
Abstract
Prominin 1 (PROM1) is considered a biomarker for cancer stem cells, although its biological role is unclear. Prominin 2 (PROM2) has also been associated with certain cancers. However, the prognostic value of PROM1 and PROM2 in cancer is controversial. Here, we performed a systematic data analysis to examine whether prominins can function as prognostic markers in human cancers. The expression of prominins was assessed and their prognostic value in human cancers was determined using univariate and multivariate survival analyses, via various online platforms. We selected a group of prominent functional protein partners of prominins by protein-protein interaction analysis. Subsequently, we investigated the relationship between mutations and copy number alterations in prominin genes and various types of cancers. Furthermore, we identified genes that correlated with PROM1 and PROM2 in certain cancers, based on their levels of expression. Gene ontology and pathway analyses were performed to assess the effect of these correlated genes on various cancers. We observed that PROM1 was frequently overexpressed in esophageal, liver, and ovarian cancers and its expression was negatively associated with prognosis, whereas PROM2 overexpression was associated with poor overall survival in lung and ovarian cancers. Based on the varying characteristics of prominins, we conclude that PROM1 and PROM2 expression differentially modulates the clinical outcomes of cancers.
Similar content being viewed by others


Introduction
Cancer is one of the leading causes of human death and it has a profound impact on global health. According to the Surveillance, Epidemiology, and End Results Program’s (SEER) Cancer Statistics Review (CSR) 1975–2014, the number of new cases of cancer in the United States of America alone for all sites combined was 442.7 per 100,000 (both men and women) per year [1]. Cancer constantly introduces new challenges to various stakeholders, including researchers, medical practitioners, industrialists, and economists attempting to implement optimal health solutions.
Cumulative genetic alterations in the form of base insertions, deletions, substitutions, duplications, and translocations can be considered as the basis of cancer. Fortunately, targeted therapies or biological therapies based on the use of either molecular medicine or nano-engineered enzymes have been relatively successful in blocking the growth of cancer and cancer stem cells (CSCs) [2]. Owing to their self-renewing, multipotent, and high proliferative capacities, CSCs play a pivotal role in tumor invasion and metastasis. Therefore, targeting CSCs is critical for achieving high therapeutic efficiencies and preventing tumor recurrence. To develop targeted therapies, it is necessary to first identify suitable markers for identifying CSCs. A member of the prominin family, prominin 1 (PROM1), is considered a valuable marker of stem cells and CSCs [3,4,5]. Prominin family homologues in mammals consist of five transmembrane domains, an N-terminal domain exposed to the extracellular space, two small cytoplasmic loops, two large glycosylated extracellular loops, and a cytoplasmic C-terminal domain [6,7,8,9]. PROM1 is commonly reported as a marker of neuronal and hematopoietic stem cells [4], but it is also expressed in CSCs and cancer cells, including in breast cancer [5], acute myeloid leukemia (AML) [10], and various pediatric brain tumors [11]. In addition, PROM1 is involved in maintaining microvillar architecture and dynamics [12]. Recent studies suggest that PROM1 is upregulated in non-small cell lung cancer tissue compared to normal lung tissue, and mutations in PROM1 are associated with poor prognosis [13, 14]. High levels of PROM1 mRNA are also associated with poor prognosis in pediatric medulloblastoma [15]. Moreover, PROM1 regulates metastasis, drug resistance, and stemness properties in various cancer cells [3, 16, 17]. It can also regulate the activation of stem cells by orchestrating ciliary dynamics, and the absence of PROM1 allows stem cells to resist the effects of sonic hedgehog (SHH) on growth stimulation, thereby disrupting stem cell activation [18]. Previous studies have shown that therapies targeting PROM1 may prevent tumor development in various human cancers [19,20,21,22]. Although PROM1 has been studied as a CSC marker and a regulator of cancer progression and prognosis over the last two decades, specific studies regarding the relationship between PROM1 expression and prognosis in certain cancers are lacking.
PROM2, the second member of the prominin family, is structurally related to PROM1, but is encoded by a separate gene [6]. PROM2 expression is limited to epithelial cells, where it may be involved in the organization of plasma membrane microdomains [6]. Furthermore, PROM2 causes cell protrusions that recruit cholesterol with the aid of lipid rafts and subsequently, increase the phosphorylation of caveolin-1 in membrane microdomains [23]. A whole-genome expression profiling study reported that PROM2 is overexpressed in endothelial cells in lung cancer [24]. Moreover, several other expression profiling studies have reported that PROM2 is upregulated in various cancers including breast, brain, lung, renal, and tongue cancers and melanoma [6, 25, 26]. Despite this research, studies on PROM2 expression in cancer and its relevance to clinical outcomes are still limited. Moreover, to the best of our knowledge, PROM1 and PROM2 genes have not yet been studied using data mining approaches. Therefore, this is the first systematic analysis of the possible role of PROM1 and PROM2 in various cancers, based on publicly available gene expression and clinical data.
Here, we aimed to identify the role of prominins in cancer progression and their value in cancer prognosis. As these proteins may exert their effects through signaling pathways, we hypothesized that a multiomics data mining approach could identify the link between prominin expression and clinical outcomes in cancer patients. Therefore, we investigated the patterns of expression, mutation, and copy number alteration of PROM1 and PROM2 genes to determine their clinical significance in human cancers through systematic data analysis. Moreover, we aimed to determine the combined prognostic significance of PROM1 and PROM2 in certain cancers using a multivariate prognosis analysis. We also analyzed the interacting partners and genes co-expressed with PROM1 and PROM2 in various cancers and subsequently, analyzed these genes to predict the probable underlying signaling pathways involved. These results provide useful information to facilitate the development of new approaches for anti-cancer therapies that target cancer stem cells.
Methods
mRNA expression analysis using Oncomine
Data regarding PROM1 and PROM2 mRNA expression in various cancer types was retrieved from the online database, Oncomine (https://www.oncomine.org/resource/login.html) [27, 28]. This database platform contains a large collection of independent datasets and expertly curated data. It can be used to identify novel targets for drug development and to interrogate gene expression profiles along with clear and consistent interpretation of results. Differences in mRNA expression between cancer tissues and their normal tissue counterparts were calculated using the following threshold parameters: p < 0.01, fold-change > 2, and gene ranking in the top 10%. The details of the analyses are summarized in Supplementary Tables 1 and 2. The co-expression profiles of prominins in different cancer types were also extracted from Oncomine and are illustrated as a heat map in Fig. 7.
mRNA expression analysis using GEPIA
Gene expression profiling interactive analysis (GEPIA, http://gepia.cancer-pku.cn/) is a newly developed interactive online platform for analyzing RNA sequencing data [29]. It provides access to a large collection of data from 9736 tumors and 8587 normal samples from The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx) project. GEPIA was used for differential expression analysis of PROM1 and PROM2 in tumor/normal tissue from various cancers. GEPIA also provides other customizable functions, including patient survival and correlation analyses.
Survival analysis using Kaplan–Meier plotter
The web-based tool, Kaplan-Meier plotter, was used to analyze the impact of 54,675 genes on survival using HGU133 Plus 2.0 array data from 10,461 cancer samples. Of these samples, 5143 were from breast cancer patients, 1816 from ovarian cancer patients, 2437 from lung cancer patients, and 1,065 from gastric cancer patients, who underwent an average follow-up period of 69, 40, 49, and 33 months, respectively. The main objective of this tool is to perform a meta-analysis-based biomarker assessment. In this study, the correlations between prominin expression and patient survival were analyzed using the Kaplan–Meier plotter (http://kmplot.com/analysis/) [30]. According to various quantiles of biomarker expression, the tool divides patient samples into pairs of groups to analyze the prognostic value of a particular gene. Kaplan–Meier survival plots were constructed to compare the two patient groups and calculate the log-rank p-value and the hazard ratio, with 95% confidence intervals.
Prognosis analysis using PrognoScan
PrognoScan (http://dna00.bio.kyutech.ac.jp/PrognoScan/) [31] is a database that is used for the meta-analysis of the prognostic value of various genes. This online platform assists in investigating the relationship between gene expression and patient prognosis across a large collection of cancer microarray datasets. The correlation between prominin expression and survival was investigated in several cancer types using this tool. The significance threshold was adjusted to a Cox p-value < 0.05. These results are briefly presented in Supplementary Tables 3 and 4.
Survival analysis using OncoLnc
OncoLnc (http://www.oncolnc.org/) is a web-based interactive tool for analyzing survival correlations and retrieving clinical data matched with expression data for mRNAs, miRNAs, and long non-coding RNAs [32]. It is a large collection of clinical data from 8,647 patients across 21 cancer studies from the TCGA and MiTranscriptome beta collections. Using this platform, Cox regression analysis data were acquired for PROM1 and PROM2 in up to 21 cancers. These data were then used to generate Kaplan-Meier plots for further analysis of RNAs of interest.
Survival analysis using SurvExpress
SurvExpress (http://bioinformatica.mty.itesm.mx/SurvExpress) is a cancer-wide gene expression database with clinical outcomes and a web-based tool for survival analysis [33]. This database contains more than 39,000 samples and 225 datasets covering tumors in more than 26 different tissues. Using this platform, survival plots were generated for specific cancer types, using TCGA and GEO microarray data. Biomarkers can be assessed in several ways using this tool. For example, specific genes can be switched on and off, samples can be framed by available clinical information, and training and test samples can be identified.
