
Abstract
Axonal injury is a common feature of central nervous system insults. Upregulation of amyloid precursor protein (APP) is observed following central nervous system neurotrauma and is regarded as a marker of central nervous system axonal injury. However, the underlying mechanism by which APP mediates neuronal death remains to be elucidated. Here, we used mouse optic nerve axotomy (ONA) to model central nervous system axonal injury replicating aspects of retinal ganglion cell (RGC) death in optic neuropathies. APP and APP intracellular domain (AICD) were upregulated in retina after ONA and APP knockout reduced Tuj1+ RGC loss. Pathway analysis of microarray data combined with chromatin immunoprecipitation and a luciferase reporter assay demonstrated that AICD interacts with the JNK3 gene locus and regulates JNK3 expression. Moreover, JNK3 was found to be upregulated after ONA and to contribute to Tuj1+ RGC death. APP knockout reduced the ONA-induced enhanced expression of JNK3 and phosphorylated JNK (pJNK). Gamma-secretase inhibitors prevented production of AICD, reduced JNK3 and pJNK expression similarly, and protected Tuj1+ RGCs from ONA-induced cell death. Together these data indicate that ONA induces APP expression and that gamma-secretase cleavage of APP releases AICD, which upregulates JNK3 leading to RGC death. This pathway may be a novel target for neuronal protection in optic neuropathies and other forms of neurotrauma.
Similar content being viewed by others


Introduction
Optic neuropathies are diseases characterized by visual loss due to damage to the optic nerve resulting in loss of retinal ganglion cells (RGCs). Optic neuropathies can result from various causes, including glaucoma, ischemia and trauma [1], but axonal injury underlies RGC death in most cases [2]. Lack of clinically applicable treatment for optic neuropathies [3] drives the need for further research into the underlying mechanisms. Axonal injury also occurs in many other forms of central nervous system insult such as stroke and traumatic brain injury. Optic nerve axotomy (ONA) offers a simplified model of CNS axonal injury that allows for reproducible injury of a relatively homogenous population of axons. Thus, ONA is a reproducible model for analyzing neuron degeneration in response to axon injury [4,5]. Additionally, ONA models characteristics of the specific kind of axonal degeneration that occurs in optic neuropathies. This model is particularly attractive because the vitreous chamber of the eye permits experimental manipulations via intraocular injections. As the ganglion cell layer is a monolayer, RGC densities can be directly quantified in flat-mounted tissue with accuracy, without the need for stereology [6]. RGC apoptosis has a characteristic time-course whereby cell death is delayed until 3–4 days post-axotomy, after which the cells rapidly degenerate. This provides a time window for experimental manipulations directed against pathways involved in apoptotic cell death [7,8].
Amyloid precursor protein (APP) is best known for its involvement in the pathogenesis of Alzheimer disease (AD). However, APP can also be detected immunocytochemically at sites of axonal injury in the brain, and has long been used as a general marker for axonal injury [9,10]. APP accumulation was also found in demyelinated axons in multiple sclerosis [11]. APP is transported by fast anterograde axonal transport [12], and is thought to accumulate in injured axons due to axonal transport failure. It was reported that high Aβ and APP levels were detected in chronic ocular hypertension glaucoma models [13]. APP intracellular domain (AICD) is derived by proteolytic processing of APP [14]. Recently, there has been considerable interest in the putative roles of AICD in the pathogenesis of AD and neurodegeneration [15]. AICD peptides were originally identified in the brains of AD patients. They have been implicated both in induction of apoptosis and in enhancement of responses to other apoptotic stimuli [14]. AICD translocates to the nucleus and acts as a transcription factor or in concert with other transcription factors signaling to the nucleus [16].
In RGCs, the JNK pathway is activated by many apoptotic stimuli [17,18]. The active phosphorylated form of JNK is detected in RGCs in human glaucoma [19]. JNK3 is the major JNK isoform expressed in neural tissue [20]. JNK3 deficiency protects neurons from insults such as excitotoxicity or ischemia [21,22]. While in a mouse model of chronic ocular hypertension, increased ocular pressure resulting in apoptosis of RGCs was associated with increased expression of JNK3 [23].
In summary, although axonal injury is known to upregulate APP expression in axons, it is not known whether this upregulation of APP occurs in RGCs and whether it mediates axon injury-associated neuronal death, which likely involves JNK3. We hypothesized that axon injury induces upregulation of APP expression in RGCs and that APP, in turn, activates JNK3-mediated neuronal death. Here we report that APP regulates JNK3 gene expression via gamma-secretase-dependent release of AICD and plays a role in RGC degeneration after ONA in the mouse.
Results
APP is upregulated and involved in RGC death after ONA
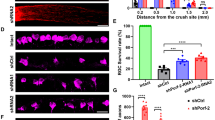
APP is upregulated on neural injury and has long been regarded as a marker for axonal degeneration [24,25]. RGC death after ONA is caused by axon injury [5,26], so we wondered whether APP plays a role in ONA-induced cell death. To identify the role of APP in RGC degeneration, we first detected APP expression after ONA. Mouse retinae were harvested 1 day, 7 days and 14 days after intraorbital ONA. The number of RGCs in ONA-injured retinae remained similar to those in control retinae until day 7 after which most of the RGCs were lost by day 14 [5]. Our results showed that 1 day after ONA, APP protein levels were upregulated by twofold and were still elevated, although slightly decreased comparing with day 1 post-ONA, 7 days and 14 days after ONA (Figs. 1a, b), and AICD production was also increased (Fig. S1a). Additional immunostaining in mouse retina sections showed that APP was upregulated in RGCs after ONA (Fig. S2). Next, we investigated whether APP contributed to RGC loss in ONA-injured eyes by counting Tuj1+ RGCs in wild-type and APP-null mice. The mice were perfused with 4% paraformaldehyde (PFA) and their retinae were harvested. The flat-mounted retinae were stained using mouse anti-Tuj1 monoclonal antibody to label RGCs (Fig. 1d), and then were analyzed by the counting method shown in Fig. 1c. The Tuj1+ RGC number in intact retinae from wild-type or APP-null mice was not significantly different (wild-type 2123 ± 85 cells/mm2, APP-null 2052 ± 171 cells/mm2, p > 0.05, n = 3 for each genotype); whereas the number of surviving Tuj1+ RGCs in APP-null mice was significantly more than that of wild-type mice 1 week or 2 weeks after ONA (Figs. 1d, e). The Tuj1+ RGC densities observed in intact retinae were consistent with previously reported Tuj1+ RGC numbers [27] although lower than RGC numbers measured by OHSt (hydroxystilbamidine methanesulfonate) retrograde labeling [28]. We also performed terminal deoxynucleotidyl transferase-mediated, dUTP nick end labeling (TUNEL) to test whether the RGC death is through the apoptosis pathway (Fig. S3). The detection of TUNEL-positive apoptotic cell death peaked (2–3 positive RGCs per section) at 6–7 days after ONA, which indicates that most of the ONA-induced cell death was through necrosis, instead of apoptosis. Nevertheless, APP deficiency also protected RGCs from apoptosis. We did not observe TUNEL-positive RGCs in intact wild-type or APP-null retina sections, which excluded a basal level difference between genotypes.

