
Abstract
Breast cancer is one of the most prevalent malignancies in women worldwide. Early-stage breast cancer is considered a curable disease; however, once distant metastasis occurs, the 5-year overall survival rate of patients becomes significantly reduced. There are four distinct metastatic patterns in breast cancer: bone, lung, liver and brain. Among these, breast cancer brain metastasis (BCBM) is the leading cause of death; it is highly associated with impaired quality of life and poor prognosis due to the limited permeability of the blood–brain barrier and consequent lack of effective treatments. Although the sequence of events in BCBM is universally accepted, the underlying mechanisms have not yet been fully elucidated. In this review, we outline progress surrounding the molecular mechanisms involved in BCBM as well as experimental methods and research models to better understand the process. We further discuss the challenges in the management of brain metastases, as well as providing an overview of current therapies and highlighting innovative research towards developing novel efficacious targeted therapies.
Similar content being viewed by others

Background
Breast cancer (BC) is the most prevalent malignancy and the second leading cause of cancer-related death among the female population worldwide. According to published cancer statistics, ~2.7 million estimated new cases of BC arose in 2020, alongside 42,170 estimated deaths, accounting for 30% of all newly diagnosed cancer cases and 15% of cancer-related deaths.1 Despite improvements in early diagnosis and effective treatment, recurrence and metastasis remain considerable threats to the survival of BC patients.2
Bone, lung, liver and brain are four common metastatic sites of BC. Breast cancer brain metastasis (BCBM), which accounts for 10–30% of cases of all metastatic breast cancer (MBC), is associated with poor prognosis and impairs both the cognitive and sensory functions of patients, leading to a seriously limited quality of life (QoL).3 Several modes of therapy are currently available for BCBM patients, including surgery, whole-brain radiation therapy (WBRT), stereotactic radiosurgery (SRS) and chemotherapy or a combination of these therapies.4 However, the life expectancy of patients with BCBM remains unsatisfactory, owing to the presence of the blood–brain barrier (BBB), which limits penetrability, and chemoresistance.5 There is therefore an urgent need to elucidate the molecular mechanisms that underlie BCBM in order to find novel therapeutic targets, enhance treatment efficacy and improve BCBM patient prognosis.
In this review, we outline the current knowledge regarding the molecular mechanisms that underlie BCBM provide an overview of the research models current used to study the process, particularly the relevance of the BBB in extravasation. Moreover, advances in targeted treatments, immunotherapy and novel drug carrier nanosystems, which might help to improve the outcomes of BC patients with brain metastasis, are also discussed in depth.
Metastatic dissemination of breast cancer to the brain
Metastasising breast cancer cells have been reported to give rise to three types of brain metastasis, depending on the anatomic features of the brain in which the secondary tumour arises: parenchymal metastasis, leptomeningeal metastasis and choroid plexus metastasis. The different microenvironments provide different conditions for the metastatic cancer cells and ultimately lead to distinct epidemiologies, and biologies, and possibly also different therapies. Metastases in the choroid plexus are rare,6 metastasis to leptomeninges accounts for 8% of cases, and parenchymal metastasis is the most common form, consisting of multiple metastases (78%) and solitary metastases (14%).7
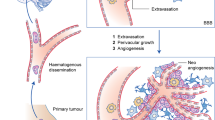
Following detachment from the primary tumour, the metastatic process comprises a sequence of steps, including cell invasion through the basement membrane into surrounding tissue, intravasation into blood vessels, survival and arrest in the circulatory system, extravasation through transendothelial migration, colonisation and the eventual formation of distant metastatic lesions (Fig. 1). Invasion and intravasation are common initial steps in distant metastasis, and involve the process of epithelial-to-mesenchymal transition (EMT), components of the tumour microenvironment and angiogenesis. Intravasation, based on the breakdown of endothelial junction proteins, can be promoted by perivascular macrophages and interactions between tumour cells and endothelial cells (ECs).8 As for extravasation, metastatic cells in circulation cross through the cell junctions between the distant endothelial cells, under the support of specific factors. Eventually, survival metastatic cells escaping from dormancy stage form micrometastatic foci along blood vessels and grow up under the interaction with local cells. Herein, we mainly discuss the specific events involved in BCBM.

a small population of cells at the primary site acquired invasive properties before gaining stem-cell-like properties and losing cell polarity through epithelial-to-mesenchymal transition (EMT). Invasive cancer cells then infiltrate the surrounding tissue by ECM remodelling, and the become circulating tumour cells (CTCs) with the help of perivascular macrophages and interaction with blood vessel endothelial cells (ECs). By virtue of the bloodstream, cancer cells then spread throughout the body and cross the blood–brain barrier (BBB) through extravasation after their adhesion with ECs in the brain. Subsequently, most cells die or enter a state of dormancy, few proliferate within this new microenvironment. Vascular co-option can be regarded as the first step of colonisation and then micrometastases forms. After metabolic reprogramming, tumour cells establish a more stable colonisation and then form brain metastasis. Besides, dormant cells tend to be reawakened in certain condition and participate in colonisation and cause tumour recurrence.
Survival, migration and adhesion of cancer cells in BCBM
The presence of circulating tumour cells (CTCs)—cells derived from a primary tumour that have entered the bloodstream—signifies that the tumour is no longer localised. Before they reach the circulation, invasive cancer cells, already obtaining mesenchymal features from EMT, remodel their surrounding extracellular matrix (ECM) and release tumour-promoting factors that enable them to infiltrate the surrounding tissue and then cross the blood vessel endothelium in the form of single cells or clusters.9Whether these changes of CTCs are related to a specific subtype like HER2+ or triple-negative breast cancer (TNBC) needs further exploration.
Survival
Once in the bloodstream, only a small percentage of CTCs can successfully survive the shear stress generated by the blood flow, surveillance by immune cells and anoikis (cell death owing to detachment from the ECM) (Fig. 2). CTCs that are clustered by platelets in the bloodstream are more resistant to shear stress and are protected from immune attack by the platelet-induced release of transforming growth factor-β (TGF-β), which can neutralise natural killer (NK) granule mobilisation, cytotoxicity and interferon-γ (IFN-γ) secretion.10 Furthermore, CTCs can express CD47 to evade host cell phagocytosis,11 and can also educate NK cells, the main protagonists of anti-tumour immunity, towards a metastasis-promoting state that shows a reduction in cytotoxic ability and favours the immune escape of CTCs.12 Although EMT endows CTCs with partial resistance to anoikis, the overexpression of TrkB, a neurotrophic tyrosine kinase receptor, also contributes to protection from apoptosis by activating the phosphatidylinositol 3-kinase (PI3K)–AKT survival pathway.13

In the bloodstream, a number of circulating tumours cells (CTCs) can successfully survive by upregulating AKT and CD47 to resist shear stress and escape clearance by immune cells such as natural killer (NK) cells and macrophages. The interaction of CXCR4 on CTCs with CXCL12 secreted by endothelial cells (ECs) facilitates chemotactic migration to the brain. CTCs undergo shear-resistant adhesion to the inside of blood vessel walls by expressing CD44 and MUC1, which interact with E-selectin on ECs. To achieve stronger adhesion, further cell adhesion molecules, such as ICAM-1, VCAM-1, ALCAM and integrin, will be expressed on CTCs and ECs.
Migration
Increasing evidence indicates that metastatic cancer cells possess organotropism at many metastatic stages, which has not been fully elucidated yet. During migration stage, circulating tumour-secreted exosomes function in the communication between cancer cells and distant organs.14 The co-expression of SEMA4D and MYC on CTCs can also help themselves to build brain metastasis in following metastasis stages (extravasation and colonisation), which will be discussed in detail later, indicating that CTC-derived markers can be early prognostic factors owing to organotropic metastases.15 Moreover, cancer-associated fibroblasts (CAFs) derived from the stroma of brain metastasis express increased levels of the chemokine ligand CXCL12, thereby enhancing the migration of breast cancer cells, which express its receptor, CXCR4, to the brain (Fig. 2).16
Attachment
The attachment of CTCs to ECs constitutes the first step before the subsequent extravasation process (Fig. 2). CTCs that are carried passively by the flow of blood within cerebral microvessels, begin to slow down and then arrest in small capillaries that have a similar diameter to CTCs, especially at vascular branch points,17 thereby forming an initial site of weak adhesion. The interaction between selectins and their ligands induces tethering and rolling of tumour cells along the vascular endothelium. E-selectin, the expression of which on the surface of ECs, is induced by tumour necrosis factor α (TNFα) under an inflammatory stimulation, which is present at high levels in brain metastases, and preferentially contributes to shear-resistant adhesion and transendothelial migration by binding the transmembrane glycoprotein CD44 on BC cells to establish heterogenous adhesion.18 MUC1, another transmembrane glycoprotein expressed by almost all BC cells, can also function as a ligand for E-selectin as well as for intercellular adhesion molecule (ICAM)-1 to establish firmer adhesion between cancer cells and the endothelium.19 Further interactions between ECs and tumour cells mediated by cell adhesion molecules, including integrin α4β1/vascular cell adhesion molecule (VCAM-1) and activated leukocyte cell adhesion molecule (ALCAM)/ALCAM, reinforce the adhesion of BC cells to ECs.20
Extravasation and the BBB
Unlike other sites of metastasis, BCBM shows a much longer latency after the initial diagnosis of BC, which can be explained by the presence of the BBB. The BBB is a highly specialised structure that comprises ECs, tight junctions (TJs), basement membrane, pericytes, astrocytes, microglia, enzymes and transporters. It controls the permeability of the brain to macromolecules as well as being involved in transmitting signals and maintaining central nervous system homeostasis.21 Thus, exploration of the biological pathways and regulatory molecules involved in movement across the BBB are pivotal for BCBM prevention.
