
Abstract
Background
Noninvasive plasma-based detection of EGFR mutations using digital PCR promises a fast, sensitive and reliable approach to predicting the efficiency of EGFR-TKI. However, the low throughput and high cost of digital PCR restricts its clinical application.
Methods
We designed a digital PCR assay, which can simultaneously detect 39 mutations of exons 18–21 of the EGFR gene. To assess overall performance, retrospective FFPE tissues from 30 NSCLC patients and plasma from 33 NSCLC patients were collected and analysed.
Results
The LoD of the EGFR mutations was as low as 0.308 copies/μL, and the linear correlation between the detected and expected values at different concentrations (0.01–10%) was low as well. Compared to ARMS-PCR in FFPE, the accuracy values of the dEGFR39 assay in plasma from 33 patients was 87.88% (29/33, 95% CI 72.67–95.18%). While monitoring the 33 patients, the EGFR mutation load as assessed by dEGFR39 was associated with the objective response to treatment. Thirteen samples from eight patients were identified by dEGFR39 to harbour the T790M mutation over time; of these patients, only nine (69%) were detected using SuperARMS.
Conclusion
Our results indicate that dEGFR39 assay is reliable, sensitive and cost-efficient. This method is beneficial for profiling EGFR mutations for precision therapy and prognosis after TKI treatment, especially in patients with insufficient tissue biopsy samples.
Similar content being viewed by others

Background
In patients with non-small-cell lung cancer (NSCLC), the possibility of treatment with tyrosine kinase inhibitor (TKI) is determined by the presence of mutations on exons 18–21 of the epidermal growth factor receptor (EGFR).1 Clinical evidence heightens the fact that EGFR-targeted therapy can significantly improve progression-free survival (PFS) and overall survival (OS) in patients.2,3 Therefore, assessing the status of EGFR mutations is important for potential EGFR-TKI therapy.
Previous methods of EGFR mutation detection have been based on invasive approaches, such as surgical resection of tumours and needle biopsies.4 However, the heterogeneity of tumour tissue usually confounds the analysis of EGFR mutation load.5 Interestingly, recent data have shown that EGFR mutations can be found in plasma-derived cell-free DNA (cfDNA) from NSCLC patients. Moreover, studies have confirmed that EGFR mutations from plasma can predict the clinical response to targeted therapy.6,7
Based on plasma-derived DNA, noninvasive detection approaches have been developed, such as super amplification refractory mutation system (superARMS) PCR, next-generation sequencing (NGS), and digital PCR.8,9,10 Although ARMS-PCR is cost-efficient, its overall performance appears to be least sensitive. The NGS method, on the other hand, is more expensive. The digital PCR method features a quick turn-around and improved sensitivity, and is ideally used for the detection of known mutation types in cancer patients, especially in those whose tissue biopsy samples are insufficient.11,12,13,14 Digital PCR is a new approach to nucleic acid detection. The PCR reaction is first pressurised to partition into 30,000 droplets, each of which contains zero, one or more copies of the target molecule, and then PCR analysis is carried out. During amplification, TaqMan chemistry with dye-labelled probes is used to detect sequence-specific targets. The droplets containing the target molecule will generate fluorescence signals, which are defined as positive, while others are negative. According to the Poisson distribution, the fraction of negative reactions is used to generate an absolute count of the number of target molecules in the sample. Despite the fact that studies have shown good consistency of EGFR detection between plasma and tissue using digital PCR,12,15,16 the low throughput and high cost of digital PCR restricts its clinical application. There is not yet a method that simultaneously assesses all the driver mutations of the EGFR gene in plasma using digital PCR.17,18
Therefore, we developed an integrated digital PCR assay, named dEGFR39, which can unambiguously distinguish multiple mutations using three different fluorescence channels. This method is based on the need for low cost and small sample input. In this report, we provide evidence that dEGFR39 has excellent performance, which is not only useful to screen EGFR-targeted mutations from plasma of NSCLC patients to guide targeted therapy, but also to evaluate prognosis after treatment. To the best of our knowledge, this is the first report regarding the introduction of digital PCR for the screening and monitoring of all EGFR mutations from patient plasma.