Correlation and survival analysis using the R2 platform
R2 (http://r2platform.com) is a genomics analysis and visualization platform with a database, coupled to a web-interface that provides a set of analysis tools [34]. R2 supports all types of survival data (e.g., overall survival) and can also be used to generate a Kaplan-Meier plot for a specific dataset. The Kaplan Scan feature was used to establish the optimum cut-off values for PROM1 and PROM2, based on the p-value from a log-rank test of the cancers of interest. Although the Kaplan Scan tool was applied, the binary heat map was also used to show clustering based on “good” vs. “bad” prognoses. The R2 software was also used to identify genes that correlated with PROM1 and PROM2, using the freely available TCGA datasets on the R2 website. To import the list of common genes that correlated with PROM1 or PROM2 for all tumors of interest, a Venn diagram was generated using the tool, Venny 2.1 [35]. To understand how these common correlated genes collectively regulate signaling pathways, gene ontology and pathway analyses were performed using the Protein Analysis Through Evolutionary Relationships (PANTHER) tool (http://pantherdb.org/) [36]. This is an online system that classifies proteins (and their genes) in terms of various ontologies, including molecular function, biological process, cellular components, and pathway.
Analysis of gene expression and mutations using cBioPortal
An integrative analysis of PROM1 and PROM2 and clinical characteristics was performed using cBioPortal for Cancer Genomics (http://www.cbioportal.org), which is an open-access and open-source resource for the interactive visualization and analysis of multidimensional cancer genomics data sets [37, 38]. At the time of this study, 56,250 tumor samples from 215 cancer studies were available online. It contains various types of data, including DNA copy number, mRNA expression, non-synonymous mutations, DNA methylation, and limited de-identified clinical data. The query interface combined with personalized data storage enables interactive investigations of genetic alterations within specific genes, across available samples. The primary search parameters included alterations (amplifications, deep deletions, and missense mutations), copy number alterations (CNAs) from GISTIC, and RNA sequencing data, using the default settings. For the secondary search, we focused on RNA sequencing data.
Protein-protein interaction (PPI) analysis using GeneMANIA
GeneMANIA (https://genemania.org/) is an online tool that provides information on hypothetical gene function, interrogates gene lists, and ranks genes based on functional evaluations [39]. It contains a large set of functional association data, including protein and gene interactions, pathways, and co-expression data. We applied the GeneMANIA analysis tool to predict PPIs using PROM1 and PROM2 as queries. The prediction output is in the form of a network that shows the relationships between genes in the list, where nodes symbolize genes and links represent networks.
Statistical analysis
Bar and forest plots were drawn using GraphPad Prism version 7 (GraphPad Software, La Jolla, CA, USA). Survival curves were constructed using PrognoScan, PROGgeneV2, OncoLnc, and Kaplan-Meier plotters. All results are displayed with p-values obtained from a log-rank test. The levels of significance (p-values) of the Oncomine and heat map data were determined by the programs used for the analyses. An unpaired t-test was performed to analyze two groups (normal vs. cancer). For multivariate survival analysis, clinical outcome data were retrieved from the TCGA database using OncoLnc. The data were then processed to generate survival curves using GraphPad Prism version 7 software. Cox regression analysis was performed for univariate and multivariate survival analysis to define the independent factors that had a significant effect on patient survival. Log-rank p-values < 0.05 were considered significant and p = ns denoted results that were not significant.
Results
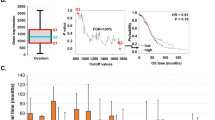
To understand the role of prominins in cancer, we compared their transcription levels in cancer tissues and normal tissues using the visualization tools provided by the Oncomine database. The various underlying threshold parameters, without altering any filter settings, were as follows: p-value, 0.01; fold change, 2; and gene ranking, 10%. Using these settings, we observed that prominins were overexpressed in some cancer tissues when compared to their expression levels in normal tissues and were underexpressed in others. These results indicate that the prominins may possess either oncogenic or anti-oncogenic characteristics, depending on the type of cancer (Fig. 1a). TCGA contains a large collection of RNA sequencing data and is useful resource for understanding the molecular basis of cancer. By accessing TCGA data via cBioPortal, we also further analyzed the mRNA levels of prominins in various types of cancer. PROM1 and PROM2 were found to be differentially expressed in many cancer types (Fig. 1b). The analyses of prominins are described in more detail below.

Transcription levels of PROM1 and PROM2 in different types of cancers (Oncomine and TCGA databases). a This graphic was generated using Oncomine, indicating the numbers of datasets with statistically significant (p < 0.01) overexpression (red) or underexpression (blue) of PROM1 and PROM2 mRNA (cancer vs. corresponding normal tissue). The threshold was designed with the following parameters: p-value = 0.01, fold change = 2, and gene ranking = 10%. The numbers in the boxes represent the number of analyses that met these thresholds. b Analysis of PROM1 and PROM2 mRNA levels in 30 types of human cancer using data from cBioPortal (http://www.cbioportal.org/index.do). Every dot represents a single study. White dots represent those without gene sequencing data, blue dots represent normal gene sequencing results (no mutation), and orange spots represent missense mutations. Flowchart: merged spots represent frame shifts, isosceles triangles represent splice sites, diamonds represent nonsense mutations, and rectangles represent others. The median and interquartile ranges are shown in each box
Prominin mRNA expression analysis
To explore the expression patterns of prominins in various cancer types, we analyzed cDNA microarray data, using the differential analysis tool of the Oncomine database. The database was queried for prominin expression in each cancer type and in respective normal tissue, individually. The analysis showed that PROM1 was overexpressed in pro-B acute lymphoblastic leukemia, and brain, esophageal, liver, testis, ovarian, and gastric cancers, but underexpressed in bladder, breast, kidney, and skin cancers, compared to that in normal tissue (Fig. 2a [i-xii)], Supplementary Fig. S1, Supplementary Table 1) [40,41,42,43,44,45,46,47,48,49]. To confirm the PROM1 expression results obtained from the Oncomine database, we performed a single-gene analysis of PROM1 using another online platform, GEPIA. These results, as shown in Fig. 2b (i-x), confirmed PROM1 overexpression in brain, esophageal, leukemia, testis, ovarian and stomach cancers and PROM1 underexpression in bladder, breast, and kidney cancers. The pattern of PROM2 expression in different types of cancers was considerably different to the expression pattern of PROM1. We observed that compared to that in normal tissues, PROM2 was significantly overexpressed in breast, lung, bone marrow, and ovarian cancers, whereas it was underexpressed in colon, esophageal, gastric, kidney, prostate and skin cancers (Fig. 3a [i-xi], Supplementary Fig. S3, Supplementary Table 2) [42, 49,50,51,52,53,54,55]. The above expression pattern of PROM2 in colon, lung, ovarian, kidney, and skin cancers has also been reported from TCGA data on the GEPIA website (Fig. 3b [i-vii]). Note that TCGA-based GEPIA results on prominin mRNA expression are mainly used for validating the expression results obtained via Oncomine-assisted analysis. When we performed expression analysis using GEPIA, we also recorded the expression patterns of for some other cancers which are not available in Oncomine platform (Supplementary Fig. S2 and S4). The systematic analysis carried out here was able to assess the mRNA expression status of prominins across a wide range of cancer types. Since expression the prominin expression status was confirmed in multiple databases, these results can be considered reliable. These results regarding PROM1 and PROM2 expression are also supported by previous studies. For example, PROM1 overexpression has been shown in esophageal and ovarian cancers [56, 57]. Moreover, PROM1+ ovarian CSCs are highly tumorigenic, chemo-resistant, and metastatic and they promote the adhesion capabilities of the ovarian cancer metastatic niche [57]. In contrast, our analysis showed that PROM2 is overexpressed in breast and lung cancers, and this result is in agreement with those of previous genome profiling studies and studies on expressed sequence tag (EST) clones deposited in the GenBank database [58, 59]. Thus, these results imply that prominins may be either oncogenes or tumor suppressor genes, depending on the type of cancer. These results showing the dysregulation of prominins can potentially be translated into clinical practice. For example, PROM1 may be a biomarker for the prediction of lung metastasis and poor prognosis in patients with osteosarcoma, in which the expression of this gene is considerably high [60, 61]. PROM1 expression may also be a predictor of poor clinical outcome in patients with ovarian cancer [62]. Therefore, we next investigated the extent to which prominin expression is associated with prognosis.

