
Abstract
Heme is required for cell respiration and survival. Nevertheless, its intracellular levels need to be finely regulated to avoid heme excess, which may catalyze the production of reactive oxygen species (ROS) and promote cell death. Here, we show that alteration of heme homeostasis in endothelial cells due to the loss of the heme exporter FLVCR1a, results in impaired angiogenesis. In vitro, FLVCR1a silencing in endothelial cells causes defective tubulogenesis and poor viability due to intracellular heme accumulation. Consistently, endothelial-specific Flvcr1a knockout mice show aberrant angiogenesis responsible for hemorrhages and embryonic lethality. Importantly, we demonstrate that impaired heme export leads to endothelial cell death by paraptosis and provide evidence that endoplasmic reticulum (ER) stress precedes heme-induced paraptosis. These findings highlight a crucial role for the cytosolic heme pool in the control of endothelial cell survival and in the regulation of the angiogenic process. Interfering with endothelial heme export represents a valuable model for a deeper understanding of the molecular mechanisms underlying heme-triggered paraptosis and, in the future, might provide a novel tool for the modulation of angiogenesis in pathophysiologic conditions.
Similar content being viewed by others


Introduction
Blood vessels originate from endothelial precursors (angioblasts), which give rise to a primitive vascular labyrinth of small capillaries, a process known as vasculogenesis [1]. During subsequent angiogenesis, the vascular plexus progressively expands through vessel sprouting [1]. The sprouting process is based on endothelial cell (EC) migration toward chemotactic and angiogenic stimuli, proliferation and tube formation. Afterward, a highly organized network of larger vessels ramifying into smaller ones, emerges. A functional circulatory system is essential to guarantee oxygen and nutrients delivery to distant organs [2]. Consistently, angiogenesis is crucial for organ growth in the developing embryo and repair of wounded tissues in the adult [1]. Many molecular players are involved in the formation and remodeling of blood vessels, including growth factors, integrins, chemokines, junctional molecules and oxygen sensors. Interestingly, metabolic demands are also thought to modulate angiogenesis [3]. Intrinsic, as well as extrinsic factors involved in the regulation of angiogenesis, show a dual role, by both stimulating vessel growth and inhibiting EC death. Indeed, excessive cell death might interfere with angiogenesis and actively lead to vessel regression [4]. Stimuli that cause endothelial injury or death include environmental stressors such as oxidative stress and nutrient deprivation, endoplasmic reticulum (ER) stress and metabolic stress [5, 6]. Here, we provide evidence that heme homeostasis in ECs controls the angiogenic process. In particular, we show that the expression of the heme exporter Feline leukemia virus subgroup C receptor 1a (FLVCR1a) on ECs is essential for proper angiogenesis. FLVCR1 is a widely conserved heme export protein, which exists as two isoforms, FLVCR1a and FLVCR1b, located at the plasma and mitochondrial membrane, respectively [7]. Heme, a complex of iron with protoporphyrin IX, is required for many biological processes, including oxygen transport, cell respiration and drug metabolism but has also cytotoxic properties as a pro-oxidant molecule [8]. Therefore, intracellular heme levels are strictly regulated through the biosynthesis, the incorporation in hemoproteins, the catabolism and the export [9]. Heme export through FLVCR1a has proved fundamental in preventing intracellular heme overload and heme-induced oxidative stress in many cellular systems, including HeLa, Caco2, primary hepatocytes and macrophages [7, 10,11,12]. Furthermore, FLVCR1a loss in mice causes embryonic lethality [7], whereas intestinal-specific deletion impairs normal proliferation in enterocytes and FLVCR1a depletion in a mouse model of ulcerative colitis is associated with reduced animal survival [12]. Consistently, FLVCR1a silencing affects cell viability in K562 cells [13] and induces programmed cell death (PCD) in neuroblastoma cells [14]. These data suggest that FLVCR1a deficiency may increase the susceptibility to PCD. Here, we show that FLVCR1a loss in ECs leads to an expansion of the intracellular heme pool and promotes cell death by paraptosis, a specific type of PCD. Paraptosis of Flvcr1a null ECs in mouse embryo prevents the formation of a functional microvascular network, thus leading to extensive hemorrhages and embryonic lethality.
Results
Silencing of FLVCR1a in human ECs results in increased heme levels and reduced viability
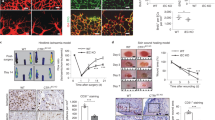
FLVCR1a has been reported to act as a heme exporter in hepatocytes [10], enterocytes [12] and erythroid cells [13]. Here, we tested whether FLVCR1a exerts a similar function in ECs. To address this point, we downmodulated FLVCR1a in two different types of human primary ECs: the human microvascular ECs (HMECs) and the human umbilical vein ECs (HUVECs), which derive from micro- and macro-vasculature, respectively. To downmodulate FLVCR1a, ECs have been infected with a lentiviral vector carrying the specific short hairpin RNA (shRNA) for FLVCR1a or a “scramble” shRNA as control. FLVCR1a mRNA levels were significantly reduced to about 40% in both HMECs (Fig. 1a) and HUVECs (Fig. 1c). Conversely, the FLVCR1b isoform was not affected (Figs. 1b, d). To study the effects of FLVCR1a loss on heme metabolism, the expression of heme-regulated genes was assessed in silenced human primary ECs. Interestingly, we found reduced levels of the δ-aminolevulinic acid synthase-1 (ALAS-1), the first and rate-limiting enzyme involved in heme biosynthetic pathway, in FLVCR1a-downregulated HMECs and HUVECs compared with scramble cells (Figs. 1e, g). The expression of heme oxygenase-1 (HO1), the enzyme mainly responsible for heme catabolism, was not altered in HMECs and slightly but not significantly increased in HUVECs after FLVCR1a silencing (Figs. 1f, h). These data suggest that the first compensatory response to the lack of heme export in ECs is the decrease of endogenous heme biosynthesis. Next, we directly measured the intracellular heme content in basal HMECs and we found a higher amount of heme after FLVCR1a silencing compared with scramble cells (Fig. 1i). In HUVECs, we did not find any difference in heme levels after FLVCR1a silencing (Fig. 1k). This is likely due to the more efficient downmodulation of ALAS1 and the slight increase in HO1 in HUVECs compared with HMECs, which might better compensate for the lack of heme export in this cell line. However, FLVCR1a-downregulated HUVECs showed increased intracellular heme levels compared with control cells, upon treatment with the heme precursor δ-Aminolevulinic acid (ALA) (Fig. 1k). Notably, the increased intracellular heme content was associated to a higher production of reactive oxygen species (ROS) in HMECs under resting conditions and in HUVECs following ALA treatment (Figs. 1j, l), as previously reported in other cell types undergoing intracellular heme accumulation [10, 12, 15]. Taken together, these data show that FLVCR1a-downregulated ECs accumulate de novo synthetized heme, which in turn gives rise to high ROS production. To investigate the effects of heme overload on ECs viability, we performed 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and flow cytometry analysis after Annexin V staining. Interestingly, FLVCR1a-silenced cells were less viable (Figs. 1m, o) and a higher percentage of Annexin V-positive cells was found in FLVCR1a-downregulated HMECs and HUVECs compared with scramble cells (Figs. 1n, p).

