
Abstract
Survival for glioma patients has shown minimal improvement over the past 20 years. The ability to detect and monitor gliomas relies primarily upon imaging technologies that lack sensitivity and specificity, especially during the post-surgical treatment phase. Treatment-response monitoring with an effective liquid-biopsy paradigm may also provide the most facile clinical scenario for liquid-biopsy integration into brain-tumour care. Conceptually, liquid biopsy is advantageous when compared with both tissue sampling (less invasive) and imaging (more sensitive and specific), but is hampered by technical and biological problems. These problems predominantly relate to low concentrations of tumour-derived DNA in the bloodstream of glioma patients. In this review, we highlight methods by which the neuro-oncological scientific and clinical communities have attempted to circumvent this limitation. The use of novel biological, technological and computational approaches will be explored. The utility of alternate bio-fluids, tumour-guided sequencing, epigenomic and fragmentomic methods may eventually be leveraged to provide the biological and technological means to unlock a wide range of clinical applications for liquid biopsy in glioma.
Similar content being viewed by others
Introduction
Gliomas are rare and deadly brain pathologies that possess an age-corrected incidence range of 4.67–5.73 per 100,000 persons [1] with glioblastoma being the most common malignant primary brain tumour [2]. The current treatment paradigm involves maximal surgical resection (where possible) followed by radiotherapy and chemotherapy (temozolomide). Complete resection of contrast-enhancing tumour as demonstrated on magnetic resonance imaging (MRI) improves average survival, which, with the addition of chemotherapy and radiotherapy, is just over 1 year [3]. The location of this cancer within the brain results in certain patients being unsuitable for ‘complete’ resection. For these cases, biopsy is performed to confirm the diagnosis if further treatment (chemotherapy and radiotherapy) is indicated, but this carries significant risk of neurological morbidity and mortality. Despite treatment, only 30% of patients survive their first year after diagnosis and only 13% survive for 5 years as patients often present late and with extensive disease.
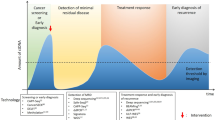
Liquid biopsy may have utility across all phases of brain-tumour investigation and management. Although screening for rare pathologies is difficult, a non-invasive, low-cost and reliable cancer- diagnostic assay could potentially benefit cancer patients and the public healthcare system. If glioma were to be included, then this may provide some benefit for certain glioma patients. Diagnosis of glioma, especially in patients for whom debulking surgery is not indicated, would benefit from minimally invasive diagnostic options such as liquid biopsy. Moreover, the ability to characterise the heterogeneous genomic landscape may be of further benefit for precision therapeutics. The expectation for disease recurrence following first-line treatment for glioma mandates the use of regular clinical follow-up associated with interval contrast-enhanced MRI. This paradigm has limitations with several radiological scenarios, such as pseudoprogression, an abnormal contrast enhancement associated with improved prognosis that affects up to one-third of patients, highlighting the need for improved detection and monitoring protocols. The frequency with which patients are currently monitored is dictated by pragmatism related to logistics and cost [4]. MR imaging mandates specialist equipment, trained individuals, comes at high cost per patient and possesses uncertainties with regard to the frequent use of gadolinium contrast. Existing radiological and clinical strategies lack sensitivity and specificity, leading to uncertainty as to when to stop therapies that lack efficacy, and when to persevere. Monitoring of tumour burden and treatment response with an effective liquid-biopsy approach may help to address these challenges [5, 6].
Multiple analytes can be detected in the bio-fluids of patients with cancer and used as liquid biopsy for a range of clinical applications: cell-free DNA (cfDNA), cell-free RNA, mitochondrial DNA, extracellular vesicles, tumour-educated platelets, proteins and metabolites among others [7,8,9]. cfDNA analysis, in particular, has emerged over the past years as potential game changer for detection, monitoring and treatment guidance in oncology [10, 11]. Despite the importance of investments from academic institutions and commercial entities, clinical validation has delayed widespread implementation [12]. Whilst initial data indicating that cfDNA may be challenging to detect using blood samples in glioma [13], several biological (e.g. impact of the blood–brain barrier) and technical reasons for this apparent difficultly exist. We aim to summarise recent progress in the application of liquid biopsy for brain-tumour characterisation and to identify future research directions.
The concentration in plasma cfDNA can be increased for gliomas
Due to the technical limitations of the time, initial studies that recovered cfDNA focused on global DNA quantification in control and diverse malignant plasma samples [14]. The overall load of cfDNA molecules in plasma (or other bio-fluids) from patients with brain tumours can be recovered quickly and at minimal cost (via spectrofluorometer methods or PCR quantification) and has recently regained interest [15]. Multiple studies have explored the difference in concentration of cfDNA between various types of gliomas and healthy controls, alone or in combination with other methods [16,17,18]. In a prospective study, glioma patients had higher plasma cfDNA concentration than age-matched healthy controls prior to initial surgery (mean 13.4 vs. 6.7 ng/mL, respectively) and this correlated with tumour burden on pre-irradiation MRI [16]. Another study identified in patients with progressive disease, a significant increase in cfDNA concentration from pretreatment to time of progression (9.7 vs. 13.1 ng/mL, p = 0.037), while no difference was observed for non-progressive patients [18]. Despite global DNA-modification evaluation, using cfDNA concentration is likely to be hampered by a lack of specificity (as cfDNA can be released by various physiological mechanisms), which may limit clinical applicability [19].