PROM1 expression analysis in different cancer types (Oncomine and TCGA databases). a The box plot comparing PROM1 expression in normal (left plot) and cancer tissues (right plot) were derived from the Oncomine database. The fold change of PROM1 in various cancer types was determined from the analyses shown in Supplementary Table 1 (i). Analysis of SBC relative to normal bladder (ii), GBM relative to normal brain (iii), MBC relative to normal breast (iv), BE relative to normal esophagus (v), CCSK relative to normal kidney (vi), PBALL relative to PBMC (vii), cirrhosis relative to normal liver (viii), TEC relative to normal testis (ix), OMA relative to normal ovary (x), BMSN relative to normal skin (xi) and GST relative to normal stomach (xii). The threshold was designed with the following parameters; p-value = 0.01, fold change = 2, and gene rank = 10%. b PROM1 expression in The Cancer Genome Atlas (TCGA) database. Box plots showing PROM1 mRNA expression in various tumor (T) and corresponding normal (N) tissues, using TCGA data from GEPIA (i-x). The threshold was designed with the following parameters: p-value = 0.01, fold change = 2. SBC superficial bladder cancer, MBC mucinous breast carcinoma, BE Barrett’s esophagus, EA esophageal adenocarcinoma, CCSK clear cell sarcoma of the kidney, CRCC chromophobe renal cell carcinoma, PBMC peripheral blood mononuclear cell, PBALL pro-B acute lymphoblastic leukemia, TEC testicular embryonal carcinoma, OMA ovarian mucinous adenocarcinoma, OCCA ovarian clear cell adenocarcinoma, BMSN benign melanocytic skin nevus, GST gastrointestinal stromal tumor, BLCA bladder urothelial carcinoma, BRCA invasive breast carcinoma, CHOL cholangiocarcinoma, COAD colon adenocarcinoma, ESCA esophageal carcinoma, GBM glioblastoma multiforme, HNSC head and neck squamous cell carcinoma, KICH kidney chromophobe, KIRC kidney renal clear cell carcinoma, LAML acute myeloid leukemia, OV ovarian serous cystadenocarcinoma, PAAD pancreatic adenocarcinoma, READ rectum adenocarcinoma, STAD stomach adenocarcinoma, TGCT testicular germ cell tumor, THCA thyroid carcinoma, UCEC uterine corpus endometrial carcinoma, UCS uterine carcinosarcoma

PROM2 expression analysis in different cancer types (Oncomine and TCGA databases). a The box plot comparing PROM2 expression in normal (left plot) and cancer tissues (right plot) was derived from the Oncomine database. The fold change of PROM2 in various cancer types was identified from our analyses presented in Supplementary Table 2 (i). Analysis of MBC relative to normal breast (ii), CC relative to normal colon (iii), EAC relative to normal esophagus (iv), DGAC relative to normal stomach (v), PRCC relative to normal kidney (vi), LAC relative to normal lung (vii), MGUC relative to normal bone marrow (viii), OSAC relative to normal peritoneum (ix), PC relative to normal prostate (x) and SBCC relative to normal skin (xi). The threshold was designed with the following parameters: p-value = 0.01, fold change = 2, and gene rank = 10%. b PROM2 expression data from the Cancer Genome Atlas (TCGA) database. Box plots showing PROM2 mRNA expression in various tumors (T) and corresponding normal (N) tissues using TCGA data from GEPIA (i-vii). The threshold was designed with the following parameters: p-value = 0.01, fold change = 2. MBC mucinous breast carcinoma, CC colon carcinoma, EAC esophageal adenocarcinoma, DGAC diffuse gastric adenocarcinoma, PRCC papillary renal cell carcinoma, LAC lung adenocarcinoma, MGUC monoclonal gammopathy of undetermined significance, OSAC ovarian serous adenocarcinoma, PC prostate carcinoma, SBCC skin basal cell carcinoma, CESC cervical squamous cell carcinoma and endocervical adenocarcinoma, CHOL cholangiocarcinoma, KIRC kidney renal clear cell carcinoma, KIRP kidney renal papillary cell carcinoma, LUAD lung adenocarcinoma, LUSC lung squamous cell carcinoma, OV ovarian serous cystadenocarcinoma, PAAD pancreatic adenocarcinoma, SARC sarcoma, SKCM skin cutaneous melanoma, THYM thymoma, UCEC uterine corpus endometrial carcinoma
Estimation of the prognostic value of prominins
To investigate the relationship between prominin gene expression and clinical prognosis, we used several online tools, namely, PrognoScan, R2, Kaplan-Meier plotter, SurvExpress, and OncoLnc. A positive correlation was observed between PROM1 overexpression and poor prognosis in brain, skin, and soft tissue cancers (Fig. 4a [iii, v, vi], Supplementary Fig. S5 [b–d], Supplementary Table 3), as analyzed using the PrognoScan database. In addition, low levels of PROM1 expression were correlated with poor overall survival (OS) in prostate and lung cancers (Fig. 4a [iv, vii], Supplementary Table 3). Therefore, these results indicated that PROM1 can be considered an oncogene for brain, skin, and soft tissue cancers, but a tumor suppressor gene for prostate and lung cancers. However, the relationship between PROM1 expression and survival in breast cancer was not clear, because of contradictory results (Fig. 4a [i, ii], Supplementary Fig. S5a, Supplementary Table 3). For example, an analysis of the GEO dataset, GSE12093, showed that the probability of long-term survival of a patient with breast cancer was higher with higher levels of PROM1 expression, whereas an analysis of dataset GSE11121, gave the opposite result. In addition, we found that high PROM1 expression was associated with poor OS in patients with esophageal cancers and mixed Ewing sarcoma (Fig. 4a [viii and ix]), whereas Ewing sarcoma patients with high PROM1 expression showed good prognosis (Supplementary Fig. S5e), as analyzed using the R2 platform. Similarly, in gastric, liver, and ovarian cancer patients, high levels of PROM1 expression were associated with poor OS (Fig. 4a [x-xii]). In kidney cancers, we observed a positive correlation between PROM1 expression and high rates of survival (Supplementary Fig. S5g). In liver cancer, mixed results were observed. Data from the SurvExpress database showed that PROM1 expression correlated positively with OS, whereas the Kaplan-Meier plotter showed that low PROM1 expression was associated with increased OS rates (Fig. 4a [xii]; Supplementary Fig. S5f). Interestingly, the Oncomine database also showed low and high expression of PROM1 in liver cancer and sarcoma (Fig. 1a) depending on the analysis. The source of the contradiction in survival results between Kaplan-Meier plotter and SurvExpress may be the inadequate number of studies and reports. Therefore, survival data for patients with liver cancer may be more precise if the merged data from the SurvExpress database and the Kaplan-Meier plotter database were analyzed.

Correlation of PROM1 and PROM2 expression with prognosis of various cancers (PrognoScan, R2: Kaplan Meier Scanner, Kaplan–Meier plotter, SurvExpress, and OncoLnc). a Survival curves comparing patients with high (red) and low (blue) PROM1 expression were plotted using breast (i and ii), brain (iii), prostate (iv), skin (v), soft tissue (vi), and lung (vii) cancer data from PrognoScan; esophageal cancer (viii) and sarcoma (ix) data from R2: Kaplan Meier Scanner; gastric (x) and ovarian (xi) cancer data from Kaplan-Meier Plotter; and liver cancer (xii) data from SurvExpress. Cox p-value threshold < 0.05. b Survival curves comparing patients with high (red) and low (blue) PROM2 expression were plotted using breast (i and ii), lung (iii), eye (iv), and blood (v) cancer data from PrognoScan; colon cancer (vi), esophageal cancer (vii), lymphoma (viii), and sarcoma (ix) data from R2: Kaplan Meier Scanner; and ovarian cancer (x) data from Kaplan-Meier plotter. Cox p-value threshold < 0.05. c Co-expression of PROM1 and PROM2 with respect to the cancer patient prognosis. Multivariate survival curves comparing the prognosis of patients with high/high (red), high/low (orange), low/high (green), and low/low (blue) expression co-expression patterns of PROM1/PROM2 in KIRC (i), KIRP (ii), and SKCM (iii). Clinical outcome data were retrieved from TCGA using OncoLnc. p < 0.05 represents statistical significance. Abbreviations: KIRC - kidney renal clear cell carcinoma; KIRP - kidney renal papillary cell carcinoma; SKCM - skin cutaneous melanoma
Analysis of data from the PrognoScan database showed a significant correlation between PROM2 overexpression and poor relapse-free survival in patients with lung cancer (Fig. 4b [iii], Supplementary Table 4). In addition, low PROM2 expression was associated with poor prognosis in eye and blood cancers (Fig. 4b [iv and v], Supplementary Table 4). The relationship between PROM2 expression and survival in breast cancer patients is also questionable because of contradictory results (Fig. 4b [i and ii], Supplementary Fig. S6a and b, Supplementary Table 4). In this regard, the analysis of data from the PrognoScan database is in agreement with the results of Kaplan-Meier plotter data analysis. According to the latter, the association of PROM2 expression with survival of breast cancer patients is not straightforward and shows some inconsistencies (Supplementary Figs. S6c–e), depending on the receptor status of breast cancer cells. For instance, low PROM2 expression was associated with poorer relapse-free survival in HER2+ breast cancer patients compared to ER+ and PR+ breast cancer patients (Supplementary Figs S6f-h). We also found that HER2+ breast cancers showed an association between low survival rates and low PROM2 expression, whereas breast cancers with mutated p53 showed an association between low survival rates and high PROM2 expression (Supplementary Fig. S6h and i). Therefore, PROM2 expression is not a reliable prognostic marker of OS in patients with breast cancer. We also observed that both low and high levels of PROM2 expression were associated with poor survival in ovarian cancer patients (Fig. 4b [x]). In contrast, low PROM2 expression was associated with poor clinical outcomes in patients with colon and esophageal cancer, lymphoma, and sarcoma, using the R2 platform (Fig. 4b [vi-ix]).
The prognostic value of PROM1 and PROM2 expression levels for different cancer patients was also determined based on data from the PrognoScan database (Supplementary Tables 3 and 4). The poor prognosis seen in lung cancer patients with higher PROM2 expression (Fig. 4b [iii]) was in agreement with the analysis of Kaplan–Meier plotter data (Supplementary Fig. S6j). While high PROM1 expression was common in esophageal and liver cancers, based on data from Oncomine, R2, and SurvExpress (see Figs. 1a, 4a [viii and xii]), this gene was underexpressed in kidney cancer, according to the analysis of data from the Oncomine and OncoLnc databases (Fig. 1a and Supplementary Fig. S5g). In contrast, data from both the Kaplan-Meier plotter and PrognoScan databases confirmed high PROM2 expression in lung cancer (Figs. 1a, 4b [iii] and Supplementary Fig. S6j). In summary, a comprehensive analysis of survival data from a range of online resources, highlighted the oncogenic role of PROM1 in brain and ovarian cancers. However, the role PROM1 in liver and breast cancers was not clear. In contrast, the oncogenic role of PROM2 in lung cancer was obvious, unlike in breast cancer.