a Representative western blots showing APP expression after ONA. Intact mouse retinae or retinae 1 day (1 d), 1 week (1 w) or 2 weeks (2 w) after ONA were harvested, lysed and immunoblotted using antibodies against APP or GAPDH (internal loading control). The location of molecular weight markers is shown in kDa on the right. b The bar chart shows quantitative analysis of relative APP expression after ONA. The density of the western blot bands was normalized to the internal loading control and then normalized to control mice. Data were analyzed by one-way ANOVA followed by post-hoc Bonferroni tests: *p < 0.05; **p < 0.01 (n = 3 for each time point). c Method for quantifying Tuj1+ RGCs in the whole-mounted retina. For each retina, images were obtained from eight 20 × fields along two perpendicular retinal diameters. The average number of the Tuj1+ RGCs was obtained by averaging the counts for the 16 fields for each retina. d Representative fluorescence microscope images showing flat-mounts of intact retinae or retinae 1 w, or 2 w after ONA from wild-type or APP-null mice. The retinae were immunostained using anti-tubulin beta III (Tuj1) antibody, a marker for RGC neurons in the retina. Scale bar, 50 μm. e The bar chart shows quantitative analysis of Tuj1+ RGC numbers from wild-type and APP-null mice after ONA (n = 3 for each genotype). Student’s t-test: ** p < 0.01. Error bars represent SEM. f Representative confocal images show immunostaining of intact and injured (14 days after ONA) mouse retina flat-mounts using anti-GPAP antibody, a marker for astrocytes. No significant difference was observed in the astrocyte number between intact and injured retina. Scale bar, 50 μm
ONA selectively injures the axons of RGCs. To investigate whether this injury led to RGC death specifically or influenced other cell types in ganglion cell layer as well, we examined the survival of astrocytes using an anti-GFAP (glial fibrillary acidic protein) antibody. No significant difference in GFAP+ cells was observed between intact retinae and retinae 14 days after ONA (Fig. 1f), which is consistent with previous studies [7,29]. Thus, we showed that APP was upregulated following ONA and that APP knockout reduced Tuj1+ RGC death induced by ONA.
Identification of JNK3 as an APP downstream effector
To identify the possible downstream effector of APP, we performed microarray and chromatin immunoprecipitation (ChIP)-seq screening using APP695-overexpressing SH-SY5Y cells or control SH-SY5Y cells. SH-SY5Y cells are a commonly used human-derived neural cell line that expresses Fe65 and Tip60, important co-factors of AICD in the AICD transcriptional complex [30]. In our microarray experiment, we identified 1291 genes that were differentially expressed in APP695-overexpressing SH-SY5Y cells vs. control. Pathway analysis revealed a number of these genes belonging to the MAPK pathway (Fig. 2a). JNKs are an important subgroup of MAPKs involved in injury-induced stress responses and apoptosis [31]. In the ChIP-seq screening, we identified interaction of AICD at the fourth intron of JNK3 in human chromosome 4 (Fig. 2b), which suggested that JNK3 was potentially regulated by AICD. We also performed an independent ChIP-PCR experiment with a highly specific anti-V5 antibody to immunoprecipitate V5-tagged AICD59 overexpressed in SH-SY5Y cells transfected with pcDNA4/V5-His or pcDNA4/V5-His-AICD59 for 48 h (Fig. 2c). Using primers targeting the fourth intron of JNK3, we validated that AICD interacts with this particular locus, which further confirmed the finding that AICD59 is sufficient to interact with the intron of JNK3. Furthermore, SH-SY5Y cells were transfected with pcDNA4/V5-His or pcDNA4/V5-His-AICD59 for 48 h, and expression of JNK3 mRNA was measured by real-time PCR. Our data confirmed that JNK3 expression was induced by AICD59 overexpression (Fig. 2d). The interacting locus of AICD in human chromosome 4 is about 7.4 kb downstream of transcription starting site of JNK3 gene (inside the fourth intron). This locus is not probably a promoter but may serve as the regulatory element where AICD regulates JNK3 expression [32,33]. To further investigate the role of AICD in JNK3 gene regulation, we cloned this AICD-interacting locus together with flanking regions in a 788 bp sequence and tested its enhancer activity. We used the pGL4.23 reporter vector containing a minimal promoter to detect enhancer activity. The luciferase assay showed that the AICD-interacting locus was only activated by the overexpression of AICD (Fig. 2e). Thus, these data confirmed that AICD interacts with the JNK3 intron region and upregulates its gene expression.

a Pathway analysis of microarray data on genes differentially expressed between control and SH-SY5Y-APP cells. b ChIP-seq screening results in SH-SY5Y cells showed that the region of AICD interaction is in the fourth intron of the JNK3 locus in chromosome 4. TSS, transcription starting site. c Validation of the ChIP-seq results. SH-SY5Y cells were transfected with either pcDNA4/V5-His or pcDNA4/V5-His-hAICD59 plasmid, and subjected to ChIP assay, using normal mouse IgG as control. Input, 5% of the sonicated chromatin. Left panel, real-time PCR results show V5, but not IgG control interacts with the JNK3 intron region (n = 3). Right panel, representative image shows the real-time PCR products resolved by agarose gel. The molecular weight of the DNA ladder shown in bp on the left. d Real-time PCR results showing the JNK3 mRNA level in pcDNA4/V5-His (control) or pcDNA4/V5-His-hAICD59 (AICD) transfected SH-SY5Y cells (n = 3). Student’s t-test: **p < 0.01. Error bars represent SEM. e Luciferase assay showing the enhancer activity of the AICD-interacting site. One-way ANOVA followed by post-hoc Bonferroni test: **p < 0.01. Error bars represent SEM
APP and JNK3 are co-expressed in RGCs
Our results show that APP may regulate JNK3 function in mouse RGCs. To investigate whether APP and JNK3 are colocalized in RGCs, we performed an immunostaining assay to detect their cellular localization in mouse retina. Adult wild-type mice were perfused with 4% PFA, and the retinae were harvested and post-fixed for 3 h. Paraffin-embedded retinae were sectioned to 5 μm thick slices longitudinally. The slices were then triple immunostained with mouse anti-APP, rabbit anti-JNK3 and chicken anti-Tuj1 antibodies. The results showed that APP and JNK3 were both expressed in most Tuj1+ RGCs (Fig. 3a). The specificity of the antibodies was confirmed by the absence of immunostaining in APP-null and JNK3-null retinal slices (Fig. 3b). To further confirm the co-expression of APP and JNK3 in RGCs, we also utilized a well-accepted method, OHSt retrograde tracing, to label RGCs [28]. As only the RGCs send axons out of the eye via the optic nerve, retrograde tracing from the optic nerve is specific to RGCs. Both APP and JNK3 were expressed in OHSt-labeled RGCs in the ganglion cell layer (Fig. 3c). These results showed that APP and JNK3 were co-expressed in mouse RGCs.