a-h qRT-PCR analysis of FLVCR1a a, c, FLVCR1b b, d, ALAS1 e, g and HO1 f, h in HMECs a, b, e, f or HUVECs c, d, g, h in which the expression of FLVCR1a was downregulated using a specific shRNA 1a. Transcript abundance, normalized to 18s mRNA expression, is expressed as a fold increase over a calibrator sample. Data represent mean ± SEM, n = 5; *p < 0.05, **p < 0.01, ***p < 0.001. i, k Heme content in HMECs i or HUVECs k in which the expression of FLVCR1a was downregulated using a specific shRNA. Intracellular heme levels in HUVECs cultured in basal condition or treated with δ-aminolevulinic acid (ALA) 5 mM for 2–4 h, are shown in k. Values are expressed as pmol heme/mg protein. Data represent mean ± SEM, n = 10; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. j, l ROS amount measured by detection of H2DCFDA fluorescence in HMECs j or HUVECs l with or without FLVCR1a in basal condition or after treatment with ALA 5 mM for 15 h. Values are expressed as FIU/mg protein. Data represent mean ± SEM, n = 4; *p < 0.05. m, o Cell viability assessed through 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay of HMECs m or HUVECs o in which the expression of FLVCR1a was downregulated using a specific shRNA. Values are expressed as percentage increase at 24, 48 h compared with T0 (time zero). Data represent mean ± SEM, n = 8 (HMECs), n = 3 (HUVECs); ***p < 0.001, ****p < 0.0001. n, p Flow cytometry assay performed on HMECs n or HUVECs p with or without FLVCR1a, after incubation with Annexin V-FITC antibody. Values are expressed as percentage of Annexin V high/PI low cells. Data represent mean ± SEM, n = 3; *p < 0.05. ALAS1 δ-aminolevulinate synthase-1; HO1 heme oxygenase-1; RQ relative quantification; qRT-PCR quantitative real-time PCR; ROS reactive oxygen species; shRNA short hairpin RNA; ALA δ-aminolevulinic acid; PI propidium iodide
These data demonstrate that ECs are highly sensitive to the absence of the cell surface heme exporter, as the loss of FLVCR1a leads to intracellular heme accumulation, oxidative stress and reduced cell viability.
Endothelial deletion of Flvcr1a leads to embryonic lethality and extensive hemorrhages in vivo
To study the role of FLVCR1a in ECs in vivo, we generated an endothelial-specific knockout (eKO) mouse model for FLVCR1a (Figs. 2a-d). To this end, we crossed FLVCR1a floxed mice with Tie2-Cre mice, expressing the Cre recombinase under the control of the Tie2 promoter, which is mainly active in the endothelial and erythroid compartments. Importantly, we did not recover any viable Flvcr1a eKO within the offspring (Table 1), thus indicating that disruption of Flvcr1a gene in ECs results in embryonic lethality. Flvcr1a eKO embryos were found at the expected Mendelian ratio from embryonic stage E11.5 to embryonic stage E14.5 (Table 1). At 11.5 days of embryonic development, Flvcr1a eKO embryos appeared normal (Fig. 2e). Starting from E12.5, massive intraembryonic bleeding was visible in the distal regions of the body, particularly the developing limbs or the tail, and in the yolk sac (Figs. 2f-h). In Flvcr1a eKO limbs, the hemorrhages were often associated to edema and skeletal malformation, as suggested by impaired digits formation (Figs. 2g, h). At E14.5 Flvcr1a eKO embryos appeared heavily damaged (Fig. 2h), whereas at E16.5 viable Flvcr1a eKO embryos were no longer found (Fig. 2i). This phenotype showed complete penetrance indicating that FLVCR1a loss in ECs leads to severe bleeding in vivo and embryonic death between E14.5 and E15.5. To better investigate the organogenesis and overall morphology in Flvcr1a eKO embryos, X-ray computed micro-computed tomography (micro-CT) was performed on E13.5 embryos (Fig. 2j). The analysis of embryonic internal organs did not reveal the presence of evident malformations in Flvcr1a eKO embryos compared with controls, except for the fetal liver that was smaller in Flvcr1a eKO embryos due to the Cre activity in erythroid precursors [13] (Supplementary Figure S1). Consistently, the histological analysis performed on E12.5 Flvcr1a eKO showed no altered cell morphology in major organs (Fig. 2k).

a Schematic representation of the knockout strategy in mice. Briefly, we named “floxed allele” the allele in which the first exon of Flvcr1 gene was flanked by loxP sites, recognizable by the Cre transgene. The excision of exon 1 results in a null allele preventing the production of FLVCR1a protein. b Representative PCR products from yolk sac DNA analysis of E13.5 embryos. Specific primers allowed distinguishing the wt (242 bp), floxed (280 bp) and null allele (320 bp) of Flvcr1a. c, d qRT-PCR analysis showing Flvcr1a mRNA levels in E13.5 whole embryo c and yolk sac d. Data represent mean ± SEM; *p < 0.05, **p < 0.01. e Phenotype of E11.5 control and Flvcr1a eKO embryos. f E13.5 embryos enfolded in their yolk sacs. At E13.5, major yolk sac vessels in Tie2-Cre conditional mutant embryos are clearly visible and hemorrhagic spots were often observed within the yolk sac (white arrows). g Representative picture of E13.5 control and Flvcr1a eKO embryos with undeveloped limbs and intraembryonic hemorrhages (white arrows). h, i Representative pictures of E14.5 h and E16.5 i embryos. At E16.5 endothelial Flvcr1a knockout, embryos begin to be re-absorbed by the endometrium. White arrows indicate hemorrhages. j X-ray computer micro-tomography analysis on E13.5 embryos showing the internal organ morphology. k Sagittal sections from E13.5 embryos stained with hematoxylin and eosin to look at the cellular morphology in tissues. RQ relative quantification; eKO, Flvcr1a endothelial knockout; E embryonic stage
Collectively, our results indicate that FLVCR1a loss in ECs impairs vascular development by leading to severe hemorrhages and late intrauterine fetal death.
