
Abstract
Background
Recently, molecular tumour boards (MTBs) have been integrated into the clinical routine. Since their benefit remains debated, we assessed MTB outcomes in the Comprehensive Cancer Center Ostbayern (CCCO) from 2019 to 2021.
Methods and results
In total, 251 patients were included. Targeted sequencing was performed with PCR MSI-evaluation and immunohistochemistry for PD-L1, Her2, and mismatch repair enzymes. 125 treatment recommendations were given (49.8%). High-recommendation rates were achieved for intrahepatic cholangiocarcinoma (20/30, 66.7%) and gastric adenocarcinoma (10/16, 62.5%) as opposed to colorectal cancer (9/36, 25.0%) and pancreatic cancer (3/18, 16.7%). MTB therapies were administered in 47 (18.7%) patients, while 53 (21.1%) received alternative treatment regimens. Thus 37.6% of recommended MTB therapies were implemented (47/125 recommendations). The clinical benefit rate (complete + partial + mixed response + stable disease) was 50.0% for MTB and 63.8% for alternative treatments. PFS2/1 ratios were 34.6% and 16.1%, respectively. Significantly improved PFS could be achieved for m1A-tier-evidence-based MTB therapies (median 6.30 months) compared to alternative treatments (median 2.83 months; P = 0.0278).
Conclusion
The CCCO MTB yielded a considerable recommendation rate, particularly in cholangiocarcinoma patients. The discrepancy between the low-recommendation rates in colorectal and pancreatic cancer suggests the necessity of a weighted prioritisation of entities. High-tier recommendations should be implemented predominantly.
Similar content being viewed by others

Background
In 2022, a total of 116 biomarker-dependent oncological therapeutic agents were listed by the Food and Drug Administration (FDA) [1], a result of remarkably dynamic development in recent years [2]. In parallel, next-generation sequencing techniques have entered the clinic, necessitating the implementation of multidisciplinary molecular tumour boards (MTB) to keep pace with these novel requirements.
While a few biomarkers have achieved tissue-agnostic approval by the FDA, such as tumour mutational burden (TMB) predictive for pembrolizumab [3] and neurotrophic tropomyosin-receptor kinase (NTRK) gene-fusions for entrectinib [4] and larotrectinib [5], most biomarker-drug combinations are tissue specific. Exemplarily, histologic context specificity has been demonstrated for alpelisib in phosphatidylinositol-4,5-biphosphate 3-kinase catalytic subunit alpha (PIK3CA)-mutated solid tumours [6], neratinib in HER2 and HER3 mutated [7], and vemurafenib in BRAFV600 altered cancers [8]. Furthermore, one should bear in mind that tissue-agnostic approvals remain controversial. Indeed, the seminal KEYNOTE-158 study, on which the FDA approved pembrolizumab in TMB-high tumours, excluded the common entity of colorectal cancer (CRC) by study design and did not recruit a single TMB-high cholangiocarcinoma [3]. Moreover, recent literature casts considerable doubt on the suitability of TMB as a predictive marker in CRC [9, 10].
These controversies exemplify the need for multidisciplinary case discussions in MTBs. Given that CRC is a frequent entity, this specific question has the potential to be addressed by future multicentric clinical trials. However, considering that a substantial proportion of cancers are rare cancers harbouring even more sporadic molecular alterations [11], structured evidence is hard to obtain. Therefore, new adaptive study designs [12] or multi-institutional collaborations through large consortia, such as the German DKTK MASTER programme [13], are mandatory. The latter programme impressed with the inclusion of 1310 patients, with 75.5% rare cancers, of whom 362 received molecularly informed therapies resulting in improved progression-free survival (PFS) compared to previous treatments (PFS2/PFS1 ratio >1.3 in 35.7% of evaluable patients) [13]. Similar clinical benefit in approximately one-third of patients has also been reported in a few additional larger-scale genomic medicine studies [12, 14]. At the same time, the objective response rates are generally low (11% in the MOSCATO-01 trial [14] and 0.9% in the ProfiLER trial [15]). Thus, despite a growing body of evidence, MTBs are often neither anchored in clinical routine (except for large cancer centres) nor represented in clinical guidelines. Major obstacles to generating more evidence include the following: late consideration of molecularly targeted therapies only at an advanced stage of disease, which can lead to non-implementation due to the patient’s death or deterioration of the general condition, and the difficulty in accessing recommended therapeutics [16]. In addition, health insurance companies play a critical role since they are responsible for the reimbursement of molecular diagnostics and the cost coverage of recommended off-label therapies.
Given the novelty of MTBs and the controversy surrounding their benefit in patients’ care, we established a longitudinal observational study at the Comprehensive Cancer Center Ostbayern (CCCO) with a large catchment area including rural regions. We present results from 2 years of expertise after MTB implementation to optimise the procedural, inclusion and outcome parameters.
Methods
Study design and patient population
The prospective observational study of the MTB of the CCCO was conducted following the Declaration of Helsinki and approved by the Ethics Committee of the University of Regensburg (protocol code 20-1682-101). This prospective registry study has been continuously recruiting since the end of 2019. All patients discussed in the MTB from 2019 to 2021 with available written consent were included in the current evaluation. For patient recruitment, the following clinical inclusion criteria have been defined: advanced disease, guidelines or standard therapy exhausted (patients should ideally just have begun the last-line therapy), sufficient life expectancy (6 months estimate), suitable tissue available, and open-mindedness to experimental treatments. In addition, rare oncological diseases lacking standard therapies were also accepted for MTB evaluation.