a Intact retinae from wild-type mice were fixed, paraffin-embedded and 5 μm thick longitudinal sections were cut. The sections were stained with anti-JNK3, anti-APP and anti-Tuj1 antibodies after antigen retrieval. The inserts show lower magnification photomicrographs illustrating the immunofluorescence signal distribution across all the cell layers of the retina. APP and JNK3 were co-expressed in Tuj1+ RGCs. b Intact retinae from wild-type, APP-null or JNK3-null mice were stained with anti-APP or anti-JNK3 antibodies after antigen retrieval. The inserts show lower magnification photomicrographs illustrating the immunofluorescence signal distribution across all the cell layers of the retina. The anti-APP and anti-JNK3 antibodies were specific as no immunofluorescence signal is seen in the APP-null and JNK3-null retinal sections, respectively. c Adult, wild-type mouse retinae were retrogradely labeled with OHSt, fixed, and 5 μm thick sections were cut. The sections were stained with anti-APP and anti-JNK3 antibodies without antigen retrieval as antigen retrieval destroys the OHSt signal. Arrow heads indicate several OHSt-labeled RGCs, which co-express both APP and JNK3. Scale bar, 20 μm
JNK3 is upregulated and involved in RGC death after ONA
There are three isoforms of JNKs expressed in RGCs (Figs. 4a, b). Each of the three JNK isoforms is thought to have independent functions [34]. To determine the JNK isoform involved in the RGC degeneration, the expression levels of the respective isoforms were detected by real-time PCR and western blot. At the RNA level, ONA-induced slight upregulation of Jnk1 and Jnk2, which returned to basal levels by day 14 after ONA (Fig. 4a). However, the upregulation of Jnk3 was more significant and persisted until 14 days after ONA (Fig. 4a). The expression profile of JNK isoforms was further confirmed by western blot (Fig. 4b). Additional immunostaining in mouse retina sections showed that JNK3 was upregulated in RGCs after ONA (Figure S2). Moreover, activated JNKs (phosphorylated JNK (pJNK)) were also upregulated until 14 days after ONA in a similar pattern to JNK3 (Fig. 4b). Thus, these results showed that JNK3 was the predominant isoform of JNK upregulated after ONA at both RNA and protein levels. We checked the survival of Tuj1+ RGCs after ONA in JNK1, JNK2 or JNK3-null mice and found that, only in JNK3-null mice, the number of surviving Tuj1+ RGCs was significantly higher than that of JNK1 or JNK2-null mice, as well as wild-type mice (Figs. 4c, d). To exclude the possibility of a larger Tuj1+ RGC population in intact retinae in JNK3-null mice, we counted the Tuj1+ RGC number of JNK3-null mice. No significant difference was found between the Tuj1+ RGC number of JNK3-null and wild-type (JNK3-null 1908 ± 178 cells/mm2, wild-type 1937 ± 61 cells/mm2, p > 0.05, n = 3 for each genotype). These results indicated that JNK3 was the predominant JNK isoform playing a role in mediating ONA-induced RGC loss.

a Quantitative analysis of relative mRNA levels of Jnks in intact retinae or retinae 1 day (1 d), 1 week (1 w) or 2 weeks (2 w) after ONA (n = 3 for each time point). b Representative western blots from at least three biological repeats showing JNK expression after ONA. Intact retina or retinae 1 d, 1 w, 2 w after ONA were harvested, lysed and immunoblotted using antibodies against JNK1, JNK2, JNK3, pJNK or GAPDH. The location of molecular weight markers is shown in kDa on the right. c Representative fluorescence microscope images show immunostaining of mouse retina flat-mounts from wild-type or JNK1, JNK2, or JNK3-null mice (n = 6 for each genotype) 14 days after ONA using an anti-Tuj1 antibody. d The bar chart shows quantitative analysis of Tuj1+ RGC numbers 14 days after ONA. Scale bar, 50 μm. One-way ANOVA followed by post-hoc Bonferroni test: **p < 0.01. Error bars represent SEM
APP upregulated JNK3 expression and JNK3 function through AICD
To further identify the role of APP in JNK3 gene regulation, we next detected JNK3 protein levels and JNK3 function in APP-overexpressing SH-SY5Y cells. Overexpression of APP upregulates JNK3 protein levels by 2.1-fold compared with normal SH-SY5Y cells (Figs. 5a, b), and also led to more AICD production (Fig. S1b). JNK activation involves dual phosphorylation at the Thr-Pro-Tyr motif [35]. JNK activation was probed in SH-SY5Y cells using a pJNK (Thr183/Tyr185) antibody, which recognizes all three isoforms of JNK upon phosphorylation. To detect JNK3-specific activity, we used anti-JNK3 antibody to immunoprecipitate the JNK3 protein and then probed with the anti-pJNK antibody. The JNK3 antibody specificity during application in immunoprecipitation is shown in Fig. S4. To measure the phosphorylative activation of JNK3 specifically, we pulled down the JNK3 from equal amounts cell lysate and detected with anti-pJNK or anti-JNK3 antibody. Furthermore, we also detected the phosphorylation of c-Jun, one of the important downstream substrates of JNK. Our results showed that overexpression of APP increased the pJNK3 level significantly, through upregulating JNK3 expression, which was manifest in the increased pc-Jun73/c-Jun ratio in the cell lysate (Figs. 5c, d). We also tested the JNK3 protein level and activity in APP-null mice. Retinae from adult APP-null mice and wild-type littermates were harvested, used to extract total proteins for western blot analysis to detect JNK3 protein level and JNK3 activity. JNK3 expression was reduced to 0.42-fold, and the pc-Jun73/c-Jun ratio was reduced to 0.50-fold in APP-null mice compared with wild-type littermates (Figs. 5e, f and Figs. 5g, h, respectively). These results indicate that APP upregulated JNK3 expression and overall JNK3 function both in vitro and in vivo.

Representative western blots show JNK3 expression in a pcDNA3-FLAG-APP695 transfected or pcDNA3 transfected SH-SY5Y cells, e retinae from wild-type or APP-null mice, or i pcDNA4/V5-His-AICD59 or pcDNA4/V5-His transfected SH-SY5Y cells. The samples were lysed, resolved in SDS-page gel, and immunoblotted using antibodies against JNK3 or GAPDH. b, f, j The bar charts show quantitative analysis of relative JNK3 expression (n = 3 for each experiment). Representative western blots show phosphorylative activation of JNK3 in c APP695-overexpressing or normal SH-SY5Y cells, g retinae from wild-type or APP-null mice, or k pcDNA4/V5-His-AICD59 or pcDNA4/V5-His transfected SH-SY5Y cells. JNK3 was immunoprecipitated with anti-JNK3 antibody and then detected for kinase activity with anti-phospho-JNK (pJNK) or anti-JNK3 antibody. Whole-cell lysate western blots show the downstream substrate pc-Jun73 or c-Jun level. d, h, l The bar charts show quantitative analysis of activated pJNK/mg lyate proteins or pc-Jun73/c-Jun ratio (n = 3 for each experiment). Student’s t-test: *p < 0.05; **p < 0.01. Error bars represent SEM
AICD is the product of APP cleavage by α- or β-secretase and γ-secretase [36] and on binding with Fe65 and Tip60 translocates to the nucleus to initiate transcription [37], and AICD production was observed to be associated with APP upregulation in vivo and in vitro (Fig. S1). To further test whether APP exerted its function through AICD, we transfected a human AICD59 construct into SH-SY5Y cells and detected JNK3 expression after 48 h. JNK3 expression was upregulated approximately twofold in AICD59-overexpressing cells compared with control vector-transfected cells (Figs. 5i, j), and the pc-Jun73/c-Jun ratio was also similarly increased by overexpression of AICD59 (Figs. 5k, l). As AICD nuclear translocation is essential for its transcriptional regulation, we also observed the subcellular localization of AICD in RGCs. We used an antibody against the APP C-terminal to detect the AICD-like localization after ONA, because it is extremely difficult to specifically immunolabel AICD in vivo rather than other APP C-terminal fragments containing AICD (such as C83 and C99). Indeed, we observed that more C-terminal fragments were accumulated in RGC nuclei on day 1 after ONA (Fig. S5). Thus, we showed that APP upregulated JNK3 expression and overall JNK3 function through AICD.
APP deficiency abolished JNK3 upregulation and overall phosphorylation of JNK after ONA
Our results above showed that JNK3 upregulation was involved in loss of RGCs after ONA and that APP regulated JNK3 expression and JNK3 function under physiological conditions. Next, we asked whether APP regulates JNK3 expression and overall phosphorylative activation of JNK under the pathological conditions of the ONA model. We used APP-null mice and their wild-type littermates to test the hypothesis. Approximately 2-month-old APP-null mice and wild-type littermates were subjected to ONA [6,38] of the left eye and then both retinae were harvested 1 day, 7 days or 14 days later. The retinae were then lysed and subjected to western blot analysis to detect JNK3 expression and overall phosphorylative activation of JNK. In the wild-type mice, JNK3 expression and phosphorylation of JNK were upregulated in the ipsilateral retina (injured) compared with the contralateral (uninjured) side 1 day, 7 days and 14 days after ONA but in the APP-null mice, the JNK3 expression and overall phosphorylation of JNK were almost the same in ipsilateral retinae compared with the contralateral retinae (Figs. 6a, b). Thus, here we showed that APP deficiency blocked JNK3 upregulation and overall phosphorylative activation of JNK after ONA in vivo.