Loss of FLVCR1a in ECs disrupts microvessel architecture
To further investigate the cause of hemorrhages, we analyzed the vasculature of Flvcr1a eKO embryos at E11.5 when hemorrhages were not detectable, by performing whole-mount staining for PECAM-1 (CD31), a pan-EC marker. At the level of developing limbs, which are the sites where hemorrhages would appear earlier, the structure of the vascular network was severely impaired in E11.5 Flvcr1a eKO embryos compared with controls (Fig. 3a). In control embryos, a well-defined network of capillaries was properly organized throughout the limb bud. Conversely, in Flvcr1a eKO mutants, microvessel architecture was compromised and large areas completely devoid of vessels were observed in both the forelimbs and the hindlimbs (Fig. 3a). Notably, when we looked at the vascular network in other regions of E11.5 Flvcr1a eKO embryo’s body, as the yolk sac and the head, vessels appeared generally intact (Supplementary Figure S2). As the limbs’ capillaries are mainly composed of microvascular cells, we used HMECs to study the ability of FLVCR1a-silenced human microvascular cells to generate tubule-like structures in vitro. Interestingly, silenced cells showed a reduced ability to form a complex capillary network compared with scramble cells (Fig. 3b). These results highlight a key role for FLVCR1a in the formation of a properly patterned capillary network, which is essential for the development of a functional circulatory system.

a Whole-mount staining with CD31/Pecam-1 antibody showing the embryonic vasculature in E11.5 embryos. Enlarged pictures of both the forelimb (left) and the hindlimb (right) are reported. b Matrigel-based tube formation assay of HMECs in which the expression of FLVCR1a was downregulated using a specific shRNA. The histograms of the number of nodes, junctions, segments and isolated segments are shown. The total segments length is also reported. Data represent mean ± SEM, n = 19; *p < 0.05. eKO Flvcr1a endothelial knockout; scr cells infected with a “scramble” shRNA; 1a, cells infected with a shRNA specific for FLVCR1a
Loss of FLVCR1a in ECs causes paraptotic cell death
In order to understand why FLVCR1a-deficient ECs fail to form a proper capillary network, we performed electron microscopy analysis on forelimb transverse sections from both Flvcr1a eKO and control E11.5 embryos (Fig. 4). Flvcr1a null ECs were characterized by the presence of several intracellular vacuoles of different sizes and shapes (Figs. 4a, b, d, e). The vacuoles often originated from an enlargement of the ER and mitochondria (Figs. 4c, f). Intracellular vacuoles can reach extended size, sometimes affecting the entire cytoplasmt (Figs. 4g-i). Interestingly, cytoplasmic vacuolation is the main feature of paraptosis, a specific type of PCD characterized by ER and mitochondria swelling and lack of apoptotic or autophagic morphology [16]. Consistently, the defining morphological hallmarks of apoptosis such as chromatin condensation, nuclear fragmentation and apoptotic body formation, were not observed in Flvcr1a null ECs (Fig. 5a). Moreover, the intracellular vacuoles observed in ECs without FLVCR1a did not contain fragments of cellular organelles and in most cases were defined by single membranes, leading us to exclude an autophagic mechanism (Fig. 5b). To confirm that Flvcr1a null ECs undergo paraptosis, we analyzed CD31+ cells isolated from Flvcr1a eKO embryos (Figs. 5c-e, Supplementary Figure S3) for biochemical markers of this type of cell death, that is, translocation of phosphatidylserine to the cell surface, no caspase activation and lack of expression of autophagic markers [16,17,18]. As shown in Figs. 5f, g, we found a higher percentage of Annexin V-positive cells in Flvcr1a null ECs at both E11.5 and E14.5. Moreover, Flvcr1a null ECs did not express activated caspase-3 (Fig. 5h) and did not show abnormal expression of the autophagic marker LC3B (Figs. 5i, j). Importantly, these morphological and biochemical features were associated to heme overload (Fig. 5k) and normal expression of HO1 (Fig. 5l) suggesting that developing ECs are fully dependent on heme export for detoxifying heme excess. Taken together, these data demonstrate that loss of FLVCR1a in ECs leads to heme accumulation which triggers paraptotic cell death.

a-i Electron microscopy analysis performed on transverse section from E11.5 embryonic forelimbs. Two capillaries from a control a, b and an eKO d, e embryo are shown. Flvcr1a null ECs display extensive cytoplasmic vacuolation d, e and an altered swollen morphology of ER and mitochondria f compared with controls c. In g, a small limb capillary made of two Flvcr1a null ECs is shown. The presence of huge intracellular vacuoles and heavily damaged mitochondria without cristae (black arrowheads) is clearly visible in the enlargements h, i. eKO Flvcr1a endothelial knockout; BC blood cell; EC endothelial cell; IEJ inter endothelial junction; V vacuole; M mitochondrion; N nucleus; ER endoplasmic reticulum

a Electron micrographs showing the nuclear morphology of a control (left) and an Flvcr1a null EC (right). The main apoptotic features as chromatin condensation and nuclear fragmentation are not observed in Flvcr1a null ECs. b Electron micrographs showing the single-membrane vacuoles (black arrowheads) in Flvcr1a null ECs from E11.5 forelimb transverse sections. c Optical imaging on CD31+ cells isolated from E13.5 control and eKO embryos. Black arrows indicate intracellular vacuoles. d, e qRT-PCR analysis of Flvcr1a d and Flvcr1b e in CD31-positive and -negative cell fractions from E13.5 embryos. Values represent mean ± SEM; n = 5; ***p < 0.001. f, g Flow cytometer analysis of CD31+ cells from E11.5 f and E14.5 g embryos after staining with Annexin V. The percentage of Annexin V/CD31 double-positive cells is reported. *p < 0.05, **p < 0.01. h Western blot analysis of full-length (35 kDa) and cleaved (17–19 kDa) caspase-3. The active form of caspase-3 was not detected in CD31+ cells from E13.5 eKO embryos. As positive control HUVEC cells treated for 3 h with 5 μM STS were used. i Western blot analysis of the autophagy protein LC3. The ratio between LC3-II (14 kDa) and LC3-I (17 kDa) does not change in CD31+ Flvcr1a null ECs. j Quantification of normalized LC3-II/I ratio. Values are expressed as fold increase. k Intracellular heme levels in CD31+ cells isolated from E13.5 embryos. Values for each sample are expressed as the ratio between the fluorescence of CD31+ cells and the total fluorescence (CD31+/-). *p < 0.05. l qRT-PCR analysis of Ho-1 in CD31+ cells from E13.5 control and eKO embryos. Values represent mean ± SEM; n = 5. eKO, Flvcr1a endothelial knockout; EC endothelial cell; N nucleus; STS Staurosporine
To further characterize the mechanism of cell death promoted by endogenously synthetized heme overload in ECs, we performed electron microscopy on FLVCR1a-silenced HMECs both under resting conditions and upon treatment with ALA to stimulate heme synthesis. A proper morphology and organization of the ER and mitochondria was observed in control cells (Fig. 6a). Conversely, the ER structure in FLVCR1a-silenced HMECs appeared disorganized and some swollen ER cisternae were observed (Fig. 6b). Moreover, a high number of small vacuoles partially containing electron-dense dots were found throughout the cytosol of FLVCR1a-silenced HMECs (Fig. 6b). Importantly, damaged mitochondria characterized by a swollen morphology were also observed in HMECs in the absence of FLVCR1a (Fig. 6b). Upon ALA treatment, the same morphological alterations appeared also in control cells, which showed a high number of small vacuoles, swollen mitochondria and disorganized ER structure (Fig. 6c). The phenotype of FLVCR1a-silenced HMECs was strongly exacerbated by ALA treatment and huge vacuoles and heavily damaged swollen mitochondria were observed in the cytoplasm (Fig. 6d). Importantly, these morphological features well resemble paraptotic cell death, similarly to the above described embryonic Flvcr1a null ECs. As paraptosis, as well as other PCDs, requires protein synthesis, we studied the effects of the protein synthesis inhibitor cycloheximide (CHX) on heme-induced paraptosis. To this aim, we co-treated control and FLVCR1a-deficient HMECs with ALA (5 mM) and CHX (5 μg/ml). Notably, the number and size of intracellular vacuoles was strongly reduced by CHX, indicating that protein synthesis is required for this process (Figs. 6e, f). Taken together, these data indicate that heme accumulation in human ECs induces a program of death with the morphological features typically observed in paraptotic cells.