Detecting mutant cfDNA is challenging for gliomas
cfDNA is commonly detected in bio-fluids by analysing tumour-derived genomic signals, such as single- nucleotide variants (SNVs). SNVs due to their binary and potentially actionable nature, were logically the first molecular targets for cfDNA liquid-biopsy assays in the brain context. Early pan-cancer studies that evaluated cfDNA tumour fraction from various technologies have revealed that gliomas are the most challenging malignancy for liquid-biopsy applications [13, 20]. Using BEAming, a PCR-based technology, Bettegowda et al. detected mutant cfDNA in <10% of the plasma from 27 patients with gliomas [13]. Both the detection rate and concentration in mutant cfDNA were lower than those of the 14 other cancer types included in this study. Table 1 summarises the results reported from previous studies using SNVs to detect cfDNA in gliomas. Claims of higher rates of detection, and higher tumour fractions, have been made by entities using proprietary-sequencing assays [20,21,22]. The high frequency of alterations resulting from clonal haematopoiesis alongside biological or technical noise may, however, have confounded these results [23]. Locus-based assays (digital PCR, targeted sequencing or CRISPR-based) exhibit some potential to detect well-defined mutations at a low tumour fraction in the circulation [24,25,26]. In a recent study, TERT promoter mutations were evaluated in plasma using a custom digital- droplet PCR assay. An overall sensitivity of 62.5% (95% CI, 52–73%) and a specificity of 90% (95% CI, 80–96%) when compared with matched tumour-tissue-based detection was identified [27]. Nevertheless, these studies highlight that more sensitive methods, or alternative approaches, for plasma cfDNA detection, are required in gliomas.
Sampling the cerebrospinal fluid as a liquid biopsy
The blood–brain barrier protects the central nervous system and has been proposed as the main reason for low cfDNA in glioma patients. Thus, a potential strategy for improving cfDNA yield may be through collecting samples ‘closer’ to the tumour. The most direct method by which to achieve this may be via cerebrospinal fluid (CSF) sampling [28,29,30]. Compared with blood plasma, CSF in glioma has shown higher relative tumour fraction although this is due, in part, to the low levels of normal DNA present within this immune-privileged anatomic location [31,32,33]. Studies examining brain-tumour patients (using whole-exome sequencing or PCR-based methods) have identified higher cfDNA tumour fraction in CSF when compared with plasma, and a higher detection rate [28, 29, 34]. The analysis of tumour-derived cfDNA in CSF from a cohort of diffuse gliomas indicated that they could be subtyped through mutational analysis with IDH1 and IDH2, ATRX, TP53, TERT, and H3F3A being suggested as having classification and prognostication potential [35]. The representation of intra-tumoural heterogeneity is biased in CSF with shared clones being overrepresented, and private clones unrepresented, mimicking plasma observations in other cancer types [34]. Nevertheless, the high tumour fractions observed in CSF permit a higher detection range with multiple technologies ranging from PCR [29], to targeted sequencing [30, 35], low-coverage whole-genome sequencing (WGS) [36] and other genome-wide approaches [28, 33, 34]. For example, by using deep sequencing of cfDNA on CSF from 57 brain-tumour patients, at least one tumour-specific mutation was detected in over 82.5% of CSF ctDNA samples (47/57) [37]. In 8 selected cases with mutation detected in CSF, mutations were detected in 3/8 (37.5%) matched plasma samples [37]. This relatively high tumour fraction in CSF has allowed preliminary clinical application and molecular characterisation of gliomas [35, 37, 38]. Unfortunately use of CSF has significant clinical limitations as CSF is obtained via lumbar puncture, an invasive and morbid procedure relatively contraindicated in brain-tumour patients [39, 40]. CSF use in monitoring for recurrence is likely to be limited as a consequence. Moreover, despite the use of advanced sequencing and bioinformatic techniques, the tumour-derived cfDNA-detection rate in CSF remains relatively low, ranging from 39% to 98% (the highest detection rate being reported in brain metastasis from other primary sites) [33, 34]. Potential factors affecting this include the position of the tumour within the brain, whether the tumour is in direct contact with the CSF or not [29], and by the high level of intra-tumour heterogeneity in gliomas [41]. The acceptance and clinical integration of liquid biopsy for the management of gliomas will always be compared with the current imaging paradigm. Therefore, truly minimally invasive sampling, like blood plasma or urine, alongside better sensitivity and specificity, will be mandatory. Several emerging concepts and technologies have the potential to unlock such applications for brain cancer.
Guiding plasma analysis using tumour-tissue DNA mutations
A practical approach to improve tumour-signal detection in cfDNA is to track multiple mutations simultaneously. Standardised panels are conceptually limited by the fraction of the genome covered, the depth of sequencing and the number of starting cfDNA molecules in the sample. By using tumour-informed (or personalised) sequencing, mutations and alterations initially identified in the tumour can be sought. This approach boosts coverage across defined loci and thus increases the chances of low tumour-fraction variants being detected at modest cost. A larger number of variants, compared with standardised panels, can also be followed, therefore increasing the sensitivity of sequencing without compromising specificity. Such an approach has been validated for multiple cancer types and clinical scenarios using PCR systems [42], targeted sequencing [43], capture sequencing or genome-wide sequencing [44,45,46]. Using tumour-guided sequencing and an algorithm called INVAR, a recent study reports that mutations were detected in 7/8 CSF, 10/12 plasma and 10/16 urine glioma samples [47]. Although tumour-guided sequencing is not suitable for diagnostic and screening purposes [44, 48], its potential to detect relapse and pseudoprogression in glioma remains unexplored. Tumour-guided sequencing could be further combined with mutational enrichment methods (via size selection or thermodynamic-based strategies), which even if they have not been tested in the glioma context, could have a strong potential for improving the monitoring of relapse in these patients [49, 50].