Additional procedural inclusion criteria for patient allocation to the MTB were: availability of the patient’s written consent; organ-specific tumour board decision stating the exhaustion of established therapeutic standards and recommending an MTB discussion; letter of referral and provision of comprehensive medical documentation (a most recent oncological record).
We held the MTB discussion weekly. Representatives from the affiliated departments and clinics were designated and actively involved in the case annotation and discussion based on a rotational schedule. The attendance of one trained specialist in oncology, one specialist in pathology, one molecular biologist with expertise in bioinformatics, and one rotational representative of the affiliated clinics and departments was the minimal requirement for the MTB meeting. In detail, the affiliated departments comprised the Department of Internal Medicine III (Hematology and Oncology), the Department of Surgery, the Department of Internal Medicine I (Gastroenterology), the Department of Neurology, the Department of Neurosurgery, the Department of Dermatology, the Department of Radiotherapy, the Department of Pediatric Hematology, Oncology and Stem Cell Transplantation, the Department of Urology, the Department of Gynecology, and the Department of Pneumology. The Institute of Human Genetics partly received selected case documents (with potential germline implications) before the discussions for additional screening without actively participating. Genetic counselling was suggested based on their assessment in addition to the criteria proposed by Mandelker et al. [17] for somatic sequencing and the input of two oncologists who had obtained a special qualification in genetic counselling. The stated aim of the MTB was to offer additional information to the treating physician to allow for genomically informed decisions. Thus, molecular and immunohistochemical analyses of the tumour were performed. The DKTK/NCT Master evidence grading was employed to stratify MTB recommendations (Supplementary Table S1) in conjunction with clinical parameters. An exception to the outlined workflow was the entity of lung cancer. Here, the thoracic cancers tumour board already discusses a broad array of molecular alterations as part of the clinical routine. Therefore, patients were referred to the MTB only in case of the detection of rare alterations without evidence-based therapeutic approaches. Molecular analysis was performed on the most relevant tissue reflecting the current state of disease progression (e.g., latest relapse or metastases). Exceptions to the defined inclusion criteria were occasionally made so that, in several cases, MTB diagnostics and discussion were conducted before the commencement of the last therapy line according to the guidelines. A decision from an organ-specific tumour board was necessary for this deviation from the protocol. Reasons for early inclusion were sparsity of tissue obtained from a biopsy (to avoid repeated cutting of the sample), an oncological assessment of a limited remaining life span, and an expected little benefit of the current or last-line therapy.
Choice of analysis methods
The employed analysis methods comprised panel-based next-generation sequencing (NGS) for DNA and RNA, polymerase chain reaction (PCR)-based fragment-sizing for microsatellite instability (MSI), immunohistochemistry (IHC) for PD-L1, Her2, and mismatch repair (MMR) proteins MLH1, MSH2, MSH6 and PMS2. In addition, in the case of the detection of copy number variations, MET or HER2 fluorescence in situ hybridisation (FISH) was performed as a confirmatory method. For most included patients, all methods mentioned above were used. The absolute numbers of employed methods are detailed in 'Results'.
Documentation, follow-up and clinical reassessment
All MTB decisions were documented, including the most relevant literature citations on which the recommendations were primarily based. Structured follow-up data were collected from the attending physicians at prespecified time points 1, 3, 6, 12, 18 and 24 months after the MTB. The data collection period started on December 1, 2019 and was cut off on April 30, 2022. Structured follow-up comprised the following items: survival status, type of MTB-recommended therapy and timepoint of initiation, on/off-label therapy, reasons for non-implementation, other oncological therapies since the MTB discussion, response to MTB or alternative therapy and adverse effects. Clinical data were pseudonymized and documented primarily in Excel® version 16.0 (Microsoft Corporation, Redmond, WA, USA). For the final data assessment, clinical data were visualised in conjunction with genomic data (see below) in a local instance of cBioportal [18,19,20,21] provided by the MIRACUM consortium (https://github.com/buschlab/MIRACUM-cbioportal; last accessed August 4, 2022).
Responses were assigned to the categories progressive disease (PD), stable disease (SD), partial response (PR), mixed response (MR), and complete response (CR) based on radiological reports and clinical documentation for both MTB-recommended therapies and alternatively administered therapies (i.e., non-genomically informed therapies after an MTB discussion). MR was defined as a disease stabilisation or size reduction of a tumour lesion with concomitant progression at another location. This response class was included as it might indicate biological drug efficacy. Note that RECIST criteria were not employed for the retrospective evaluation of radiological responses. We determined the progression-free survival for the preceding therapeutic therapy line and the MTB-recommended or alternative therapies (time from MTB to progression or death). The PFS2/1-value indicates the ratio of the PFS of MTB or alternative treatment (PFS2) over the PFS of the preceding therapy (PFS1). PFS2/1 > 1.3 has been defined by previous precision oncology clinical trials to indicate intra-patient benefit and hence was adopted in the present study [13, 14]. In addition to the clinical follow-up, the patients’ overall survival (OS) relative to the date of the MTB was obtained from the cancer registry of the Regensburg Tumour Centre. Here clinical reports, death certificates issued by the local public health departments, and the registration offices of the respective residential districts are collected. Patients with no information on survival, patients lost to follow-up, or alive at the last medical visit were censored at the latest record date.