To investigate the relationship between prognosis and co-expression of PROM1 and PROM2, we retrieved clinical prognosis data from patients with various types of cancers, including breast, kidney, brain, ovarian, lung, and skin cancers, using OncoLnc, which accesses data from TCGA (Fig. 4c and Supplementary Fig. S7). The clinical prognosis data were then used to prepare a multivariate survival plot to assess the effect of high/high, high/low, low/high, and low/low expression of PROM1 and PROM2 in each cancer. The primary endpoint for this analysis was OS. The expression levels of both PROM1 and PROM2 were higher in ovarian cancer tissues than in their corresponding normal tissues, leading to poor prognosis (Fig. 4a [xi] and 4b [x]). Based on this expression pattern, we performed a multivariate survival analysis of PROM1/PROM2 co-expression in ovarian cancer. We did not observe a significant effect on survival probability among the high/high, high/low, low/high, and low/low groups (Supplementary Fig. S7). A similar result was also observed in several other cancers, including breast, brain, and lung cancers (Supplementary Fig. S7). The multivariate survival analysis revealed a significant association between low/high expression of PROM1/PROM2 and poorer prognosis of patients with kidney renal clear cell carcinoma (KIRC) compared to patients with high/high, low/low, or high/low expression patterns (Fig. 4c [i]). This result suggested that the partial-co-expression of PROM1 and PROM2 may regulate cancer prognosis. In the case of kidney renal papillary cell carcinoma (KIRP), the low/low expression pattern of PROM1/PROM2 was associated with poor prognosis compared to the high/high, high/low, and low/high patterns (see Fig. 4c [ii]). We next focused on skin cancer, where the high expression of both PROM1 and PROM2 was associated with poorer prognosis than the high/low, low/high, and low/low PROM1/PROM2 expression patterns (Fig. 4c [iii]). Thus, our multivariate survival analyses showed that the pattern of PROM1 and PROM2 co-expression modulated the clinical outcomes of patients with certain types of cancers, which may help our understanding of the underlying mechanism of cancer prognosis with respect to prominin expression. Furthermore, the interaction between these two prominins may be associated with the progression of various types of cancers.
Predicting PPIs of prominins
Studies have shown that PPIs are crucial events in cellular mechanisms that process downstream signaling and subsequently, affect cellular processes, including cell growth and division [63,64,65]. Accumulating evidence suggests that several factors, including DNA hypomethylation and hypoxia, affect PROM1 expression in cancer cells [66, 67]. However, unlike PROM1, the factors that affect PROM2 expression are still mostly unknown. As reported previously [68], PROM1 may play a role in cell differentiation, proliferation, and apoptosis. It also plays a major role as the principal regulator of disk morphogenesis and MAPK and AKT signaling pathways [69,70,71]. Several studies have shown that mutations in PROM1 are associated with photoreceptor degeneration in mice [69, 72, 73] and this photoreceptor degeneration is regulated through interactions between PROM1 and PCDH21 [69]. It has also been reported that PROM1 directly interacts with actin filaments (β-actin) in the protrusions of cells [69]. However, the factors associated with the expression of PROM2 remain largely unidentified. In silico studies suggest that PROM2 transcription may be regulated by several proteins, including E74A, HFH-2, Snail, and Spz1 [74, 75]. Moreover, PROM2 has been reported to be a testosterone-regulated gene in the rat ventral prostate [76], which suggests the possible hormone-mediated regulation of PROM2 expression. Another study has also reported that PROM2 is regulated by the androgen receptor (AR) [77]. Therefore, regulation of prominins by other interacting partners warrants further investigation. To identify the PPIs involving prominins, we used GeneMANIA, which compiles data on co-expression, co-localization, genetic interactions, pathways involved, physical interaction predictions, and shared protein domains. We selected the prominent functional protein partners of prominins from this analysis, as mentioned below, for further investigation. The predicted protein partners of PROM1 along with their respective genes were: prominin 2 (PROM2), gastrin-releasing peptide (GRP), zinc finger protein 157 (ZNF157), frizzled related protein (FRZB), claudin 10 (CLDN10), phosphatidylinositol-4-phosphate 5-kinase type 1 beta (PIP5K1B), cadherin-related family member 1 (CDHR1), C-X3-C motif chemokine ligand 1 (CX3CL1), and PDZ domain-containing protein (PDZD2, Fig. 5a [i]). The predicted protein partners of PROM2 along with their corresponding genes were: prominin 1 (PROM1), ETS homologous factor (EHF), family with sequence similarity 110 member C (FAM110C), sphingosine-1-phosphate phosphatase 2 (SGPP2), adhesion G protein-coupled receptor G1 (ADGRG1), PGAP2-interacting protein (CWH43), proline-rich gla protein 2 (PRRG2), tetraspanin 1 (TSPAN1), and E74-like ETS transcription factor 3 (ELF3, Fig. 5a [ii]). Thus, these predicted interacting partners of prominins may be involved in the regulation of prominin-mediated cancer progression and prognosis.

Identification of known and predicted structural proteins essential for PROM1 and PROM2 function (GeneMANIA) and frequency of mutations and copy number alterations (CNAs) in various types of cancer (cBioPortal web). a Interacting nodes are displayed in circles using GeneMANIA. Predicted functional partners of PROM1 (i) and PROM2 (ii) are shown after considering co-expression, co-localization, genetic interactions, pathways, physical interactions, and predicted shared protein domains. b In total, 331 mutation sites were identified and were located between amino acids 0 and 865 of PROM1. PROM1 mutations mainly occurred in uterine cancer and existed in a hotspot in the prominin domain (i). In total, 405 mutation sites were detected and were located between amino acids 0 and 834 of PROM2. PROM2 mutations mainly occurred in melanoma and also existed in a hotspot in the prominin domain (ii). c The alteration frequency of a ten-gene signature (PROM1, PROM2, GRP, ZNF157, FRZB, CLDN10, PIP5K1B, CDHR1, CX3CL1, and PDZD2) was determined using cBioPortal. Only data sets containing >100 samples per cancer type and an alteration frequency of >25% are shown. The alterations included mutations (green), amplifications (red), deep deletions (blue), or multiple alterations (grey) (i). The alteration frequency of a ten-gene signature (PROM2, PROM1, EHF, FAM110C, SGPP2, ADGRG1, CWH43, PRRG2, TSPAN1, and ELF3) was determined using cBioPortal. Only data sets containing >100 samples per cancer type and an alteration frequency of >20% are shown. The alteration frequency included mutations (green), fusions (brown), amplifications (red), deep deletions (blue), or multiple alterations (grey) (ii)
Cross-cancer analysis of prominin mutations and copy number alterations
We analyzed genetic alterations of PROM1 in different cancers using cBioPortal and compared the results with those of other genes of interest mentioned in the preceding subsection. The database was first queried for PROM1 gene mutations in 56,250 samples from 215 studies that covered the entire set of available cancers. The gene set or pathway was altered in 310 of the queried samples, with a somatic mutation frequency of 0.6%. As shown in Fig. 5b (i), 331 mutations, including 118 duplications were detected in patients with multiple samples. The mutation sites were located between amino acids 0 and 865. Of these mutations, 243 missense mutations and 88 truncating mutations were detected. We also observed that PROM1 mutations primarily occurred in uterine cancer and spanned the prominin domain, with a hotspot in N566Ifs*29/Kfs*2. The database was also queried for PROM2 using the same settings as for PROM1. In this case, the gene set or pathway was altered in 372 of the queried samples. Therefore, the somatic mutation frequency was 0.7%, which was slightly higher than the frequency for PROM1. In total, 405 mutations, including 114 duplications, were detected, which were located between amino acids 0 and 834. Thus, the mutations in PROM2 were slightly denser than those in PROM1. We found 344 missense mutations, 55 truncations, and 6 in-frame mutations among these mutations. Unlike PROM1, PROM2 mutations primarily occurred in skin cancer. They also spanned the prominin domain, with hotspots in Q508R and R582Q/W (Fig. 5b [ii]).
Next, we analyzed mutations and CNAs in a set of genes (corresponding to functional protein partners) centered around PROM1 (Fig. 5a [i]) in 215 different cancer studies. The query was customized to select 20 different cancer studies, representing 5796 samples that contained an alteration frequency > 25%, with at least 100 samples in each dataset (Fig. 5c [i], Supplementary Table 5). Fig. 5c (i) shows that the alteration frequency ranged from 25.27 to 52.34%. The alterations occurred mostly in neuroendocrine prostate cancer (NEPC). Similarly, an analysis of mutations and CNAs in the PROM2-centered functional partner genes (Fig. 5a [ii]) showed alteration frequencies ranging from 20.2 to 44.9% (Fig. 5c [ii], Supplementary Table 6). Similar to PROM1, the PROM2-centered gene set also showed that, with the exception of deep deletions, alterations occurred mainly in NEPC (Fig. 5c, Supplementary Tables 5 and 6).
Next, we applied the OncoPrint sub-tool of cBioPortal to investigate how the genomic alterations in NEPC are distributed over various genes corresponding to the functional protein partners of prominins. For PROM1, alterations in the gene set of PROM1, PROM2, GRP, ZNF157, FRZB, CLDN10, PIP5K1B, CDHR1, CX3CL1, and PDZD2 are shown in Fig. 6a. For PROM2, alterations in the gene set of PROM2, PROM1, EHF, FAM110C, SGPP2, ADGRG1, CWH43, PRRG2, TSPAN1, and ELF3 are shown in Fig. 6b. In the PROM1 and PROM2 gene sets, the alteration percentages varied in the range of 8–39% and 6–32%, respectively, for individual genes. In both cases, alterations largely occurred due to amplifications. Notably, ZNF157 was predominantly amplified in the PROM1-centered gene set (Fig. 6a) and this gene has been shown to be epigenetically regulated in medulloblastoma [78]. In the PROM2-centered gene set, however, ELF3, a member of the E-twenty-six family of transcription factors, was predominantly amplified (Fig. 6b). It plays a key role in β-catenin signaling in colorectal cancer and thus, has potential prognostic and therapeutic significance [79]. The genomic alterations in other cancer types were distributed over various genes corresponding to functional protein partners of prominins. These are summarized in Supplementary Tables 7 and 8.