Leveraging the epigenome for the analysis of glioma-derived cfDNA
Mutation analysis utilises few, highly specific, mutated regions within the genome of cancer patients for signal detection and analysis. This strategy relies heavily upon the presence of loci that are present in few DNA fragments and so, inevitably, suffers from a reduction in sensitivity, especially for cancers where relatively few copies of DNA exist within a liquid-biopsy sample (e.g. brain tumours). Sequencing the cfDNA epigenome, for example, methylated (mC) DNA, is advantageous because signatures exist across multiple, less specific, regions of difference [51, 52]. By deriving a positive signal from several characteristic differentially methylated regions, the specificity required to identify a sample as having features suggestive of a positive result is putatively generated. Importantly, the presence of multiple different DNA fragments (with no specific fragment being essential) improves sensitivity. This is also of particular relevance when dealing with scenarios whereby the number of DNA molecules is limiting (e.g. brain tumours or screening/early detection/minimal residual disease detection). Recently interest has also been directed towards alternate stable epigenomic marks such as hydroxymethylation (hmC) [53, 54] and histone modifications [55] as potential circulating epigenomic biomarkers. Moreover, the combination of multiple modified DNA and histone sites across the genome may prove additive for further boosting-test sensitivity and specificity. Currently, however, separate techniques are required for these analyses, and so, DNA quantity and total number of molecules of interest become limiting.
Rather than purely detecting the presence of cancer DNA, epigenomic marks associated with the cell of origin could be also leveraged to improve detection and classification of pathologies [32, 56]. A recent study examining CpG sites identified up to 2% of brain-derived cfDNA in the circulation of healthy individuals [57]. CpG-site analysis can also be applied to other bio-fluids (e.g. CSF) [58]. Successful strategies might therefore need to exploit cancer- and organ-specific epigenomic changes, perhaps even with identification of epigenomic marks related to treatment response and treatment failure adding benefit.
cfDNA epigenome analysis using bisulfite conversion
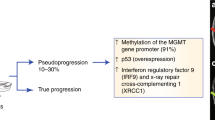
Epigenomic modification of DNA can be detected through either direct or indirect means, sometimes requiring only small quantities of DNA [51]. The most common indirect technique utilises bisulfite ions to deaminate unmethylated cytosines to uracil, which are then read as thymine in the resultant amplified DNA. PCR, array and sequencing approaches can be utilised for subsequent processing. If sequencing the converted samples, computation of uracil/cytosine signal at each loci of interest provides a continuous variable of mC at that base. Bisulfite conversion provides a cheap, fast and reproducible method for mC analysis in glioma cfDNA. Lavon et al demonstrated excellent specificity (100%) although modest sensitivity (59% astrocytoma, 47% oligodendroglioma) when using methylation at the MGMT promoter as a serum marker for primary brain tumours [59]. Following bisulfite conversion of cfDNA, mC- specific PCR was performed at the MGMT locus using different primers for methylated/unmethylated fragments [59]. Estival et al. used a similar technique in matched tumour and blood samples with variable results, depending upon the assay used and the presence or absence of MGMT methylation in the primary sample [60]. Gong et al. showed benefit by analysing Alu hypomethylation (AUC 0.904) in conjunction with MGMT hypermethylation (AUC 0.962) across a cohort of 124 glioma patients [61]. Recently, Sabedot et al. used bisufite conversion of plasma-derived cfDNA followed by Epic array processing across a cohort of 149 glioma patients to discriminate with machine-learning classifier between tumour and normal samples with a sensitivity of 100% and specificity of 97.78% [62]. The same study suggested a potential utility of this methylation-based approach for tracking clinicopathological changes (e.g. progression, pseudoprogression or response to standard or experimental treatment). A recent novel approach used MR-guided focused ultrasound to ‘open’ the blood–brain barrier [63]. Meng et al. noted an increase in the amount of cfDNA and used bisulfite conversion to identify neural-lineage cells via gene-set enrichment of hypomethylated probes. In 1/9 cases (with IDH mutation), they identified the mutation through targeted dPCR [63].
However, bisulfite conversion presents limitations for cfDNA analysis. First, it necessitates harsh conditions of pH and temperature that can degrade the DNA, which when dealing with low-input quantities of cfDNA, can be limiting [64]. Moreover, the conversion can be incomplete, or there can be over-conversion, resulting in subsequent interpretation difficulties. Finally, the inability to distinguish mC from hmC, both being resistant to bisulfite-induced deamination, can further confound analysis. Despite these limitations, protocols exist that mandate only small amounts of DNA and seem to provide reliable data for use in liquid biopsy. Whole-genome bisulfite sequencing (WGBS) requires high-depth sequencing and whole-genome coverage, thus rendering it expensive. This has been partially circumvented by applying the adapter tags post bisulfite conversion (PBAT) with a resultant reduction in sequencing burden [65]. PBAT generates a PCR-free approach that can determine methylation status from as little as 125 pg of DNA and provides utility for single-cell bisulfite sequencing [66].