a Representative western blots show JNK3 expression and overall phosphorylative activation of JNK in retinae from wild-type or APP-null mice 1 day (1 d), 1 week (1 w) and 2 weeks (2 w) after ONA (n = 3 for each time point). Anti-JNK3, anti-pJNK and anti-GAPDH antibodies were used to detect the respective protein expression. b The bar charts show quantitative analysis of relative JNK3 expression or overall phosphorylative activation of JNK in the ipsilateral-injured retinae vs. the contralateral-uninjured retina. One-way ANOVA followed by post-hoc Bonferroni test: **p < 0.01. Error bars represent SEM
γ-Secretase inhibitors suppressed JNK3 expression and inhibited overall phosphorylation of JNK
The results above showed that AICD-regulated JNK3 expression. AICD is the product of APP cleavage by α- or β-secretase and γ-secretase [36]. Therefore, compounds that reduce AICD production would be expected to suppress JNK3 expression and overall phosphorylative activation of JNK. To test the hypothesis, we used γ-secretase inhibitors to reduce AICD production, and detected JNK3 expression and overall phosphorylation of JNK. We first detect the effect of γ-secretase inhibitors on AICD expression. L-685, 458 is a selective γ-secretase inhibitor (IC50 = 17 nM) that displays > 50-fold selectivity over a range of aspartyl, serine and cysteine proteases [39]. BMS 299897 is an oral active, potent γ-secretase inhibitor (IC50 = 12 nM), which exhibits no Notch toxicity and is brain penetrant [40]. We used the γ-secretase inhibitor L-685, 458 or BMS 299897 to treat APP-overexpressing SH-SY5Y cells for 48 h, and then the cells were washed and lysed for western blot analysis. We detected AICD production, JNK3 expression and overall phosphorylation of JNK. The γ-secretase inhibitors efficiently reduced AICD production, which lead to the decrease of JNK3 expression and overall phosphorylative activation of JNK (Figs. 7a–d). These results showed that γ-secretase inhibitors suppressed JNK3 expression and inhibited phosphorylative activation of JNK in SH-SY5Y cells.

a–d Representative western blots show JNK3 expression and overall phosphorylative activation of JNK in SH-SY5Y-APP cells treated with vehicle or γ-secretase inhibitors a L-685,458 (5 μM) or c BMS 299897 (1 μM). b, d The bar charts show quantitative analysis of relative JNK3 expression and overall phosphorylative activation of JNK normalized to the GAPDH (n = 3 for each treatment). e Representative western blots show phosphorylative activation of JNK3 in retinae from vehicle-treated or γ-secretase inhibitor BMS 299897 treated mice 2 weeks after ONA. f Representative fluorescence microscope images show immunostaining of mouse retina flat-mounts from vehicle or γ-secretase inhibitor BMS 299897 treated mice 2 weeks after ONA using anti-Tuj1 antibody. Scale bar, 50 μm. g The bar chart shows quantitative analysis of the surviving Tuj1+ RGC number in injured mouse retinae 14 days after ONA (n = 4 for each treatment). Student’s t-test: **p < 0.01. Error bars represent SEM
BMS 299897 suppressed phosphorylative activation of JNK and promoted RGC survival after ONA
We next tested the effect of BMS 299897 on RGC survival in mice in vivo. After ONA, BMS 299897 was administrated twice a day by oral gavage for 14 days and then the mice were euthanized. For biochemical analysis, the retinae were fresh harvested. For RGC staining, the mice were perfused with 4% PFA and the retinae were harvested and subjected to Tuj1 staining. BMS 299897 suppressed the elevation of JNK3 phosphorylation by ONA on day 14 after axotomy (Fig. 7e). The Tuj1 staining results showed that BMS 299897 protected RGCs against cell death induced by ONA on day 14 (Figs. 7f, g). Thus, we showed that BMS 299897 suppressed phosphorylative activation of JNK3 and promoted Tuj1+ RGC survival after ONA, which suggests a potential drug target for neuroprotection via inhibiting JNK3 signal cascades.
Discussion
APP is upregulated in neural injury and has long been utilized as a marker for axonal degeneration [24,25]. According to the well-known amyloid hypothesis, accumulation of Aβ in the brain is the primary influence driving neural injury in AD pathogenesis [41]. Recent studies strongly suggest that Aβ is not the sole product of APP processing that may play a role in development of AD. AICD is another product of APP processing by α- or β-secretases and γ-secretase cleavage [36]. AICD binds with Fe65 and Tip60, and translocates to the nucleus and initiates transcription [37]. AICD/Fe65 double transgenic mice showed alterations in cell signaling [42], impairments in neuronal activity [43], memory deficits and neurodegeneration [15]. Interestingly, transcription of the GSK3B gene, coding for an enzyme strongly implicated in neurodegeneration, is activated by AICD [43]. Here we show that JNK3 is a new target regulated by AICD. JNK3 is important in axon injury-induced RGC death [44]. In the CNS, JNK activation is involved in ischemia, stroke, AD, Parkinson's disease and neurotrauma [45], suggesting that JNKs play a pivotal role in neurodegeneration. AICD has previously been reported to be involved in the regulation of JNK activity via interactions with the JNK-interacting protein-1 [46]. In contrast, we found a new mechanism by which AICD regulate JNK function via regulating JNK3 gene expression (Fig. S6). Our study supplies a new mechanism that explains how APP can exert functions in axon injury-induced neurodegeneration by upregulating JNK function.
To find the downstream effectors of APP, we performed microarray and ChIP-seq screening. Among these genes, we identify JNK3 as an important downstream effector of APP. It has been reported that both JNK2 and JNK3 are major regulators of axonal injury-induced RGC death in the controlled optic nerve crush model [44]. However, after ONA, we only observed marked upregulation of JNK3 and JNK3 deficiency alone was sufficient to provide protection against RGC loss after ONA. This discrepancy may be due to differences between partial optic nerve injury in the nerve crush model and complete optic nerve injury in the axotomy model we employed. Our results indicate that JNK3 plays important roles in axonal injury-induced neuronal death, which is thought to be one of the major causes of RGC loss in optic neuropathies, as well as neuronal death in other central nervous system diseases [45]. As JNK3 is predominantly restricted to central nervous system neurons [47], it is regarded as a potential target for drug screening for neurodegenerative diseases [48]. Drugs that specifically inhibit JNK3 activity are not available so far, due to the high similarity among JNK1, JNK2 and JNK3. Here we found that APP is the upstream regulator of JNK3, and showed that γ-secretase inhibitors suppress JNK3 expression and overall phosphorylation of JNK (a measure thought to reflect phosphorylative activation of JNK), and protect against RGC death after ONA (Fig. 8). As APP is unlikely the only regulator of JNK3, APP deletion or blockade should have weaker effects than JNK3 deletion in RGC protection. This can explain why the JNK3 deletion protected RGCs better than APP deletion or inhibitors of APP cleavage in our experiment.