a-f Electron microscopy analysis performed on control a, c, e and FLVCR1a-silenced b, d, f HMECs. a Control HMECs displayed a well-organized ER and normal mitochondria. b FLVCR1a-deficient HMECs showed some swollen ER cisternae (black arrows) and many vacuoles partially containing electron-dense dots (white arrowheads). Some damaged mitochondria (*) were also observed. Interestingly, electron-dense deposits (black arrowheads) were found in the cytosol of some FLVCR1a-deficient HMECs. c Control HMECs treated with ALA 5 mM for 16 h. ALA treatment induced the same morphological changes observed in the absence of FLVCR1a. Intracellular vacuoles (white arrowheads) and swollen mitochondria (*), are shown. d FLVCR1a-silenced HMECs treated with ALA displayed huge intracellular vacuoles (V) and heavily damaged swollen mitochondria (*), indicating a mechanism of paraptotic cell death. e, f HMECs co-treated with ALA and CHX (5 μg/ml) for 16 h, are shown. The inhibition of protein synthesis by CHX strongly reduced the number and size of intracellular vacuoles. Electron-dense dots in FLVCR1a-silenced cells f are indicated by black arrowhead. ER endoplasmic reticulum; G Golgi apparatus; M mitochondrion; N nucleus; V vacuole; CHX Cycloheximide; ALA Aminolevulinic acid
Elevated intracellular ROS have been reported in several models of paraptosis [19, 20]. As heme is known to promote the formation of ROS, as reported in FLVCR1a-silenced human ECs (Figs. 1j, l), we studied their possible involvement in heme-induced paraptosis. To do this, we directly measured ROS production in ECs isolated from control and Flvcr1a eKO embryos. As shown in Supplementary Figure S4, we found no differences in generic ROS as well as in mitochondrial superoxide production between control and Flvcr1a null CD31+ cells (Supplementary Figures S4A-B). Moreover, the expression levels of superoxide dismutase 1 (Sod1) in CD31+ cells did not change between Flvcr1a eKO and controls (Supplementary Figure S4C). Although we cannot exclude that a mild production of ROS can occur, these data suggest that ROS are rather a contributory cause than a driving force in the induction of heme-triggered endothelial paraptosis in vivo.
As proteasome dysfunction and accumulation of unfolded proteins have been described in several paraptotic cells [19, 21, 22], we investigate the possible involvement of ER stress in heme-induced endothelial paraptosis. To this aim, we evaluated the expression of main ER stress markers in ECs without FLVCR1a. Interestingly, we found a significant induction of PERK, GRP78/BIP and GRP94 transcripts in Flvcr1a null embryonic ECs, as well as in human ECs silenced for FLVCR1a (Figs. 7a-c, e-g). Another evidence of ER stress derived from the electron micrographs of Flvcr1a null ECs, widely characterized by the presence of swollen ER cisternae (Fig. 7d). Finally, a time-course experiment with the heme precursor ALA on human ECs correlated de novo synthetized heme accumulation with increasing expression of GRP78/BIP protein (Figs. 7h, i). These data strongly indicate that heme-induced paraptosis might be preceded by induction of ER stress.

a-c qRT-PCR analysis of Perk (protein kinase R (PKR)-like endoplasmic reticulum kinase) a, Grp78/Bip (binding immunoglobulin protein) b and Grp94 c in CD31+ cells isolated from E13.5 Flvcr1a eKO embryos. Data represent mean ± SEM, n = 5; *p < 0.05, ***p < 0.001. d Electron micrographs from limb transverse section of E13.5 Flvcr1a eKO embryo showing the swollen morphology of endoplasmic reticulum (black arrowheads). e-g qRT-PCR analysis of PERK e, GRP78/BIP f and GRP94 g in FLVCR1a-silenced HMECs. Data represent mean ± SEM, n = 5; *p < 0.05, **p < 0.01. h Western blot analysis of GRP78/BIP protein expression in HMECs treated with ALA 5 mM for the indicated times. eKO Flvcr1a endothelial knockout; EC endothelial cell; M mitochondrion
Discussion
In the present study, we show that heme accumulation in ECs due to heme export failure, triggers paraptotic cell death responsible for aberrant angiogenesis (Fig. 8). These findings might have important implications for physiological, as well as pathological angiogenesis.