Methods
Sample collection and processing
The plasmid DNA of EGFR L858R, 19Del and T790M were prepared using the Apexbio plasmid DNA extraction kit, according to the manufacturer’s instructions (Apexbio, Suzhou, China). The human genomic DNA standard HD802 was obtained from Horizon (Horizon, Cambridge, UK). This study was approved by the ethics committee of Cangzhou Central Hospital, Hebei, China and all patients provided written informed consent. Patients were diagnosed with NSCLC and ARMS-PCR was performed to assess the mutation status of EGFR. For patient-derived FFPE tissues, we collectively obtained 63 samples, including 30 retrospective samples. Thirty-three patients were enrolled from May 2014 to June 2019. Plasma samples and CT images were collected from the participants every 2 months. The scheme of the clinical study design is shown in Fig. 1. Patient-derived FFPE tissues were cut into three pieces of 10-μm thick paraffin. FFPE DNA was extracted according to the procedure of the Qiagen FFPE DNA kit. Ten millilitre of peripheral blood was collected into a BCT DNA tube (Streck, La Vista, USA) and mixed by gentle inversion 10 times immediately. To separate plasma, we centrifuged the whole blood sample at 1600 × g for 10 min to obtain the supernatant, then centrifuged the supernatant at 16,000 × g for 10 min according to manufacturer’s instructions. Two millilitre of plasma was used to isolate DNA using the Qiagen circulating nucleic acid kit following the manufacturer’s instructions (Qiagen, Hilden, Germany). DNA quantification was obtained by the Colibri microvolume spectrophotometer (Titertek-Berthold, Pforzheim, Germany) and Qubit Fluorometer 2.0 (Invitrogen, California, USA).

The scheme of the clinical study design.
Detection of EGFR mutations using dEGFR39 assay
Oligonucleotides were synthesised by Sangon Biotech (Shanghai, China). Detection of the EGFR mutation using dEGFR39 assay (Apexbio, Suzhou, China) was carried out in three tubes on the Naica digital PCR system (Stilla Technologies, Villejuif, France) with Sapphire chips (Stilla Technologies, Villejuif, France). The list of 39 mutations found by the dEGFR39 assay is provided in Supplementary Table S1. Twenty-five microlitre of reaction mix in each tube contained 1X PerFecTa Multiplex qPCR ToughMix, 40 nM FITC (Saint Louis, MO, USA), 1 μl of a multiplex mix of primers and probes and 3 μl of DNA. The chip was loaded into the Naica Geode thermocycler to compartmentalise the droplets and to perform the PCR reaction. PCR consisted of 10 min at 95 °C, followed by 45 cycles of 95 °C for 20 s and 60 °C for 30 s. After amplification, the Sapphire chips were imaged using the Naica Prism3 reader.
Limit of blank and limit of detection
The specificity and accuracy of dEGFR39 assay were assessed by wild-type (WT) and mutant DNA of EGFR. The limit of blank (LoB) and limit of detection (LoD) were determined as previously reported.19 To determine LoB, twenty replicates of a blank sample within two independent runs were carried out. The LoB was set as the highest mutant concentration that might be found when replicates of a blank sample are tested. The LoD was set as the lowest concentration that could be distinguished from the LoB with 95% certainty.20 The LoB and LoD were calculated with the following formulas:
Accuracy was assessed by testing the DNA Reference Standards and calculating the coefficient of variation and statistical differences.
Data analysis
Raw data from digital PCR were analysed by Crystal Miner software (Stilla Technologies, Villejuif, France) according to the principle of Poisson distribution. In general, when there are λ targets per droplets, the fraction of positive droplets (P) is:
For each analysis involving data from two channels, where λc1 and λc2 are the average number of loading molecules in these two channels, with the total number of droplets N, the number of double-positive droplets in multiple digital PCR is:
NTC and EGFR Gene-Specific Multiplex Reference Standard genomic DNA HD802 were used as negative and positive controls, respectively. Negative and positive droplets were also used to check the fluorescence spill-over compensation. The paired t-test, Bland–Altman, and McNemar’s test were used to compare the consistency of different groups with PPA (sensitivity), NPA (specificity), accuracy (OPA) and Kappa values. The cut-off for statistical significance was p < 0.05. All statistical data were analysed by IBM SPSS statistics software 22.0 (IBM, Armonk, NY, USA) and GraphPad Prism 5 (GraphPad, La Jolla, CA, USA).