Increasing the permeability of the BBB
The specific route by which tumour cells cross the BBB remains unclear. Most cancer cells seem to use a paracellular mode, just as BC cells crossing BBB, whereby they squeeze through ECs by disrupting their intercellular junctions. Before or during the process of extravasation, elongated metastatic cells round up and form cytoplasmic protrusions to expand the surrounding vascular wall; this process occurs exclusively in capillaries and/or post-capillary venules that lack smooth muscle cells.22 Interestingly, the disruption to EC junctions seems to be repaired after paracellular extravasation, without significant damage to the BBB.23 As BC cells gather along brain endothelial cells, the previously intact BBB transforms into a permeable blood–tumour barrier (BTB) via vascular remodelling of pre-existing brain vessels, including downregulation of basement collagen membrane part IV and laminin α2. Meanwhile, a change in pericyte phenotype involving the altered expression of desmin and CD13, which affect the permeability of the barrier, have been observed in BCBM. These changes enable MBC cells to more easily cross the BBB.24,25
Components of the BBB can also be directly targeted by several factors during the process of BCBM. The highly selective permeability of the BBB largely depends on the presence of continuous TJs that connect ECs, and so it follows that dysregulation of the expression of TJ proteins, such as occludin, claudin-5 and ZO-1, can critically influence BBB permeability. Vascular endothelial growth factor (VEGF) functions as a strong promotor of angiogenesis. Under the stimulation of integrins like αvβ3 and β4, elevated VEGF derived from BC cells increases vascular permeability by disrupting TJs and adherens junctions in endothelial monolayers.26 As a result, VEGF acts as an accomplice in penetration of BC cells through BBB, and the new vessels support the growth of metastatic foci. Matrix metalloproteinases (MMPs) secreted by BC cells, such as MMP-1, MMP-2 and MMP-9, also contribute to disruption of the BBB by mediating the degradation of TJ proteins, such as claudin-5.27 Angiopoietin-2, the expression of which is elevated in brain microvascular endothelial cells (BMECs) under the stimulation of VEGF secreted by not only BC cells, but also their brain-seeking variants, also increases BBB permeability by disrupting TJ proteins such as zonula, occludin-1 and claudin-5 in BCBM.28 This process can be enhanced by substance P secreted from BC cells.29 Moreover, cathepsin S generated by tumour cells and macrophages mediates BBB transmigration through proteolytic processing of the junctional adhesion molecule, JAM-B, providing a rationale for the use of the cathepsin S-specific inhibitor VBY-999 in reducing experimental brain metastasis.30 A role for non-coding RNAs in metastasis has attracted attention, with MBC cells being demonstrated to secrete exosomes containing miR-105, which increases the permeability of ECs by disrupting ZO-1.31 Similarly, BCBM-cell-derived exosomes bearing lncRNA GS1–600G8.5 were shown to enhance the permeability of BBB by downregulating the levels of ZO-1, claudin-5 and N-cadherin in BMECs.32
Enhancing the penetration of BC cells
Not only can the permeability of the BBB be increased, but a number of mediators can also enhance the ability of BC cells to penetrate the BBB. The interaction between CXCL12, a chemokine secreted by CAFs and epithelial BC cells, and its receptor CXCR4 on MDA-MB-231 breast cancer cells promotes the penetration by MDA-MB-231 cells of a monolayer of human brain microvascular endothelial cells (HBMECs), as well as increasing the permeability of these cells.33 The expression by BC cells of cyclo-oxygenase-2 (COX-2), the epidermal growth factor receptor (EGFR) ligand HBEGF, and α2,6-sialyltransferase (ST6GALNAC5) has been shown to mediate passage across the BBB;34 unlike COX-2 and HBEGF, however, ST6GALNAC5 is regarded as a specific mediator that contributes to metastasis towards the brain rather than to any other organ.34 Other genes overexpressed in BCBM cells, such as ADAM8 and SEMA4D, have also been shown to be regulators of BBB transmigration. ADAM8 upregulates MMP-9,35 while SEMA4D interacts with overexpressed Plexin-B1 in brain ECs, which contributes to the increased penetration of CTCs.15
Additional cell types involved in BBB transmigration
As well as the interaction of tumour cells with brain ECs, additional cell types are also likely to be involved during migration across the BBB. For example, T lymphocytes upregulates the expression of guanylate-binding protein 1 in BC cells to facilitate the BBB transmigration.36 CAFs were proved to enhance the BBB permeability.37 In addition, novel pathways such as endothelial–mesenchymal transition (EndEMT) are involved in BBB transmigration.38 These provide additional perspectives on the mechanism of extravasation during BCBM.
Colonisation of BCBM cells in the brain
Once the cancer cells have exited the bloodstream, they begin the process of founding a metastatic colony. The colonisation of BCBM cells includes a series of steps, including reawakening from dormancy state, vascular co-option for micrometastases formation, and metabolic reprogramming. After extravasation, a considerable proportion of metastatic cells are likely to die owing to the body’s active anti-tumour response; others enter dormancy, with the risk of recurrence, and a few enter a proliferative state.
Dormancy and reawakening
Metastatic dormancy is likely to be induced when extracellular signal-regulated kinase (ERK) is inhibited by activation of p38 signalling, and to end when the ratio of ERK-to-p38 increases.39 It has been shown in a hydrogel model that the cyclin-dependent kinase (CDK) inhibitors p21 and p27, which are dormancy-associated markers that are regulated by ERK activity, tend to cluster in the cell nucleus in dormant cells as opposed to in the cytoplasm in proliferative BCBM cells.40 Several stimuli, such as VCAM, TGF-β1 and periostin, are required to awake dormant cells, and ECM stiffness can also contribute to the escape from dormancy.41 Active metastatic cells tend to form micrometastases along blood vessels through vascular co-option, preparing for continuous growth of metastatic lesions.17
Vascular co-option
Vascular co-option, during which BC cells maintain physical contact with the abluminal side of pre-existing blood vessels, is regarded as the first step of colonisation after extravasation. The successful growth of tumour cells in the brain has been shown to require adhesion to the surface of capillaries and stretching to form elongated clusters around the vessels along with capillary loop formation. The absence of vascular co-option can cause a failure of metastasis;17,22 thus, interfering with this step is an attractive therapeutic strategy.
Various key molecules and cells that are involved in promoting vascular co-option have been identified. For instance, BCBM cells secrete CCL2 to recruit macrophages, which subsequently produce oncostatin M (OSM) and interleukin (IL)-6 as promoting factors to activate lncRNA associated with BCBM (known as lnc-BM). This long non-coding RNA enhances vascular co-option through the Janus kinase 2 (JAK2)–signal transducer and activator of transcription 3 (STAT3)–ICAM pathway as well as antagonising apoptosis pathways in metastatic cells.5 The nervous system development-related factor L1 cell adhesion molecule (L1CAM), which is expressed in neurons and tumour cells, has been proven to facilitate the spread of BCBM cells to the parenchymal side of capillaries and eventually to establish metastatic outgrowth by activating downstream neuroserpin, which prevents the production of brain-metastasis-suppressing plasmin by astrocytes (see later).42 β1 Integrin on tumour cells can also enhance vascular co-option by mediating adhesion to the vascular basement membrane (BM).43
Metabolic reprogramming
For stable colonisation to occur, tumour cells need to alter their metabolic pattern to better adapt to the new microenvironments in which they find themselves. An increased level of fatty-acid-binding protein 7 (FABP7) in BCBM cells enables them to thrive in hostile environments through an enhanced “Warburg effect” and fatty-acid utilisation.44 In the event of glucose deprivation, the overexpression of GRP94 in BCBM cells leads to upregulation of the anti-apoptotic proteins B-cell lymphoma 2 (BCL2) and inhibitor of apoptosis proteins (IAPs) and engagement of autophagy to promote cancer cell survival.45 In parallel, gluconeogenesis is enhanced through the use of glutamine and branched chain amino acids for energy production.46 To counteract potentially abnormal oxygen metabolism and the consequent generation of reactive oxygen species (ROS), the transcription factor lymphoid enhancer-binding factor-1 (LEF1) supports brain metastatic colonisation by boosting the levels of glutathione to hinder ROS-mediated destruction in epithelial BC cells.47
Interactions between cancer cells and resident cells
As the survival and growth of metastases depend on adaptation to distant organ microenvironments, the interactions between BC cells and host cells merits further investigation (Fig. 3)—they could be considered as potential therapy targets for improving treatment efficacy.

The main host cells that interact with tumour cells in the brain include neurons, astrocytes and microglia. a Breast cancer cells that metastasis to the brain can develop a GABAergic or glutamatergic phenotype to take up GABA and glutamate for proliferation. b Interactions between astrocytes and cancer cells through gap-junction communication can activate the STING pathway in astrocytes as well as STAT1, NF-κB and AKT–MAPK pathway in cancer cells, and also upregulate GSTA5, BCL2L1 and TWIST1 in BC cells to enhance tumour growth and chemoresistance. Exosome-mediated secretion miRNAs downregulate the tumour suppressor PTEN. Other key molecule such as serpins, PPARγ, EGFR ligands and vital processes such as actin stress fibre organisation also promote BC cell colonisation. c Interactions between cancer cells, microglia and T cells can promote BCBM immune escape. Exosomal miR-503 regulated by lncRNA XIST in cancer cells prompted microglia M2 polarisation. M2-like phenotype promote BCBM invasion and colonisation in a Wnt-dependent manner and suppress T-cell proliferation and cytotoxic effect by immune-suppressive cytokines secretion.