Genomic, FISH, MSI, immunohistochemical and statistical analyses
The exact procedure is described in the Supplementary Methods section.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Performed molecular analyses and patient population
This prospective registry study includes a total of 251 patients managed by the MTB of the CCCO over the indicated 2-year period.
For 262 case discussions of 251 patients, 147 biopsies (56.1%), 92 primary resection specimens (35.1%), 9 cytologies (3.4%), 6 pooled samples (2.3%) were analysed. In eight cases (3.1%), a denotation as either biopsy or resection is not applicable. In total, 149 analyses were performed on metastatic tumour samples (56.6%), 96 on primary tumour tissue (36.6%), 5 on pooled metastatic and primary tumour tissue (1.9%), and for 12 cases a distinction was not possible (4.6%, e.g., carcinoma of unknown primary [CUP]). The median age of analysed tumour specimens respective to the time of enrolment was 89 days (minimum age 0 days, maximum age 2931 days).
Overall, 237 out of 257 cases (92.2%) were evaluable for DNA and RNA alterations. Initially, in 22 cases, panel sequencing had failed and not reached sufficient coverage for analysis, 13 of which had been repeated, resulting in 2 additional evaluable cases.
In absolute numbers, the TSO500® panel was used in 214 (81.7%), the QIAseq Tumour Mutational Burden Panel® in 17 (6.5%), the Human Actionable Solid Tumour Panel Kit® in 12 (4.6%), a combination of the latter two panels in 3 (1.2%), the nNGMv2 custom panel in 5 (1.9%) and other DNA panels in 4 (1.5%) out of a total of 262 case discussions, while DNA panel sequencing was unavailable in 7 cases (2.7%).
For RNA panel sequencing, the TSO500® panel was used in 213 (81.3%), the FusionPlexLung-panel® in 37 (14.1%), a combination of the latter two panels in 1 (0.4%), and other RNA panels in 3 (1.2%) out of 262 case discussions.
Her2 IHC was available in 171 (65.5%) and confirmatory FISH analyses in 24 (9.2%) case discussions. MMR enzymes were assessed 173 times (66.0%) and MSI by PCR-based fragment-sizing 143 times (54.6%). PD-L1 scores (TPS, ICS and CPS) were determined in 197 cases (75.2%).
Across the entire cohort, 125 patients (49.8%) received treatment recommendations categorised into tiers according to the evidence of the given medication/ genetic alteration. Across all tiers, a total of 219 recommendations were given. 62 patients obtained more than one treatment recommendation (mean 1.74; maximum 4). Among all 251 patients, 47 (18.7%) received a genomically justifiable therapy. In contrast, 53 patients (21.1%) received alternative treatments, which comprised any systemic therapy administered without considering MTB recommendations (including in-label and off-label treatments) (Table 1). The remaining patients did not receive additional anticancer therapy. The alternative treatment cohort was a reference cohort to compare the patients receiving an MTB-recommended therapy. Gender and mean age of the patients and ECOG performance status at the MTB time revealed no differences between the two groups. The frequency of MTB recommendations was 35.9% for patients receiving alternative therapies. In the whole cohort, the highest evidence level of m1A was most frequent (24.8%), followed by m1C, (18.2%), m1B (16.5%), m2A (14.9%), and m2C (10.7%). m3 (5.8%), m2B (5.0%) and m4 (4.1%) were comparatively rare. In the subcohort analysis, m1A-evidence recommendations were more abundant in the MTB therapy cohort than in the alternative therapy cohort (36.2% versus 5.3%; P = 0.0133). Thus, recommendations with this high evidence level are implemented comparatively more often. In contrast, no significant difference could be demonstrated for recommendation levels below m1A.
The median line of preceding therapies was identical between groups (n = 2).
The overall distribution of cancer types was similar between the MTB and alternative therapy group, without significant differences among entities. However, on descriptive evaluation, CRC, pancreatic ductal adenocarcinoma, and breast cancer appeared more frequently in the alternative therapy group.
Qualitative and quantitative analysis of MTB recommendations
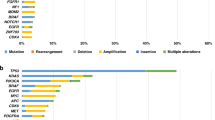
We assessed the frequency of a given biomarker that had been used for molecular evidence-level- assigned treatment recommendations (Fig. 1). The highest percentage of tier 1 recommendations was based on PD-L1 immunohistochemistry (12.8% of patients with tier 1/2.4% lower tier recommendation), i.e., immune-oncological therapies with PD-L1/PD-1 inhibitors. This was followed by recommendations based on the biomarkers PIK3CA (8.8% of patients with tier 1/4% lower tier recommendations), molecularly confirmed ERBB2 alterations (8.8%/0%), and FGFR2 (7.2%/0%). Note that one patient with a novel FGFR2-NDC80 fusion, who received a therapy recommendation for FGFR inhibitors, has been described previously as a case study [22]. MSI (also comprising two patients with only dMMR, in whom MSI PCR could not be conducted due to the unavailability of normal control tissue) was the next most frequently underlying biomarker for treatment recommendations (6.4%/0%). MET (4.8%/1.6%), EGFR (4.8%/0%), BRAF (4.8%/0%), KRAS (4%/0%), IDH1 (3.2%/0.8%), and KIT (3.2%/0%) were used for recommendations in ≥4 patients each. Importantly, also Her2 IHC was relevant for recommendations (without confirmed concomitant molecular alterations) (4%/1.6%), and a high TMB (defined as >10 mutations/MB) was occasionally used as a basis for MTB recommendations (4%/0.8%). Finally, many more recommendations in individual patients were based on biomarkers that occurred with low frequency (Supplementary Fig. S1). A detailed list of individual recommendations (in the order of patients in Fig. 1) can be found in Supplementary Table S3. The complete information on clinically reported underlying mutations is depicted in Supplementary Table S4, on reported CNVs in Supplementary Table S5, and on detected genetic rearrangements in Supplementary Table S6.