PROM1 and PROM2 gene signatures are predominantly amplified and significantly co-expressed in neuroendocrine prostate cancer (NEPC) and bladder cancer. a We used the Oncoprint feature of cBioPortal to determine the copy number alteration frequency of each individual gene of the PROM1 (a) cluster (PROM1, PROM2, GRP, ZNF157, FRZB, CLDN10, PIP5K1B, CDHR1, CX3CL1, and PDZD2) and the PROM2 (b) cluster (PROM2, PROM1, EHF, FAM110C, SGPP2, ADGRG1, CWH43, PRRG2, TSPAN1, and ELF3) within selected cancer subtypes. The alterations included missense mutations (green), amplifications (red), deep deletions (blue), or no alterations (grey). c and d Mutual exclusivity panel analysis revealed the co-occurrence of alterations of PROM1 gene signatures and correlations between PROM1 gene copy number and mRNA expression. The correlation between PROM1 CNAs and mRNA levels was investigated using the cBioPortal for Cancer Genomics. Data are shown for the 107 NEPC samples in which CNAs were available. The search criteria were: “neuroendocrine prostate cancer (NEPC), mutation, and putative copy-number alterations adjusted by ploidy and purity with CLONET.” The x-axis is divided according to the copy number status of the tumor and the Y-axis represents PROM1 mRNA levels. e and f Mutual exclusivity panel analysis revealed the co-occurrence of alterations of PROM2 gene signatures and correlation between PROM2 gene copy number and mRNA expression. The correlation between PROM2 CNAs and mRNA levels was investigated using cBioPortal for Cancer Genomics. Data are shown from the 402 bladder cancer (TCGA PanCan Atlas) samples in which CNAs were available. The search criteria were “Bladder cancer (TCGA PanCan Atlus), mutation, putative copy-number alterations from GISTIC, and mRNA expression z-Scores (U133 microarray only).” The x-axis is divided according to the copy number status of the tumor and the Y-axis represents PROM2 mRNA level
To determine whether each member in the set of genes corresponding to the functional protein partners of PROM1 were significantly correlated, we used the co-occurrence analysis sub-tool of cBioPortal, which is based on Fisher’s exact test. This analysis confirmed the statistical significance of the co-occurrence of alterations in PROM1 and each gene in the associated set (Fig. 6c). A similar analysis was performed for PROM2, which also shows the statistically significant co-occurrence of alterations of each pair of PROM2 and its partner genes (Fig. 6e). An analysis of the corresponding levels of expression associated with the mutation status of prominins showed that deletions of both PROM1 and PROM2 correlated with increased mRNA expression (Figs. 6d and f).
The Oncomine database provides an important list of co-expressed genes, which may help to identify the pathways involved. Co-expression analysis showed that PROM1 was significantly co-expressed with ProSAPiP1, FAAH, and LTA in esophageal cancer (Fig. 7a). In liver cancer PROM1 was more strongly co-expressed with a larger number of genes, including CFTR, KRT7, ANXA3, TACSTD2, and FZD1 (Supplementary Fig. S8). ANXA3 encodes the annexin 3 protein, which can interact with acidic phospholipids in a calcium-dependent manner. Its dysregulation has a potential role in tumorigenesis [80]. Another study reported that the upregulation of TACSTD2 regulates breast cancer invasiveness and subsequently, correlates with poor prognosis [81]. In contrast, PROM2 was significantly co-expressed with LAD1, C1ORF106, PVRL4, and KCNK5 in lung cancer (Fig. 7b). Previous studies have reported that LAD1, C1ORF106, PVRL4, and KCNK5 are upregulated in cancer cells [82,83,84,85]. Furthermore, LAD1 has been reported as a filament-binding regulator and also regulates EGF signaling-mediated breast cancer tumorigenesis [82]. In ovarian cancer, PROM2 is slightly co-expressed with CARD14 (Supplementary Fig. S9). Thus, the findings presented here, in combination with those of previous studies, provide ample evidence that prominin expression may be involved in cancer progression and prognosis by associating with co-expressed genes.

Co-expression profile of PROM1 and PROM2 in esophageal and lung adenocarcinoma. a PROM1 is co-expressed with the indicated genes across a panel of nine esophageal adenocarcinomas, 19 Barrett’s esophagus samples, and 24 normal samples. b PROM2 is co-expressed with the indicated genes across a panel of 58 lung adenocarcinomas and 58 normal samples. Bar length represents the significance and negative logarithm of the enrichment p-value
Genes correlated with prominin genes and their functional GO and pathways
To identify genes that correlate with PROM1 and PROM2 expression in selected cancers, we performed a systematic analysis using the R2 platform, as outlined below. Two different sets were selected, each containing four tumor types for both PROM1 and PROM2, based on their high levels of expression in those cancers. From the advanced dataset selection panel of R2, we first selected the TCGARS platform as the expression data for a particular cancer. Based on the degree of expression, we individually considered the following four tumor types for PROM1: esophageal, liver, pancreatic, and prostate tumors. Next, we made a query to identify the list of genes that correlated with PROM1 in these tumor types, individually using Bonferroni correction and a p-value < 0.01 for each case. We observed that 2513, 1598, 3507, and 4206 genes positively correlated with PROM1 expression in esophageal (ESCA), pancreatic (PAAD), liver (LIHC), and prostate (PRAD) cancers, respectively. One hundred and ten genes (hereafter referred to as “PROM1-correlated gene cluster”) that positively correlated with PROM1 were common among all four cancer types considered in this analysis (Fig. 8a [i]). There were no common genes detected that negatively correlated with PROM1 in these four tumors (Supplementary Fig. S10). We performed a similar analysis for PROM2, assessing cervical (CESE), lung (LUAD), ovarian (OV), and pancreatic (PAAD) tumors. As discussed below, we analyzed a different set of cancer types for PROM2 because high expression levels of this gene were detected in these cancers. Compared to PROM1, fewer genes correlated positively with PROM2 in each cancer type and 94 genes (hereafter referred to as “PROM2-correlated gene cluster”) were common in all four cancers analyzed (Fig. 8b [i]). In addition, a short list of three genes was identified that negatively correlated with PROM2 in all cancers analyzed (Supplementary Fig. S10). Finally, as we compared the genes that correlated with PROM1 or PROM2 in each individual tumor type, we observed a substantial number of commonly correlated genes with the same correlation direction and we observed no commonly correlated genes with the reverse correlation direction. This suggested that PROM1 and PROM2 participate in many common processes, depending on the tissue.

Analysis of genes that positively correlated with PROM1 and PROM2 and their predicted pathway analysis using PANTHER. a Venn diagram of genes that positively correlated with PROM1, showing coincident genes in ESCA, PAAD, LIHC, and PRAD (i). Pathway analysis using PANTHER and subsequent classification based on their pathways (ii). b Venn diagram of genes that positively correlated with PROM2, showing coincident genes in CESE, LUAD, OV, and PAAD (i). Pathway analysis using PANTHER and subsequent classification based on their pathways (ii). c Summary of predictive value of PROM1 and PROM2 in different cancers, based on systematic genetic analyses
Next, we performed GO and pathway analyses for the PROM1/PROM2-correlated gene clusters using the web-based classification system, PANTHER. We made a separate query for the functional classification of each of the correlated genes and presented the four important ontologies associated with them. Each of the ontologies consisted of multiple sub-ontologies. We showed that binding, catalytic, and transporter activities were the main molecular functions associated with the PROM1-correlated gene cluster (Supplementary Fig. S11a [i]), whereas binding and catalytic activity were the major molecular functions associated with the PROM2-correlated gene cluster (Supplementary Fig. S11b [i]). In addition, the major biological process associated with both PROM1 and PROM2-correlated gene clusters was “Cellular process” (Supplementary Figs S11a [ii], S9b [ii]). “Cell part” and “Membrane” were the two key cellular components associated with both PROM1- and PROM2-correlated gene clusters (Supplementary Figs S11a [iii], S11b [iii]). The PROM1-correlated gene cluster affected 20 pathways, whereas the PROM2-correlated gene cluster affected 36 pathways and was involved in more diverse roles (see Figs. 8a [ii], 8b [ii]). The Alzheimer disease-amyloid secretase pathway was the most significant pathway associated with the PROM1-correlated gene, whereas the EGF receptor signaling pathway was the key player in the PROM2-correlated gene cluster. However, seven common pathway classes occurred in both clusters, including those involved in 5-hydroxytryptamine degradation, Alzheimer disease-presenilin, chemokine and cytokine signaling, integrin signaling, interleukin signaling, and PDGF signaling. In summary, our results suggested that, although PROM1 and PROM2 participate in the regulation of different pathways, they demonstrate similar correlations in certain signaling pathways, depending on the tissue.