Neurons
Neurons are regarded as the fundamental structural and functional units of the central nervous system (CNS). Neman et al.48 reported that BCBM cells and tissue derived from patients undergoing neurosurgical resection acquired brain-like properties, displaying a GABAergic phenotype similar to that of resident neurons, with high levels of the GABAA receptor and GABA transporter. The enhanced uptake and catabolism of GABA in BCBM cells promote the formation of NADH as a biosynthetic source and thus can confer a proliferative advantage on these cells. In another report, Zeng et al. observed that BC cells can take up glutamate for activation of N-methyl-D-aspartate (NMDA) receptors—required for metastatic colonisation of the brain and associated with poor prognosis—without destroying intrinsic connections between neurons through the formation of pseudo-tripartite synapses between cancer cells and glutamatergic neurons.49
Astrocytes
Astrocytes are the most abundant glial cells in the brain and are integral cells of the BBB. They ensheath the entire surface of microvessels and neuronal processes by extending polarised end-feet that contain a set of functional proteins, such as dystroglycan, dystrophin and aquaporin-4, and maintain BBB function by producing laminin α1 and α2 as well as secreting retinoic acid and sonic hedgehog.50 Interestingly, astrocytes initially seem to attack metastatic cells as a defensive mechanism; plasmin derived from the reactive brain stroma cleaves Fas ligand (FasL) on the surface of astrocytes, mobilising it to interact with its receptor, Fas, to induce apoptosis in tumour cells. However, plasminogen activator (PA) inhibitory serpins (e.g. neuroserpin and serpin B2), which are highly expressed on BCBM cells, subvert this process.42 The surviving BC cells can then exploit astrocytes through communication via gap junctions composed of connexin 43 (Cx43) and protocadherin 7 (PCDH7) to gain benefits in terms of growth, chemoresistance, stemness and autophagy.51 For instance, the movement of the second messenger cGAMP from tumour cells to astrocytes activates the STING pathway, triggering the secretion of interferon-α (IFN-α) and TNF-α by astrocytes; IFN-α and TNF-α then activate the STAT1 and nuclear factor κB (NF-κB) pathways in brain metastatic cells to promote tumour growth and chemoresistance.52 Gap-junction-mediated signalling between astrocytes and MDA-MB-231 cells also upregulates the expression of IL-6 and IL-8, leading to an increase in the production of endothelin-1 (ET-1) from astrocytes and the expression of the ET receptor on the tumour cells. The ET-1–ET receptor interaction activates the AKT–mitogen-activated protein kinase (MAPK) pathway in cancer cells to confer survival against taxol.53 Through gap-junction-mediated intercellular contact, astrocytes are also able to induce the upregulation of certain pro-survival genes, such as GSTA5, BCL2L1 and TWIST1, in BC cells to enhance chemoresistance.54 As well as gap junctions, exosome-mediated secretion by astrocytes has been proven to facilitate brain metastasis. Zhang et al.55 reported that exosomes derived from astrocytes transfer microRNAs that target the tumour suppressor phosphatase and tensin homologue (PTEN) into brain metastatic tumour cells. Furthermore, astrocytes can release polyunsaturated fatty acids to activate peroxisome proliferator-activated receptor γ (PPARγ) in BCBM cells and thereby promote proliferation.56 Moreover, astrocytes can alter the morphology of BCBM cells by remodelling their cytoskeleton, enhancing actin stress fibre organisation and inducing cell elongation to increase the migration potential.57 And, finally, in response to oestrogens in the brain, oestrogen-receptor (ER)+ astrocytes upregulate the production and secretion of EGFR ligands to promote EGFR activation in BMBC cells, increasing their proliferation, migration and invasion.58
Microglia
Microglia are unique resident macrophages in the brain parenchyma that play a vital role in various CNS diseases such as stroke, cancer and Alzheimer’s disease. Microglia have two activation states that have opposing roles in brain metastases; an M1-like phenotype, which promotes BBB opening to facilitate leukocyte extravasation; and an M2-like phenotype, which participates in angiogenesis and immunosuppression to promote tumour progression.59 When activated after physical contact with tumour cells, microglia promote the accumulation in these cells of ROS, leading to ROS-induced apoptosis or other oxidative damage as an immune defence; however, these effects can be mitigated by the expression in CTCs of high levels of MYC, which induces the expression of the antioxidant glutathione peroxidase 1 (GPX1).15 Similar to other immune cells, microglia also carry out cytotoxic functions; however, the elevated secretion of neurotrophin-3 (NT-3) from brain metastatic cells can protect them from this attack, as well as promoting reversion of EMT to mesenchymal-to-epithelial transition (MET) to enable metastatic growth by upregulating E-cadherin at intercellular junctions.60 On the other hand, microglia can promote BCBM invasion and colonisation in a Wnt-dependent manner.61 Moreover, significant downregulation of the brain metastatic lncRNA suppressor X-inactive-specific transcript (XIST) in metastatic tumour cells leads to augmented secretion by these cells of exosomal miRNA-503, which triggers M2 polarisation and the accumulation in microglia of immune-suppressive cytokines, subsequently inhibiting T-cell proliferation.62
Research models for Bcbm
The paucity of optimal and clinically relevant models presents a huge obstacle to the study of the mechanisms underlying BCBM and thus the development of possible treatments. However, several models, including cell, murine and PDX models (summarised in Table 1), have been widely implemented, providing a theoretical basis and methodological guides for further BCBM studies.
Cell models
HER2+ breast cancer and TNBC (ER−PR−HER2−) phenotypes are prone to metastasis to the brain; thus, cell models with these phenotypes have been widely used in BCBM research. TBCP-1 cells, derived from a spontaneous BALB/c mouse mammary tumour, naturally overexpress HER2; these cells maintain HER2+ status in vivo, and show a high tendency toward spontaneous and experimental brain metastasis in a mouse model.63 The human HER2+ BT474 (BT474BR) cell lines is also been widely used.64 In terms of TNBC cell lines, 4T1Br4 is a mouse-derived cell line that displays a higher selective and spontaneous brain-metastasis tendency compared to parental 4T1 cell lines.65 The human-derived TNBC brain-metastasis cell line, MDA-MB-231-BR, was constructed via repeated rounds of intraarterial injection and in vivo selection.34 A new brain metastatic cell model, known as BR-eGFP-CMV/Luc-V5 (BRV5), based on a human-derived 435-Br1 model, which uses a brain-metastasis nude mouse inoculated with MDA-MB-435 cells,66 was developed by passing 435-Br1 cells expressing the enhanced green fluorescent protein (eGFP) gene and Photinus luciferase (PLuc) gene through five rounds of in vivo and in vitro selection. These cells have been shown to undergo brain metastasis within 60.8 ± 13.8 days after intracarotid injection in all mice.67
Murine models
Three main types of BCBM murine models exist: orthotopic models, intracarotid/intraventricular models and stereotactic intracranial injection models.
Orthotopic models
Orthotopic models, created by the orthotopic injection of breast cancer cell lines into the mammary fat pad or mammary gland, can imitate the natural course of BCBM.28,68 However, spontaneous brain metastasis in these models is often accompanied by multiorgan metastases. Therefore, intracarotid/intraventricular models are now commonly used in the study of BCBM study.
Intracarotid/intraventricular models
Intracarotid/intraventricular models are constructed by injecting mammary carcinoma cell lines into the carotid artery or left cardiac ventricle of 6–8-week-old BALB/c nude or CB17/SCID female mice, images of isolated whole brains are obtained or samples are harvested from the brain at different time points after inoculation.68,69,70 However, intracardiac injection can also cause cancer cells to systemically spread to other organs, whereas injection to the intracarotid artery can reduce systematic distribution.
Stereotactic intracranial injection models
In 2019, Delaney et al. reported the generation of a novel murine model in which cancer cells are seeded into different sites of nude mouse brains, such as the hemisphere or cortex, by stereotactic injection.71 The advantage of this model is that the phenotypes and biologic features of the metastatic foci are consistent with those of the injected cells. However, strong microsurgery skills and experience are required for this approach.
PDX models
Patient-derived xenograft (PDX) models, which can better mimic the heterogeneity of human tumours, have been successfully used in BCBM research. These PDX models, which can be orthotopic or ectopic, depending on the transplant site, can retain the genetic, transcriptional and phenotypic properties of primary tumours even after long-term continuous passage in vivo.72 Oshi et al. reported three approaches for constructing orthotopic PDX models: the forceps method, the needle method and the pipette method. The pipette method, in which a gently crushed tumour fragment is implanted through a burr hole using a 10 μl plastic tip connected to a pipette, appears to be the more favoured among the three.72 Generating ectopic PDX models is relatively simple: freshly resected brain metastases from consenting donors are partitioned into small pieces and implanted into the mammary fat pads of immunocompromised mice.73
Current therapeutic strategies and challenges in the management of Bcbm
Traditional therapeutic strategies for BCBM involve multimodality approaches, such as whole-brain radiotherapy (WBRT), stereotactic radiosurgery (SRS), surgery, chemotherapy and palliative therapy,4,74 while treatment options for intra-cerebrospinal fluid (CSF) administration for leptomeningeal metastases are limited to methotrexate, cytarabine and thio-TEPA; these agents show poor efficacy, and survival after diagnosis is disappointingly short (2–3 months).75 Multidisciplinary comprehensive therapy has become the current preferred treatment. For example, results from a randomised open-label Phase 3 study (NCT01645839) suggested that intrathecal liposomal cytarabine plus systemic therapy significantly decreased the risk of leptomeningeal metastases progression or death, and prolonged progression free survival (PFS) from 2.2 to 3.8 months and median overall survival (OS) from 4 to 7.3 months.76 However, the prognosis of patients with BCBM is far from satisfactory. This frustrating outcome can be partly attributed to our incomplete understanding of the brain structure as well as difficulties in generating research models during BCBM investigation. Several additional challenges also limit the success of current BCBM treatment approaches.