Graphic representation of wild-type a and FLVCR1a null b endothelial cells. FLVCR1a exports the excess of heme derived from heme biosynthesis, thus contributing to maintain heme homeostasis. The loss of FLVCR1a leads to cytosolic heme accumulation and triggers paraptotic cell death characterized by extensive cytoplasmic vacuolation, swollen morphology of endoplasmic reticulum and large mitochondria without cristae. The effects of FLVCR1a loss in ECs in vivo cause deficient angiogenesis, hemorrhages and embryonic lethality. M mitochondrion; ER endoplasmic reticulum; V vacuoles; N nucleus
Heme toxicity on ECs has been widely studied in the context of hemolytic diseases. During hemolysis, hemoglobin derived from lysed erythrocytes, releases free heme that accumulates in plasma and becomes a major source of ROS [11, 23]. The endothelium is the first tissue facing the heme-related oxidative insult due to intravascular hemolysis. The exposure of the vessel wall to heme leads to ECs activation, which involves increased expression of adhesion molecules, leukocytes recruitment and cytokines production [23, 24]. In this context, HO1 induction represents a valuable mechanism for counteracting heme toxicity [25]. Consistently, several articles reported the therapeutic effects of HO1 overexpression in the vascular system [26, 27]. Here, we show that ECs are also highly sensitive to the accumulation of heme derived by intracellular metabolism. Indeed, heme overload in FLVCR1a-deficient ECs is associated with poor cell viability both in vitro and in vivo. Moreover, in Flvcr1a-silenced cells heme overload further increases upon the stimulation of heme biosynthesis. Interestingly, in Flvcr1a-deficient ECs, expanded intracellular heme pool triggers a specific compensatory response that is mainly mediated by the inhibition of heme biosynthesis, rather than by the induction of heme catabolism. Thus, heme catabolism (HO1) and export (FLVCR1a) are involved in the management of heme excess derived from extracellular or intracellular sources, respectively. Collectively, these data suggest the existence of distinct heme pools on specific cell compartments, likely accessible to different mechanisms of heme detoxification. This conclusion is supported by data obtained in other cell types and in liver- and intestine-specific Flvcr1a null mice, where Ho1 induction cannot reduce heme overload caused by FLVCR1a loss [10, 12].
In vivo, heme overload in Flvcr1a null ECs results in increased cell death responsible for disrupted vascularization, extensive hemorrhages and, consequently, embryonic lethality. Reduced viability and defective tubulogenesis demonstrated in FLVCR1a-silenced HMECs strongly supports this conclusion.
We identified the mechanism of EC death induced by endogenously synthetized heme accumulation as paraptosis, a specific type of PCD characterized by extensive cytoplasmic vacuolation that begins with a progressive swelling of ER and mitochondria [16, 17, 28]. The term “paraptosis” was first introduced by Sperandio et al. [16] to describe a form of PCD morphologically and biochemically distinct from apoptosis. Indeed, paraptosis does not involve the activation of caspases nor the classical morphological changes found in cells undergoing apoptosis [16, 17]. Similarly to other PCDs, paraptosis is known to require de novo protein synthesis. Consistently, inhibition of protein synthesis by CHX is able to rescue heme-induced cytoplasmic vacuolation in ALA treated FLVCR1a-silenced HMECs. The molecular mechanisms through which heme triggers paraptosis in ECs is unclear. ER stress and accumulation of unfolded proteins are known to be involved in paraptosis [19, 21, 22, 29, 30]. Notably, several indications of ER stress were found in both Flvcr1a null embryonic ECs and FLVCR1a-silenced human ECs, suggesting that ER stress might contribute to the cell death process. In particular, dilation of the ER-lumen and increased expression of ER stress markers were observed in ECs without FLVCR1a both in vivo and in vitro. Moreover, ALA treatment was shown to increase the expression of GRP78/BIP protein in human ECs, further correlating heme accumulation and ER stress. These data taken together strongly suggest that heme-induced vacuolation can be preceded by induction of ER stress. Consistently, heme excess has been shown to inhibit the proteasome leading to accumulation of misfolded proteins and ER stress [31]. Furthermore, protein aggregation has been recently reported as a cellular response to oxidative stress induced by heme [32]. Nevertheless, we cannot exclude a contribution of intracellular ROS to the mechanism of heme-induced EC death. Heme is known to promote the formation of ROS as previously demonstrated in other models of Flvcr1a deficiency [10, 12]. Consistently, FLVCR1a-silenced ECs displayed enhanced ROS production. Although we were not able to measure a significant increase of ROS in CD31+ cells from Flvcr1a eKO embryos, we think that a mild production of ROS in Flvcr1a null ECs might sensitize the cells to paraptosis as previously reported in other systems [19,20,21, 33]. Finally, the swollen morphology of ER and mitochondria observed in Flvcr1a null ECs and FLVCR1a-silenced human ECs indicates an imbalance in ions homeostasis (e.g., Ca2+ and K+). Accordingly, alterations in ions exchange between ER and mitochondria are involved in paraptosis [21, 34, 35].
Interestingly, some natural and synthetic compounds used as anticancer agents, exert a pro-paraptotic effect, which is applied as an alternative way to kill cancer cells resistant to apoptosis [29, 30, 36, 37]. As evasion of cell death is one of the hallmarks of tumorigenesis, the elucidation of the mechanisms promoting non-canonical types of cell death can provide novel therapeutic options [36]. In this context, the finding that intracellular heme accumulation promotes paraptosis opens new perspectives based on the manipulation of intracellular heme pool for reducing tumor growth. In particular, angiogenesis is an important rate-limiting step in tumor growth and anti-angiogenesis therapy represents a valuable opportunity for cancer treatment [38]. Our data suggest that heme-promoted paraptosis might be exploited as a way for reducing tumor angiogenesis. We can speculate that agents aimed at increasing cytosolic heme by promoting heme synthesis, blocking heme export and/or heme catabolism might have anti-angiogenic effects [39]. These drugs could be used in combination with other drugs to improve antitumor efficacy by killing apoptosis-resistant cells.
Cell death through paraptosis has been reported also in other pathological conditions. For instance, paraptosis has been described in neural development and neurodegeneration (e.g., amyotrophic lateral sclerosis and Huntington’s disease), in the ischemic damage and in the context of retina pathophysiology [36]. Interestingly, Flvcr1 gene is mutated in posterior column ataxia with retinitis pigmentosa and hereditary sensory and autonomic neuropathy type 2, both characterized by degeneration of specific neuronal cell types [14, 40, 41]. We hypothesize that impaired FLVCR1 function in these patients might promote paraptosis of specific cell types.
In conclusion, our data unravel the critical role of the physiologic regulation of the intracellular heme pool for the maintenance of ECs homeostasis and their angiogenic functions. We speculate that acting on the endothelial heme pool might provide novel therapeutic options for controlling neo-angiogenesis in cancer and other pathologies characterized by enhanced/aberrant vascularization.