Discussion
Prominins have been shown to contribute to the generation and development of various cancers [4, 6, 86]. PROM1 is reported to be a regulator of stem cell activation by modifying microvillar architecture and dynamics and ciliary dynamics [12, 18]. PROM2 can cause cellular protrusions and enhance phosphorylation of caveolin-1 to promote endocytosis [23]. In addition, several studies have shown that PROM1 and PROM2-targeted therapies are promising for the prevention of tumor development [14, 87]. However, the role of these prominin family members in the development of human cancers is still not understood. To determine the utility of PROM1 and PROM2 as markers of cancer prognosis, we performed a systematic data analysis of numerous gene expression datasets with clearly defined distinguishing parameters between cancer and normal tissues. PROM1 and PROM2 were found to be differentially expressed in cancer and normal tissues. The extent of their expression also differed depending on the tissue, according to Oncomine and GEPIA-based transcription analysis. Analysis of Oncomine data showed that PROM1 was upregulated in leukemia and esophageal, liver, and ovarian cancers, but downregulated in other cancer types, including kidney cancer. In contrast, PROM2 was upregulated in myeloma and breast, lung, and ovarian cancers and downregulated in colon, esophageal, gastric, kidney, and prostate cancers.
To improve our understanding of the value of prominins as prognostic markers, we next investigated the association between their expression levels and OS in various cancers. The prognostic value of PROM1 and PROM2 mRNA expression was assessed using PrognoScan, R2, Kaplan-Meier plotter, SurvExpress, and OncoLnc. In general, high PROM1 expression was associated with poor survival in mixed Ewing sarcoma and brain, esophageal, gastric, liver, and ovarian cancers. However, owing to contradictory results from different analyses, its role in liver cancer was not clear. Our observations are in agreement with the results of several previous studies. For example, PROM1 upregulation has been reported in ovarian cancer [88, 89] and targeting this gene retards ovarian cancer development in an in vivo model [90]. Furthermore, PROM1 has been identified as a stem cell marker in esophageal and breast cancers [5, 56]. Next, we observed that PROM2 upregulation was associated with poor OS in lung cancer; however, owing to contradictory results, its role in breast cancer was not clear and the receptor status of breast cancer cells also had some effect on patient survival. Any significant deviation (either high or low) in PROM2 expression can be considered as a valid prognostic marker of low survival rates in patients with ovarian cancer. Thus, based on mRNA expression and clinical data from Oncomine and TCGA, we conclude that PROM1 has as an oncogenic role in brain, esophageal, ovarian, and gastric cancers and melanoma and PROM2 shows oncogenic behavior in lung and ovarian cancers (Fig. 8c). To assess the impact of PROM1/PROM2 co-expression on various cancers, we performed several multivariate survival analyses on OncoLnc, using TCGA data. The co-expression (high/high), partial co-expression (low/high), and lack of co-expression (low/low) of PROM1/PROM2 were associated with skin, KIRC, and KIRP cancers, respectively. The results suggested that the co-expression of PROM1 and PROM2 may impact clinical outcomes in patients with certain types of cancers.
Tumorigenesis is a multi-step process that leads to the development of a tumor. It is primarily influenced by four major factors - somatically acquired genetic, epigenetic, transcriptomic, and proteomic alterations [91, 92]. Somatic loss-of-function or gain-of-function alterations occur in particular genomic regions involved in potential inhibitory or carcinogenic effects, respectively [93, 94]. Therefore, we used cBioPortal to determine human cancers with CNAs and mutations in prominin genes. Missense and truncating mutations predominantly occurred within protein-coding sequences. Mutations in PROM1 were mainly missense and truncating mutations, including p.R373C, p.Y452fsX12, p.G614fsX626, and p.Q576X in the prominin domain, which are associated with human inherited diseases [95,96,97,98]. A recent study reported the identification of a point mutation at p.S281R (serine [S] changed to arginine [R] due the mutation of thymine [T] to guanine [G]) in the prominin domain of the PROM1 protein in 8/555 lung cancer patients [99]. However, mutations in the PROM2 protein have not been well studied. In our systematic analysis, we also found several missense and truncating mutations within PROM1 and PROM2 protein-coding sequences, especially in the prominin domain, in various cancer types. These results are yet to be experimentally validated. Specifically, PROM1 mutations mainly occurred in uterine pan-cancer and consisted of several frame-shift insertion or deletion mutations in a hotspot at position p.N566Ifs*29/Kfs*2 in the prominin domain. PROM2 mutations were mainly found in skin cancers and predominantly consisted of point mutations of a hotspot at position p.Q508R and p.R582Q/W in the prominin domain. These mutations have a potential role in the regulation of cancer progression and prognosis, but this is yet to be confirmed. PPIs trigger the majority of biological processes, including signaling and disease development [100]. Thus, understanding the factors that modulate PPIs is important. Therefore, we first identified the top ten significantly correlated functional protein partners of PROM1 and PROM2 and constructed their interaction network using GeneMANIA. With a cross-cancer viewpoint for both, as Subsequent analysis using cBioPortal showed that genetic alterations of the ten genes in the PROM1 and PROM2 signatures, mainly occurred in NEPC, with alteration frequencies of 25.27–52.34% and 20.2–44.9%, respectively. Genomic alterations in NEPC were differentially distributed over the set of genes, with individual gene alterations occurring at frequencies of 8 to 39% for PROM1-related genes and 6–32% for PROM2-related genes.
A high degree of correlation between PROM1 and PROM2 gene expression indicates that they have either have comparable roles or are involved in the same biochemical processes and may be co-regulated [101]. Therefore, the R2 genomics platform was used to identify genes correlated with PROM1 and PROM2 in certain cancers in which these prominins are highly expressed. For PROM1, a large number of positively correlated genes were detected in esophageal, pancreatic, liver, and prostate cancers. Of those genes, 110 were common in all cancers. For PROM2, a relatively fewer number of correlated genes were detected in cervical, lung, ovarian, and pancreatic tumors, among which 94 genes were common in all cancers. Next, to determine the shared role of these correlated genes in the above cancers, we used the online tool, PANTHER, to perform GO and pathway analyses. Despite the existence of seven common pathways, PROM2-correlated genes demonstrated more varied characteristics than PROM1-correlated genes (20 vs. 36 pathways). This correlation analysis suggested that PROM1 and PROM2 perform different functions with respect to pathway regulation; however, they may have some similar functions in certain signaling pathways, in certain cancer types.
In summary, we investigated the expression, mutation, and copy number alteration patterns of PROM1 and PROM2 genes and assessed their prognostic significance through a systematic data analysis, using publicly available expression and clinical data. This analysis was able to predict the expression status of prominins for various cancer types. These data suggest that prominin expression may be translated into clinical practice and that their co-expression may impact clinical outcomes in patients with certain types of cancers. In addition, this study found that some functional protein partners of prominins show high frequencies of genomic alterations in certain cancer types. For example, amplifications were predominantly found in ZNF157 and ELF3 genes among the PROM1- and PROM2-centered gene sets, respectively, in NEPC. Therefore, investigation of the combined roles of PROM1 and ZNF157 and PROM2 and ELF3 are important topics for future research. Moreover, we propose that effects of PROM1 and PROM2 on cancer progression and prognosis, may be more prominent when associated with their respective co-expressed genes, such as ANXA3 and TACSTD2 for PROM1 and LAD1 and C1ORF106 for PROM2. Furthermore, in this study, we demonstrated that PROM1 and PROM2 perform different functions with respect to pathway regulation.
Concluding remarks
In this study, we focused on data mining approaches to investigate the expression, mutation, and CNA of prominins with respect to clinical outcomes in various cancers. Our results demonstrated that PROM1 and PROM2 expression differentially modulate the clinical outcomes of cancer patients. While the former can act as a therapeutic target for certain cancers, including esophageal, liver, and ovarian cancer, the latter can be considered as an oncogenic marker for ovarian and lung cancers. Meanwhile, both PROM1 and PROM2 are potential targets for skin and kidney cancers. PPI and co-expression analyses of prominins may also predict the probable underlying signaling mechanisms associated with prominins in certain cancers. In summary, these multiomics findings are expected to improve our understanding of the relationship between prominin expression and clinical prognosis. They also provide new insights into the molecular mechanisms involved in cancer and thus, will assist in transforming genomic knowledge into clinical practice. Hence, the specific roles, detailed molecular mechanisms, and clinical significance of prominins in cancer progression and prognosis deserve further investigation.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA: Cancer J Clin. 2017;67:7–30.
Zhang C-L, Huang T, Wu B-L, He W-X, Liu D. Stem cells in cancer therapy: opportunities and challenges. Oncotarget. 2017;8:75756–66.
Liou G-Y. CD133 as a regulator of cancer metastasis through the cancer stem cells. Int J Biochem Cell Biol. 2019;106:1–7.
Florek M, Haase M, Marzesco AM, Freund D, Ehninger G, Huttner WB, et al. Prominin-1/CD133, a neural and hematopoietic stem cell marker, is expressed in adult human differentiated cells and certain types of kidney cancer. Cell Tissue Res. 2005;319:15–26.
Tume L, Paco K, Ubidia-Incio R, Moya J. CD133 in breast cancer cells and in breast cancer stem cells as another target for immunotherapy. Gac Mex Oncol. 2016;15:22–30.
Fargeas CA, Florek M, Huttner WB, Corbeil D. Characterization of prominin-2, a new member of the prominin family of pentaspan membrane glycoproteins. J Biol Chem. 2003;278:8586–96.
Miraglia S, Godfrey W, Buck D. A Response to AC133 hematopoietic stem cell antigen: human homologue of mouse kidney prominin or distinct member of a novel protein family? Blood. 1998;91:4390–1.
Corbeil D, Fargeas CA, Huttner WB. Rat Prominin, like its mouse and human orthologues, is a pentaspan membrane glycoprotein. Biochem Biophys Res Commun. 2001;285:939–44.