Heterogeneity between primary BC and BCBM
Heterogeneity between the primary tumour and the brain metastasis is a major obstacle for effective treatment. Traditionally, various aspects of the heterogeneity of BC, such as histological subtype, treatment sensitivity and clinical outcome, have been documented, as have aspects of the heterogeneity of their resulting brain metastases. BCBM also has its own hormone receptor (HR) and HER2 expression statuses that are not completely consistent with those of the primary tumour. Moreover, molecular subtype discordances and changes, as well as genetic heterogeneity between the primary BC and BCBM, can significantly influence treatment and prognosis,77 limiting the potential efficacy of targeted therapies that are based on the analysis of the primary tumour. HR switching rates—whether positive-to-negative or negative-to-positive—were reported to be higher than HER2 switching rates. ER discordance is statistically more prevalent, while progesterone receptor discordance is less common in CNS metastases than other sites. By contrast, HR−/HER2+ primary tumours have the lowest probability of subtype switching, probably owing to their strong neurotropic properties,78 since HER2+ phenotype itself is reported to retain high brain-metastasis tendency. Genetic mutations—mostly in cancer-related genes such as TP53, FGFR2, MAP2K4, ATR and PIK3CA—can be specific to or enriched in BCBM. Furthermore, amplifications of clinically actionable target genes such as ERBB2 and myeloid leukaemia cell differentiation protein (MCL1) are also common in brain-tropic metastatic cells. These molecular features make the cells sensitive to corresponding inhibitors, and preclinical research on the application of HER2/EGFR inhibitors, CDK inhibitors, and MAPK inhibitors in suppressing brain metastatic cells is underway.79,80 The heterogeneity of genomic alterations can therefore be clinically targetable.
Presence of the BBB and additional barriers
Another critical limitation in BCBM treatment is largely attributable to the existence of the BBB, which selectively inhibits agents of high molecular weight, strong negative charge and low lipid solubility.81 In parallel, the expression of ATP-binding cassette (ABC) transporters, especially P-gp (ABCB1), MRP1 (ABCC1) and BCRP (ABCG2), expressed both on tumour cells and in the BBB, as drug efflux pumps has been proven to play a crucial role in multidrug-resistance.82 Along with the BBB, the blood-CSF (BCSFB) is another gatekeeper of the CNS system; its function is principally a result of the properties of the choroidal epithelial cells.83 The BCSFB has been reported to express P-gp and MRP1, which thus brings challenges in anti-metastatic therapy.83,84 The BTB also, presents a challenge for BCBM treatment, displaying non-uniform permeability and active efflux of molecules.85 The aforementioned factors account for the limited curative effect of current therapeutic agents for BCBM compared to metastases of peripheral organs. Below, we discuss novel therapeutic strategies such as targeted therapies, immunotherapy and nanotherapy, and the prospects of these strategies in treating BCBM.
Novel therapies and prospects for BCBM
An urgent need exists for novel therapeutic options for BCBM patients due to their poor QoL and prognosis. Our increasing understanding of the molecular mechanisms underlying BCBM is providing potential opportunities for targeted therapies. Several key molecules and pathways identified as potential new targets have entered into clinical trials with the aim of achieving translation from basic study to clinical practice (Fig. 4). Moreover, the emergence of novel strategies such as immunotherapy and nanotherapy has changed the treatment approach and the formulation of existing compounds, respectively, leading to clinical improvements in the treatment of BCBM.

Increased research into the molecular mechanisms of breast cancer brain metastasis has identified that, key molecule inhibitors such as PARP inhibitors, cathepsin S inhibitors, HER2/ERBB inhibitors, EGFR inhibitors; and pathway inhibitors such as VEGF pathway inhibitors, PI3K–Akt–mTOR pathway inhibitors and CDK4/6 pathway inhibitors, play vital roles and that have become target of study in preclinical research or clinical trials.
Targeted therapy
HER2/ERBB2 inhibitors
As noted above, HER2+ BC patients are at great risk of BCBM. Numerous anti-HER2 drugs have been explored for clinical use: monoclonal antibodies (e.g. trastuzumab, pertuzumab); antibody–drug conjugates (e.g. T-DM1); or small molecule tyrosine kinase inhibitors (e.g. lapatinib, neratinib, tucatinib, pyrotinib). The advantages of trastuzumab, a standard therapeutic option in clinical practice, in improving HER2+ BCBM outcomes have been widely verified, although it is unclear whether or not the benefits are due to the improvement in extracranial disease control rather than a direct effect on brain metastases.86 Pertuzumab has also been approved for the treatment of patients with advanced HER2+ BC. Significant efficacy of the combination of pertuzumab, trastuzumab and the antimitotic drug docetaxel has been reported in the CLEOPATRA study group, although neutropenia occurred in 49% of the experimental group as a common serious adverse event.87 Studies on novel HER2-targeted therapies have reported promising outcomes. Trastuzumab emtansine (T-DM1) performed better than capecitabine plus lapatinib in terms of efficacy and safety, in a Phase 3 study of 991 patients with previously treated HER2+ MBC,88 leading to the FDA approval of T-DM1 in 2013. A single-arm Phase 3b clinical trial (NCT01702571) evaluating T-DM1 in patients with HER2+ MBC and brain metastases revealed that T-DM1 is efficacious and well-tolerated.89
With regard to small molecule tyrosine kinase inhibitors, several agents are under exploration or in use in clinical application. Lapatinib seems to inhibit efflux transporters on BBB,90 which makes it more efficient when combined with chemotherapy agents. A Phase 1b/2 single-arm trial verified the efficacy of a combination of lapatinib and capecitabine as a first-line therapy for untreated HER2+ BCBM, although nearly half of patients in the experimental treatment group experienced grade 3–4 adverse events such as diarrhoea and hand-foot syndrome, and 31% suffered severe side effects.91 Neratinib plus capecitabine has recently shown efficacy against refractory HER2+ BCBM in a Phase 2 trial.92 Moreover, tucatinib, an oral HER2-specific tyrosine kinase inhibitor, conferred favourable effects on the treatment of brain metastases when combined with trastuzumab and capecitabine;93 adverse effects included nausea, diarrhoea, fatigue and elevated aminotransferase levels. Pyrotinib, an oral irreversible pan-ERBB inhibitor targeting HER1, HER2 and HER4, has emerged as a promising agent for the treatment of HER2+ MBC, following a series of clinical trials. A first-in-patient study (NCT01937689) of 38 HER2+ MBC patients determined the maximum tolerated dose of pyrotinib to be 400 mg once per day; the overall response rate was higher (87.5%) for the 400-mg dose cohort than other low-dose cohorts, and the median PFS was also longer (59.4 weeks) for the 400-mg dose cohort compared with other, lower-dose groups.94 In a Phase 3 trial, the median PFS for the pyrotinib plus capecitabine group was 11.1 months, which was significantly longer than that of only 4.1 months for the placebo group.95 Based on these positive results, pyrotinib received conditional approval in combination with capecitabine in 2018 in China for the treatment of HER2+ MBC patients who previously received anthracycline or taxane chemotherapy.96 Adverse events such as gastrointestinal reactions, skin reaction, blood system diseases and hepatobiliary diseases were also observed in these clinical trials.94 In 2020, investigators reported that the novel biparatopic anti-HER2 antibody–tubulysin conjugate (bHER2-ATC) inhibited tumour growth in a murine BCBM model; increased uptake of the agent was shown to be mediated by endocytosis, suggesting a potential new mechanism of brain penetration.97
Overall, the combination of HER2-targeted inhibitors and chemotherapeutic cytotoxic agents has achieved some success in clinical studies. However, the instability of drug effects and frequent adverse events are a reminder that more attention and efforts are needed to improve drug efficacy and reduce toxicity.
EGFR inhibitors
There are two main groups of EGFR-targeted drugs—EGFR-targeted monoclonal antibodies (e.g. cetuximab, panitumumab) and tyrosine kinase inhibitors (e.g. erlotinib, gefitinib, afatinib). However, EGFR-targeted treatment has not shown a clear clinical benefit for BC and much less so for BCBM; this lack of clinical benefit might be explained by the presence of alternate mechanisms for EGFR pathway activation. A randomised Phase 2 study suggested that treatment with afatinib does not confer therapeutic benefits, and resulted in even more frequent adverse events than the investigator’s choice of treatment for HER2+ BCBM patients.98
PARP inhibitors
Poly (ADP-ribose) polymerase (PARP) inhibitors, including olaparib, rucaparib, niraparib, veliparib and talazoparib, have been approved for the treatment of BRCA-mutated BC, and several clinical trials (Phase 1: NCT01853306; Phase 2: NCT01506609; Phase 3: NCT02032277) have shown the efficacious anti-tumour effect of PARP inhibitors on BC.99,100,101 A study analysing 21 brain metastases and their matched primary breast cancer showed that BCBM tends to have high genomic-aberration-based-homologous recombination deficiency (HRD) scores compared with their primary tumour counterparts, which indicates that brain metastases might be more sensitive to PARP inhibitors than their corresponding primary tumours.69
VEGF pathway inhibitors
VEGF and its receptors (VEGFR-1 and VEGFR-2) are highly relevant owing to the involvement of angiogenesis in primary BC and BCBM. The expression level of VEGF has been associated with poor prognosis of BC and poor response to endocrine therapy and chemotherapy,102 indicating the potential use of VEGF inhibitors. However, the efficacy and usage of bevacizumab, a humanised monoclonal antibody against VEGF-A, in BCBM is controversial. Despite the removal of Food and Drug Administration (FDA) approval of bevacizumab for the treatment of MBC in November 2011, the Centers for Medicare and Medicaid Services still indicate the use of bevacizumab as a first-line treatment for metastatic BC patients, on the basis of controversial results of clinical trials.103 Hence, more appropriately designed research strategies are needed to prove the effectiveness of bevacizumab. Ramucirumab, a human immunoglobulin G1 antibody that binds to VEGFR-2, is used to treat some advanced digestive malignancies. However, it did not confer significant benefit when added to docetaxel in the treatment of advanced MBC in a Phase 3 trial.104 Interestingly, variants of the antibody herceptin that can bind not only to HER2 but also to VEGF have been isolated, providing new opportunities for antibody-based therapy;105 a ‘four-in-one’ antibody approach targeting EGFR, HER2, HER3 and VEGF106 has also proven efficacious in inhibiting the growth of anti-HER-resistant cancer cells. These experiments indicate that further preclinical explorations and clinical trials are needed to find more effective and safe strategies for anti-VEGF therapy in BCBM treatment.