Results
Characteristics of dEGFR39 assay
In this study, dEGFR39 assay was developed to detect up to 39 mutations along exons 18–21 of the EGFR gene, including common mutation sites such as L858R, 19Del and T790M, as well as rare mutation sites such as L861Q, S768I, G719X, C797S and 20ins.
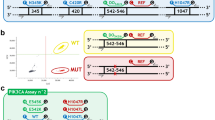
To develop dEGFR39 assay, we designed digital PCR into three consecutive reactions. In the first reaction, 19DelREF was labelled with HEX, L858RMU and L861QMU were labelled with FAM, and 19DelWT and S768IMU were labelled with CY5 (Fig. 2). In the presence of 19Del, probe 19DelWT could cross-react with the neighbouring nondeleted WT sequences; in the meantime, probe 19DelREF was used to detect the EGFR gene (including 19DelWT and 19DelMU). As a result, 19DelWT molecules were double-positive (CY5+/HEX+), whereas the rest of the signals in the HEX and CY5 channels were only from 19DelMU (CY5−/HEX+) and S768IMU (CY5+/HEX−), respectively. The signals in the FAM channel were generated by L858RMU and L861QMU. Similarly, in the second reaction, G719XWT and 20insREF were labelled with FAM and CY5, respectively, while 20insWT and G719XMU were labelled with HEX. 20insWT molecules were consequently double-positive (CY5+/HEX+), and the rest of the signals in the HEX and CY5 channels were only from G719XMU (CY5−/HEX+) and 20insMU (CY5+/HEX−), respectively. Given that the presence of C797S and T790M, whether in the trans or cis form, would determine the efficacy of TKI treatment,21 the third reaction was specifically designed to characterise this genotype. T790MMU and C797SMU were labelled with FAM and HEX, respectively. Since the amount of plasma-derived DNA was exceedingly low, the positive signal from the HEX channel could only be from C797SMU in the trans configuration with T790MMU.

a 19DelWT molecules are double positive (CY5+/HEX+), and the rest of the signals in the HEX and CY5 channels are generated by 19DelMU (CY5−/HEX+) and S768IMU (CY5+/HEX−), respectively. The signals in the FAM channel are generated by L858RMU. b 20insWT molecules are double positive (CY5+/HEX+), and the rest of the signals in the HEX and CY5 channels are generated by G719XMU (CY5−/HEX+) and 20insMU (CY5+/HEX−), respectively. c The blue and green dots represent the signal of T790M and C797S in trans configuration with T790M, respectively. The double positive represents C797S in cis configuration with T790M.
Notably, our assay included the C797S site, a critical mutation recently identified after TKI treatment.22 We did not detect C797S in patients enrolled in our study, which is consistent with previous reports that C797S does not occur in TKI-naïve NSCLC.23
The specificity and sensitivity of dEGFR39
To assess the specificity of the dEGFR39 assay, both WT and mutant (MU) DNA of EGFR were synthesised. Notably, only a specific fluorescent signal could be detected that corresponded to WT and mutant forms (Supplementary Figs. S1–3), indicating the great specificity of the assay. To evaluate the accuracy, we performed the dEGFR39 reaction with a standard DNA HD802 (expected mutant ratio 12.5%). Our data showed the mutation loads of 19Del, T790M and L858R as being 12.63%, 12.19% and 12.13%, respectively. This assessment was in concordance with the expected values (p > 0.05) (Table 1), strongly suggesting that dEGFR39 assay has good accuracy. Additionally, we tested the EGFR mutations with abundance from 0.01 to 10%, and the slope between measured and expected abundance was close to 1, indicating that the correlation was strikingly significant (Fig. 3 and Supplementary Figs. S4–7). For L858R/L861Q, S768I, 19Del, T790M and C797S, the LoB was 0.2 copies, while the LoB of 20ins and G719X was 0.3 copies (Supplementary Table S2–S4). Next, we assessed the LoD using serial dilutions of mutant DNA following the CLSI EP17 method. The LoD of L858R/L861Q, 19Del and T790M was 0.339, 0.305 and 0.333 copies/μL, respectively. For rare mutations, the LoD of S768I, G719X, 20ins and C797S was 0.311, 0.434, 0.457 and 0.349 copies/μL, respectively (Supplementary Table S5).