In order to circumvent the large quantities of DNA lost through bisulfite conversion, techniques have been developed or adopted that reduce the need for large quantities of input DNA (e.g. as required for WGBS). Reduced-representation bisulfite sequencing (RRBS) enriches for CpG islands that are putatively most representative of cell methylation status. This approach reduces sequencing costs but at the cost of omitting non-CpG methylation (e.g. intergenic and enhancer regions) [67]. It also requires high-quality DNA due to the restriction-enzyme sequence utilised to identify target regions, thus potentially limiting its efficacy in fragmented cfDNA. Whilst bisulfite conversion enables distinction between mC and C, it is unable to discriminate between mC and hmC. Given our improved understanding of the often-opposing action of hmC (when compared with mC) across the genome, it has become important to discriminate between the two modifications. Alternate techniques are involving additional base-conversion steps alongside bisulfite-conversion OxBS seq [68], TAB-seq [69] or the use of enzymatic methods of deamination [70, 71]. Moreover, recent appropriation of pyridine borane- reductive decarboxylation and deamination chemistry has provided a direct means of reading mC and hmC [72].
cfDNA epigenome analysis using bisulfite-free methods
Bisulfite-free methods are also used and these can loosely be divided between affinity-enrichment and restriction-enzyme-mediated technologies. Affinity-enrichment methods have been developed for both methylation (MeDIP) [73, 74] and hydroxymethylation analysis [75]. MeDIP involves the use of antibody- based mC-sensitive pulldown of fragmented genomic DNA for the analysis of mC by fragmented region via PCR or sequencing [76]. Recently, this was adapted for use with cfDNA present in brain tumours and AUC was >0.71 when defining a broad range of brain tumours [77]. These plasma cfDNA methylomes combined with machine-learning analysis can discriminate common intracranial tumours with similar cells of origin, for example, by classifying IDH-mutant glioma (mean AUC = 0.82), or low-grade glioma (mean AUC = 0.93) from a group of 161 cases [77]. A similar method for hmC uses click chemistry to biotinylate glucosylated hmC residues with similar PCR or sequencing outputs [75]. These techniques are well adapted for cfDNA applications as they require low-starting input of cfDNA molecules (~5 ng). However, these techniques only determine that a modified base exists at some point along the fragment. Methylation-restriction enzymes are able to cleave unmethylated but not methylated regions. Subsequent PCR or sequencing can be performed; however, the fragmented nature of cfDNA with limited CpG-containing recognition sites can hamper methylome coverage [78].
Direct analysis of DNA without the need for conversion or amplification is a ‘holy grail’ of methylation sequencing. Long-read sequencing may also enable the phasing of data providing contextual information that may further boost sensitivity whilst reducing epigenomic noise. Recent improvements in nanopore-sequencing analysis pipelines provide an interesting counterpoint to the short-read narrative and may deliver value for analysis where DNA molecules are limited, such as those in cfDNA for brain tumours [79]. In addition, nanopore sequencing could enable the integration of genomic with epigenomic data in a fast turn-around time, which could be well adapted to liquid-biopsy application pending an increase in sensitivity [80].
Epigenetics beyond methylation: integrating fragmentomics for liquid-biopsy applications
cfDNA is fragmented in plasma around 167 bp and multiple thereof, a size corresponding to the wrapping of DNA helix around the nucleosome. Plasma cfDNA from a variety of cancer cells tends to be shorter than the bulk of cfDNA derived from hematopoietic cells by 20–30 bp [81,82,83]. The sources and mechanisms of release of cfDNA in the bloodstream are multiple, including apoptosis, necrosis, senescence and other forms of cell death and active secretion [84, 85]. Beyond the size of the cfDNA fragments, more information can also be retrieved from where they are located in the genome [52], and how these fragments end [86]. For example, cfDNA fragments tend to be depleted in the promoter/TSS regions of highly expressed genes because the nucleosomes protecting them from degradation have to be moved away to enable transcription [87, 88]. As the size and position of cfDNA are not random but biologically regulated and/or altered in cancer, this is opening the development of applications focused on analysing such patterns or fragmentomics.
The methods focusing on fragmentomics, based on genome-wide paired-end sequencing, could improve existing sequencing methods, as well as mutation analysis. First, by leveraging the 20–30-bp size differences between tumour and non-tumour DNA, it is possible to either enrich for cancer signal, or filter out the false-positive signal (e.g. mutant DNA from clonal haematopoiesis). Indeed, clonal haematopoiesis-derived mutations should on average appear on longer fragments than mutations from tumour-derived cfDNA. Incorporating such information could reinforce the confidence in mutation calling when tumour fraction in plasma is extremely low (e.g. in gliomas). The potential of such a strategy has been demonstrated in a small cohort of glioma samples using INVAR [44, 48], as well as in other cancer types [89]. It is also possible to use fragment-size information to directly enrich for true positives. In vitro and in silico size selection of specific size ranges has highlighted potential enrichment in multiple cancer types and with different sequencing technologies [83, 90]. However, although size selection increases the sensitivity of detection, there is a reciprocal loss of cfDNA for analysis. Moreover, the relative median genome-wide enrichment is ~2-fold, and therefore direct application to glioma, with their low tumour fraction, might be challenging. In addition to plasma, the cfDNA fragmentation is also altered in the CSF from patients with high tumour fraction, revealing a potential for other bio-fluids [36].