Materials and methods
Generation and analysis of Flvcr1a-conditional mutant mice
Endothelial-specific conditional Flvcr1a knockout mice were generated by crossing Flvcr1afl/+; Tie2-Cre male mice with Flvcr1afl/fl female mice [10] to generate EC-specific deletion of Flvcr1a gene (Flvcr1afl/fl; Tie2-Cre). Upon Cre-mediated recombination, excision of the first exon would fully inactivate the gene in ECs, creating a null Flvcr1a allele (Flvcr1a null). Mice were genotyped by PCR analyses using DNA from yolk sacs or tail biopsies. To detect the Cre allele, primers Cre-Fw (5ʹ-GGACATGTTCAGGGATCGCCAGGCG-3ʹ) and Cre-Rev (5ʹ-GCATAACCAGTGAAACAGCATTGCT-3ʹ) were used. To detect the Flvcr1a null allele, primers ILox-Fw (5ʹ-TCTAAGGCCCAGTAGGACCC-3ʹ) and IILox-Rev (5ʹ-AGAGGGCAACCTCGGTGTCC-3ʹ) were used, given a 320-bp fragment. To analyze the LoxP sites on Flvcr1 gene, primers ILox-Fw (5ʹ-TCTAAGGCCCAGTAGGACCC-3ʹ) and ILox-Rev (5ʹ-GAAAGCATTTCCGTCCGCCC-3ʹ) were used, given a 280-bp band for the floxed allele and a 242-bp band for the wild-type allele (Fig. 2a). Embryos were obtained from timed pregnant females with the morning of vaginal plug considered E0.5. All experiments were approved by the animal studies committee of the University of Torino (Italy).
Whole-mount immunostaining
Whole-mount staining was performed as previously described [42], using anti-mouse CD31 (Pecam-1) monoclonal antibody (BD Biosciences, Franklin Lakes, NJ, USA). 3,3′-diaminobenzidine (DAB)/hydrogen peroxide was used for visualization.
CD31+ cells isolation from mouse embryo
E13.5–14.5 embryos were isolated from Tie2-Cre transgenic mice and minced into 1–2 mm fragments with a scalpel. After rinsing in phosphate-buffered saline (PBS), the tissue pieces were incubated for 45 min at 37 °C in 10 ml of pre-warmed PBS with collagenase (5 mg/ml, Collagenase Type II, Worthington, CLS-2) with shaking until a single-cell suspension was obtained. During this incubation, the cells were dissociated at 10-min intervals by pipetting. To stop the collagenase activity, Dulbecco’s modified Eagle’s medium (Invitrogen) containing 10% fetal bovine serum (FBS; Invitrogen) was added to the cell suspension, gently pelleted and rinsed with PBS. The cells in PBS were then filtered through a cell strainer (40 mm mesh, BD Falcon). After centrifugation, cells were re-suspended in 1 ml NH4Cl 154 mM, overlaid onto 5 ml of FBS and centrifuged. If red blood cells were not fully removed, this step was repeated. Cells were washed and re-suspended in PBS. Then, embryonic cells were magnetically labeled with MicroBeads conjugated to monoclonal anti‑mouse CD31 antibody (Miltenyi Biotec) and loaded onto a MACS® Column, which was placed in the magnetic field of a MACS Separator. After removing the column from the magnetic field, the magnetically retained CD31+ cells were eluted as the positively selected cell fraction.
RNA extraction and real-time PCR analysis
Total RNA was extracted from tissue and cell samples using PureLink® RNA Mini Kit (ThermoFisher Scientific). For quantitative real-time polymerase chain reaction (qRT-PCR), 0.5–1 μg total RNA was transcribed into complementary DNA by M-MLV reverse transcriptase (Invitrogen) and random primers (Invitrogen). qRT-PCR was performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems, Monza, Italy). Primers and probes were designed using the Universal ProbeLibrary Assay Design Center software (www. lifescience.roche.com). For FLVCR1a, specific primers and the probe were designed using Primer Express Software Version 3.0 (Applied Biosystems). Transcript abundance, normalized to 18s mRNA expression (for mouse tissues) or to beta-actin mRNA expression (for human cells), is expressed as a fold increase over a calibrator sample.
Western blotting
For western blot analysis, 20 μg of CD31+ cells protein extracts were loaded on 15% sodium dodecyl sulfate-polyacrylamide gels and analyzed by western blotting as previously described [43]. Cleaved caspase-3 (Asp175) antibody (Cell Signaling) was used to recognize the activated caspase-3 resulting from cleavage adjacent to Asp175 (17–19 kDa). Full-length caspase-3 (Cell Signaling) antibody was used to detect the full-length protein (35 kDa). Rabbit polyclonal antibody against LC3 (TA301542, Origene) was used to detect conversion of LC3-I (cytosolic) to LC3-II (autophagic vacuole membrane bound). Purified mouse anti-Grp78/Bip (610979, BD Transduction Laboratories) was used to detect the protein expression in HMECs.
Cell culture
HUVECs were propagated in M199 medium (Invitrogen) with 20% FBS (Invitrogen), 100 U/ml penicillin, 100 μg/ml streptomycin, 20 U/ml Heparin sodium salt from porcine intestinal mucosa (Sigma), 10 ng/ml recombinant human Fibroblast Growth Factor (FGF)-basic (PeproTech). HUVECs were used up to passages 6. Human adult dermal microvascular ECs were purchased by Lonza and propagated in EndoGRO MV-VEGF medium (Millipore, Merck, Italy) and used up to passages 12. Cells were maintained at 37 °C under a 5% CO2 atmosphere. To induce endogenous heme biosynthesis, 5-aminolevulinic acid hydrochloride (A3785, Sigma) was added to the cell medium. To inhibit protein synthesis, CHX (5 μg/ml, Sigma) was added to the cell medium.
FLVCR1a silencing
A set of five pLKO.1 HIV-based lentiviral vectors targeting human FLVCR1 gene were purchased (RHS4533-NM_014053, Dharmacon). A shRNA against the first exon of human FLVCR1 gene was used to specifically downregulate FLVCR1a expression. The lentiviruses pLKO.1-scr (expressing a “scramble” shRNA as control) and pLKO.1-1a (expressing the shRNA specific for FLVCR1a) were produced in HEK293FT cells. HMEC and HUVEC cells were infected with the lentiviruses in the presence of Sequa-brene. Following lentiviral infection, cells were selected with 0.02 μg/ml puromycin.
Measurement of heme concentration
Intracellular heme concentration was measured using a fluorescence assay, as previously reported [44]. Briefly, HMEC and HUVEC cells or CD31+/- embryonic cells were collected and 2 M oxalic acid was added to them. Samples were heated at 95 °C for 30 min leading to iron removal from heme. Fluorescence (wavelengths: excitation 400 nm – emission 662 nm) of the resultant protoporphyrin was assessed on a Glomax Multi Detection System (Promega Corporation). The endogenous protoporphyrin content (measured in parallel unheated samples in oxalic acid) was subtracted. Data were normalized to total protein concentration in each sample.