EGFR mutations were detected on a series of DNA with the mutant ratio of 10%, 1%, 0.1%, 0.05%, and 0.01%. Regression plot for the dilutions shows linearity and a good correlation for expected and measured values.
The consistence between the dEGFR39 method and ARMS-PCR in FFPE tissues
To compare the performance between dEGFR39 assay and ARMS-PCR, 30 FFPE tissues from NSCLC patients were collected. As revealed by the dEGFR39 assay, the individual mutation load ranged from 0.04 to 43.7%. Among these mutations, a large number had an abundance of less than 1% (ranging from 0.04 to 0.79%) (Supplementary Fig. S8).
In this study, we set the respective LoD of mutations as the cut-off value for dEGFR39 assay. Mutations with a concentration less than the LoD value were defined as negative. Consequently, the overall predictive agreement (OPA), positive predictive value (PPV) and negative predictive agreement (NPA) were analysed. In addition, we summarised the detection results of all sites. Mutations with a lower abundance could not be correctly detected using the ARMS-PCR method. In contrast, dEGFR39 was able to detect that the PPV, NPV and OPA were 94.44% (95% CI 74.24–99.01%), 25% (95% CI 8.89–53.23%) and 66.67% (95% CI 48.78–80.77%), respectively (Table 2 and Supplementary Table S6–8). Altogether, these results demonstrate that dEGFR39 can effectively detect low-abundance EGFR mutations in patients, and is more sensitive than the ARMS-PCR method.
Evaluation of dEGFR39 for detecting EGFR mutations in plasma
To evaluate the performance of dEGFR39 in patient-derived plasma, we analysed 33 matched plasma samples from patients with advanced NSCLC. The diagnosis was previously based on the ARMS-PCR method from FFPE tissues, in which 16 patients were positive for EGFR mutations (six for L858R/L861Q, eight for 19Del and three for T790M, including compound mutation) and 17 were negative for EGFR mutations. The histopathological characteristics of the patients are summarised in Supplementary Table S9. In this analysis, dEGFR39 detected 17 positives and 16 negatives from patient-matched plasma. The PPV, NPV and OPA of EGFR mutations were analysed and showed a lower NPA and OPA due to the higher sensitivity of the dEGFR39 assay. Take T790M as an example: two more positive cases were detected by dEGFR39 than ARMS-PCR, reducing its OPA to 93.94% (95% CI 80.39–98.32%), while the OPA of other mutations was 96.97% (Table 3 and Supplementary Table S10–12). This experiment indicates that the dEGFR39 assay is sensitive and specific enough to detect more EGFR mutations than other methods.
Correlation between EGFR mutations detected by dEGFR39 and response to treatment
We analysed the abundance of EGFR mutations before and after TKI treatment. Prior to treatment, multiple plasma samples were collected for the initial assessment. dEGFR39 assay was subsequently performed to monitor the EGFR mutation undergoing multiple lines of different treatment (Fig. 4 and Supplementary Table S13–15). Although 20ins and L858R were detected by both dEGFR39 assay and superARMS PCR in patient P-07 at the baseline, there was still a great benefit in taking gefitinib, consistent with the dEGFR39 result at 450 days (Fig. 4c). Patient P-04 received first-line treatment with Icotinib, and was diagnosed as PD at 45 days after treatment, and the subsequent combination therapy was not effective. The mutant abundance of patient P-12, who initially harboured a 19Del mutation, slowly decreased with the treatment of Icotinib, but increased significantly at 444 days (15 months) with the radiographic evidence of PD (Fig. 4a). A steady increase in mutation abundance might indicate poor efficacy of TKI treatment (Fig. 4d). Nevertheless, this data implicate that mutation load as examined by dEGFR39 correlates with disease progression.