Leveraging other biological properties of cfDNA-fragmentation profiles could enhance the detection of tumour-derived cfDNA. By combining copy number alterations with specific fragment-size patterns using random forest classifier, cancer can be classified with a high accuracy (AUC = 0.91 for glioma cases) [83]. Recently, Mouliere et al. demonstrated that the cfDNA-fragmentation profile from urine samples could also be leveraged using machine learning to improve the classification of gliomas from controls and other brain pathologies [47]. Beyond specific fragment-size ranges, more patterns could be inferred from cfDNA fragmentation. Various approaches using regional information [91], nucleosomal densities [87], chromatin compaction [92] and the composition in bases of fragment ends [93] have revealed potential to extract more tumour signal from genome-wide sequencing in other cancer types. Despite prior studies highlighting the prevalence of short cfDNA in plasma, longer circulating DNA also exists, some of which may represent extrachromosomal (circular) DNA (ecDNA) [94]. Despite the relative importance of ecDNA in glioma oncogenesis [95], ecDNA remains underexplored in liquid biopsy and may benefit from the use of long-read technologies.
Discussion
The cfDNA field has benefited from a fast-growing portfolio of technologies and from a dynamic entrepreneurial ecosystem with a desire to improve non-invasive cancer analysis. Each cfDNA technology could be well adapted to one of the clinical applications of interest for CNS cancers: screening, diagnosis/classification, treatment guidance, monitoring progression and detecting relapse earlier (Table 2). However, technical advances do not always translate into the clinic for applications related to gliomas and other CNS pathologies. Despite clear progress to improve the sensitivity and specificity of cfDNA sequencing, significant improvements are required at a conceptual, technical and analytical level before clinical implementation of liquid biopsy in the CNS can be realised. In recent years, new technologies and concepts, such as epigenetic or fragmentomic characterisation alongside tumour-guided sequencing, have strong clinical potential [52]. In addition, methods leveraging the epigenome, due to their non-cancer-specific nature, have the potential to be applied to CNS pathologies beyond cancer [57]. Evidence of neuron-derived cfDNA and cerebellum cfDNA within acute neurotrauma and chronic neurodegeneration has also been generated [96]. The potential of disruptive cfDNA technologies and concepts for analysing neurodegenerative disease beyond cancer has barely started to be explored.
Beyond cfDNA, multiple other tumour components (or components influenced by cancer cells) circulate within a cancer patient. Exosomes [97, 98], mitochondria [31], tumour-educated platelets [99, 100] and circulating tumour cells [7] have all been identified in the bio-fluids from patients with brain tumours. Combining with machine learning, these multiple analytes and signals could unlock some of the most challenging applications for liquid biopsy in the CNS context [9, 52]. Integrating multiple analytes with machine-learning classifier has led to an increase in detection and classification performance in other cancer types, but the potential of such approach remains underexplored in brain cancer [101, 102]. Bayesian integration of multi-omics and imaging data exhibits a potential to improve the predictive performance on longitudinal data, as demonstrated in other cancer types [103, 104]. The impact of new cfDNA technologies for the management of brain cancer using liquid biopsy can be boosted by the rise of disruptive computational and machine-learning strategies.
Conclusion
The utility of liquid biopsy in glioma remains relatively underexplored at the view of its clinical potential. At every stage of the analysis paradigm, novel approaches to cfDNA have incrementally improved sensitivity and specificity. The advent of improved biobanking approaches has further improved substrate accessibility and reliability. We, therefore, hope that the past difficulties associated with translating cfDNA analysis in brain-tumour populations will not discourage further investigation amongst the liquid-biopsy community.
Data availability
Not applicable.
References
Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, et al. The epidemiology of glioma in adults: A state of the science review. Vol. 16, Neuro-Oncology. Oxford University Press; 2014. p. 896–913.
Davis FG, Smith TR, Gittleman HR, Ostrom QT, Kruchko C, Barnholtz-Sloan JS. Glioblastoma incidence rate trends in Canada and the United States compared with England, 1995–2015. Neuro Oncol. 2020;22:301–2.
Sanai N, Berger MS. Glioma extent of resection and its impact on patient outcome. Neurosurg Oxf Academic. 2008;62:753–64.
Wesseling P. The ABCs of molecular diagnostic testing of CNS tumors: acceptance, benefits, costs. Neuro Oncol. 2019;21:559–61.
Kros JM, Mustafa DM, Dekker LJM, Smitt PAES, Luider TM, Zheng PP. Circulating glioma biomarkers. Vol. 17, Neuro-Oncology. Oxford University Press; 2015. p. 343–60.
Westphal M, Lamszus K. Circulating biomarkers for gliomas. Nat Rev Neurol Nat Publ Group. 2015;11:556–66.
Alix-Panabières C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6:479–91.
Anfossi S, Babayan A, Pantel K, Calin GA. Clinical utility of circulating non-coding RNAs—an update. Nat Rev Clin Oncol. 2018;15:541–63.
Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20:71–88.
Pantel K, Alix-Panabières C. Liquid biopsy and minimal residual disease—latest advances and implications for cure. Nat Rev Clin Oncol. 2019;16:409–424.
Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–38.
Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol. 2018;36:1631–41.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24.
Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the Serum of Cancer Patients and the Effect of Therapy. Cancer Res. 1977;37:646-50.
Nørøxe DS, Østrup O, Yde CW, Ahlborn LB, Nielsen FC, Michaelsen SR, et al. Cell-free DNA in newly diagnosed patients with glioblastoma – a clinical prospective feasibility study. Oncotarget 2019;10:4397–406.
Bagley SJ, Ali Nabavizadeh S, Mays JJ, Till JE, Ware JB, Levy S, et al. Clinical utility of plasma cell-free DNA in adult patients with newly diagnosed glioblastoma: A pilot prospective study. Clin Cancer Res. 2020;26:397–407.