Oncoprint representation of alterations bearing relevance for MTB recommendations in ≥3.2% of patients (see Supplementary Fig. S1 for all alterations). Blue frames highlight individual alterations on which MTB recommendations were based for first-priority recommendations and orange frames for lower-priority recommendations. The percentage of first- and lower-priority recommendations is given on the right. The order of patients can be translated to PatID using Supplementary Table S3. The row “others” subsumes individual cases with BCL2 loss by IHC, androgen receptor expression, and FGF CNV. MSI microsatellite stability status, TMB tumour mutational burden in mutations per megabase, CUP carcinoma of unknown primary, Ca cancer, CCA cholangiocarcinoma, GIST gastrointestinal stromal tumour, HCC hepatocellular carcinoma, HNSCC head and neck squamous cell cancer, CRC colorectal cancer, NEC neuroendocrine carcinoma, RCC renal cell cancer.
Corresponding to these biomarkers, the most commonly recommended drug classes were: immune checkpoint inhibitors (38/219 [17.4%]), HER2 inhibitors (31/219 [14.2%]), combination therapies (21/219 [9.6%]), PARP inhibitors (20/219 [9.1%]), FGFR inhibitors (17/219 [7.8%]), PI3K inhibitors (13/219 [5.9%]), multi tyrosine kinase inhibitors (13/219 [5.9%]), MEK inhibitors (8/219 [3.7%]), MET inhibitors (8/219 [3.7%]), ALK inhibitors (6/219 [2.7%]), EGFR inhibitors (6/219 [2.7%]), IDH1 inhibitors (5/219 [2.3%]), mTOR inhibitors (5/219 [2.3%]), CDK4/6 inhibitors (4/219 [1.8%]), androgen blockade (3/219 [1.4%]) and others (21/219 [9.6%]).
We compared the recommendation rates between entities to address whether certain cancer types benefit more from MTB discussions (Fig. 2a). Focusing on the ten most abundant entities in our cohort, we can demonstrate that colorectal cancer (CRC) and pancreatic ductal adenocarcinoma (PDAC) yielded only low-recommendation rates of 25% and 16%, respectively. Intermediate-recommendation frequencies were observed for sarcoma (50%), carcinoma of unknown primary (CUP) (46%), breast cancer (40%), and gallbladder carcinoma (37%). In contrast, comparatively high-recommendation rates were achieved for intrahepatic cholangiocarcinoma (iCCA; [60%]), perihilar cholangiocarcinoma (pCCA, [70%]), extrahepatic cholangiocarcinoma (eCCA; [55%]), and gastric cancer (62%). Concerning the less frequently included entities, the high-recommendation frequency for NSCLC should be interpreted cautiously, since only selected cases were discussed in our MTB. It is noteworthy that a high frequency of therapy recommendations (62%) was yielded for rarely included entities summarised as “others” with n ≤ 3. Concordantly, the entities with higher recommendations frequencies were also more likely to obtain multiple recommendations per case discussion (Fig. 2b), and the distribution of evidence levels (Fig. 2c) favoured higher evidence levels in the entities with high- and intermediate-recommendation frequencies (except for CUP, where m1 evidence is not possible per definition).

a Recommendation frequency by cancer type. The percentage of patients receiving treatment recommendations by the MTB is indicated. b Percentage of patients receiving either single or multiple MTB recommendations. c Evidence level of primary treatment recommendation by entity as NCT/DKTK grade. CRC colorectal cancer, iCCA intrahepatic cholangiocarcinoma, PDAC pancreatic ductal adenocarcinoma, Ca cancer, CUP carcinoma of unknown primary, pCCA perihilar cholangiocarcinoma, NSCLC non-small cell lung cancer, HNSCC head and neck squamous cell cancer, HGSC high-grade serous carcinoma, HCC hepatocellular carcinoma, NEC neuroendocrine carcinoma.
Clinical outcome of MTB-guided versus alternative therapies
In total, 47 patients (18.73%) received therapies recommended by the MTB, while 53 (21.12%) received alternative treatments following the MTB discussion. An assessment by a board-certified oncologist demonstrated that MTB-guided treatments were off-label in 96.6%. (according to the European Medicines Agency [EMA] approval status) at the time of the MTB decision. In contrast, only 39.6% of the alternatively administered therapies were off-label at the latter timepoint (see Table 2 for details on MTB therapies and Supplementary Table S7 for alternative therapies). The analysis of drug classes yields the following distribution for MTB-based treatments: immune checkpoint inhibitors were predominantly employed (14/47 [29.8%]), followed by combination therapies (6/47 [12.8%]), Her2 inhibitors (4/47 [8.5%]), FGFR inhibitors and inhibitors of multiple tyrosine kinases (3/47 [6.4%] each), ALK inhibitors, androgen blockade, CDK4/6 inhibitors, EGFR inhibitors and IDH1 inhibitors (2/47 [4.3%] each) as well as a single case of administration of cytostatic chemotherapy, a JAK inhibitor, a MEK inhibitor, a nucleoside analogue, a PARP inhibitor, a PI3K and a RET inhibitor (1/47 [2.1%] each). Alternatively, administered therapies were very heterogenous, including immune checkpoint inhibitors and classical chemotherapy (see Supplementary Table S7 for details).
