs were subjected to histological analysis.
Statistical analyses
Statistical analyses were performed with the two-tailed Student’s t-test for two-group comparisons, or one-way ANOVA and post-hoc multiple comparisons with Bonferroni correction for three- or four-group comparisons. The levels of significance are represented as *P < 0.05; **P < 0.01; ***P < 0.005 or n.s. non-significant (P > 0.05), and data are shown as mean ± SEM unless noted otherwise.
References
Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–42.
Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–55.
Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791–801.
Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–8.
Khosla S, Westendorf JJ, Oursler MJ. Building bone to reverse osteoporosis and repair fractures. J Clin Invest. 2008;118:421–8.
Georgess D, Machuca-Gayet I, Blangy A, Jurdic P. Podosome organization drives osteoclast-mediated bone resorption. Cell Adh Migr. 2014;8:191–204.
Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13:376–85.
Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12:413–26.
Jurdic P, Saltel F, Chabadel A, Destaing O. Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol. 2006;85:195–202.
Saltel F, Chabadel A, Bonnelye E, Jurdic P. Actin cytoskeletal organisation in osteoclasts: a model to decipher transmigration and matrix degradation. Eur J Cell Biol. 2008;87:459–68.
Biosse Duplan M, Zalli D, Stephens S, Zenger S, Neff L, Oelkers JM, et al. Microtubule dynamic instability controls podosome patterning in osteoclasts through EB1, cortactin, and Src. Mol Cell Biol. 2014;34:16–29.
Ito Y, Teitelbaum SL, Zou W, Zheng Y, Johnson JF, Chappel J, et al. Cdc42 regulates bone modeling and remodeling in mice by modulating RANKL/M-CSF signaling and osteoclast polarization. J Clin Invest. 2010;120:1981–93.
Takegahara N, Kang S, Nojima S, Takamatsu H, Okuno T, Kikutani H, et al. Integral roles of a guanine nucleotide exchange factor, FARP2, in osteoclast podosome rearrangements. FASEB J: Off Publ Fed Am Soc Exp Biol. 2010;24:4782–92.
Vives V, Cres G, Richard C, Busson M, Ferrandez Y, Planson AG, et al. Pharmacological inhibition of Dock5 prevents osteolysis by affecting osteoclast podosome organization while preserving bone formation. Nat Commun. 2015;6:6218.
Bai X, Jiang Y. Key factors in mTOR regulation. Cell Mol Life Sci. 2010;67:239–53.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93.
Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15:155–62.
Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24.
Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57.
Oh JM. Bone changes in a patient with tuberous sclerosis complex. J Clin Rheumatol. 2012;18:161.
Huang B, Wang Y, Wang W, Chen J, Lai P, Liu Z, et al. mTORC1 prevents preosteoblast differentiation through the notch signaling pathway. PLoS Genet. 2015;11:e1005426.
Riddle RC, Frey JL, Tomlinson RE, Ferron M, Li Y, DiGirolamo DJ, et al. Tsc2 is a molecular checkpoint controlling osteoblast development and glucose homeostasis. Mol Cell Biol. 2014;34:1850–62.
Glantschnig H, Fisher JE, Wesolowski G, Rodan GA, Reszka AA. M-CSF, TNFalpha and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ. 2003;10:1165–77.
Indo Y, Takeshita S, Ishii KA, Hoshii T, Aburatani H, Hirao A, et al. Metabolic regulation of osteoclast differentiation and function. J Bone Mineral Res: Off J Am Soc Bone Mineral Res. 2013;28:2392–9.
Smink JJ, Begay V, Schoenmaker T, Sterneck E, de Vries TJ, Leutz A. Transcription factor C/EBPbeta isoform ratio regulates osteoclastogenesis through MafB. EMBO J. 2009;28:1769–81.
Sugatani T, Hruska KA. Akt1/Akt2 and mammalian target of rapamycin/Bim play critical roles in osteoclast differentiation and survival, respectively, whereas Akt is dispensable for cell survival in isolated osteoclast precursors. J Biol Chem. 2005;280:3583–9.
Pellegatti P, Falzoni S, Donvito G, Lemaire I, Di Virgilio F. P2X7 receptor drives osteoclast fusion by increasing the extracellular adenosine concentration. FASEB J: Off Publ Fed Am Soc Exp Biol. 2011;25:1264–74.