Nabavizadeh SA, Ware JB, Guiry S, Nasrallah MP, Mays JJ, Till JE, et al. Imaging and histopathologic correlates of plasma cell-free DNA concentration and circulating tumor DNA in adult patients with newly diagnosed glioblastoma. Neuro-Oncol Adv. 2020;2:1–9.
Fontanilles M, Marguet F, Beaussire L, Magne N, Pépin LF, Alexandru C, et al. Cell-free DNA and circulating TERT promoter mutation for disease monitoring in newly-diagnosed glioblastoma. Acta Neuropathol Commun. 2020;8:179.
Meddeb R, Dache ZAA, Thezenas S, Otandault A, Tanos R, Pastor B, et al. Quantifying circulating cell-free DNA in humans. Sci Rep. 2019;9:5220.
Zill OA, Banks KC, Fairclough SR, Mortimer SA, Vowles JV, Mokhtari R, et al. The landscape of actionable genomic alterations in cell-free circulating tumor DNA from 21,807 advanced cancer patients. Clin Cancer Res. 2018;24:3528–38.
Schwaederle M, Husain H, Fanta PT, Piccioni DE, Kesari S, Schwab RB, et al. Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget. 2016;7:9707–16.
Piccioni DE, Achrol AS, Kiedrowski LA, Banks KC, Boucher N, Barkhoudarian G, et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. 2019;8:CNS34.
Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med. 2019;25:1928–37.
Perkins G, Lu H, Garlan F, Taly V. Droplet-Based Digital PCR: Application in Cancer Research. In: Advances in Clinical Chemistry. Academic Press Inc.; 2017. p. 43–91.
Newman AM, Bratman SV, To J, Wynne JF, Eclov NCW, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548–54.
Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356:438–42.
Muralidharan K, Yekula A, Small JL, Rosh ZS, Kang KM, Wang L, et al. TERT promoter mutation analysis for blood-based diagnosis and monitoring of gliomas. Clin Cancer Res. 2021;27:169–78.
De Mattos-Arruda L, Mayor R, Ng CKY, Weigelt B, Martínez-Ricarte F, Torrejon D, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun. 2015;6:8839.
Wang Y, Springer S, Zhang M, McMahon KW, Kinde I, Dobbyn L, et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci USA. 2015;112:9704–9.
Pan W, Gu W, Nagpal S, Gephart MH, Quake SR. Brain tumor mutations detected in cerebral spinal fluid. Clin Chem. 2015;61:514–22.
Mair R, Mouliere F, Smith CG, Chandrananda D, Gale D, Marass F, et al. Measurement of plasma cell-free mitochondrial tumor DNA improves detection of glioblastoma in patient-derived orthotopic xenograft models. Cancer Res. 2019;79:220–30.
Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci USA. 2016;113:E1826–34.
Seoane J, De Mattos-Arruda L, Rhun ELE, Bardelli A, Weller M. Cerebrospinal fluid cell-free tumour DNA as a liquid biopsy for primary brain tumours and central nervous system metastases. Ann Oncol. 2019;30:211–8.
Miller AM, Shah RH, Pentsova EI, Pourmaleki M, Briggs S, Distefano N, et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature. 2019;565:654–8.
Martínez-Ricarte F, Mayor R, Martínez-Sáez E, Rubio-Pérez C, Pineda E, Cordero E, et al. Molecular diagnosis of diffuse gliomas through sequencing of cell-free circulating tumor DNA from cerebrospinal fluid. Clin Cancer Res. 2018;24:2812–9.
Mouliere F, Mair R, Chandrananda D, Marass F, Smith CG, Su J, et al. Detection of cell‐free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Mol Med. 2018;10:e9323.
Pan C, Diplas BH, Chen X, Wu Y, Xiao X, Jiang L, et al. Molecular profiling of tumors of the brainstem by sequencing of CSF-derived circulating tumor DNA. Acta Neuropathol. 2019;137:297–306.
Pentsova EI, Shah RH, Tang J, Boire A, You D, Briggs S, et al. Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. J Clin Oncol. 2016;34:2404–15.
Engelborghs S, Niemantsverdriet E, Struyfs H, Blennow K, Brouns R, Comabella M, et al. Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimer’s Dement Diagnosis. Assess Dis Monit. 2017;8:111–26.
Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N. Engl J Med. 2001;345:1727–33.
Sottoriva A, Spiteri I, Piccirillo SGM, Touloumis A, Collins VP, Marioni JC, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. 2013;110:4009–14.
Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017;545:446–51.
McDonald BR, Contente-Cuomo T, Sammut SJ, Odenheimer-Bergman A, Ernst B, Perdigones N, et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci Transl Med. 2019;11:eaax7392.
Wan JCM, Heider K, Gale D, Murphy S, Fisher E, Mouliere F, et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci Transl Med. 2020;12:eaaz8084.
Zviran A, Schulman RC, Shah M, Hill STK, Deochand S, Khamnei CC, et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat Med. 2020;26:1114–24.
Smith CG, Moser T, Mouliere F, Field-Rayner J, Eldridge M, Riediger AL, et al. Comprehensive characterization of cell-free tumor DNA in plasma and urine of patients with renal tumors. Genome Med. 2020;12:23.
Mouliere F, Smith CG, Heider K, Su J, Pol Y van der, Thompson M, et al. Fragmentation patterns and personalized sequencing of cell-free DNA in urine and plasma of glioma patients. EMBO Mol Med. 2021;13:e12881.