Weigmann A, Corbeil D, Hellwig A, Huttner WB. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci USA. 1997;94:12425–30.
Horn PA, Tesch H, Staib P, Kube D, Diehl V, Voliotis D, et al. Expression of AC133, a novel hematopoietic precursor antigen, on acute myeloid leukemia cells. Blood. 1999;93:1435–7.
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8.
Thamm K, Šimaitė D, Karbanová J, Bermúdez V, Reichert D, Morgenstern A, et al. Prominin‐1 (CD133) modulates the architecture and dynamics of microvilli. Traffic. 2019;20:39–60.
Li Y, He Y, Qiu Z, Zhou B, Shi S, Zhang K, et al. CRTC2 and PROM1 expression in non-small cell lung cancer: analysis by Western blot and immunohistochemistry. Tumor Biol. 2014;35:11719–26.
Qiu Z-X, Zhao S, Mo X-M, Li W-M. Overexpression of PROM1 (CD133) confers poor prognosis in non-small cell lung cancer. Int J Clin Exp Pathol. 2015;8:6589–95.
Raso A, Mascelli S, Biassoni R, Nozza P, Kool M, Pistorio A, et al. High levels of PROM1 (CD133) transcript are a potential predictor of poor prognosis in medulloblastoma. Neuro-Oncol. 2011;13:500–8.
Gupta VK, Sharma NS, Kesh K, Dauer P, Nomura A, Giri B, et al. Metastasis and chemoresistance in CD133 expressing pancreatic cancer cells are dependent on their lipid raft integrity. Cancer Lett. 2018;439:101–12.
Huang J, Fu Y, Cai Z. CD133 correlation with chemoradiotherapy resistance in rectal cancer. Biomed Res. 2018;29:252–6.
Singer D, Thamm K, Zhuang H, Karbanová J, Gao Y, Walker JV, et al. Prominin‐1 controls stem cell activation by orchestrating ciliary dynamics. EMBO J. 2019;38:e99845.
Rappa G, Fodstad O, Lorico A. The stem cell‐associated antigen CD133 (Prominin‐1) is a molecular therapeutic target for metastatic melanoma. Stem Cells. 2008;26:3008–17.
Smith LM, Nesterova A, Ryan MC, Duniho S, Jonas M, Anderson M, et al. CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br J Cancer. 2008;99:100.
Ferrandina G, Petrillo M, Bonanno G, Scambia G. Targeting CD133 antigen in cancer. Expert Opin Ther Targets. 2009;13:823–37.
Hori Y. Prominin-1 (CD133) reveals new faces of pancreatic progenitor cells and cancer stem cells: current knowledge and therapeutic perspectives. In: Denis Corbeil, editor. Prominin-1 (CD133): new insights on stem & cancer stem cell biology. New York, NY, USA: Springer; 2013. pp. 185–96.
Singh RD, Schroeder AS, Scheffer L, Holicky EL, Wheatley CL, Marks DL, et al. Prominin-2 expression increases protrusions, decreases caveolae and inhibits Cdc42 dependent fluid phase endocytosis. Biochem Biophys Res Commun. 2013;434:466–72.
Zhuang X, Herbert JMJ, Lodhia P, Bradford J, Turner AM, Newby PM, et al. Identification of novel vascular targets in lung cancer. Br J Cancer. 2014;112:485.
Rohan S, Tu JJ, Kao J, Mukherjee P, Campagne F, Zhou XK, et al. Gene expression profiling separates chromophobe renal cell carcinoma from oncocytoma and identifies vesicular transport and cell junction proteins as differentially expressed genes. Clin Cancer Res. 2006;12:6937–45.
Winnepenninckx V, Lazar V, Michiels S, Dessen P, Stas M, Alonso SR, et al. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J Natl Cancer Inst. 2006;98:472–82.
Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6.
Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–80.
Tang Z, Li C, Kang B, Gao G, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102.
Lanczky A, Nagy A, Bottai G, Munkacsy G, Szabo A, Santarpia L, et al. miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat. 2016;160:439–46.
Mizuno H, Kitada K, Nakai K, Sarai A. PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC Med Genom. 2009;2:18.
Anaya J. OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput Sci. 2016;2:e67.
Aguirre-Gamboa R, Gomez-Rueda H, Martinez-Ledesma E, Martinez-Torteya A, Chacolla-Huaringa R, Rodriguez-Barrientos A, et al. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE. 2013;8:e74250.
Koster J, Molenaar JJ, Versteeg R. Abstract A2-45: R2: Accessible web-based genomics analysis and visualization platform for biomedical researchers. Cancer Res. 2015;75:A2-45. [Online Available] http://r2.amc.nl. Accessed 12 Jan 2019.
Oliveros JC. VENNY. An interactive tool for comparing lists with Venn diagrams. 2007; http://bioinfogp.cnb.csic.es/tools/venny/index.html. Accessed 15 Jan 2019.
Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–66.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao JJ, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:p11.
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–W220.
Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou M-F, et al. Stem cell–related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26:3015–24.
Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346.
Kim SM, Park Y-Y, Park ES, Cho JY, Izzo JG, Zhang D, et al. Prognostic biomarkers for esophageal adenocarcinoma identified by analysis of tumor transcriptome. PLoS One. 2010;5:e15074.
Cutcliffe C, Kersey D, Huang CC, Zeng Y, Walterhouse D, Perlman EJ. Clear cell sarcoma of the kidney: up-regulation of neural markers with activation of the sonic hedgehog and Akt pathways. Clin Cancer Res. 2005;11:7986–94.
Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–37.
Wurmbach E, Chen YB, Khitrov G, Zhang W, Roayaie S, Schwartz M, et al. Genome‐wide molecular profiles of HCV‐induced dysplasia and hepatocellular carcinoma. Hepatol. 2007;45:938–47.
Skotheim RI, Lind GE, Monni O, Nesland JM, Abeler VM, Fosså SD, et al. Differentiation of human embryonal carcinomas in vitro and in vivo reveals expression profiles relevant to normal development. Cancer Res. 2005;65:5588–98.
Hendrix ND, Wu R, Kuick R, Schwartz DR, Fearon ER, Cho KR. Fibroblast growth factor 9 has oncogenic activity and is a downstream target of Wnt signaling in ovarian endometrioid adenocarcinomas. Cancer Res. 2006;66:1354–62.
Talantov D, Mazumder A, Jack XY, Briggs T, Jiang Y, Backus J, et al. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 2005;11:7234–42.
Cho JY, Lim JY, Cheong JH, Park Y-Y, Yoon S-L, Kim SM, et al. Gene expression signature–based prognostic risk score in gastric cancer. Clin Cancer Res. 2011;17:1850–7.
Skrzypczak M, Goryca K, Rubel T, Paziewska A, Mikula M, Jarosz D, et al. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One. 2010;5:e13091.
Yusenko MV, Kuiper RP, Boethe T, Ljungberg B, van Kessel AG, Kovacs G. High-resolution DNA copy number and gene expression analyses distinguish chromophobe renal cell carcinomas and renal oncocytomas. BMC Cancer. 2009;9:152.
Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012;22:1197–211.
Zhan F, Barlogie B, Arzoumanian V, Huang Y, Williams DR, Hollmig K, et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood. 2007;109:1692–1700.
Yoshihara K, Tajima A, Komata D, Yamamoto T, Kodama S, Fujiwara H, et al. Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis. Cancer Sci. 2009;100:1421–8.
Grasso CS, Wu Y-M, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239.
Sui YP, Jian XP, Ma LI, Xu GZ, Liao HW, Liu YP, et al. Prognostic value of cancer stem cell marker CD133 expression in esophageal carcinoma: a meta-analysis. Mol Clin Oncol. 2016;4:77–82.
Roy L, Bobbs A, Sattler R, Kurkewich JL, Dausinas PB, Nallathamby P, et al. CD133 promotes adhesion to the ovarian cancer metastatic niche. Cancer Growth Metastas-. 2018;11:1179064418767882.
Fargeas CA. Prominin-2 and other relatives of CD133. In: Corbeil D, editors. Prominin-1 (CD133): new insights on stem & cancer stem cell biology. New York: Springer; 2013. pp. 25–40.
Zhuang X, Herbert JM, Lodhia P, Bradford J, Turner AM, Newby PM, et al. Identification of novel vascular targets in lung cancer. Br J Cancer. 2015;112:485–94.
Xie YR, Huang JH, Wu MH, Zhou YF. Expression of CD133 protein in osteosarcoma and its relationship with the clinicopathological features and prognosis. J Cancer Res Ther. 2018;14:892–5.
He AN, Qi WX, Huang YJ, Feng T, Chen J, Sun YJ, et al. CD133 expression predicts lung metastasis and poor prognosis in osteosarcoma patients: a clinical and experimental study. Exp Ther Med. 2012;4:435–41.
Zhang J, Guo XQ, Chang DY, Rosen DG, Mercado-Uribe I, Liu JS. CD133 expression associated with poor prognosis in ovarian cancer. Mod Pathol. 2012;25:456–64.
Pawson T, Nash P. Protein–protein interactions define specificity in signal transduction. Genes Dev. 2000;14:1027–47.
Batada NN, Shepp LA, Siegmund DO. Stochastic model of protein–protein interaction: Why signaling proteins need to be colocalized. Proc Natl Acad Sci USA. 2004;101:6445–9.
Brugge JS. Protein: protein interactions involved in intracellular signal transduction. Chem Biol. 1994;1:xii–xiii.
Sun B, Wan Z, Shen J, Ni L, Chen J, Cui M, et al. DNA hypomethylation of CD133 promoter is associated with recurrent glioma. Oncol Rep. 2016;36:1062–8.