ONA induces upregulation of APP expression. APP is sequentially cleaved by α- or β-secretase and γ-secretase, producing several fragments and AICD, which is inhibited by APP deficiency or BMS 299897. AICD is translocated to the nucleus and upregulates JNK3 expression (probably functioning with other partners). Phosphorylated JNK3 then contributes to RGC death
In summary, we show for the first time that ONA leads to upregulation of APP protein expression as previously described for other forms of axonal injury [24,25]. γ-Secretase-dependent cleavage of APP then releases AICD, which translocates to the nucleus and upregulates JNK3 expression. Subsequently, phosphorylated JNK3 contributes to RGC death, likely by the cell death mechanisms previously described [17,18]. Our study provides a link between upregulation of APP and JNK3-mediated cell death on axonal injury. Although we have in vitro data to show that AICD overexpression increased JNK3 expression both at the mRNA level and the protein level, and increased its function, we cannot exclude the contribution of Aβ or other peptides released by γ-secretase activity in the in vivo animal model. Nevertheless, our results suggest an alternative way to protect against axon injury-induced RGC death via inhibiting JNK3 signal cascades.
Materials and methods
Mice and surgery
APP-null [49] mice were as previously described [50]. JNK1, JNK2 and JNK3-null mice were from the Jackson Laboratory. The generation of JNK1, JNK2 and JNK3-null mice has been reported previously [22,51,52]. The mice were housed under a 12:12-h light–dark cycle at 22–26 °C, with free access to water and food. The ONA model was as previously described [8]. Briefly, adult mice of either sex (n = 4–6 in each group) were anesthetized by intraperitoneal injection of ketamine (75 mg/kg) and xylazine (10 mg/kg). After anesthetization, the animal was place on the stage of an operating microscope with the left eye facing up. A 0.5 cm incision was made in the skin above the left orbit with a surgical knife. After that, the soft tissue and superior rectus were separated by forceps. The optic nerve, which was observable as a bundle of white tissue connecting with the eyeball, was exposed. Using fine eye scissors, the optic nerve was cut 1 mm behind the nerve head, avoiding injury to the ophthalmic artery. The vascular integrity of the retina was verified by fundoscopic examination. The incision was closed with a 5–0 suture and wiped with 70% ethanol. For RGC labeling, a retrogradely transported tracer, hydroxystilbamidine bis(methanesulfonate) (OHSt, also known as FluoroGold, Molecular Probes) was used. OHSt was diluted at 5% in sterile 0.9% NaCl/10% dimethyl sulfoxide (DMSO). A strip of gelatin sponge (GELITA-tampon, B. Braun Singapore) soaked in the OHSt solution was applied to the cut optic nerve and the mice were allowed to recover for 2 days [53]. Analgesic and antibiotic were administered subcutaneously after the surgery. BMS 299897 was purchased from AdooQ BIOSCIENCE (catalog no. A12444) and dissolved in 100% polyethylene glycol 400 (PEG400) to make a 20 mg/ml solution. The drug was administered (100 mg/kg) by oral gavage in a dosing volume of 5 ml/kg once before surgery and twice a day for 14 days after surgery [40]. All procedures on animals were approved by the Institutional Animal Care and Use Committee (IACUC), National University of Singapore, and were carried out in accordance with the Guidelines on the Care and Use of Animals for Scientific Purposes developed by the National Advisory Committee For Laboratory Animal Research, Singapore; the European Communities Council Directive of 24 November 1986 (86/609/EEC); and the Guidelines laid down by the National Institute of Health (NIH) in the USA regarding the care and use of animals for experimental procedures.
Immunofluorescence staining
The mice were given a lethal overdose of anesthesia and were transcardially perfused with saline, followed by 4% paraformaldehyde. For paraffin-embedded sectioning, retinae were dissected away from connective tissue, post-fixed for 3 h and dehydrated in ethanol. The tissue was immersed in xylene followed by xylene/paraffin (1:1) mixture and then submerged in paraffin for 2 h at 60 °C. Embedding was performed with a Modular Tissue Embedding Center EC 350 (Thermo Scientific). Paraffin sections of retina (5 μm) were cut longitudinally on a microtome (Microm HM 325, Thermo Scientific) and mounted onto coated glass slides. The slices were deparaffinized and rehydrated before staining. For immunolabeling, antigen retrieval was performed with an antigen-retriever (2100, Aptum Biologics, UK) before antibody incubation. The sections were incubated overnight with mouse antibody against APP (1:200, catalog no. MAB343, Millipore) [16], rabbit antibody against JNK3 (1:200, catalog no. 2305, Cell Signaling Technology) [54], or chicken antibody against Tuj1 (1:200, catalog no. NB100–1612, Novus Biologicals) [55] in primary antibody dilution buffer (1% v/v BSA, 0.3% v/v Triton × 100 and 0.01% w/v NaN3 in phosphate-buffered saline (PBS)) at 4 °C. After three washes with PBS, the sections were incubated with a mixture of Alexa Fluor 488 donkey anti-mouse IgG antibody (1:200; Invitrogen), Alexa Fluor 555 donkey anti-rabbit IgG antibody (1:200; Invitrogen) and Alexa Fluor 647 donkey anti-chicken IgG antibody (1:200; Invitrogen) for 40 min at room temperature. After three washes with PBS, the sections were mounted in FluorSave reagent (Merk Millipore) and dried overnight in a dark place. For the whole-mounted retina staining, after perfusion and post-fixation, the retinae were washed three times in PBS with 0.3% Triton X-100 for 10 min each, and then blocked in 10% normal goat serum and 0.3% Triton for 1 h. Primary mouse antibody against Tuj1 (1:400, catalog no. MMS435P, Covance) [56] or sheep antibody against GFAP (1:200, catalog no. AF2594, R&D Systems) was added into the blocking solution and incubated over two nights at 4 °C. After three 10-min washes in PBS, the retinae were incubated with Alexa Fluor 488 donkey anti-mouse IgG antibody (1:200; Invitrogen) or Alexa Fluor 647 donkey anti-sheep IgG antibody (1:200; Invitrogen) overnight at 4 °C. After three washes with PBS, the retinae were mounted onto the slides in FluorSave reagent (Merk Millipore) and slides are subjected to RGC cell number counting. RGC density varies greatly with respect to location within the retina. Therefore, for each retina, images were obtained from four 20 × fields in each quadrant along two perpendicular retinal diameters (Fig. 1c). The numbers of RGCs immunolabelled with Tuj1 in each image were quantified manually using the cell counter tool in ImageJ. For each individual retina, the total count of surviving RGCs was obtained by averaging the 16 counts for each retina. RGC survival data from each group of animals are presented as the mean number and SEM for n = 3–6 retinae. The slices were visualized using a confocal microscope system (LSM 510, Carl Zeiss Microimaging GmbH, Germany) or a normal fluorescence microscope.
TUNEL detection
TUNEL assay was performed according to the manufacturer’s protocol (catalog no. 11684795910, Roche). Briefly, 8 µm-thick frozen retina sections were rinsed in Tris-buffered saline (TBS; 10 mM Tris-HCl, 150 mM NaCl, pH 8.0), followed by 15-min permeablization in 0.3% Triton in TBS. Sections were then washed for 5 min in TBS and incubated with TUNEL reaction mixture in a humidified dark chamber for 60 min at 37 °C. Then sections were washed three times in TBS, coverslipped with anti-fade mounting medium with DAPI (ProLong Gold; Invitrogen), and the fluorescent signal visualized using an Olympus FV3000 confocal microscope. The negative control for the TUNEL assay was incubating permealblized sections without terminal transferase in reaction mixture, and the positive control was inducing DNA-strand breaks in the permealblized sections by DNase I prior to labeling procedures.