PI3K–Akt–mTOR pathway inhibitors
Activation of the PI3K pathway is the most common aberration during BC development and occurs in ~70% of BCBM cases, indicating that inhibition of this pathway could be a promising therapeutic option for BCBM patients.107 Accordingly, the mammalian target of rapamycin (mTOR) inhibitor everolimus in combination with the PI3K inhibitor buparlisib is being evaluated in the clinical management of BCBM.108 GDC-0084, a dual PI3K–mTOR inhibitor that can penetrate the BBB, showed a potential role in BCBM treatment when evaluated in in vitro and in vivo experiments.107 Another preclinical study of a pan-Akt inhibitor, GDC-0068, showed encouraging efficacy results in BCBM treatment.109 Moreover, a Phase 1b/2 multicentre clinical trial (NCT01783756) supported the anti-tumour effect of the combination of everolimus, lapatinib and capecitabine in HER2+ BCBM patients.99 These promising data reflect the requirement for further future clinical trials investigating PI3K–Akt–mTOR pathway inhibitors in BCBM.
CDK4/6 pathway inhibitors
Cyclin D1 is frequently overexpressed in human BC, and the vital role of the cyclin D–CDK4/6–retinoblastoma protein pathway in BC cell proliferation provides a strong rationale for the use of CDK4/6 inhibitors.110 Three CDK4/6 inhibitors have been used in ER+ MBC treatment, including palbociclib, ribociclib and abemaciclib. Among these inhibitors, abemaciclib has shown unique advantages, including single agent activity in ER+ refractory MBC, efficient ability to cross the BBB, and capacity for continuous administration due to the low frequency of myelosuppression.111 A Phase 1 clinical trial (NCT01394016) revealed no significant differences in the concentration of abemaciclib between the CSF and plasma,112 indicating effective penetrance of the BBB. Furthermore, Tolaney et al. report on the results of a Phase 2 clinical trial (NCT02308020) that further assesses the efficacy of abemaciclib in brain metastasis in patients who were heavily treated previously. Therapeutic concentrations of abemaciclib were achieved in the brain and outdistanced those necessary for CDK4 and CDK6 inhibition. And an intracranial clinical benefit rate (iCBR) of 24% was observed.113 Far fewer, clinical trials have assessed the efficacy of palbociclib and ribociclib in BCBM, indicating that more investigations and further explorations are needed.
Immunotherapy
Immunotherapy has shown considerable success in a number of different types of cancer. The immune content of metastases is reported to be lower than that of primary tumours, but BCBM with a higher TIL content show improved prognosis compared with BCBM with a lower TIL content. Thus, improving the TIL contents by the use of stereotactic radiosurgery (SRS) has become a new approach used in conjunction with immune checkpoint inhibitors.4 Several ongoing Phase 2 trials are underway to examine the efficacy of combining SRS with atezolizumab (NCT03483012), pembrolizumab (NCT03449238), and nivolumab (NCT03807765).114 In the meantime, chimeric antigen receptor (CAR)-modified immune cells and oncolytic viruses are also under investigation. For example, combinatorial therapy using EGFR–CAR NK cells and oncolytic herpes simplex virus 1 (oHSV-1) resulted in efficient killing of BC cells and significantly lengthened the survival of tumour-bearing mice.115 HER2–CAR T cells containing the 4–1BB costimulatory domain optimised by Priceman et al. conferred enhanced tumour targeting with a reduced T-cell exhaustion phenotype and enhanced proliferative capacity compared with the CD28 costimulatory domain, and showed potent anti-tumour activity in BCBM orthotopic xenograft models.116 As for leptomeningeal metastasis, a single-arm, open-label Phase 2 trial (NCT02886585) of pembrolizumab demonstrated the safety and feasibility of this immune checkpoint inhibitor as well as its prospective activity.117 Although immunotherapy has good prospects, this approach is still in its infancy for treating BCBM and requires further exploration.
Nanotherapy
As mentioned above, systematic treatment for BCBM is not as efficacious as it is for peripheral metastases owing to the presence of the BBB. Thus, looking for ways to improve the penetration of existing medicines has become an area of active research. Nanotherapy, an emerging approach for intravenous drug delivery whereby nanoparticles are conjugated to therapeutic agents targeting overexpressed antigens or receptors, might be an optimal system with a promising future. Nanocarriers have unique advantages for drug delivery: they have a high drug-loading capacity and they can protect the enclosed agents from overly rapid clearance. Consequently, a concentration gradient is formed, allowing the drugs to diffuse passively through tumour microvessels—this is called the enhanced permeability and retention (EPR) effect of nanoparticles. Moreover, nanocarriers help to circumvent the effect of efflux transporters.118
Five nanoparticles currently exist for brain cancer therapy: liposomes (60–500 nm), micelles (10–60 nm), dendrimers (5–250 nm), polymers (10–60 nm) and SPIO (iron oxide oleic acid amphiphilic polymers, 5–30 nm).118 Among them, polymer nanoparticles are commonly used in BCBM research. Chunsheng He and colleagues developed a polysorbate (PS)-80-based amphiphilic polymer–lipid nanoparticle system to carry docetaxel to brain metastatic lesions, and this prolonged median survival in mouse models of BCBM. These PS-80-coated nanoparticles can mimic low-density lipoprotein (LDL) particles to participate in LDL-receptor-mediated transcytosis, a feasible drug delivery approach via the binding between the antibody–drug conjugates and their respective receptors. They can facilitate BBB crossing by undergoing endogenous lipidation in cooperation with apolipoprotein E.119 In a preclinical study published in 2020, Zhang et al. synthesised a shrinkable polymer nanoparticle loaded with lexiscan and conjugated with the specific CXCR4 antagonist AMD3100. It efficiently crossed and pharmacologically disrupted the BBB in tumours through size alteration as well as lexiscan-mediated autocatalysis.120 Autocatalysis is a nanoparticle delivery strategy in brain tumour treatment. In this strategy, a small number of nanoparticles primarily penetrate the BBB via transcytosis or through the BBB gaps, and then release BBB modulators to enables more nanoparticles to be transported, creating a positive feedback loop for increased delivery.121 Loaded with lexiscan, an adenosine receptor agonist, this nanoparticle can pharmacologically modulate permeability of the BBB, and it specifically targeted tumour tissues via AMD3100-mediated recognition of CXCR4 on tumour cells. When this nanoparticle was used to encapsulate doxorubicin, an improvement in survival outcome was achieved in BCBM-bearing mice.120 However, no clinical data yet support the idea that nanotherapy is superior to current treatment regimens, and clinical trials of nanotherapy in BCBM treatment are rare, so the adverse effects are unknown—indicating that the journey to find optimal nanocarriers to facilitate BBB permeability will be arduous.
Conclusions
In this review, we have comprehensively discussed the current knowledge of BCBM. This knowledge has grown exponentially over the past 5 years, benefiting from emerging research experiments and models. The process of BCBM is very complicated, and includes CTCs adhesion, BBB crossing, tumour colonisation and proliferation, as well as involving cellular BBB structures, diverse molecules and pathways; consequently, a multitude of targets exist for future therapeutic strategies. However, based on the complexity of the BBB and the diversity of the molecules involved, combination therapies and optimal drug carriers, such as nanocarriers, are likely to be required for the efficacious treatment of BCBM, although further clinical trials will obviously be needed.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30 (2020).
Liang, Y., Zhang, H., Song, X. & Yang, Q. Metastatic heterogeneity of breast cancer: molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 60, 14–27 (2020).
Kanchan, R. K., Siddiqui, J. A., Mahapatra, S., Batra, S. K. & Nasser, M. W. microRNAs orchestrate pathophysiology of breast cancer brain metastasis: advances in therapy. Mol. Cancer 19, 29 (2020).
Ren, D., Cheng, H., Wang, X., Vishnoi, M., Teh, B. S., Rostomily, R. et al. Emerging treatment strategies for breast cancer brain metastasis: from translational therapeutics to real-world experience. Ther. Adv. Med. Oncol. 12, 1758835920936151 (2020).
Wang, S., Liang, K., Hu, Q., Li, P., Song, J., Yang, Y. et al. JAK2-binding long noncoding RNA promotes breast cancer brain metastasis. J. Clin. Invest. 127, 4498–515. (2017).
Custodio-Santos, T., Videira, M. & Brito, M. A. Brain metastasization of breast cancer. Biochim. Biophys. Acta Rev. Cancer 1868, 132–47. (2017).
Wilhelm, I., Molnár, J., Fazakas, C., Haskó, J. & Krizbai, I. Role of the blood-brain barrier in the formation of brain metastases. Int. J. Mol. Sci. 14, 1383–1411 (2013).