Saigusa S, Tanaka K, Toiyama Y, Yokoe T, Okugawa Y, Koike Y, et al. Clinical significance of CD133 and hypoxia inducible factor-1α gene expression in rectal cancer after preoperative chemoradiotherapy. Clin Oncol. 2011;23:323–32.
Mak AB, Pehar M, Nixon AM, Williams RA, Uetrecht AC, Puglielli L, et al. Post-translational regulation of CD133 by ATase1/ATase2-mediated lysine acetylation. J Mol Biol. 2014;426:2175–82.
Yang Z, Chen Y, Lillo C, Chien J, Yu Z, Michaelides M, et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Invest. 2008;118:2908–16.
Jang J-W, Song Y, Kim S-H, Kim J-s, mo Kim K, Choi EK, et al. CD133 confers cancer stem-like cell properties by stabilizing EGFR-AKT signaling in hepatocellular carcinoma. Cancer lett. 2017;389:1–10.
Wang Y, Zhu Y, Qiu F, Zhang T, Chen Z, Zheng S, et al. Activation of Akt and MAPK pathways enhances the tumorigenicity of CD133 + primary colon cancer cells. Carcinogenesis. 2010;31:1376–80.
Permanyer J, Navarro R, Friedman J, Pomares E, Castro-Navarro J, Marfany G, et al. Autosomal recessive retinitis pigmentosa with early macular affectation caused by premature truncation in PROM1. Invest Ophthalmol Vis Sci. 2010;51:2656–63.
Zacchigna S, Oh H, Wilsch-Bräuninger M, Missol-Kolka E, Jászai J, Jansen S, et al. Loss of the cholesterol-binding protein prominin-1/CD133 causes disk dysmorphogenesis and photoreceptor degeneration. J Neurosci. 2009;29:2297–308.
Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89.
Hsu S-H, Shyu H-W, Hsieh-Li H-M, Li H. Spz1, a novel bHLH-Zip protein, is specifically expressed in testis. Mech Dev. 2001;100:177–87.
Zhang Q, Haleem R, Cai X, Wang Z. Identification and characterization of a novel testosterone-regulated prominin-like gene in the rat ventral prostate. Endocrinol. 2002;143:4788–96.
Cunha GR, Hayward SW, Wang YZ. Role of stroma in carcinogenesis of the prostate. Differentiation. 2002;70:473–85.
Anderton JA, Lindsey JC, Lusher ME, Gilbertson RJ, Bailey S, Ellison DW, et al. Global analysis of the medulloblastoma epigenome identifies disease-subgroup-specific inactivation of COL1A2. Neuro-Oncol. 2008;10:981–94.
Wang JL, Chen ZF, Chen HM, Wang MY, Kong X, Wang YC, et al. Elf3 drives β-catenin transactivation and associates with poor prognosis in colorectal cancer. Cell Death Dis. 2014;5:e1263–e1263.
Du RK, Liu BJ, Zhou L, Wang D, He XY, Xu XJ, et al. Downregulation of annexin A3 inhibits tumor metastasis and decreases drug resistance in breast cancer. Cell Death Dis. 2018;9:126.
Lin H, Huang JF, Qiu JR, Zhang HL, Tang XJ, Li H, et al. Significantly upregulated TACSTD2 and Cyclin D1 correlate with poor prognosis of invasive ductal breast cancer. Exp Mol Pathol. 2013;94:73–78.
Roth L, Srivastava S, Lindzen M, Sas-Chen A, Sheffer M, Lauriola M, et al. SILAC identifies LAD1 as a filamin-binding regulator of actin dynamics in response to EGF and a marker of aggressive breast tumors. Sci Signal. 2018;11:eaan0949.
Mohanan V, Nakata T, Desch AN, Levesque C, Boroughs A, Guzman G, et al. C1orf106 is a colitis risk gene that regulates stability of epithelial adherens junctions. Science. 2018;359:1161–6.
Pavlova NN, Pallasch C, Elia AE, Braun CJ, Westbrook TF, Hemann M, et al. A role for PVRL4-driven cell–cell interactions in tumorigenesis. Elife. 2013;2:e00358.
Dookeran KA, Zhang W, Stayner L, Argos M. Associations of two-pore domain potassium channels and triple negative breast cancer subtype in The Cancer Genome Atlas: systematic evaluation of gene expression and methylation. BMC Res Notes. 2017;10:475.
Feng HL, Liu YQ, Yang LJ, Bian XC, Yang ZL, Gu B, et al. Expression of CD133 correlates with differentiation of human colon cancer cells. Cancer Biol Ther. 2010;9:216–23.
Bao L, Zhang Y, Wang J, Wang H, Dong N, Su X, et al. Variations of chromosome 2 gene expressions among patients with lung cancer or non-cancer. Cell Biol Toxicol. 2016;32:419–35.
Bapat SA. Human ovarian cancer stem cells. Reproduction. 2010;140:33–41.
Kryczek I, Liu S, Roh M, Vatan L, Szeliga W, Wei S, et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int J Cancer. 2012;130:29–39.
Skubitz AP, Taras EP, Boylan KL, Waldron NN, Oh S, Panoskaltsis-Mortari A, et al. Targeting CD133 in an in vivo ovarian cancer model reduces ovarian cancer progression. Gynecol Oncol. 2013;130:579–87.
Kim P, Cheng FX, Zhao JF, Zhao ZM. ccmGDB: a database for cancer cell metabolism genes. Nucleic Acids Res. 2016;44:D959–D968.
Xie S, Shen C, Tan M, Li M, Song X, Wang C. Systematic analysis of gene expression alterations and clinical outcomes of adenylate cyclase-associated protein in cancer. Oncotarget. 2017;8:27216–39.
Cui XR, Jing X, Yi Q, Long CL, Tan B, Li X, et al. Systematic analysis of gene expression alterations and clinical outcomes of STAT3 in cancer. Oncotarget. 2018;9:3198–213.
Klonowska K, Czubak K, Wojciechowska M, Handschuh L, Zmienko A, Figlerowicz M, et al. Oncogenomic portals for the visualization and analysis of genome-wide cancer data. Oncotarget. 2016;7:176–92.
Liang J, She X, Chen J, Zhai Y, Liu Y, Zheng K, et al. Identification of novel PROM1 mutations responsible for autosomal recessive maculopathy with rod-cone dystrophy. Graefe’s Arch Clin Exp Ophthalmol. 2019;257:619–28.
Imani S, Cheng J, Shasaltaneh MD, Wei C, Yang L, Fu S, et al. Genetic identification and molecular modeling characterization reveal a novel PROM1 mutation in Stargardt4-like macular dystrophy. Oncotarget. 2017;9:122–41.
Michaelides M, Gaillard M-C, Escher P, Tiab L, Bedell M, Borruat F-X, et al. The PROM1 mutation p. R373C causes an autosomal dominant Bull’s eye maculopathy associated with rod, rod–cone, and macular dystrophy. Invest Ophthalmol Vis Sci. 2010;51:4771–80.
Arrigoni FI, Matarin M, Thompson PJ, Michaelides M, McClements ME, Redmond E, et al. Extended extraocular phenotype of PROM1 mutation in kindreds with known autosomal dominant macular dystrophy. Eur J Hum Genet. 2011;19:131–7.
He YQ, Li YL, Qiu ZX, Zhou B, Shi SQ, Zhang K, et al. Identification and validation of PROM1 and CRTC2 mutations in lung cancer patients. Mol Cancer. 2014;13:19.
Laraia L, McKenzie G, Spring DR, Venkitaraman AR, Huggins DJ. Overcoming chemical, biological, and computational challenges in the development of inhibitors targeting protein-protein interactions. Chem Biol. 2015;22:689–703.
Reynier F, Petit F, Paye M, Turrel-Davin F, Imbert PE, Hot A, et al. Importance of correlation between gene expression levels: application to the type i interferon signature in rheumatoid arthritis. PLoS ONE. 2011;6:e24828.
Acknowledgements
This study was supported by grants from the National Research Foundation (NRF) funded by the Korean government (2015R1A5A1009701). This research was also supported by the KU-Research Professor Program of Konkuk University, and in part by the Sejong University Research Faculty Program (2019–2021).
Author information
Authors and Affiliations
Contributions
conceptualization, SKS; formal analysis, investigation, and data curation, SKS and SMRI; formal analysis, KSK, MSR, and SGC; funding acquisition, SGC; writing—original draft preparation, SMRI; writing—review and editing, SKS.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saha, S.K., Islam, S.M.R., Kwak, KS. et al. PROM1 and PROM2 expression differentially modulates clinical prognosis of cancer: a multiomics analysis. Cancer Gene Ther 27, 147–167 (2020). https://doi.org/10.1038/s41417-019-0109-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-019-0109-7
This article is cited by
-
Single-cell analysis reveals the stromal dynamics and tumor-specific characteristics in the microenvironment of ovarian cancer
Communications Biology (2024)
-
Identification of cholesterol metabolism-related subtypes in nonfunctioning pituitary neuroendocrine tumors and analysis of immune infiltration
Lipids in Health and Disease (2023)
-
Regulation of tumorigenesis and ferroptosis in non-small cell lung cancer by a novel BBOX1-AS1/miR-326/PROM2 axis
Molecular and Cellular Biochemistry (2023)
-
LncRNA RP11-89 facilitates tumorigenesis and ferroptosis resistance through PROM2-activated iron export by sponging miR-129-5p in bladder cancer
Cell Death & Disease (2021)
-
Hybrid structure based high performance SPR sensor: a numerical approach of structure optimization for DNA hybridization
Optical and Quantum Electronics (2021)