Plasmids, cell culture and transfection
pcDNA3-flag-hAPP695 was as previously described [50]. pcDNA4/V5-His-hAICD59 expressing the C-terminal 59 amino acid was subcloned from full-length hAPP695. SH-SY5Y cells were obtained from ATCC and grown in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) FBS. SH-SY5Y cells were transfected with various plasmids using the Lipofectamine 2000 reagent (Invitrogen) according to the instructions of the manufacturer. Two days after transfection, the cells were used for experiments. The SH-SY5Y cell line-overexpressing APP (SH-SY5Y-APP) was generated by transfection with a pcDNA4 plasmid construct (Invitrogen) carrying APP695 and a Zeocin resistance gene followed by selection for Zeocin resistance. To inhibit AICD degradation, the cells were treated with 10 mM NH4Cl overnight in the culture medium [57]. L-685,458 (5 μM final concentration; Tocris, catalog no. 2627) or BMS 299897 (1 μM final concentration; Tocris, catalog no. 2870) were administered to the cells in culture medium for 24 h.
Real-time PCR
RNA from ONA retinae was isolated using an RNeasy Mini kit (Qiagen). The reverse transcription kit, the assay probes and other related reagents were ordered from TaqMan. The real-time PCR procedures were based upon the manual protocol provided by the company. The primer sequences to detect the gene expression in mice were: Jnk1 F 5′-AAACAGGCCTAAATACGCTGGA-3′, R 5′-GACGGCTGCCCTCTTATGAC-3′; Jnk2 F 5′-GAGCTGGTGAAAGGTTGTGTGATATTCCA-3′,R 5′-AACAGTAATATACGGGTGGCGCAAG-3′; Jnk3 F 5′-AACAATCGCTACACCTCCAAAGAC-3′, R 5′-TGGCAATAGATGACACATCCAGG-3′; Gapdh F 5′-ATGGTGAAGGTCGGTGTGAACG-3′, R 5′-CGCTCCTGGAAGATGGTGATGG-3’. The primer sequences to detect the gene expression in SH-SY5Y were: JNK3 F 5′-GGGAGAAATGGTTCGCCACA-3′, R 5′-GGTGAGTCCCGCATACTTGG-3′; GAPDH F 5′-CATGTTCGTCATGGGTGTGAACCA-3′, R 5′-AGTGATGGCATGGACTGTGGTCAT-3’.
Microarray
Briefly, SH-SY5Y and SH-SY5Y-APP cells were harvested, processed and read on an Illumina microarray platform. Total RNA was extracted from three biological replicates. Cells were washed twice, scraped in ice-cold PBS and centrifuged for 5 min at 500 g, 4 °C. The cell pellet obtained was resuspended in TRIzol Reagent (Invitrogen) for RNA extraction, followed by purification using the RNeasy Mini kit (Qiagen) according to the instructions of the manufacturer. DNA contamination was removed by DNase treatment using the RNase-Free DNase Set (Qiagen) during the RNA purification step. RNA quantification was carried out using a Nanodrop spectrophotometer. Biotinylated complementary RNA was then generated from 400 ng of the harvested RNA using the Illumina TotalPrep RNA Amplification Kit, and hybridized to the HumanHT-12 v4 Expression BeadChips (Illumina), which contains 47 231 probes against known genes. Data collection was carried out by scanning in an Illumina BeadStation array reader. The data were processed and controlled for quality using BeadStudio 3.2 (Illumina), and subsequently imported into GeneSpring GX 11.5 (Agilent) for analysis. Differential gene lists were generated based on a fold change of >1.5. Statistical significance was established at p-value < 0.05 according to the unpaired Student’s t-test with the Benjamini–Hochberg multiple testing correction.
Functional annotation clustering analysis
A total of 1291 differentially expressed genes were identified in microarray analysis, with fold change > 1.5, p-value < 0.05. This list of genes was used as the input for analysis by the David Functional Annotation Clustering website (http://david.abcc.ncifcrf.gov/home.jsp), using the default setting of medium classification stringency. The results of the clustering analysis were replotted in a pie graph.
ChIP and deep sequencing (ChIP-seq)
ChIP-seq was performed as described previously [58], and was done in biological duplicates with independent experiments for each replicate. Briefly, for cross-linking and chromatin extract preparation, SH-SY5Y-APP cells were treated with 20 mM NH4Cl for 18–24 h prior to harvesting. Cells were harvested by rinsing with PBS. Cross-linking was performed by incubating the cells in 2 mM dithiobis(succinimidylpropionate) (Thermo Scientific) in PBS for 45 min, followed by a brief rinse in PBS, and a second cross-linking step with 1% (v/v) formaldehyde for 10 min. Glycine (final concentration of 0.2 M) was then added to quench the reaction. After rinsing twice with PBS, the cells were collected by scraping. The nuclear fraction was isolated by washing the cells in cell lysis buffer (10 mM Tris pH 8, 0.25% Triton × 100, 10 mM EDTA, 0.1 M NaCl), lysing the nuclei in high sodium dodecyl sulfate (SDS) lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 1% SDS), and washing the chromatin twice in low SDS lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS). Complete protease inhibitor (Roche) was added to all lysis buffers. Chromatin extracts were then sheared by sonication for eight cycles (30 s on, 30 s off) using a Bioruptor, to obtain chromatin fragments of 500 bp and below. The sonicated chromatin extract was pre-cleared with protein G-sepharose beads for 3 h at 4 °C. Six μg of rabbit anti-AICD antibody (BR188), a generous gift from Dr M. Goedert (Cambridge, UK), was incubated with protein G-sepharose beads for 3 h at room temperature. On the next day, the beads were washed and the chromatin– protein–antibody complexes eluted from the beads at 68 °C for 30 min, with shaking at 1400 rpm. The cross-links were reversed and protein was degraded using pronase (0.8 mg/ml) by incubating the samples at 42 °C for 2 h, followed by 68 °C overnight. The eluted ChIP DNA was then purified by phenol–chloroform extraction and resuspended in 30 μl of nuclease-free water. ChIP-Seq was performed on an Illumina Solexa platform. ChIP-Seq libraries were prepared using the ChIP-seq DNA Sample Prep Kit (Illumina), according to instructions of the manufacturer. ChIP DNA was quantified using the Quant-iT PicoGreen dsDNA assay (Invitrogen) and 6.5 ng of ChIP DNA was used as input for each sample library preparation. In brief, DNA was subjected to end repair and adaptor ligation, followed by DNA size selection by running the DNA on agarose gel and cutting out the area corresponding to 200–300 bp DNA fragments. The DNA was then extracted and purified using a Qiagen gel extraction kit, and PCR-amplified for 18 cycles. The quality of the ChIP-seq library was then validated using a Bioanalyzer 2100 (Agilent) and 5 nM was submitted for sequencing. Short reads of 36 bp were sequenced and aligned to the hg19 Human Genome Assembly (GRCh37, February 2009). AICD interaction sites, represented by peaks, were then identified by the MACS (Model-based Analysis for ChIP-seq) peak-calling algorithm.
ChIP and PCR
Cells were cross-linked by addition of formaldehyde to the culture medium at a final concentration of 1% for 10 min followed by quenching with 0.125 M glycine for 5 min. After cell lysis, chromatin extracts were fragmented by sonication (40% amplitude, 20 s on, 40 s off for 10 cycles, Vibra cell sonicator) to fragments of ~ 200–500 bp and then pre-cleared with protein G agarose beads. The pre-cleared chromatin extracts were subsequently incubated with anti-V5 antibody (catalog no. R96025, Invitrogen) or normal mouse IgG (catalog no. sc-2025, Santa Cruz Biotechnology) overnight at 4 °C, and then incubated with pre-washed Protein G for 4 h at 4 °C. The beads were then washed, and the chromatin–protein–antibody complexes were eluted. After treatment with proteinase K (Sigma-Aldrich), crosslink reversal was done by incubating at 65 °C for 8 h, the eluted DNA was purified by phenol:chloroform extraction and ethanol precipitation, and analyzed by RT-PCR with primers specific to the JNK3 locus (F 5′-GGGTCATACCAACTGCCTATTC-3′, R 5′-GCTCCCACTGAGAAGAATGAAA-3′). Quantitative PCR reaction mixes were assembled using SYBR Green master mix (Kapa Biosystems). The reactions were performed on an ABI7500 System (Applied Biosystems) using the following cycling parameters: 95 °C for 10 min, and then 40 cycles of 95 °C (10 s), 60 °C (10 s) and 72 °C (10 s) followed by a melting curve analysis. All reactions were performed with three technical replicates, and at least three biological repeats were done. Fold change values were normalized against input DNA and compared with the mouse IgG control.