EGFR frequency of activated mutations decreases initial reception of TKI treatment and subsequently increases (a, e); T790M mutation was not observed (c); emergence of resistant mutation was detected 2 months and 8 months prior to clinical PD, respectively (b, f), and the activated mutation was never cleared at the beginning of TKI treatment (d). The arrow refers to the result of significant changes in the patientʼs tissue samples by ARMS or imaging tests.
dEGFR39 can predict clinical prognosis after TKI treatment earlier than superARMS PCR
In NSCLC patients with resistance to TKI treatment, an important mechanism of primary resistance involves a mutation in T790M that blocks the binding of TKI with the adenosine triphosphate domain of EGFR.24,25 Of the 33 enrolled patients with NSCLC, 13 samples from eight patients were identified by dEGFR39 to harbour the T790M mutation over time; of these, only nine (69%) were detected using SuperARMS. In the plasma of patient P-15, L858R and T790M mutations were simultaneously detected after 368 days of Icotinib treatment. SuperARMS PCR, however, failed to detect the T790M mutation; moreover, there was no significant progress evident in imaging until 121 days (Fig. 4b). Similarly, in patient P-25, dEGFR39 results showed an increase in mutant abundance in 19Del and T790M after 251 days of TKI treatment, which was 43 days ahead of that of imaging progression (Fig. 4e). In patient P-23, the emergence of L858R was initially observed and dropped upon Icotinib treatment. Meanwhile, T790M was detected by dEGFR39, which gradually increased with Icotinib as well as Apatinib plus chemotherapy treatment. In this case, detection of T790M was comparatively delayed by superARMS PCR and CT imaging (Fig. 5). Taken together, this data supports the conclusion that detection of T790M mutations by dEGFR39 occurs relatively earlier than by superARMS PCR and CT imaging, which can play a role in diagnosis and prognosis (Fig. 4f).

CT imaging scans performed at the start of Icotinib treatment (day 0, 83), at the change of treatment to Icotinib and Apatinib treatment (day 187, 340), and at the change of treatment to TKI and chemotherapy (day 412). Lesions identified in the lung (blue arrow), liver lobe (yellow arrow), and pleura (red arrow) are indicated.
Discussion
Early detection of EGFR mutations promises more precise therapy for patients with NSCLC. Using digital PCR, we developed a plasma-based noninvasive method, named dEGFR39, that detects multiple mutations of EGFR. To our knowledge, this is the first report regarding the utilisation of digital PCR for this purpose.
Since digital PCR usually has two channels of fluorescence, the system is primarily used for allele mutations in duplex assays because each specialised fluorescent probe can only recognise single allele mutations.26 Based on labelling the same fluorescent probe for each mutation, seven common KRAS mutations were consistently detected in plasma from patients with colorectal cancer (CRC).27 Moreover, multiple mutations can be recognised by different clusters, which depend on different corresponding concentrations of input primer and probe.28,29 However, it remains challenging to identify the cross region of clusters or define the mutation type when the DNA abundance is low.30 In contrast, the dEGFR39 assay is ideal for detecting samples with low DNA content, such as plasma. When the template content is too high in the reaction, some additional double-positive signals will appear, which can lead to mistakes in calculating mutation abundance. As shown in Supplementary Fig. S5, due to the large amount of template added, a part of S768IMU and 19DelMU are in one droplet, and the fluorescence intensity of these double-positive signals is different from that of true 19DelWT. Therefore, it is clearly divided into two regions, wherein the signal enclosed by the red dotted line is considered to be generated by S768IMU and 19DelMU template amplification. Furthermore, according to the formula provided in this method, the lower the concentration within the confidence range, the lower the frequency of false double-positive signals. The dEGFR39 assay takes advantage of the low abundance of DNA in plasma, making the test results more accurate.