Sonoshita, M., Aoki, M., Fuwa, H., Aoki, K., Hosogi, H., Sakai, Y. et al. Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell 19, 125–137 (2011).
Friedl, P. & Alexander, S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009 (2011).
Leblanc, R. & Peyruchaud, O. Metastasis: new functional implications of platelets and megakaryocytes. Blood 128, 24–31 (2016).
Papadaki, M., Koutsopoulos, A., Tsoulfas, P., Lagoudaki, E., Aggouraki, D., Monastirioti, A. et al. Clinical relevance of immune checkpoints on circulating tumor cells in breast cancer. Cancers. 12, 376 (2020).
Chan I. S., Knutsdottir, H., Ramakrishnan, G., Padmanaban, V., Warrier, M., Ramirez, J. C. et al. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J. Cell Biol. 219, 9 (2020).
Douma, S., Van Laar, T., Zevenhoven, J., Meuwissen, R., Van Garderen, E. & Peeper, D. S. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature 430, 1034–1039 (2004).
Hoshino, A., Costa-Silva, B., Shen, T. L., Rodrigues, G., Hashimoto, A., Tesic Mark, M. et al. Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335 (2015).
Klotz, R., Thomas, A., Teng, T., Han, S. M., Iriondo, O., Li, L. et al. Circulating tumor cells exhibit metastatic tropism and reveal brain metastasis drivers. Cancer Discov. 10, 86–103 (2020).
Chung, B., Esmaeili, A., Gopalakrishna-Pillai, S., Murad, J., Andersen, E., Kumar Reddy, N. et al. Human brain metastatic stroma attracts breast cancer cells via chemokines CXCL16 and CXCL12. npj Breast Cancer 3, 6 (2017).
Kienast, Y., von Baumgarten, L., Fuhrmann, M., Klinkert, W. E., Goldbrunner, R., Herms, J. et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 16, 116–122 (2010).
Kang, S. A., Hasan, N., Mann, A. P., Zheng, W., Zhao, L., Morris, L. et al. Blocking the adhesion cascade at the premetastatic niche for prevention of breast cancer metastasis. Mol. Ther. 23, 1044–1054 (2015).
Geng, Y., Yeh, K., Takatani, T. & king, M. R. Three to tango: MUC1 as a ligand for both E-selectin and ICAM-1 in the breast cancer metastatic cascade. Front. Oncol. 2, 76 (2012).
Soto, M. S., Serres, S., Anthony, D. C. & Sibson, N. R. Functional role of endothelial adhesion molecules in the early stages of brain metastasis. Neuro Oncol. 16, 540–551 (2014).
Daneman, R. & Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 7, a020412 (2015).
Lorger, M. & Felding-Habermann, B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am. J. Pathol. 176, 2958–2971 (2010).
Winger, R. C., Koblinski, J. E., Kanda, T., Ransohoff, R. M. & Muller, W. A. Rapid remodeling of tight junctions during paracellular diapedesis in a human model of the blood-brain barrier. J. Immunol. (Baltimore, Md: 1950) 193, 2427–2437 (2014).
Lyle, L., Lockman, P., Adkins, C., Mohammad, A., Sechrest, E., Hua, E. et al. Alterations in pericyte subpopulations are associated with elevated blood-tumor barrier permeability in experimental brain metastasis of breast cancer. Clin. Cancer Res. 22, 5287–5299 (2016).
Lockman, P., Mittapalli, R., Taskar, K., Rudraraju, V., Gril, B., Bohn, K. et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res. 16, 5664–5678 (2010).
Pedrosa, R., Mustafa, D. A., Soffietti, R. & Kros, J. M. Breast cancer brain metastasis: molecular mechanisms and directions for treatment. Neuro Oncol. 20, 1439–49. (2018).
Yang, Y., Estrada, E. Y., Thompson, J. F., Liu, W. & Rosenberg, G. A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 27, 697–709 (2007).
Avraham, H. K., Jiang, S., Fu, Y., Nakshatri, H., Ovadia, H. & Avraham, S. Angiopoietin-2 mediates blood-brain barrier impairment and colonization of triple-negative breast cancer cells in brain. J. Pathol. 232, 369–381 (2014).
Rodriguez, P., Jiang, S., Fu, Y. & Avraham, S. The proinflammatory peptide substance P promotes blood-brain barrier breaching by breast cancer cells through changes in microvascular endothelial cell tight junctions. Int. J. Cancer 134, 1034–1044 (2014).
Sevenich, L., Bowman, R. L., Mason, S. D., Quail, D. F., Rapaport, F., Elie, B. T. et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat. Cell Biol. 16, 876–888 (2014).
Zhou, W., Fong, M. Y., Min, Y., Somlo, G., Liu, L., Palomares, M. R. et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 25, 501–515 (2014).
Lu, Y., Chen, L. & Li, L. Exosomes derived from brain metastatic breast cancer cells destroy the blood-brain barrier by carrying lncRNA GS1-600G8.5. BioMed Research International 2020,1-10 (2020).
Lee, B. C., Lee, T. H., Avraham, S. & Avraham, H. K. Involvement of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1alpha in breast cancer cell migration through human brain microvascular endothelial cells. Mol. Cancer Res. 2, 327–338 (2004).
Bos, P. D., Zhang, X. H., Nadal, C., Shu, W., Gomis, R. R., Nguyen, D. X. et al. Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009 (2009).
Conrad, C., Gotte, M., Schlomann, U., Roessler, M., Pagenstecher, A., Anderson, P. et al. ADAM8 expression in breast cancer derived brain metastases: functional implications on MMP-9 expression and transendothelial migration in breast cancer cells. Int. J. Cancer 142, 779–791 (2018).
Mustafa, D., Pedrosa, R., Smid, M., van der Weiden, M., de Weerd, V., Nigg, A. et al. T lymphocytes facilitate brain metastasis of breast cancer by inducing Guanylate-Binding Protein 1 expression. Acta Neuropathol. 135, 581–599 (2018).
Choi, Y., Lee, J., Gao, M., Kim, B., Kang, S., Kim, S. et al. Cancer-associated fibroblast promote transmigration through endothelial brain cells in three-dimensional in vitro models. Int. J. Cancer 135, 2024–2033 (2014).
Krizbai, I. A., Gasparics, A., Nagyoszi, P., Fazakas, C., Molnar, J., Wilhelm, I. et al. Endothelial-mesenchymal transition of brain endothelial cells: possible role during metastatic extravasation. PLoS ONE 10, e0119655 (2015).
Sosa, M., Avivar-Valderas, A., Bragado, P. & Wen, H. ERK1/2 and p38α/β signaling in tumor cell quiescence: opportunities to control dormant residual disease. Clin. Cancer Res. 17, 5850–5857 (2011).
Narkhede, A., Crenshaw, J., Crossman, D. & Shevde, L. An in vitro hyaluronic acid hydrogel based platform to model dormancy in brain metastatic breast cancer cells. Acta Biomater. 107, 65–77 (2020).
Sosa, M. & Bragado, P. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat. Rev. Cancer 14, 611–622 (2014).
Valiente, M., Obenauf, A. C., Jin, X., Chen, Q., Zhang, X. H., Lee, D. J. et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156, 1002–1016 (2014).
Carbonell, W. S., Ansorge, O., Sibson, N. & Muschel, R. The vascular basement membrane as “soil” in brain metastasis. PLoS ONE 4, e5857 (2009).
Cordero, A., Kanojia, D., Miska, J., Panek, W. K., Xiao, A., Han, Y. et al. FABP7 is a key metabolic regulator in HER2+ breast cancer brain metastasis. Oncogene 38, 6445–60. (2019).
Santana-Codina, N., Muixí, L., Foj, R., Sanz-Pamplona, R., Badia-Villanueva, M., Abramowicz, A. et al. GRP94 promotes brain metastasis by engaging pro-survival autophagy. Neuro Oncol. 22, 652–64. (2020).
Chen, J., Lee, H. J., Wu, X., Huo, L., Kim, S. J., Xu, L. et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 75, 554–565 (2015).
Blazquez, R., Rietkotter, E., Wenske, B., Wlochowitz, D., Sparrer, D., Vollmer, E. et al. LEF1 supports metastatic brain colonization by regulating glutathione metabolism and increasing ROS resistance in breast cancer. Int. J. Cancer. 146, 3170–3183 (2019).
Neman, J., Termini, J., Wilczynski, S., Vaidehi, N., Choy, C., Kowolik, C. M. et al. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl Acad. Sci. USA 111, 984–989 (2014).
Zeng, Q., Michael, I. P., Zhang, P., Saghafinia, S., Knott, G., Jiao, W. et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 573, 526–31. (2019).
Sweeney, M. D., Zhao, Z., Montagne, A., Nelson, A. R. & Zlokovic, B. V. Blood-brain barrier: from physiology to disease and back. Physiol. Rev. 99, 21–78 (2019).
Kaverina, N., Borovjagin, A. V., Kadagidze, Z., Baryshnikov, A., Baryshnikova, M., Malin, D. et al. Astrocytes promote progression of breast cancer metastases to the brain via a KISS1-mediated autophagy. Autophagy 13, 1905–23. (2017).
Chen, Q., Boire, A., Jin, X., Valiente, M., Er, E. E., Lopez-Soto, A. et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 (2016).
Kim, S. W., Choi, H. J., Lee, H. J., He, J., Wu, Q., Langley, R. R. et al. Role of the endothelin axis in astrocyte- and endothelial cell-mediated chemoprotection of cancer cells. Neuro Oncol. 16, 1585–1598 (2014).