Mouliere F, Heider K, Smith CG, Su J, Morris J, Wan JCM, et al. Integrated clonal analysis reveals circulating tumor DNA in urine and plasma of glioma patients. BioRxiv. 2019; Preprint at https://www.biorxiv.org/content/10.1101/758441v1.
Gydush G, Nguyen E, Bae JH, Rhoades J, Reed SC, Shea D, et al. MAESTRO affords ‘breadth and depth’ for mutation testing. bioRxiv. 2021 Jan 24;2021.01.22.427323; Preprint at https://doi.org/10.1101/2021.01.22.427323.
Song P, Chen SX, Yan YH, Pinto A, Cheng LY, Dai P, et al. Selective multiplexed enrichment for the detection and quantitation of low-fraction DNA variants via low-depth sequencing. Nat Biomed Eng. 2021;5:690–701.
Dor Y, Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet. 2018;392:777–86.
van der Pol Y, Mouliere F. Toward the early detection of cancer by decoding the epigenetic and environmental fingerprints of cell-free DNA. Cancer Cell. 2019;36:350–68.
Li W, Zhang X, Lu X, You L, Song Y, Luo Z, et al. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res. 2017;27:1243–57.
Song CX, Yin S, Ma L, Wheeler A, Chen Y, Zhang Y, et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res. 2017;27:1231–42.
Sadeh R, Sharkia I, Fialkoff G, Rahat A, Gutin J, Chappleboim A, et al. ChIP-seq of plasma cell-free nucleosomes identifies gene expression programs of the cells of origin. Nat Biotechnol. 2021;39:586–98.
Wells M, Asmaro KP, Sabedot TS, Malta TM, Mosella MS, Nelson K, et al. Detection of circulating tumor-specific DNA methylation markers in the blood of patients with pituitary tumors. medRxiv. medRxiv; 2020.2020.05.29.20116202;Preprint at https://doi.org/10.1101/2020.05.29.20116202.
Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun. 2018;9:5068.
Pan L, McClain L, Shaw P, Donnellan N, Chu T, Finegold D, et al. Non-invasive epigenomic molecular phenotyping of the human brain via liquid biopsy of cerebrospinal fluid and next generation sequencing. Eur J Neurosci. 2020;52:4536–45.
Lavon I, Refael M, Zelikovitch B, Shalom E, Siegal T. Serum DNA can define tumor-specific genetic and epigenetic markers in gliomas of various grades. Neuro Oncol. 2010;12:173–80.
Estival A, Sanz C, Ramirez JL, Velarde JM, Domenech M, Carrato C, et al. Pyrosequencing versus methylation-specific PCR for assessment of MGMT methylation in tumor and blood samples of glioblastoma patients. Sci Rep. 2019;9:11125.
Gong M, Shi W, Qi J, Shao G, Shi Z, Wang J, et al. Alu hypomethylation and MGMT hypermethylation in serum as biomarkers of glioma. Oncotarget. 2017;8:76797–806.
Sabedot T, Malta T, Snyder J, Nelson K, Wells M, DeCarvalho A, et al. A serum-based DNA methylation assay provides accurate detection of glioma. Neuro Oncol. 2021;23:1494–1508.
Meng Y, Pople CB, Suppiah S, Llinas M, Huang Y, Sahgal A, et al. MR-guided focused ultrasound liquid biopsy enriches circulating biomarkers in patients with brain tumors. Neuro Oncol. 2021;23:1789–97.
Tanaka K, Okamoto A. Degradation of DNA by bisulfite treatment. Bioorg Med Chem Lett. 2007;17:1912–5.
Miura F, Enomoto Y, Dairiki R, Ito T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012;40:e136–e136.
Miura F, Ito T Post-bisulfite adaptor tagging for PCR-free whole-genome bisulfite sequencing. In: Methods in Molecular Biology. Humana Press Inc.; 2018. p. 123–36.
Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008;454:766–70.
Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–7.
Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–80.
Schutsky EK, Denizio JE, Hu P, Liu MY, Nabel CS, Fabyanic EB, et al. Nondestructive, base-resolution sequencing of 5-hydroxymethylcytosine using a DNA deaminase. Nat Biotechnol. 2018;36:1083–90.
Vaisvila R, Chaithanya Ponnaluri VK, Sun Z, Langhorst BW, Saleh L, Guan S, et al. EM-seq: Detection of DNA methylation at single base resolution from picograms of DNA. bioRxiv. bioRxiv; 2019. 2019.12.20.884692; Preprint at https://www.biorxiv.org/content/10.1101/2019.12.20.884692v1.
Liu Y, Hu Z, Cheng J, Siejka-Zielińska P, Chen J, Inoue M, et al. Subtraction-free and bisulfite-free specific sequencing of 5-methylcytosine and its oxidized derivatives at base resolution. Nat Commun. 2021;12:1–8.
Taiwo O, Wilson GA, Morris T, Seisenberger S, Reik W, Pearce D, et al. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat Protoc. 2012;7:617–36.
Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–62.
Han D, Lu X, Shih AH, Nie J, You Q, Xu MM, et al. A highly sensitive and robust method for genome-wide 5hmC profiling of rare cell populations. Mol Cell. 2016;63:711–9.
Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563:579–83.
Nassiri F, Chakravarthy A, Feng S, Shen SY, Nejad R, Zuccato JA, et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat Med. 2020;26:1044–7.