Another study reported a drop-off method to achieve multiple detections; although this method can detect mutations located together, it fails to simultaneously detect other mutation types.26 To overcome these challenges, we have devised a three step reaction by optimising annealing temperature, combined probe concentrations, and modifying system configuration with fluorescent compensation (Supplementary Fig. S9). After these improvements, dEGFR39 exhibits superior performance to profile the driver mutations of the EGFR gene. It is important to note that our data show that accuracy for dEGFR39 from plasma and ARMS from FFPE is 87.88%, which is similar to previous studies (80.8–94.19%).31,32,33
Importantly, it has been previously reported that digital PCR has technical issues such as the phenomenon of “rain”, which ranges between explicit positive and negative droplets. This issue eventually hinders the correct setting of threshold and consequently leads to failure of the digital PCR experiment.34,35 In this study, we induced a “double positive” discrimination method in the detection of 19Del, 20ins and C797S, which made the distribution of rain primarily in the diagonal area in the 2D plot diagram, thus avoiding interference with the identification of positive droplets. This approach greatly facilitated multiplex detection and led to less background noise (Fig. 2). Although it was minimal, the rain phenomenon still occurred. While detecting 20ins mutant locus from patient-derived FFPE tissues, we observed an increase in LoB and low correlation with the accuracy of 66.67% (95% CI 48.78–80.77%). Interestingly, while using patient-derived plasma, we observed a significant improvement in the ratio of signal-to-noise. We reason that FFPE, but not plasma, might potentially contain an inhibitor for digital PCR analysis.
Several studies have demonstrated that mutant abundance of EGFR is closely associated with the response to treatment.36 More importantly, dEGFR39 is more sensitive than superARMS PCR and CT imaging, thus allowing early detection of EGFR mutations. Of note, some patients, although positive for EGFR driver mutations, received no clinical benefit from EGFR-TKI treatment (Fig. 4). In these patients, we observed an obvious trend of increasing mutation load.
Another comparative advantage of the dEGFR39 assay is low sample input. Whereas the commercial EGFR kit (ARMS method) requires detection of all EGFR mutations in eight tubes37, dEGFR39 uses three reactions to sufficiently characterise the EGFR mutation status even from plasma-derived DNA. In future studies, assessment of EGFR, ALK, ROS1 and perhaps other oncogenes can be streamlined in the same digital PCR platform. Although NGS can parallelly analyse multiple variations, including unknown variations, digital PCR is still the most suitable method in clinical testing, because of its higher sensitivity, easier-to-understand results, low turn-around time and low cost.
In conclusion, we developed a noninvasive plasma-based digital PCR method, hereby named dEGFR39, that allows simultaneous detection of multiple mutation sites of the EGFR gene. In this report, we provide evidence that this method is highly sensitive, reliable and cost-efficient, promising efficacy for clinical diagnosis and treatment assessment for patients with NSCLC.
References
Keedy, V. L., Temin, S., Somerfield, M. R., Beasley, M. B., Johnson, D. H., McShane, L. M. et al. American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) Mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J. Clin. Oncol. 29, 2121–2127 (2011).
Mok, T. S., Wu, Y. L., Thongprasert, S., Yang, C. H., Chu, D. T., Saijo, N. et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957 (2009).
Cheng, Y., Murakami, H., Yang, P. C., He, J., Nakagawa, K., Kang, J. H. et al. Randomized phase II trial of Gefitinib with and without pemetrexed as first-line therapy in patients with advanced nonsquamous non-small-cell lung cancer with activating epidermal growth factor receptor mutations. J. Clin. Oncol. 34, 3258–3266 (2016).
Tiseo, M., Rossi, G., Capelletti, M., Sartori, G., Spiritelli, E., Marchioni, A. et al. Predictors of gefitinib outcomes in advanced non-small cell lung cancer (NSCLC): study of a comprehensive panel of molecular markers. Lung Cancer 67, 355–360 (2010).
Fenizia, F., De Luca, A., Pasquale, R., Sacco, A., Forgione, L., Lambiase, M. et al. EGFR mutations in lung cancer: from tissue testing to liquid biopsy. Future Oncol. 11, 1611–1623 (2015).
Seki, Y., Fujiwara, Y., Kohno, T., Yoshida, K., Goto, Y., Horinouchi, H. et al. Circulating cell-free plasma tumour DNA shows a higher incidence of EGFR mutations in patients with extrathoracic disease progression. ESMO Open 3, e000292 (2018).
Vallee, A., Marcq, M., Bizieux, A., Kouri, C. E., Lacroix, H., Bennouna, J. et al. Plasma is a better source of tumor-derived circulating cell-free DNA than serum for the detection of EGFR alterations in lung tumor patients. Lung Cancer 82, 373–374 (2013).