Luciferase assay
The putative AICD-interacting locus on the human JNK3 gene was identified by ChIP-seq. The DNA fragment flanking this locus was amplified and cloned into the PGL4.23 vector (Promega) to test potential enhancer activity. Primers (F 5′-ATATCTCGAG TTCCACCTAAAGAAGCA-3′, R 5′-TATAAAGCTTGGCGGATTACTTGAGGTCAG-3′) were used to amplify the DNA fragment, and all cloned sequences were verified by DNA sequencing. SH-SY5Y cells were transfected with pGL4.23, or pGL4.23-JNK3 enhancer luciferase reporter vectors, pcDNA4-AICD59 (or pcDNA4B empty vector), and pCMV-LacZ vectors using Lipofectamine 2000 (Invitrogen). After 48 h, the cells were harvested for firefly luciferase activity assay using the One-Glo Luciferase Assay System and β-Galactosidase Enzyme Assay System (Promega). Each experiment was carried out at least in triplicate and repeated at least three times. The efficiency of transfection was normalized to the β-galactosidase activity.
Western blot analysis
For preparation of mouse retina lysates, mouse retinae were harvested, briefly rinsed in PBS and put into lysis buffer (150 mM NaCl, 30 mM HEPES, 10 mM NaF, 1% v/v Triton X-100, 0.01% w/v SDS and complete protease inhibitor cocktails; pH 7.5), homogenized and rotated at 4 °C for 1.5 h. To prepare total cell lysates, cultured cells were rinsed with PBS, and lysed in the lysis buffer. After centrifugation (16 000 g, 4 °C, 10 min), the supernatants were collected, mixed with 4× protein loading buffer and stored at −20 °C for future use. Equal amounts of protein were resolved in acrylamide gels and transferred onto nitrocellulose membranes. For AICD detection, the proteins were transfer to PVDF membranes [16]. Rabbit antibodies against APP (Y188, Abcam) [50], JNK1 (catalog no. MAB17761, R&D Systems), JNK2 (catalog no. MAB1846, R&D Systems) [59], JNK3 (catalog no. 2305, Cell Signaling Technology), p-cJun73 (catalog no. 9164, Cell Signaling Technology) and c-Jun (catalog no. 9165, Cell Signaling Technology); mouse antibodies against GAPDH (catalog no. ab8245, Abcam) [50], V5 (catalog no. R96025, Invitrogen), pJNK (catalog no. 9255, Cell Signaling Technology) [60] and JNK3 (catalog no. CP10162, Cell Applications) were used. AICD was detected using an anti-APP C-terminal antibody (catalog no. A8717, Sigma). Either anti-mouse IgG or anti-rabbit IgG peroxidase-conjugated secondary antibodies were applied at 1:10 000 and blots were visualized with West Pico (Thermo Scientific) or Luminata Forte (Millipore) ECL detection kits.
Immunoprecipitation
The mouse retina or cell lysate was incubated overnight at 4 °C with rabbit anti-JNK3 antibody (Cell Signaling Technology) or normal rabbit IgG control (Cell Signaling Technology), followed by incubation with pre-washed protein G-Sepharose 4 Fast Flow (GE Healthcare) for 3 h at 4 °C. The immunoprecipitates were washed efficiently with lysis buffer and analyzed by western blotting. Each experiment was repeated at least three times.
Statistics
Data are presented as mean ± SEM. The densities of the western blot bands were normalized to the internal loading controls and then normalized to control plasmids or treatments. Student’s t-test was used for two group comparisons, and one-way analysis of variance (ANOVA) was used for multigroup comparisons, followed by appropriate post-hoc tests.
References
Osborne NN, Chidlow G, Layton CJ, Wood JP, Casson RJ, Melena J. Optic nerve and neuroprotection strategies. Eye (Lond) 2004;18(11):1075–84.
Levin LA. Axonal loss and neuroprotection in optic neuropathies. Can J Ophthalmol 2007;42(3):403–8.
Chaon BC, Lee MS. Is there treatment for traumatic optic neuropathy? Curr Opin Ophthalmol 2015;26(6):445–9.
Bahr M. Live or let die - retinal ganglion cell death and survival during development and in the lesioned adult CNS. Trends Neurosci 2000;23(10):483–90.
Berkelaar M, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J Neurosci. 1994;14(7):4368–74.
Villegas-Perez MP, Vidal-Sanz M, Rasminsky M, Bray GM, Aguayo AJ. Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. J Neurobiol 1993;24(1):23–36.
Nadal-Nicolas FM, Sobrado-Calvo P, Jimenez-Lopez M, Vidal-Sanz M, Agudo-Barriuso M. Long-term effect of optic nerve axotomy on the retinal ganglion cell layer. Invest Ophthalmol Vis Sci. 2015;56(10):6095–112.
Sanchez-Migallon MC, Valiente-Soriano FJ, Nadal-Nicolas FM, Vidal-Sanz M, Agudo-Barriuso M. Apoptotic retinal ganglion cell death after optic nerve transection or crush in mice: delayed RGC loss with BDNF or a caspase 3 inhibitor. Invest Ophthalmol Vis Sci. 2016;57(1):81–93.
Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160(2):139–44.
Sherriff FE, Bridges LR, Gentleman SM, Sivaloganathan S, Wilson S. Markers of axonal injury in post mortem human brain. Acta Neuropathol. 1994;88(5):433–9.
Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A 2004;101(21):8168–73.
Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K,, et al Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A 1990;87(4):1561–5.
Kipfer-Kauer A, McKinnon SJ, Frueh BE, Goldblum D. Distribution of amyloid precursor protein and amyloid-beta in ocular hypertensive C57BL/6 mouse eyes. Curr Eye Res. 2010;35(9):828–34.
Passer B, Pellegrini L, Russo C, Siegel RM, Lenardo MJ, Schettini G,, et al Generation of an apoptotic intracellular peptide by gamma-secretase cleavage of Alzheimer’s amyloid beta protein precursor. J Alzheimers Dis. 2000;2(3-4):289–301.
Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009;106(43):18367–72.
Ma QH, Futagawa T, Yang WL, Jiang XD, Zeng L, Takeda Y,, et al A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat Cell Biol 2008;10(3):283–94.
Kwong JM, Caprioli J. Expression of phosphorylated c-Jun N-terminal protein kinase (JNK) in experimental glaucoma in rats. Exp Eye Res 2006;82(4):576–82.
Lukas TJ, Wang AL, Yuan M, Neufeld AH. Early cellular signaling responses to axonal injury. Cell Commun Signal. 2009;7:5.
Tezel G, Chauhan BC, LeBlanc RP, Wax MB. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Invest Ophthalmol Vis Sci 2003;44(7):3025–33.
Martin JH, Mohit AA, Miller CA. Developmental expression in the mouse nervous system of the p493F12 SAP kinase. Brain Res Mol Brain Res 1996;35(1-2):47–57.
Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C,, et al A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci U S A 2003;100(25):15184–9.
Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, et al Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997;389(6653):865–70.
He Y, Chen J, Zhang SG, Yuan YS, Li Y, Lv HB,, et al c-Jun N-terminal kinase 3 expression in the retina of ocular hypertension mice: a possible target to reduce ganglion cell apoptosis. Neural regeneration research 2015;10(3):432–7.