A total of 38 MTB-based treatments and 47 alternative therapies were clinically evaluated for response (Fig. 3a). The clinical benefit rate (defined here as complete response [CR] + partial response [PR]+ mixed response [MR]+ stable disease [SD]) was 50.0% for MTB and 63.8% for alternative treatments. In detail, 7 PR (29.8%), 1 MR (2.6%), 11 SD (29.0%), and 19 PD (50.0%) were observed for MTB-based therapies, while 1 CR (2.1%), 14 PR (29.8%), 3 MR (6.4%), 12 SD (25.5%) and 17 PD (36.2%) were described for alternative therapies. Next, the distribution of responses concerning the biomarkers underlying the recommendations was analysed (Fig. 3b). PR were obtained for treatments based on BRAF, EGFR, Her2, JAK2, ALK and MSI. The MR was established on an activating KIT mutation, and SD were yielded by therapies founded on Her2, MSI, KIT, AR, IDH1, BRCA2, PD-L1 and POLE. Treatments established on CDK4, PIK3CA, POLD1, and RET alterations led to PD. Interestingly, among the patients experiencing clinical benefit from MTB therapies, 63.2% had evidence levels of m1A (12/19), 15.8% had m1C evidence (3/19), and 5.3% had m1B, m2A, m2C and m3 evidence (1/19 each). Altogether, 84.2% of patients (16/19) had m1 evidence from the same entity. This distribution differs among patients who did not experience clinical benefit from MTB-recommended therapies, where only 52.6% of patients (10/19) had m1 evidence, while 47.4% (9/19) had evidence level of m2 or below (Fig. 3b).

a Stacked bar chart of best-observed responses to MTB-recommended therapies as opposed to alternative therapies. b Alluvial plot displaying the best-observed responses in relation to alteration type and NCT/DKTK evidence level.
To further analyse the efficacy of MTB-recommended therapies, we looked at treatment durations (Fig. 4a). In total, 11 patients received MTB-recommended treatments for more than 200 days. Among these, 10 had documented responses (3 PR and 6 SD), indicating that a certain proportion of MTB patients can benefit significantly from the recommended therapies. The recommendations for these patients were based on MSI and FGFR2-fusions (two cases each), IDH1, POLE, ERBB2, BRAF mutations, and PD-L1 expression. Also, ten treatments were still ongoing at the data cut-off.

a Swimmer plot of 43 patients receiving an MTB-recommended therapy with available follow-up data. Blue bars indicate the duration of MTB-recommended therapy. b Comparison of the frequency of PFS2/1 ratios ≥1.3 and <1.3 between MTB-recommended and alternative therapies in evaluable patients. Progression-free survival (c) and overall survival (d) over the entire cohort comparing patients receiving MTB-recommended therapies, alternative therapies after the MTB, and patients without further therapies. Progression-free survival (e) and overall survival (f) achieved by tier m1A-based MTB therapies as opposed to all alternative non-MTB-recommended therapies. The P value was calculated by the log-rank test.
Due to the heterogeneity of the cohort, PFS2/1 ratios were studied for intra-patient comparisons. 26 patients were evaluable for PFS2/1 in the MTB cohort and 31 in the alternative treatment cohort. PFS2/1 ratios over 1.3 were 34.6% and 16.1%, respectively (Fig. 4b; Supplementary Tables S8 and S9 for details).
Altogether, a median PFS of 4.20 months was obtained for MTB therapies as opposed to 2.83 months for alternative treatments, which did not reach statistical significance (95% CI of ratio 0.8762 to 2.508; P = 0.1509; Fig. 4c). An encouraging difference in OS for the MTB therapy (median 11.90 months) compared to no treatment (median 6.90 months; 95% CI of ratio 1.1114 to 2.671; P = 0.0368; Fig. 4d) was detectable. A significant difference could be observed in the OS of alternative therapy (median 13.07 months) versus no treatment (median 6.90 months; 95% CI of ratio 11.200 to 2.987; P = 0.0020; Fig. 4d), while MTB therapy and alternative therapy were not significantly different in terms of OS (P = .4219; Fig. 4d). Given that clinical benefit was rather achieved in the case of higher evidence levels, we also compared m1A-based MTB therapies to alternative therapies. For this selected group, PFS was significantly longer for m1A-MTB therapies (median 6.30 months) than alternative therapies (median 2.83 months; 95% CI of ratio 1.027 to 4.814; P = 0.0278; Fig. 4e). Despite the difference in PFS, OS was not different between these two groups (Fig. 4f).
OS and PFS were also compared in the CCA cohort, since a comparatively high number of MTB therapies had been administered in this entity, and a particular emphasis was placed on CCA by the ESMO guideline NGS recommendation [23] (Supplementary Fig. S2). While these comparisons were not statistically significant (median OS MTBCCA 12.57 months; median OS alternativeCCA 11.33 months, median OS no therapyCCA 8.43 months), a trend was detected for PFS in favour of the MTB therapy in CCA (median PFS MTBCCA 9.90 months, median PFS alternativeCCA 2.70 months; 95% CI of ratio 1.06 to 12.67; P = 0.1170). In this analysis, a CCA harbouring an IDH1 mutation with long-term survival of >22 months is particularly interesting (PatID 26). However, due to the low sample size and the heterogeneity of cohorts, propensity score matching between cohorts was not possible for all survival analyses. Thus, the results should be interpreted cautiously, since confounders cannot be controlled.