Brunner AL, Johnson DS, Si WK, Valouev A, Reddy TE, Neff NF, et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009;19:1044–56.
Yuen ZWS, Srivastava A, McNevin D, Jack C, Eyras E. Systematic benchmarking of tools for CpG methylation detection from Nanopore sequencing. bioRxiv. bioRxiv; 2020. 2020.10.14.340315; Preprint at https://www.biorxiv.org/content/10.1101/2020.10.14.340315v2.
Euskirchen P, Bielle F, Labreche K, Kloosterman WP, Rosenberg S, Daniau M, et al. Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol. 2017;134:691–703.
Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, et al. Fragment length of circulating tumor DNA. PLoS Genet. 2016;12:e1006162.
Mouliere F, Robert B, Peyrotte E, Del Rio M, Ychou M, Molina F, et al. High fragmentation characterizes tumour-derived circulating DNA. Lee T, editor. PLoS ONE. 2011;6:e23418.
Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018;10:eaat4921.
Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016;35:347–76.
Rostami A, Lambie M, Yu CW, Stambolic V, Waldron JN, Bratman SV. Senescence, necrosis, and apoptosis govern circulating cell-free DNA release kinetics. Cell Rep. 2020;31:107830.
Chandrananda D, Thorne NP, Bahlo M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med Genomics. 2015;8:29.
Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell. 2016;164:57–68.
Ulz P, Thallinger GG, Auer M, Graf R, Kashofer K, Jahn SW, et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet. 2016;48:1273–8.
Chabon JJ, Hamilton EG, Kurtz DM, Esfahani MS, Moding EJ, Stehr H, et al. Integrating genomic features for non-invasive early lung cancer detection. Nature. 2020;580:245–51.
Hellwig S, Nix DA, Gligorich KM, O’Shea JM, Thomas A, Fuertes CL, et al. Automated size selection for short cell-free DNA fragments enriches for circulating tumor DNA and improves error correction during next generation sequencing. PLoS ONE. 2018;13:e0197333.
Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019;570:385–9.
Sun K, Jiang P, Cheng SH, Cheng THT, Wong J, Wong VWS, et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. 2019;29:418–27.
Jiang P, Sun K, Tong YK, Cheng SH. Cheng THTT, Heung MMSS, et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc Natl Acad Sci USA. 2018;115:E10925–33.
Sin STK, Jiang P, Deng J, Ji L, Cheng SH, Dutta A, et al. Identification and characterization of extrachromosomal circular DNA in maternal plasma. Proc Natl Acad Sci USA. 2020;17:1658–65.
Decarvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, Cherba D, et al. Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat Genet. 2018;50:708–17.
Chatterton Z, Mendelev N, Chen S, Raj T, Walker R, Carr W, et al. Brain-derived circulating cell-free DNA defines the brain region and cell specific origins associated with neuronal atrophy. bioRxiv. bioRxiv; 2019:538827. Preprint at https://www.biorxiv.org/content/10.1101/538827v1.
Xu R, Rai A, Chen M, Suwakulsiri W, Greening DW, Simpson RJ. Extracellular vesicles in cancer—implications for future improvements in cancer care. Vol. 15, Nature Rev Clin Oncol. 2018;15:617–38.
Ebrahimkhani S, Vafaee F, Hallal S, Wei H, Lee MYT, Young PE, et al. Deep sequencing of circulating exosomal microRNA allows non-invasive glioblastoma diagnosis. npj Precis. Oncol 2018;2:1–9.
Best MG, Sol N, Kooi I, Tannous J, Westerman BA, Rustenburg F, et al. RNA-Seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell. 2015;28:666–76.
Sol N, In’t Veld GJG, Vancura A, Tjerkstra M, Leurs C, Rustenburg FO, et al. Tumor-educated platelet RNA for the detection and (pseudo)progression monitoring of glioblastoma. Cell Rep. Med. 2020;1:100101.
Cohen JD, Javed AA, Thoburn C, Wong F, Tie J, Gibbs P, et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci USA. 2017;114:10202–7.
Wan N, Weinberg D, Liu T-Y, Niehaus K, Ariazi EA, Delubac D, et al. Machine learning enables detection of early-stage colorectal cancer by whole-genome sequencing of plasma cell-free DNA. BMC Cancer. 2019;19:832.
Kurtz DM, Esfahani MS, Scherer F, Soo J, Jin MC, Liu CL, et al. Dynamic risk profiling using serial tumor biomarkers for personalized outcome prediction. Cell 2019;178:699–713.e19.
Khan KH, Cunningham D, Werner B, Vlachogiannis G, Spiteri I, Heide T, et al. Longitudinal liquid biopsy and mathematical modeling of clonal evolution forecast time to treatment failure in the prospect-c phase ii colorectal cancer clinical trial. Cancer Disco. 2018;8:1270–85.
Acknowledgements
FM is funded by the Amsterdam UMC Liquid Biopsy Center, an initiative made possible through the Stichting Cancer Center Amsterdam and by a KWF grant (KWF-12822). RM is jointly funded by Cancer Research UK, the University of Cambridge and Cambridge University Hospitals NHS Foundation Trust.
Funding
FM is funded by a KWF grant (KWF-12822).
Author information
Authors and Affiliations
Contributions
RM and FM wrote and supervised the work.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mair, R., Mouliere, F. Cell-free DNA technologies for the analysis of brain cancer. Br J Cancer 126, 371–378 (2022). https://doi.org/10.1038/s41416-021-01594-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01594-5