Wang, Z., Cheng, Y., An, T., Gao, H., Wang, K., Zhou, Q. et al. Detection of EGFR mutations in plasma circulating tumour DNA as a selection criterion for first-line gefitinib treatment in patients with advanced lung adenocarcinoma (BENEFIT): a phase 2, single-arm, multicentre clinical trial. Lancet Respir. Med. 6, 681–690 (2018).
Zhao, J., Zhao, J., Huang, J., Chen, Y., Jiang, J., Wu, W. et al. A novel method for detection of mutation in epidermal growth factor receptor. Lung Cancer 74, 226–232 (2011).
Li, Y., Lv, J., Wan, S., Xin, J., Xie, T., Li, T. et al. High sensitive and non-invasive ctDNAs sequencing facilitate clinical diagnosis and clinical guidance of non-small cell lung cancer patient: a time course study. Front. Oncol. 8, 491 (2018).
Li, X., Liu, Y., Shi, W., Xu, H., Hu, H., Dong, Z. et al. Droplet digital PCR improved the EGFR mutation diagnosis with pleural fluid samples in non-small-cell lung cancer patients. Clin. Chim. Acta 471, 177–184 (2017).
Jiang, X. W., Liu, W., Zhu, X. Y. & Xu, X. X. Evaluation of EGFR mutations in NSCLC with highly sensitive droplet digital PCR assays. Mol. Med. Rep. 20, 593–603 (2019).
Feng, W. N., Gu, W. Q., Zhao, N., Pan, Y. M., Luo, W., Zhang, H. et al. Comparison of the superARMS and droplet digital PCR for detecting EGFR mutation in ctDNA from NSCLC patients. Transl. Oncol. 11, 542–545 (2018).
Wang, L., Guo, Q., Yu, W., Qiao, L., Zhao, M., Zhang, C. et al. Quantification of plasma EGFR mutations in patients with lung cancers: Comparison of the performance of ARMS-Plus and droplet digital PCR. Lung Cancer 114, 31–37 (2017).
Sacher, A. G., Paweletz, C., Dahlberg, S. E., Alden, R. S., O’Connell, A., Feeney, N. et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol. 2, 1014–1022 (2016).
Zhang, X., Chang, N., Yang, G., Zhang, Y., Ye, M., Cao, J. et al. A comparison of ARMS-Plus and droplet digital PCR for detecting EGFR activating mutations in plasma. Oncotarget 8, 112014–112023 (2017).
Quan, P. L., Sauzade, M. & Brouzes, E. dPCR: a technology review. Sensors 18, 1271 (2018).
Perkins, G., Lu, H., Garlan, F. & Taly, V. Droplet-based digital PCR: application in cancer research. Adv. Clin. Chem. 79, 43–91 (2017).
Armbruster, D. A. & Pry, T. Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev. 29, 49–52 (2008).
Milosevic, D., Mills, J. R., Campion, M. B., Vidal-Folch, N., Voss, J. S., Halling, K. C. et al. Applying standard clinical chemistry assay validation to droplet digital PCR quantitative liquid biopsy testing. Clin. Chem. 64, 1732–1742 (2018).
Oxnard, G. R., Hu, Y., Mileham, K. F., Husain, H., Costa, D. B., Tracy, P. et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 4, 1527–1534 (2018).
Hofman, V. & Hofman, P. Resistances to EGFR tyrosine kinase inhibitors in lung cancer-how to routinely track them in a molecular pathology laboratory? J. Thorac. Dis. 11, 65–70 (2019).
Oscorbin, I. P., Shadrina, A. S., Kozlov, V. V., Voitsitsky, V. E. & Filipenko, M. L. Absence of EGFR C797S mutation in tyrosine kinase inhibitor-naive non-small cell lung cancer tissues. Pathol. Oncol. Res. 26, 1229–1234 (2020).
Morgillo, F., Della Corte, C. M., Fasano, M. & Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: lung cancer. ESMO Open 1, e000060 (2016).
Su, K. Y., Chen, H. Y., Li, K. C., Kuo, M. L., Yang, J. C., Chan, W. K. et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J. Clin. Oncol. 30, 433–440 (2012).
Decraene, C., Silveira, A. B., Bidard, F. C., Vallee, A., Michel, M., Melaabi, S. et al. Multiple hotspot mutations scanning by single droplet digital PCR. Clin. Chem. 64, 317–328 (2018).