Discussion
In the presented MTB observational study, 49.8% of the 251 included patients received treatment recommendations based on IHC and NGS results. This number per se indicates that the methods employed are sufficient to detect relevant alterations for the intention of off-label treatment in a cohort of patients with advanced cancers. In 18.7% of patients, the treatment recommendation was implemented. Different factors influence this figure: due to the observational cut-off, several patients remained under last-line therapy and did not yet continue with the MTB-recommended therapy, treating physicians opted against the treatment administration, health insurers denied therapy cost coverage, or the patients deceased soon after the MTB. When comparing treatment recommendation and implementation frequencies with other genomically informed trials, similar results were obtained: the MTB in Baltimore reached 24% and 16% [24], in Vienna 54% and 23% [25], in Cleveland 49% and 11% [26], and 54% and 28% in Freiburg [16]. Of note, the DKTK/MASTER trial yielded much higher recommendation rates of 88%, while the implementation frequency was 31.8% [13]. A lower threshold could partly explain these high-recommendation rates. In the latter study, 45.3% of recommendations were based only on preclinical data (m3) and theoretical reasoning (m4), contrasting to 9.9% in our study. Moreover, in the DKTK/MASTER trial, more complex diagnostic methods were employed, such as whole genome/ exome and RNA sequencing, which could account for a higher detection rate of alterations. Nonetheless, the authors purport that our study’s targeted DNA and RNA sequencing methods were well suited to uncover off-label treatment options for about half of the recruited patients while being comparatively affordable methods. Of note, the addition of IHC for Her2, MMR proteins and PD-L1 accounted for 17.60% of primary recommendations, and we thus consider them an integral part of our MTB diagnostics. Moreover, MSI PCR yielded another 4.8% of primary recommendations, and in conjunction with MMR proteins, MSI was detected in 6.4% of patients. The authors advise against relying exclusively on panel-based MSI analysis. Not only did the NGS-based MSI analysis commonly fail (unevaluable in 78 out of 214 TSO panels [36.5%]), but also two cases of panel MSI were false negative, and one was false positive compared to PCR MSI. Beyond MSI/MMR, the addition of PD-L1 remains disputable. While 12.8% of primary recommendations were based on PD-L1 scoring (TPS, ICS and CPS), only one of five patients (PatID 40; a thymoma with a CPS of 91) reached SD on single agent immune checkpoint blockade, while the other four were progressive. While in some centres, PD-L1, MMR proteins, and Her2 immunohistochemistry are considered routine diagnostic, the authors believe these analyses an essential part of MTBs given the dynamic development of clinical studies and the fact that the resulting therapies are often off-label and should therefore be closely monitored in a study setting.
In addition to these insights on the chosen methods, we also noted different recommendation frequencies depending on the cancer type. This finding could help develop a weighted prioritisation system based on the entity in the future to optimise precision oncology workflows. This is highlighted by the relatively low-recommendation frequencies for CRC, breast cancer and PDAC, whose incidences are high. Other relatively frequent tumours like biliary tract and gastric cancer have high-recommendation rates and are thus more suited for comprehensive molecular analysis. Our real-world data align with observations made in the DKTK cohort [13] and MOSCATO-01 trial [14], emphasising the utility of molecularly informed therapies in selected, mostly rare, tumour entities. The 2020 ESMO guidelines also favour NGS in CCA, which is clearly mirrored by our cohort, while the use for gastric cancer is discouraged [23]. Regarding gastric cancer, our cohort yielded high-priority target recommendations for targeted therapy, such as MSI, Her2 expression, and PD-L1, while also uncovering rare but promising alterations such as ROS1 fusion and MET-amplifications. Although some of these alterations are already part of the standard therapy for gastric cancer, we consider a holistic reassessment of these parameters throughout the disease as reasonable and would assign a high priority to this entity. NGS for CRC and PDAC should only be implemented for research purposes according to the ESMO guidelines [23], a conclusion supported by this study. Furthermore, the ESMO guidelines recommend the routine use of NGS for lung cancer, CUP and prostate cancer. Our cohort offers limited insight into these entities due to low case numbers. However, we found it fruitful to refer selected cases that showed non-standard genetic aberrations from our thoracic tumour board to the MTB. The MTB offers an additional discussion platform for potentially actionable alterations in lung cancer patients and, due to its nature, can dedicate more time to the individual patient. This allows for comprehensive therapy recommendations such as concomitant HER2/MET-amplifications as a potential primary resistance mechanism and MET and BRAF fusions that cannot be as easily assessed in the thoracic tumour board. In addition, our data show that recommendations with high evidence levels were implemented more often and had better chances of attaining clinical responses. Thus, lower-priority evidence recommendations should be regarded with caution, because success rates are low and often clinically challenging to realise. Clinical benefit could more often be obtained from high evidence levels in the same entity (m1) and not from evidence derived from other entities or preclinical studies (m2 or below). On top of that, PFS was significantly longer for tier m1A-based MTB therapies than for alternative non-MTB therapies.