Rantakokko J, Aro HT, Savontaus M, Vuorio E. Mouse cathepsin K: cDNA cloning and predominant expression of the gene in osteoclasts, and in some hypertrophying chondrocytes during mouse development. FEBS Lett. 1996;393:307–13.
Drake FH, Dodds RA, James IE, Connor JR, Debouck C, Richardson S, et al. Cathepsin K, but not cathepsins B, L, or S, is abundantly expressed in human osteoclasts. J Biol Chem. 1996;271:12511–6.
Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genes. 2007;45:593–605.
Zhu L, Yang T, Li L, Sun L, Hou Y, Hu X, et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat Commun. 2014;5:4696.
Ory S, Brazier H, Pawlak G, Blangy A. Rho GTPases in osteoclasts: orchestrators of podosome arrangement. Eur J Cell Biol. 2008;87:469–77.
Touaitahuata H, Blangy A, Vives V. Modulation of osteoclast differentiation and bone resorption by Rho GTPases. Small GTPases. 2014;5:e28119.
Larson Y, Liu J, Stevens PD, Li X, Li J, Evers BM, et al. Tuberous sclerosis complex 2 (TSC2) regulates cell migration and polarity through activation of CDC42 and RAC1. J Biol Chem. 2010;285:24987–98.
Ohsawa M, Kobayashi T, Okura H, Igarashi T, Mizuguchi M, Hino O. TSC1 controls distribution of actin fibers through its effect on function of Rho family of small GTPases and regulates cell migration and polarity. PLoS ONE. 2013;8:e54503.
Wei Q, Adelstein RS. Pitx2a expression alters actin-myosin cytoskeleton and migration of HeLa cells through Rho GTPase signaling. Mol Biol Cell. 2002;13:683–97.
Cook JA, Albacker L, August A, Henderson AJ. CD28-dependent HIV-1 transcription is associated with Vav, Rac, and NF-kappa B activation. J Biol Chem. 2003;278:35812–8.
Benard V, Bohl BP, Bokoch GM. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J Biol Chem. 1999;274:13198–204.
Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009;29:5657–70.
Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–8.
Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–77.
Guillemot L, Guerrera D, Spadaro D, Tapia R, Jond L, Citi S. MgcRacGAP interacts with cingulin and paracingulin to regulate Rac1 activation and development of the tight junction barrier during epithelial junction assembly. Mol Biol Cell. 2014;25:1995–2005.
Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22.
Mertens AE, Pegtel DM, Collard JG. Tiam1 takes PARt in cell polarity. Trends Cell Biol. 2006;16:308–16.
Otani T, Ichii T, Aono S, Takeichi M. Cdc42 GEF Tuba regulates the junctional configuration of simple epithelial cells. J Cell Biol. 2006;175:135–46.
Wells CD, Fawcett JP, Traweger A, Yamanaka Y, Goudreault M, Elder K, et al. A Rich1/Amot complex regulates the Cdc42 GTPase and apical-polarity proteins in epithelial cells. Cell. 2006;125:535–48.
Avila NA, Dwyer AJ, Rabel A, Darling T, Hong CH, Moss J. CT of sclerotic bone lesions: imaging features differentiating tuberous sclerosis complex with lymphangioleiomyomatosis from sporadic lymphangioleiomymatosis. Radiology. 2010;254:851–7.
Helfrich MH. Osteoclast diseases and dental abnormalities. Arch Oral Biol. 2005;50:115–22.
Castaneda B, Simon Y, Jacques J, Hess E, Choi YW, Blin-Wakkach C, et al. Bone resorption control of tooth eruption and root morphogenesis: Involvement of the receptor activator of NF-kappaB (RANK). J Cell Physiol. 2011;226:74–85.
Alfaqeeh S, Oralova V, Foxworthy M, Matalova E, Grigoriadis AE, Tucker AS. Root and Eruption Defects in c-Fos Mice Are Driven by Loss of Osteoclasts. J Dent Res. 2015;94:1724–31.
Shui C, Riggs BL, Khosla S. The immunosuppressant rapamycin, alone or with transforming growth factor-beta, enhances osteoclast differentiation of RAW264.7 monocyte-macrophage cells in the presence of RANK-ligand. Calcif Tissue Int. 2002;71:437–46.
Cejka D, Hayer S, Niederreiter B, Sieghart W, Fuereder T, Zwerina J, et al. Mammalian target of rapamycin signaling is crucial for joint destruction in experimental arthritis and is activated in osteoclasts from patients with rheumatoid arthritis. Arthritis Rheum. 2010;62:2294–302.