Kim, S. J., Kim, J. S., Park, E. S., Lee, J. S., Lin, Q., Langley, R. R. et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia 13, 286–298 (2011).
Zhang, L., Zhang, S., Yao, J., Lowery, F. J., Zhang, Q., Huang, W. C. et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527, 100–104 (2015).
Zou, Y., Watters, A., Cheng, N., Perry, C. E., Xu, K., Alicea, G. M. et al. Polyunsaturated fatty acids from astrocytes activate PPARgamma signaling in cancer cells to promote brain metastasis. Cancer Discov. 9, 1720–35. (2019).
Shumakovich, M. A., Mencio, C. P., Siglin, J. S., Moriarty, R. A., Geller, H. M., Stroka, K. M. Astrocytes from the brain microenvironment alter migration and morphology of metastatic breast cancer cells. FASEB J. 31, 5049–5067 (2017).
Sartorius, C. A., Hanna, C. T., Gril, B., Cruz, H., Serkova, N. J., Huber, K. M. et al. Estrogen promotes the brain metastatic colonization of triple negative breast cancer cells via an astrocyte-mediated paracrine mechanism. Oncogene 35, 2881–2892 (2016).
Dudvarski Stankovic, N., Teodorczyk, M., Ploen, R., Zipp, F. & Schmidt, M. H. H. Microglia-blood vessel interactions: a double-edged sword in brain pathologies. Acta Neuropathol. 131, 347–363 (2016).
Louie, E., Chen, X. F., Coomes, A., Ji, K., Tsirka, S. & Chen, E. I. Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene 32, 4064–4077 (2013).
Pukrop, T., Dehghani, F., Chuang, H. N., Lohaus, R., Bayanga, K., Heermann, S. et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia 58, 1477–1489 (2010).
Xing, F., Liu, Y., Wu, S. Y., Wu, K., Sharma, S., Mo, Y. Y. et al. Loss of XIST in breast cancer activates MSN-c-Met and reprograms microglia via exosomal miRNA to promote brain metastasis. Cancer Res. 78, 4316–30. (2018).
Nagpal, A., Redvers, R. P., Ling, X., Ayton, S., Fuentes, M., Tavancheh, E. et al. Neoadjuvant neratinib promotes ferroptosis and inhibits brain metastasis in a novel syngeneic model of spontaneous HER2(+ve) breast cancer metastasis. Breast Cancer Res. 21, 94 (2019).
Contreras-Zarate, M. J., Ormond, D. R., Gillen, A. E., Hanna, C., Day, N. L., Serkova, N. J. et al. Development of novel patient-derived xenografts from breast cancer brain metastases. Front. Oncology 7, 252 (2017).
Kim, S. H., Redvers, R. P., Chi, L. H., Ling, X., Lucke, A. J., Reid, R. C., et al. Identification of brain metastasis genes and therapeutic evaluation of histone deacetylase inhibitors in a clinically relevant model of breast cancer brain metastasis. Dis. Models Mech. 11, DMM034850 (2018).
Price, J., Fabra, A., Zhang, R., Radinsky, R. & Pathak, S. Characterization of variants of a human breast-cancer cell-line isolated from metastases in different organs of nude-mice. Int. J. Oncol. 5, 459–467 (1994).
Martinez-Aranda, A., Hernandez, V., Picon, C., Modolell, I. & Sierra, A. Development of a preclinical therapeutic model of human brain metastasis with chemoradiotherapy. Int. J. Mol. Sci. 14, 8306–8327 (2013).
Malin, D., Strekalova, E., Petrovic, V., Deal, A., Al Ahmad, A., Adamo, B. et al. αB-crystallin: a novel regulator of breast cancer metastasis to the brain. Clinical cancer research: an official journal of the American Association for. Cancer Res. 20, 56–67 (2014).
Diossy, M., Reiniger, L., Sztupinszki, Z., Krzystanek, M., Timms, K. M., Neff, C. et al. Breast cancer brain metastases show increased levels of genomic aberration-based homologous recombination deficiency scores relative to their corresponding primary tumors. Ann. Oncol. 29, 1948–54. (2018).
Kodack, D. P., Askoxylakis, V., Ferraro, G. B., Fukumura, D. & Jain, R. K. Emerging strategies for treating brain metastases from breast cancer. Cancer Cell 27, 163–175 (2015).
Delaney, L. J., Ciraku, L., Oeffinger, B. E., Wessner, C. E., Liu, J. B., Li, J. et al. Breast cancer brain metastasis response to radiation after microbubble oxygen delivery in a murine model. J. Ultrasound Med. 38, 3221–3228 (2019).
Oshi, M., Okano, M., Maiti, A., Rashid, O. M., Saito, K., Kono, K. et al. Novel breast cancer brain metastasis patient-derived orthotopic xenograft model for preclinical studies. Cancers (Basel) 12, 444 (2020).
Zhang, X., Claerhout, S., Prat, A., Dobrolecki, L. E., Petrovic, I., Lai, Q. et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 73, 4885–4897 (2013).
Ryken, T. C., McDermott, M., Robinson, P. D., Ammirati, M., Andrews, D. W., Asher, A. L. et al. The role of steroids in the management of brain metastases: a systematic review and evidence-based clinical practice guideline. J. Neuro Oncol. 96, 103–114 (2010).
Jaeckle, K. A., Dixon, J. G., Anderson, S. K., Moreno-Aspitia, A., Colon-Otero, G., Hebenstreit, K. et al. Intra-CSF topotecan in treatment of breast cancer patients with leptomeningeal metastases. Cancer Med. 9, 7935–7942 (2020).
Le Rhun, E., Wallet, J., Mailliez, A., Le Deley, M., Rodrigues, I., Boulanger, T. et al. Intrathecal liposomal cytarabine plus systemic therapy versus systemic chemotherapy alone for newly diagnosed leptomeningeal metastasis from breast cancer. Neuro Oncol. 22, 524–538. (2020).
Sperduto, P. W., Mesko, S., Li, J., Cagney, D., Aizer, A., Lin, N. U. et al. Estrogen, progesterone and HER2 receptor discordance between primary tumor and brain metastases in breast cancer and its effect on treatment and survival. Neuro Oncol. 22, 1359–1367 (2020).
Schrijver, W., Suijkerbuijk, K. P. M., van Gils, C. H., van der Wall, E., Moelans, C. B. & van Diest, P. J. Receptor conversion in distant breast cancer metastases: a systematic review and meta-analysis. J. Natl Cancer Inst. 110, 568–80. (2018).
De Mattos-Arruda, L., Ng, C. K. Y., Piscuoglio, S., Gonzalez-Cao, M., Lim, R. S., De Filippo, M. R. et al. Genetic heterogeneity and actionable mutations in HER2-positive primary breast cancers and their brain metastases. Oncotarget 9, 20617–30. (2018).
Brastianos, P., Carter, S., Santagata, S., Cahill, D., Taylor-Weiner, A., Jones, R. et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 5, 1164–1177 (2015).
Dong, X. Current strategies for brain drug delivery. Theranostics 8, 1481–93. (2018).
Robey, R. W., Pluchino, K. M., Hall, M. D., Fojo, A. T., Bates, S. E. & Gottesman, M. M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 18, 452–64. (2018).
de Lange, E. Potential role of ABC transporters as a detoxification system at the blood-CSF barrier. Adv. Drug Delivery Rev. 56, 1793–1809 (2004).
Ghersi-Egea, J. & Strazielle, N. Brain drug delivery, drug metabolism, and multidrug resistance at the choroid plexus. Microscopy Res. Tech. 52, 83–88 (2001).
Arvanitis, C. D., Ferraro, G. B. & Jain, R. K. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 20, 26–41 (2020).
Park, Y. H., Park, M. J. Ji, S. H., Yi, S. Y. Lim, D. H. Nam, D. H. et al. Trastuzumab treatment improves brain metastasis outcomes through control and durable prolongation of systemic extracranial disease in HER2-overexpressing breast cancer patients. Br. J. Cancer. 100, 894–900 (2009).
Swain, S. M., Baselga, J., Kim, S. B., Ro, J., Semiglazov, V., Campone, M. et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 372, 724–734 (2015).
Dieras, V., Miles, D., Verma, S., Pegram, M., Welslau, M., Baselga, J. et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 18, 732–42. (2017).
Montemurro, F., Delaloge, S., Barrios, C. H., Wuerstlein, R., Anton, A., Brain, E., et al. Trastuzumab emtansine (T-DM1) in patients with HER2-positive metastatic breast cancer and brain metastases: exploratory final analysis of cohort 1 from KAMILLA, a single-arm phase IIIb clinical trial. Ann. Oncol.31, 1350–1358 (2020).
Radic-Sarikas, B., Halasz, M., Huber, K. V. M., Winter, G. E., Tsafou, K. P., Papamarkou, T. et al. Lapatinib potentiates cytotoxicity of YM155 in neuroblastoma via inhibition of the ABCB1 efflux transporter. Sci. Rep. 7, 3091 (2017).
Bachelot, T., Romieu, G., Campone, M., Dieras, V., Cropet, C., Dalenc, F. et al. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): a single-group phase 2 study. Lancet Oncol. 14, 64–71 (2013).
Freedman, R. A., Gelman, R. S., Anders, C. K., Melisko, M. E., Parsons, H. A., Cropp, A. M. et al. TBCRC 022: A phase II trial of neratinib and capecitabine for patients with human epidermal growth factor receptor 2-positive breast cancer and brain metastases. J. Clin. Oncol. 37, 1081–1089 (2019).
Murthy, R. K., Loi, S, Okines, A, Paplomata, E., Hamilton, E., Hurvitz, S. A. et al. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N. Engl. J. Med. 382, 597–609 (2020).