Regarding outcome measures, our study reports a clinical benefit rate (CBR; CR + PR + MR + SD) of 50.0% for MTB-recommended and 63.8% for alternatively administered treatments. In contrast, the PFS2/1 ratios were 34.6% and 16.1% for MTB and alternative treatments, thus favouring the MTB cohort. This discrepancy between CBR and PFS ratios might partly be explained by the fact that MTB-recommended therapies were off-label in 96.6%, while alternative therapies were off-label only in 39.6%. This distribution implies that in several cases, available in-label treatments were still available, so the MTB was not always initiated after the last-line treatment had begun. This contradicts the defined inclusion criteria and reflects that strict criteria cannot always be sufficiently met in clinical reality. Possible reasons include fast progression and considerable processing times for molecular diagnostics from enrolment until the MTB discussion (median 28 days).
The current observational study achieved an objective response rate (ORR) of 2.8% over the entire cohort (7 PR among 251 patients analysed) or 14.9% considering only those patients who were administered an MTB therapy (7/47). Again, this figure could potentially improve after a longer observation time since several patients were not yet evaluable for response at the data cut-off. While we acknowledge that an ORR of 14.9% is low, one should consider that this has been achieved in a heavily pretreated cohort. Moreover, disease control is sometimes just as beneficial for these incurably ill patients. This assumption is highlighted by a patient with CCA (PatID 26) who achieved SD under ivosidenib therapy for 22 months.
In the meantime, EMA has approved several MTB-recommended therapies in the respective genomic contexts, such as pembrolizumab for microsatellite instable [27] or pemigatinib for FGFR2 fusion-positive CCA [28]. With the benefit of hindsight, this justifies several MTB recommendations. Moreover, these cases clearly indicate that MTBs can facilitate allocating promising new unapproved therapeutic options to patients.
In our study, we observed significantly improved OS for MTB therapies compared to no treatment, which was also the case for alternative therapies, while PFS slightly favoured MTB therapies. We consider this additional encouraging evidence to use the MTB as a valuable clinical instrument for treatment allocation beyond the last-line therapy. At the same time, these results should be interpreted with caution due to the heterogeneity of cohorts. Finally, our study demonstrates the feasibility of establishing an MTB with a diffuse rural catchment area [29], providing its benefits also to spatially dispersed patients in Eastern Bavaria.
Data availability
The datasets generated during and/or analysed during the study are not publicly available due to restrictions of the research ethics protocol but are available (in anonymized form) from the corresponding author upon reasonable request.
References
Division of Translational and Precision Medicine (DTPM). Table of Pharmacogenomic Biomarkers in Drug Labeling. 2022. https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling.
Keeling P, Clark J, Finucane S. Challenges in the clinical implementation of precision medicine companion diagnostics. Expert Rev Mol Diagn. 2020;20:593–9.
Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353–65.
Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21:271–82.
Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl J Med. 2018;378:731–9.
Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, et al. Phosphatidylinositol 3-kinase a–selective inhibition with alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J Clin Oncol. 2018;36:1291–9.
Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature. 2018;554:189–94.
Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay J-Y, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl J Med. 2015;373:726–36.
Friedman CF, Hainsworth JD, Kurzrock R, Spigel DR, Burris HA III, Sweeney CJ, et al. Atezolizumab treatment of Tumors with high tumor mutational burden from mypathway, a multicenter, open-label, phase IIa multiple basket study. Cancer Disco. 2022;12:654–69.
Rousseau B, Foote MB, Maron SB, Diplas BH, Lu S, Argilés G, et al. The spectrum of benefit from checkpoint blockade in hypermutated tumors. N Engl J Med. 2021;384:1168–70.
Duijts SFA, van der Zwan JM. Rare cancers and cancer of unknown primary: here’s what you should know. Eur J Cancer Care. 2021;30:e13508.
van der Velden DL, Hoes LR, van der Wijngaart H, van Berge Henegouwen JM, van Werkhoven E, Roepman P, et al. The Drug Rediscovery protocol facilitates the expanded use of existing anticancer drugs. Nature. 2019;574:127–31.
Horak P, Heining C, Kreutzfeldt S, Hutter B, Mock A, Hüllein J, et al. Comprehensive genomic and transcriptomic analysis for guiding therapeutic decisions in patients with rare cancers. Cancer Discov. 2021;11:2780–95.
Massard C, Michiels S, Ferté C, Le Deley M-C, Lacroix L, Hollebecque A, et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov. 2017;7:586–95.
Trédan O, Wang Q, Pissaloux D, Cassier P, De La Fouchardière A, Fayette J, et al. Molecular screening program to select molecular-based recommended therapies for metastatic cancer patients: analysis from the ProfiLER trial. Ann Oncol. 2019;30:757–65.
Hoefflin R, Lazarou A, Hess ME, Reiser M, Wehrle J, Metzger P, et al. Transitioning the molecular tumor board from proof of concept to clinical routine: a German single-center analysis. Cancers. 2021;13:1–18.
Mandelker D, Donoghue M, Talukdar S, Bandlamudi C, Srinivasan P, Vivek M, et al. Germline-focussed analysis of tumour-only sequencing: recommendations from the ESMO Precision Medicine Working Group. Ann Oncol J Eur Soc Med Oncol. 2019;30:1221–31.
Unberath P, Knell C, Prokosch H-U, Christoph J. Developing new analysis functions for a translational research platform: extending the cBioPortal for cancer genomics. Stud Health Technol Inf. 2019;258:46–50.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
Reimer N, Unberath P, Busch H, Börries M, Metzger P, Ustjanzew A, et al. Challenges and experiences extending the cBioPortal for cancer genomics to a molecular tumor board platform. Stud Health Technol Inf. 2021;287:139–43.