Measurement of intracellular ROS content in vitro
Accumulation of ROS in HMEC and HUVEC cells was assessed using the oxidant-sensitive fluorescent dye 29,79-dichlorodihydrofluoroscein diacetate (H2DCFDA, ThermoFisher Scientific). Cells were incubated with 5 mM H2DCFDA in cell medium for 30 min at 37 °C under 5% CO2 atmosphere. Then, cells were washed twice with PBS and lysed in TBS 1 × 1% Triton. A quantity of lysate correspondent to 10 μg protein was analyzed. Fluorescence was recorded at excitation and emission wavelengths of 485 and 530, respectively, on a Glomax Multi Detection System (Promega Corporation). Background fluorescence of cells untreated with H2DCFDA was subtracted from the total fluorescence. Results are expressed as arbitrary fluorescence unit.
MTT assay and Annexin V affinity assay
In all, 5×103 HMEC or HUVEC cells were plated on 96 multiwells, and every 24 h cell growth was evaluated by MTT assay (Roche) according to the manufacturer’s instructions. For Annexin V affinity assay, 5×105 cells were collected, washed in PBS and labeled with Annexin V (BD Biosciences) for 20 min. Then, 2 μl of propidium iodide (1 mg/ml) (Sigma-Aldrich) were added. Annexin V emission was detected in the green channel (525 nm) and propidium iodide in the red channel (575 nm) on a FACSCalibur (BD Biosciences) using Cell Quest Pro Software (BD Biosciences).
Tubulogenesis assay
In vitro formation of capillary-like structures was studied on growth factor-reduced Matrigel (BD Biosciences) in 24-well plates. Cells (3.5 × 104] cells per well) were seeded onto Matrigel-coating in EndoGRO MV-VEGF (Millipore). Cell organization onto Matrigel was observed with a Nikon Eclipse Ti E microscope using a Nikon Plan 10X/0,10 objective and cells were kept at 37 °C and 5% CO2 during the experiment. Images were acquired after 8 h. At least three independent experiments were done for each experimental condition. ImageJ’s Angiogenesis Analyzer was used to analyze: master segments, master junctions, isolated segments, total length, total master segments length and nodes.
Micro-computed tomography
Micro-CT analysis was performed on E13.5 mouse embryos using a Bruker Skyscan 1172 micro-CT. Embryos were fixed in formalin and then stained for 6 days with a soft tissue contrast agent (phosphotungstic acid 2.5% dissolved in water). Acquisition were performed at 80 kV using a 0.5 mm Al filter at a resolution of 3 µm, 0.4° of rotation step, 360° scan, 4× frame averaging.
Electron microscopy
The transmission electron micrographs were obtained at the Advanced Light and Electron Microscopy BioImaging Center (ALEMBIC), San Raffaele, Milano, Italy.
Statistics
Results were expressed as mean ± SEM. Statistical analyses were performed using one-way or two-way analysis of variance or Student’s t-test. A p-value of <0.05 was considered significant.
References
Carmeliet P. Angiogenesis in life, disease and medicine. Nature 2005;438(7070):932–6.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011;473(7347):298–307.
Risau W. Mechanisms of angiogenesis. Nature 1997;386(6626):671–4.
Dimmeler S, Zeiher AM. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ Res 2000;87(6):434–9.
Pober JS, Min W, Bradley JR. Mechanisms of endothelial dysfunction, injury, and death. Annu Rev Pathol. 2009;4:71–95.
Galán M, Kassan M, Kadowitz PJ, Trebak M, Belmadani S, Matrougui K. Mechanism of endoplasmic reticulum stress-induced vascular endothelial dysfunction. Biochim Biophys Acta. 2014;1843(6):1063–75.
Chiabrando D, Marro S, Mercurio S, Giorgi C, Petrillo S, Vinchi F, et al The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J Clin Invest. 2012;122(12):4569–79.
Chiabrando D, Mercurio S, Tolosano E. Heme and erythropoieis: more than a structural role. Haematologica. 2014;99(6):973–83.
Chiabrando D, Vinchi F, Fiorito V, Mercurio S, Tolosano E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front Pharmacol. 2014;5:61.
Vinchi F, Ingoglia G, Chiabrando D, Mercurio S, Turco E, Silengo L, et al Heme exporter FLVCR1a regulates heme synthesis and degradation and controls activity of cytochromes P450. Gastroenterology. 2014;146(5):1325–38.
Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, et al Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood. 2016;127(4):473–86.
Fiorito V, Forni M, Silengo L, Altruda F, Tolosano E. Crucial role of flvcr1a in the maintenance of intestinal heme homeostasis. Antioxid Redox Signal. 2015;23(18):1410–23.
Mercurio S, Petrillo S, Chiabrando D, Bassi ZI, Gays D, Camporeale A, et al The heme exporter Flvcr1 regulates expansion and differentiation of committed erythroid progenitors by controlling intracellular heme accumulation. Haematologica. 2015;100(6):720–9.
Chiabrando D, Castori M, di Rocco M, Ungelenk M, Gießelmann S, Di Capua M, et al mutations in the heme exporter FLVCR1 cause sensory neurodegeneration with loss of pain perception. PLoS Genet. 2016;12(12):e1006461.
Ryter SW, Tyrrell RM. The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med. 2000;28(2):289–309.
Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci U S A. 2000;97(26):14376–81.
Sperandio S, Poksay K, de Belle I, Lafuente MJ, Liu B, Nasir J, et al Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 2004;11(10):1066–75.
Bröker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res. 2005;11(9):3155–62.
Yoon MJ, Kim EH, Lim JH, Kwon TK, Choi KS. Superoxide anion and proteasomal dysfunction contribute to curcumin-induced paraptosis of malignant breast cancer cells. Free Radic Biol Med. 2010;48(5):713–26.
Wang Y, Xu K, Zhang H, Zhao J, Zhu X, Wu R. Retinal ganglion cell death is triggered by paraptosis via reactive oxygen species production: a brief literature review presenting a novel hypothesis in glaucoma pathology. Mol Med Rep. 2014;10(3):1179–83.
Yoon MJ, Kim EH, Kwon TK, Park SA, Choi. Simultaneous mitochondrial Ca(2 + ) overload and proteasomal inhibition are responsible for the induction of paraptosis in malignant breast cancer cells. Cancer Lett. 2012;324(2):197–209.