Ma, F., Li, Q., Chen, S., Zhu, W., Fan, Y., Wang, J. et al. Phase I study and biomarker analysis of pyrotinib, a novel irreversible Pan-ErbB receptor tyrosine kinase inhibitor, in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J. Clin. Oncol. 35, 3105–3112 (2017).
Xuhong, J. C., Qi, X. W., Zhang, Y. & Jiang, J. Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer. Am. J. Cancer Res. 9, 2103–19. (2019).
Blair, H. A. Pyrotinib: First global approval. Drugs 78, 1751–1755 (2018).
Gril, B., Wei, D., Zimmer, A. S., Robinson, C., Khan, I., Difilippantonio, S. et al. A HER2 antibody drug conjugate controls growth of breast cancer brain metastases in hematogenous xenograft models, with heterogeneous blood-tumor barrier penetration unlinked to a passive marker. Neuro Oncol. 22, 1625–1636 (2020).
Cortés, J., Dieras, V., Ro, J., Barriere, J., Bachelot, T., Hurvitz, S. et al. Afatinib alone or afatinib plus vinorelbine versus investigator’s choice of treatment for HER2-positive breast cancer with progressive brain metastases after trastuzumab, lapatinib, or both (LUX-Breast 3): a randomised, open-label, multicentre, phase 2 trial. Lancet Oncol. 16, 1700–1710 (2015).
Han, H. S., Dieras, V., Robson, M., Palacova, M., Marcom, P. K., Jager, A. et al. Veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in patients with BRCA1/2 locally recurrent/metastatic breast cancer: randomized phase II study. Ann. Oncol. 29, 154–161 (2018).
Loibl, S., O’Shaughnessy, J., Untch, M., Sikov, W. M., Rugo, H. S., McKee, M. D. et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol. 19, 497–509 (2018).
Werner, T. L., Sachdev, J., Swisher, E. M., Gutierrez, M., Kittaneh, M., Stein, M. N. et al. Safety and pharmacokinetics of veliparib extended-release in patients with advanced solid tumors: a phase I study. Cancer Med. 7, 2360–2369 (2018).
Li, Y., Wei, X., Zhang, S. & Zhang, J. Prognosis of invasive breast cancer after adjuvant therapy evaluated with VEGF microvessel density and microvascular imaging. Tumour Biol. 36, 8755–8760 (2015).
Hey, S. P., Gyawali, B., D’Andrea, E., Kanagaraj, M., Franklin, J. M. & Kesselheim, A. S. A systematic review and meta-analysis of bevacizumab in first-line metastatic breast cancer: lessons for the research and regulatory enterprises. J. Natl Cancer Inst. 112, 335–342 (2019).
Mackey, J. R., Ramos-Vazquez, M., Lipatov, O., McCarthy, N., Krasnozhon, D., Semiglazov, V. et al. Primary results of ROSE/TRIO-12, a randomized placebo-controlled phase III trial evaluating the addition of ramucirumab to first-line docetaxel chemotherapy in metastatic breast cancer. J. Clin. Oncol. 33, 141–148 (2015).
Bostrom, J., Yu, S. F., Kan, D., Appleton, B. A., Lee, C. V., Billeci, K. et al. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science (New York, NY) 323, 1610–1614 (2009).
Hu, S., Fu, W., Xu, W., Yang, Y., Cruz, M., Berezov, S. D. et al. Four-in-one antibodies have superior cancer inhibitory activity against EGFR, HER2, HER3, and VEGF through disruption of HER/MET crosstalk. Cancer Res. 75, 159–170 (2015).
Ippen, F. M., Alvarez-Breckenridge, C. A., Kuter, B. M., Fink, A. L., Bihun, I. V., Lastrapes, M. et al. The dual PI3K/mTOR pathway inhibitor GDC-0084 achieves antitumor activity in PIK3CA-mutant breast cancer brain metastases. Clin. Cancer Res. 25, 3374–83. (2019).
Venur, V. A. & Leone, J. P. Targeted therapies for brain metastases from breast cancer. Int. J. Mol. Sci. 17, 1543 (2016).
Ippen, F. M., Grosch, J. K., Subramanian, M., Kuter, B. M., Liederer, B. M., Plise, E. G. et al. Targeting the PI3K/Akt/mTOR pathway with the pan-Akt inhibitor GDC-0068 in PIK3CA-mutant breast cancer brain metastases. Neuro-oncology 21, 1401–11. (2019).
Pernas, S., Tolaney, S. M., Winer, E. P. & Goel, S. CDK4/6 inhibition in breast cancer: current practice and future directions. Ther. Adv. Med. Oncol. 10, 1758835918786451 (2018).
Chong, Q. Y., Kok, Z. H., Bui, N. L., Xiang, X., Li-Ann, Wong A., Peng Yong, W. et al. A unique Cdk4/6 inhibitor: current and future therapeutic strategies of abemaciclib. Pharmacol. Res. 156, 104686 (2020).
Patnaik, A., Rosen, L. S., Tolaney, S. M., Tolcher, A. W., Goldman, J. W., Gandhi, L. et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 6, 740–753 (2016).
Tolaney, S. M., Sahebjam, S., Le Rhun, E., Bachelot, T., Kabos, P., Awada, A. et al. A phase 2 study of abemaciclib in patients with brain metastases secondary to hormone receptor positive breast cancer. Clin. Cancer Res. 26, 5310–5319 (2020).
Mills, M., Figura, N., Arrington, J., Yu, H., Etame, A., Vogelbaum, M. et al. Management of brain metastases in breast cancer: a review of current practices and emerging treatments. Breast Cancer Res. Treat. 180, 279–300 (2020).
Chen, X., Han, J., Chu, J., Zhang, L., Zhang, J., Chen, C. et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 7, 27764–27777 (2016).
Priceman, S., Tilakawardane, D., Jeang, B., Aguilar, B., Murad, J., Park, A. et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2 breast cancer metastasis to the brain. Clin. Cancer Res. 24, 95–105 (2018).
Brastianos, P. K., Lee, E. Q., Cohen, J. V., Tolaney, S. M., Lin, N. U., Wang, N. et al. Single-arm, open-label phase 2 trial of pembrolizumab in patients with leptomeningeal carcinomatosis. Nat. Med. 26, 1280–1284 (2020).
Zhang, L. & Zhao, D. Applications of nanoparticles for brain cancer imaging and therapy. J. Biomed. Nanotechnol. 10, 1713–1731 (2014).
He, C., Cai, P., Li, J., Zhang, T., Lin, L., Abbasi, A. Z. et al. Blood-brain barrier-penetrating amphiphilic polymer nanoparticles deliver docetaxel for the treatment of brain metastases of triple negative breast cancer. J. Control. Release 246, 98–109 (2017).
Zhang, S., Deng, G., Liu, F., Peng, B., Bao, Y., Du, F. et al. Autocatalytic delivery of brain tumor-targeting, size-shrinkable nanoparticles for treatment of breast cancer brain metastases. Adv. Funct. Mater. 30, 1910651 (2020).
Han, L., Kong, D. K., Zheng, M. Q., Murikinati, S., Ma, C., Yuan, P. et al. Increased nanoparticle delivery to brain tumors by autocatalytic priming for improved treatment and imaging. ACS Nano 10, 4209–4218 (2016).
Sierra, A., Price, J. E., Garcia-Ramirez, M., Mendez, O., Lopez, L., Fabra, A. Astrocyte-derived cytokines contribute to the metastatic brain specificity of breast cancer cells. Lab Invest. 77, 357–68 (1997).
De Meulenaere, V., Neyt, S., Vandeghinste, B., Mollet, P., De Wever, O., Decrock, E. et al. Species-dependent extracranial manifestations of a brain seeking breast cancer cell line. PLoS One. 13, e0208340 (2018).
Acknowledgements
We wish to acknowledge the support of all our research group members.
Author information
Authors and Affiliations
Contributions
Y.J.W. and F.Z.Y. are co-first authors who wrote drafts of the manuscript and generated the figures and tables together. Y.R.L. revised and polished the manuscripts. Y.R.L. and Q.F.Y. provided overall supervision and guidance. All authors read and approved the final version of manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not Applicable.
Consent to publish
Not Applicable.
Data availability
Not Applicable.
Competing interests
The authors declare no competing interests.
Funding information
This work was supported by National Key Research and Development Program (No. 2020YFA0712400), Special Foundation for Taishan Scholars (No. ts20190971), National Natural Science Foundation of China (No. 81874119; No. 82072912), Special Support Plan for National High Level Talents (Ten Thousand Talents Program W01020103), National Key Research and Development Program (No. 2018YFC0114705), Foundation from Clinical Research Center of Shandong University (No.2020SDUCRCA015), Qilu Hospital Clinical New Technology Developing Foundation (No. 2018–7; No. 2019–3).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, Y., Ye, F., Liang, Y. et al. Breast cancer brain metastasis: insight into molecular mechanisms and therapeutic strategies. Br J Cancer 125, 1056–1067 (2021). https://doi.org/10.1038/s41416-021-01424-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01424-8
This article is cited by
-
Cell-cell communication characteristics in breast cancer metastasis
Cell Communication and Signaling (2024)
-
A promising target for breast cancer: B7-H3
BMC Cancer (2024)
-
Breast cancer brain metastasis: from etiology to state-of-the-art modeling
Journal of Biological Engineering (2023)
-
A novel feedback regulated loop of circRRM2-IGF2BP1-MYC promotes breast cancer metastasis
Cancer Cell International (2023)
-
Crosstalk between small-cell lung cancer cells and astrocytes mimics brain development to promote brain metastasis
Nature Cell Biology (2023)