Taly, V., Pekin, D., Benhaim, L., Kotsopoulos, S. K., Le Corre, D., Li, X. et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin. Chem. 59, 1722–1731 (2013).
Whale, A. S., Huggett, J. F. & Tzonev, S. Fundamentals of multiplexing with digital PCR. Biomol. Detect Quantif. 10, 15–23 (2016).
Alcaide, M., Yu, S., Bushell, K., Fornika, D., Nielsen, J. S., Nelson, B. H. et al. Multiplex droplet digital PCR quantification of recurrent somatic mutations in diffuse large B-cell and follicular lymphoma. Clin. Chem. 62, 1238–1247 (2016).
Yu, Q., Huang, F., Zhang, M., Ji, H., Wu, S., Zhao, Y. et al. Multiplex picoliter-droplet digital PCR for quantitative assessment of EGFR mutations in circulating cell-free DNA derived from advanced non-small cell lung cancer patients. Mol. Med. Rep. 16, 1157–1166 (2017).
Ishii, H., Azuma, K., Sakai, K., Kawahara, A., Yamada, K., Tokito, T. et al. Digital PCR analysis of plasma cell-free DNA for non-invasive detection of drug resistance mechanisms in EGFR mutant NSCLC: Correlation with paired tumor samples. Oncotarget 6, 30850–30858 (2015).
Lee, J. Y., Qing, X., Xiumin, W., Yali, B., Chi, S., Bak, S. H. et al. Longitudinal monitoring of EGFR mutations in plasma predicts outcomes of NSCLC patients treated with EGFR TKIs: Korean Lung Cancer Consortium (KLCC-12-02). Oncotarget 7, 6984–6993 (2016).
Taniguchi, K., Okami, J., Kodama, K., Higashiyama, M. & Kato, K. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci. 99, 929–935 (2008).
Lievens, A., Jacchia, S., Kagkli, D., Savini, C. & Querci, M. Measuring digital PCR quality: performance parameters and their optimization. PLoS ONE 11, e0153317 (2016).
Gerdes, L., Iwobi, A., Busch, U. & Pecoraro, S. Optimization of digital droplet polymerase chain reaction for quantification of genetically modified organisms. Biomol. Detect Quantif. 7, 9–20 (2016).
Zhou, Q., Zhang, X. C., Chen, Z. H., Yin, X. L., Yang, J. J., Xu, C. R. et al. Relative abundance of EGFR mutations predicts benefit from gefitinib treatment for advanced non-small-cell lung cancer. J. Clin. Oncol. 29, 3316–3321 (2011).
Cui, S., Ye, L., Wang, H., Chu, T., Zhao, Y., Gu, A. et al. Use of superARMS EGFR mutation detection kit to detect EGFR in plasma cell-free DNA of patients with lung adenocarcinoma. Clin. Lung Cancer 19, 313–322 (2018).
Author information
Authors and Affiliations
Contributions
Study design: X.S., J.G., L.C. Patient enrolment and patient data collection: X.S., X.L.Z. Performing experiments and data analysis: J.G., X.S., L.C., M.G., X.Y.F., H.H. Manuscript preparation: J.G., X.S., L.C. All authors discussed the results and implications, and critically revised and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved (cch-BOC-1800020) by the ethics committee of Cangzhou Central Hospital, Hebei, China, and all patients provided written informed consent. This study was conducted in accordance with the Declaration of Helsinki.
Consent to publish
Not applicable.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Funding information
This work was funded by CIP program from Stilla technologies Co., Ltd.
Additional information
Note This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, X., Gong, J., Zhang, X. et al. Plasma-based early screening and monitoring of EGFR mutations in NSCLC patients by a 3-color digital PCR assay. Br J Cancer 123, 1437–1444 (2020). https://doi.org/10.1038/s41416-020-1024-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-020-1024-2
This article is cited by
-
New insights into the biology and development of lung cancer in never smokers—implications for early detection and treatment
Journal of Translational Medicine (2023)
-
Current and Emerging Applications of Droplet Digital PCR in Oncology: An Updated Review
Molecular Diagnosis & Therapy (2022)