Yoon MJ, Kang YJ, Lee JA, Kim IY, Kim MA, Lee YS, et al Stronger proteasomal inhibition and higher CHOP induction are responsible for more effective induction of paraptosis by dimethoxycurcumin than curcumin. Cell Death Dis. 2014;5:e1112.
Vinchi F, De Franceschi L, Ghigo A, Townes T, Cimino J, Silengo L, et al Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation. 2013;127(12):1317–29.
Belcher JD, Nath KA, Vercellotti GM. Vasculotoxic and Proinflammatory effects of plasma heme: cell signaling and cytoprotective responses. ISRN Oxidative Med. 2013; 2013.
Vinchi F, Tolosano E. Therapeutic approaches to limit hemolysis-driven endothelial dysfunction: scavenging free heme to preserve vasculature homeostasis. Oxid Med Cell Longev. 2013;2013:396-527.
Belcher JD, Vineyard JV, Bruzzone CM, Chen C, Beckman JD, Nguyen J, et al Heme oxygenase-1 gene delivery by Sleeping Beauty inhibits vascular stasis in a murine model of sickle cell disease. J Mol Med (Berl). 2010;88(7):665–75.
Belcher JD, Mahaseth H, Welch TE, Otterbein LE, Hebbel RP, Vercellotti GM. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J Clin Invest 2006;116(3):808–16.
Sperandio S, Poksay KS, Schilling B, Crippen D, Gibson BW, Bredesen DE. Identification of new modulators and protein alterations in non-apoptotic programmed cell death. J Cell Biochem. 2010;111(6):1401–12.
Lee WJ, Chien MH, Chow JM, Chang JL, Wen YC, Lin YW, et al Nonautophagic cytoplasmic vacuolation death induction in human PC-3M prostate cancer by curcumin through reactive oxygen species -mediated endoplasmic reticulum stress. Sci Rep. 2015;5:104-20.
Wang WB, Feng LX, Yue QX, Wu WY, Guan SH, Jiang BH, et al Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J Cell Physiol. 2012;227(5):2196-2206.
Vallelian F, Deuel JW, Opitz L, Schaer CA, Puglia M, Lönn M, et al Proteasome inhibition and oxidative reactions disrupt cellular homeostasis during heme stress. Cell Death Differ 2015;22(4):597–611.
Vasconcellos LR, Dutra FF, Siqueira MS, Paula-Neto HA, Dahan J, Kiarely E, et al Protein aggregation as a cellular response to oxidative stress induced by heme and iron. Proc Natl Acad Sci U S A. 2016;113:E7474–E7482.
Kar R, Singha PK, Venkatachalam MA, Saikumar P. A novel role for MAP1 LC3 in nonautophagic cytoplasmic vacuolation death of cancer cells. Oncogene. 2009;28(28):2556–68.
Yoon MJ, Lee AR, Jeong SA, Kim YS, Kim JY, Kwon YJ, et al Release of Ca2+from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget. 2014;5(16):6816–31.
Yumnam S, Hong GE, Raha S, Saralamma VV, Lee HJ, Lee WS, et al Mitochondrial dysfunction and Ca(2+) overload contributes to hesperidin induced paraptosis in hepatoblastoma cells, HepG2. J Cell Physiol. 2016;231(6):1261–8.
Lee D, Kim IY, Saha S, Choi KS. Paraptosis in the anti-cancer arsenal of natural products. Pharmacol Ther. 2016;162:120–33.
Diederich M, Cerella C. Non-canonical programmed cell death mechanisms triggered by natural compounds. Semin Cancer Biol. 2016;40-41:4–34.
Leite de Oliveira R, Hamm A, Mazzone M. Growing tumor vessels: more than one way to skin a cat - implications for angiogenesis targeted cancer therapies. Mol Aspects Med. 2011;32(2):71–87.
Fraisl P, Mazzone M, Schmidt T, Carmeliet P. Regulation of angiogenesis by oxygen and metabolism. Dev Cell. 2009;16(2):167–79.
Shaibani A, Wong LJ, Wei Zhang V, Lewis RA, Shinawi M. Autosomal recessive posterior column ataxia with retinitis pigmentosa caused by novel mutations in the FLVCR1 gene. Int J Neurosci. 2015;125(1):43–49.
Castori M, Morlino S, Ungelenk M, Pareyson D, Salsano E, Grammatico P, et al Posterior column ataxia with retinitis pigmentosa coexisting with sensory-autonomic neuropathy and leukemia due to the homozygous p.Pro221Ser FLVCR1 mutation. Am J Med Genet B Neuropsychiatr Genet. 2017;174:732–9.
Chae SS, Paik JH, Allende ML, Proia RL, Hla T. Regulation of limb development by the sphingosine 1-phosphate receptor S1p1/EDG-1 occurs via the hypoxia/VEGF axis. Dev Biol. 2004;268(2):441–7.
Marro S, Barisani D, Chiabrando D, Fagoonee S, Muckenthaler MU, Stolte J, et al Lack of haptoglobin affects iron transport across duodenum by modulating ferroportin expression. Gastroenterology. 2007;133(4):1261–71.
Sinclair PR, Gorman N, Jacobs JM. Measurement of heme concentration. Curr Protoc Toxicol 2001;Chapter 8:Unit 8.3.
Acknowledgements
The authors are grateful to Dr A. Camporeale for FACS analysis and to Prof MF Brizzi (Department of Medical Sciences, University of Turin) for the helpful discussion. The authors thank Dr Maria Carla Panzeri (Advanced Light and Electron Microscopy BioImaging Center—San Raffaele Scientific Institute) for expert assistance in electron microscopic imaging. This work was supported by the Italian Association for Cancer Research (AIRC) IG14599 and IG18857 to ET, and by the Italian Ministry of University and Research (MIUR) to ET. SP was supported by the Italian Foundation “Giovanni Goria” in collaboration with CRT Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Additional information
Edited by P. Salomoni
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Petrillo, S., Chiabrando, D., Genova, T. et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ 25, 573–588 (2018). https://doi.org/10.1038/s41418-017-0001-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-017-0001-7
This article is cited by
-
Paraptosis: a non-classical paradigm of cell death for cancer therapy
Acta Pharmacologica Sinica (2024)
-
Targeting paraptosis in cancer: opportunities and challenges
Cancer Gene Therapy (2024)
-
Unearthing FLVCR1a: tracing the path to a vital cellular transporter
Cellular and Molecular Life Sciences (2024)
-
Mfsd7b facilitates choline transport and missense mutations affect choline transport function
Cellular and Molecular Life Sciences (2024)
-
Endothelial cells require functional FLVCR1a during developmental and adult angiogenesis
Angiogenesis (2023)