Scheiter A, Keil F, Lüke F, Grosse J, Verloh N, Opitz S, et al. Identification and in-depth analysis of the novel FGFR2-NDC80 fusion in a cholangiocarcinoma patient: implication for therapy. Curr Oncol. 2021;28:1161–9.
Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2020;31:1491–505.
Dalton WB, Forde PM, Kang H, Connolly RM, Stearns V, Gocke CD, et al. Personalized medicine in the oncology clinic: implementation and outcomes of the Johns Hopkins Molecular Tumor Board. JCO Precis Oncol. 2017;2017. https://doi.org/10.1200/PO.16.00046.
Kieler M, Unseld M, Bianconi D, Waneck F, Mader R, Wrba F, et al. Interim analysis of a real-world precision medicine platform for molecular profiling of metastatic or advanced cancers: MONDTI. ESMO Open. 2019;4:e000538–e000538.
Sohal DPS, Rini BI, Khorana AA, Dreicer R, Abraham J, Procop GW, et al. Prospective clinical study of precision oncology in solid tumors. J Natl Cancer Inst. 2015;108. https://doi.org/10.1093/jnci/djv332.
Merck. European Commission approved Merck’s Keytruda (pembrolizumab) for patients with microsatellite instability-high (MSI-H) or deficient mismatch repair (dMMR) tumors in five different types of cancer. News Release. https://bit.ly/3waOTJz.
Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21:671–84.
Lüke F, Haller F, Utpatel K, Krebs M, Meidenbauer N, Scheiter A, et al. Identification of disparities in personalized cancer care—a joint approach of the German WERA consortium. Cancers. 2022;14. https://doi.org/10.3390/cancers14205040.
Gerthofer V, Scheiter A, Lüke F, Keil F, Utpatel K, Pöhmerer L-M-G, et al. STRN-ALK Fusion in a case of malignant peritoneal mesothelioma: mixed response to crizotinib, mode of resistance, and brigatinib sequential therapy. JCO Precis Oncol. 2021:1507–13. https://doi.org/10.1200/PO.21.00184.
Acknowledgements
The authors would like to thank Mr. Armin Pauer (Tumour Center—Institute for Quality Management and Health Services Research, University of Regensburg) for his support in obtaining patient survival and treatment data. Moreover, we would like to acknowledge our student assistant Janina Herold’s help with the MTB procedural process. The great support of all involved colleagues from the Immunohistochemistry and Molecular Pathology Department of the Institute of Pathology Regensburg should also be mentioned. We especially thank the numerous contributing physicians for vigorously recruiting patients amenable to tumour molecular profiling and MTB discussions. At last, we are thankful to all patients who consented to participate in this observational study.
Funding
The authors received no specific funding for this work. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualisation: KU, ME, Alexander Scheiter and FH; methodology: KU, FH, ME, Alexander Scheiter, WD, FK and FL; software: FH, PU, WD, Alexander Scheiter, MK and Andrea Scheiter; validation: KU, Alexander Scheiter and FH; formal analysis: FH, Alexander Scheiter and Andrea Scheiter; investigation: Alexander Scheiter, FH, FK, FL, DH, SE, MK, NK, AK, SS, LC, HT, AW, MG, EB, PH, MP, KE, FW, FS, AT, SH, LR, MS, CS, KD, MH, StS, MK-S, DC, TP, WD, ME and KU; resources: ME, TP and MK-S; data curation: Alexander Scheiter, FH, LR and FK; writing—original draft preparation: Alexander Scheiter and KU; writing—review and editing, all authors; visualisation: Alexander Scheiter and FH; supervision, KU, DC and ME; project administration: ME and TP; funding acquisition: ME and TP.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The prospective observational study of the molecular tumour board MTB of the Comprehensive Cancer Center Ostbayern (CCCO) was conducted following the Declaration of Helsinki and approved by the Ethics Committee of the University of Regensburg (protocol code 20-1682-101). All included patients gave their written consent to participate in this observational study.
Consent to publish
Written consent for publication was obtained from all patients included in the MTB observational study.
Competing interests
The authors declare the following competing interests: Alexander Scheiter: Travel/expenses: Roche. StS: Honoraria: AstraZeneca, lovis, GE, Gilead, GSK, MSD, Novartis, Pfizer, Roche, and Lilly. MJS: Advisory board: Bayer, Bristol Myers Squibb, Ipsen, Merck & Co, Pfizer. Honoraria: Bayer, Bristol Myers Squibb, GlaxoSmithKline, Ipsen, Medac, Merck, MSD, Pfizer. Travel/accommodation/expenses: Apogepha, Janssen, Ipsen, Pfizer. Institutional grants/contracts: Ipsen, Janssen, AstraZeneca, QED Therapeutics Inc. SH: Honoraria: BMS, Pierre Fabre, MSD, Novartis. KU: Advisory Board: BMS. Lecture fees: Roche.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scheiter, A., Hierl, F., Lüke, F. et al. Critical evaluation of molecular tumour board outcomes following 2 years of clinical practice in a Comprehensive Cancer Centre. Br J Cancer 128, 1134–1147 (2023). https://doi.org/10.1038/s41416-022-02120-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-022-02120-x
This article is cited by
-
The role of molecular tumor boards in neuro-oncology: a nationwide survey
BMC Cancer (2024)
-
Personalisierte Medizin in der Onkologie
Die Pathologie (2024)
-
Molekulare Tumorboards
Forum (2024)
-
Sondersituation der Daten in der Onkologie
Die Onkologie (2024)
-
Präzisionsmedizin in der Onkologie
Die Innere Medizin (2024)