Kneissel M, Luong-Nguyen NH, Baptist M, Cortesi R, Zumstein-Mecker S, Kossida S, et al. Everolimus suppresses cancellous bone loss, bone resorption, and cathepsin K expression by osteoclasts. Bone. 2004;35:1144–56.
Ferrer I, Mohan P, Chen H, Castellsague J, Gomez-Baldo L, Carmona M, et al. Tubers from patients with tuberous sclerosis complex are characterized by changes in microtubule biology through ROCK2 signalling. J Pathol. 2014;233:247–57.
Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106:852–9.
Wang Y, Lebowitz D, Sun C, Thang H, Grynpas MD, Glogauer M. Identifying the relative contributions of Rac1 and Rac2 to osteoclastogenesis. J Bone Mineral Res: Off J Am Soc Bone Mineral Res. 2008;23:260–70.
Lee NK, Choi HK, Kim DK, Lee SY. Rac1 GTPase regulates osteoclast differentiation through TRANCE-induced NF-kappa B activation. Mol Cell Biochem. 2006;281:55–61.
Han SY, Lee NK, Kim KH, Jang IW, Yim M, Kim JH, et al. Transcriptional induction of cyclooxygenase-2 in osteoclast precursors is involved in RANKL-induced osteoclastogenesis. Blood. 2005;106:1240–5.
Croke M, Ross FP, Korhonen M, Williams DA, Zou W, Teitelbaum SL. Rac deletion in osteoclasts causes severe osteopetrosis. J Cell Sci. 2011;124:3811–21.
Liu L, Luo Y, Chen L, Shen T, Xu B, Chen W, et al. Rapamycin inhibits cytoskeleton reorganization and cell motility by suppressing RhoA expression and activity. J Biol Chem. 2010;285:38362–73.
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68.
He Y, Li D, Cook SL, Yoon MS, Kapoor A, Rao CV, et al. Mammalian target of rapamycin and Rictor control neutrophil chemotaxis by regulating Rac/Cdc42 activity and the actin cytoskeleton. Mol Biol Cell. 2013;24:3369–80.
Wu CY, Carpenter ES, Takeuchi KK, Halbrook CJ, Peverley LV, Bien H, et al. PI3K regulation of RAC1 is required for KRAS-induced pancreatic tumorigenesis in mice. Gastroenterology. 2014;147:1405–16.e1407.
Zhu G, Fan Z, Ding M, Zhang H, Mu L, Ding Y, et al. An EGFR/PI3K/AKT axis promotes accumulation of the Rac1-GEF Tiam1 that is critical in EGFR-driven tumorigenesis. Oncogene. 2015;34:5971–82.
Brufsky A, Mathew A. Bisphosphonates, bone, and breast cancer recurrence. Lancet. 2015;386:1319–20.
Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130:811–23.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8.
Acknowledgements
We thank Dr. Shigeaki Kato (University of Tokyo) and Dr. Xu Jiake (West Australia University) for CTSK-Cre mice. This research was supported by Grant Nos. 31701033, 31600964, U1301222, 81625015, 81530070, 31529002 from the National Natural Sciences Foundation of China, 2016A030310400 from Natural Science Foundation of Guangdong Province of China, 2015CB55360 from the State Key Development Program for Basic Research of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Song Xu, Yue Zhang, and Jian Wang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Xu, S., Zhang, Y., Wang, J. et al. TSC1 regulates osteoclast podosome organization and bone resorption through mTORC1 and Rac1/Cdc42. Cell Death Differ 25, 1549–1566 (2018). https://doi.org/10.1038/s41418-017-0049-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-017-0049-4
This article is cited by
-
Abr, a Rho-regulating protein, modulates osteoclastogenesis by enhancing lamellipodia formation by interacting with poly(ADP-ribose) glycohydrolase
Molecular Biology Reports (2023)
-
Pro-angiognetic and pro-osteogenic effects of human umbilical cord mesenchymal stem cell-derived exosomal miR-21-5p in osteonecrosis of the femoral head
Cell Death Discovery (2022)
-
Role of TSC1 in physiology and diseases
Molecular and Cellular Biochemistry (2021)
-
TIMELESS regulates sphingolipid metabolism and tumor cell growth through Sp1/ACER2/S1P axis in ER-positive breast cancer
Cell Death & Disease (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}