Selkoe DJ. Alzheimer disease: mechanistic understanding predicts novel therapies. Ann Intern Med. 2004;140(8):627–38.
Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci 2005;6(11):889–98.
Magharious MM, D’Onofrio PM, Koeberle PD. Optic nerve transection: a model of adult neuron apoptosis in the central nervous system. J Vis Exp. 2011 May 12;(51):e2241.
Haenold R, Weih F, Herrmann KH, Schmidt KF, Krempler K, Engelmann C,, et al NF-kappaB controls axonal regeneration and degeneration through cell-specific balance of RelA and p50 in the adult CNS. J Cell Sci 2014;127(Pt 14):3052–65.
Vidal-Sanz M, Salinas-Navarro M, Nadal-Nicolas FM, Alarcon-Martinez L, Valiente-Soriano FJ, de Imperial JM,, et al Understanding glaucomatous damage: anatomical and functional data from ocular hypertensive rodent retinas. Prog Retin Eye Res 2012;31(1):1–27.
Misantone LJ, Gershenbaum M, Murray M. Viability of retinal ganglion cells after optic nerve crush in adult rats. J Neurocytol 1984;13(3):449–65.
Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293(5527):115–20.
Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004;23(16):2838–49.
Lee TI, Young RA. Transcription of eukaryotic protein-coding genes. Annu Rev Genet 2000;34:77–137.
Struhl K. Yeast transcriptional regulatory mechanisms. Annu Rev Genet 1995;29:651–74.
Brecht S, Kirchhof R, Chromik A, Willesen M, Nicolaus T, Raivich G,, et al Specific pathophysiological functions of JNK isoforms in the brain. Eur J Neurosci 2005;21(2):363–77.
Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000;103(2):239–52.
Kim D, Tsai LH. Bridging physiology and pathology in AD. Cell 2009;137(6):997–1000.
Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ. The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem 2001;276(43):40288–92.
Mansour-Robaey S, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc Natl Acad Sci U S A 1994;91(5):1632–6.
Shearman MS, Beher D, Clarke EE, Lewis HD, Harrison T, Hunt P,, et al L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid beta-protein precursor gamma-secretase activity. Biochemistry 2000;39(30):8698–704.
Anderson JJ, Holtz G, Baskin PP, Turner M, Rowe B, Wang B, et al Reductions in beta-amyloid concentrations in vivo by the gamma-secretase inhibitors BMS-289948 and BMS-299897. Biochem Pharmacol 2005;69(4):689–98.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002;297(5580):353–6.
Ryan KA, Pimplikar SW. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J Cell Biol 2005;171(2):327–35.
Ghosal K, Pimplikar SW. Aging and excitotoxic stress exacerbate neural circuit reorganization in amyloid precursor protein intracellular domain transgenic mice. Neurobiol Aging. 2011;32(12):2320. e1-9
Fernandes KA, Harder JM, Fornarola LB, Freeman RS, Clark AF, Pang IH, et al JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol Dis 2012;46(2):393–401.
Repici M, Borsello T. JNK pathway as therapeutic target to prevent degeneration in the central nervous system. Adv Exp Med Biol 2006;588:145–55.
Margevicius DR, Bastian C, Fan Q, Davis RJ, Pimplikar SW. JNK-interacting protein 1 mediates Alzheimer’s-like pathological features in AICD-transgenic mice. Neurobiol Aging 2015;36(8):2370–9.
Mohit AA, Martin JH, Miller CA. p493F12 kinase: a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron. 1995;14(1):7–78.
Borsello T, Forloni G. JNK signalling: a possible target to prevent neurodegeneration. Curr Pharm Des 2007;13(18):1875–86.
Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW,, et al beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 1995;81(4):525–31.
Liu C, Tan FC, Xiao ZC, Dawe GS. Amyloid precursor protein enhances Nav1.6 sodium channel cell surface expression. J Biol Chem 2015;290(19):12048–57.
Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Defective T cell differentiation in the absence of Jnk1. Science 1998;282(5396):2092–5.
Yang DD, Conze D, Whitmarsh AJ, Barrett T, Davis RJ, Rincon M,, et al Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity 1998;9(4):575–85.
Fu QL, Hu B, Wu W, Pepinsky RB, Mi S, So KF. Blocking LINGO-1 function promotes retinal ganglion cell survival following ocular hypertension and optic nerve transection. Invest Ophthalmol Vis Sci 2008;49(3):975–85.
Christensen DP, Ejlerskov P, Rasmussen I, Vilhardt F. Reciprocal signals between microglia and neurons regulate alpha-synuclein secretion by exophagy through a neuronal cJUN-N-terminal kinase-signaling axis. J Neuroinflammation 2016;13(1):59.
Arnhold M, Dening Y, Chopin M, Arevalo E, Schwarz M, Reichmann H,, et al Changes in the sympathetic innervation of the gut in rotenone treated mice as possible early biomarker for Parkinson’s disease. Clinical autonomic research: official journal of the Clinical Autonomic Research Society 2016;26(3):211–22.
Chintala SK, Putris N, Geno M. Activation of TLR3 promotes the degeneration of retinal ganglion cells by upregulating the protein levels of JNK3. Invest Ophthalmol Vis Sci 2015;56(1):505–14.
Asai M, Yagishita S, Iwata N, Saido TC, Ishiura S, Maruyama K. An alternative metabolic pathway of amyloid precursor protein C-terminal fragments via cathepsin B in a human neuroglioma model. Faseb J 2011;25(10):3720–30.
Johnson R, Teh CH, Kunarso G, Wong KY, Srinivasan G, Cooper ML,, et al REST regulates distinct transcriptional networks in embryonic and neural stem cells. PLoS Biol 2008;6(10):e256.
Kato T, Noma H, Kitagawa M, Takahashi T, Oshitani N, Kitagawa S. Distinct role of c-Jun N-terminal kinase isoforms in human neutrophil apoptosis regulated by tumor necrosis factor-alpha and granulocyte-macrophage colony-stimulating factor. J Interferon Cytokine Res 2008;28(4):235–43.
Choi WS, Abel G, Klintworth H, Flavell RA, Xia Z. JNK3 mediates paraquat- and rotenone-induced dopaminergic neuron death. J Neuropathol Exp Neurol 2010;69(5):511–20.
Acknowledgements
This work was supported by the National Medical Research Council, Ministry of Health, Singapore, with grant NMRC/1059/2006 and with NMRC NUHS Centre Grant—Memory, Ageing, and Cognition Centre Seed Funding, NMRC/CG/013/2013. We thank Miss Maying Tsemay Tse for the APP-null mice required for the additional experiments necessary for revision of the manuscript. We thank Dr M. Goedert (Cambridge, UK) for the rabbit anti-AICD antibody (BR188). We thank Mr Woon Fei Ho for excellent technical and administrative assistance. We thank Dr Zhong-Can Chen for assisting in generating the SH-SY5Y-APP cell line and Ms Jayakody Pathirannehelage Tharindunee Jayakody for technical assistance in detecting JNK3 expression in SH-SY5Y cells. We thank Dr Huck-Hui Ng and Dr Yun-Shen Chan for helpful advice on luciferase assays.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Edited by D. Rubinsztein
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, C., Zhang, CW., Zhou, Y. et al. APP upregulation contributes to retinal ganglion cell degeneration via JNK3. Cell Death Differ 25, 663–678 (2018). https://doi.org/10.1038/s41418-017-0005-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-017-0005-3
This article is cited by
-
Prion-induced photoreceptor degeneration begins with misfolded prion protein accumulation in cones at two distinct sites: cilia and ribbon synapses
Acta Neuropathologica Communications (2021)
-
Cdk5-mediated Drp1 phosphorylation drives mitochondrial defects and neuronal apoptosis in radiation-induced optic neuropathy
Cell Death & Disease (2020)
