
Abstract
Background
Germ cell tumours (GCTs) are the most common solid malignancies in young men. Although high cure rates can be achieved, metastases, resistance to cisplatin-based therapy and late toxicities still represent a lethal threat, arguing for the need of new therapeutic options. In this study, we analysed the potential of cyclin-dependent kinase 4/6 (CDK4/6) inhibitors palbociclib and ribociclib (PaRi) as molecular drugs to treat cisplatin-resistant and -sensitive paediatric and adult GCTs.
Methods
Ten GCT cell lines, including cisplatin-resistant subclones and non-malignant controls, were treated with PaRi and screened for changes in viability (triphenyl tetrazolium chloride (XTT) assay), apoptosis rates (flow cytometry, caspase assay), the cell cycle (flow cytometry), the transcriptome (RNA-sequencing, quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) and on protein level (western blot). Expression profiling was performed on paediatric and adult GCT tissues (expression microarrays, qRT-PCR, immunohistochemistry, ‘The Cancer Genome Atlas’ database).
Results
We demonstrate that adult GCTs highly express CDK4, while paediatric GCTs strongly express CDK6 instead. Thus, both GCT types are potentially treatable by PaRi. GCTs presented as highly sensitive towards PaRi, which caused a decrease in viability, cell cycle arrest and apoptosis. Although GCTs mainly arrested in the G1/G0 phase, some embryonal carcinoma cell lines were able to bypass the G1/S checkpoint and progressed to the G2/M phase. We found that upregulation of CDK3 and downregulation of many mitosis regulation factors, like the HAUS genes, might be responsible for bypassing the G1/S checkpoint and termination of mitosis, respectively. We postulate that GCT cells do not tolerate these alterations in the cell cycle and eventually induce apoptosis.
Conclusion
Our study highlights PaRi as therapeutic options for cisplatin-resistant and -sensitive paediatric and adult GCTs.
Similar content being viewed by others


Background
Testicular type II germ cell tumours (GCTs), which are sub-divided into seminomas and non-seminomas, arise from the precursor lesion germ cell neoplasia in situ (GCNIS), which itself is the result of a defective primordial germ cell development.1,2,3,4 Type II GCTs represent the most common tumour of young men of age 17–45 years and thus are of pubertal or post-pubertal type.1,2,4 Especially in Western countries, incidence rates are rising steadily.5 Additionally, GCTs can be found in newborns and infants, where these pre-pubertal tumours are termed type I GCTs and present mainly as teratomas or yolk-sac tumours.1,2,4 In contrast to type II GCTs, type I GCTs rather develop from a flawed or misrouted early primordial germ cell and not from a GCNIS precursor lesion.
In general, type II GCTs are treated by orchiectomy followed by chemo- or radiotherapy. Despite high cure rates exceeding 90% over all stages, ~10–15% of patients with advanced disease relapse despite adequate multimodal first-line and first-salvage treatments and are deemed treatment refractory.6,7,8 Such patients are usually incurable and face an awful prognosis with a life expectancy of a few months only.6,7,8
Cyclin-dependent kinases (CDK) have been defined as key drivers of cell cycle progression in several cancer entities. An arrest in the cell cycle leads to suppressed tumour growth as well as metastasis. Thus, targeting the cell cycle via selective pharmacological inhibition is a promising therapeutic option. Previous studies proved the CDK4/6 pathway as being active in solid tumours, such as liposarcoma, rhabdomyosarcoma, non-small-cell lung cancer, glioblastoma, oesophageal cancer, melanoma and breast cancer.9 The role of the most prominent CDK4/6 inhibitors palbociclib (Ibrance), ribociclib (Kisqali) and abemaciclib (Verzenio) in monotherapy and combined therapy is under evaluation in several phase 1, 2 or 3 studies. Palbociclib and ribociclib (PaRi) are reversible orally active pyridopyrimidine, which bind to the adenosine triphosphate binding site of the two CyclinD (CCND)-dependent kinases.10 Trials observed nearly indistinguishable clinical results between palbociclib and ribociclib.11 In 2017, the Food and Drug Administration approved PaRi in combination with aromatase inhibitors and abemaciclib plus fulvestrant as initial endocrine-based therapy for women with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced or metastatic breast cancer. These inhibitors eventually block the CCND–CDK4/6 complex, resulting in hypophosphorylation of the retinoblastoma (RB) protein.12 As a consequence, the transcription factor E2F cannot be cleaved from the RB complex and transcription of genes involved in cell cycle progression diminishes, resulting in a cell cycle arrest in the G1/G0 phase.10,13 In GCTs, an ‘unorthodox’ spectrum of defects within the CDK4/6-RB1 pathway has been described, that is, expression of RB1 on the messenger RNA (mRNA) level in all seminomatous and non-seminomatous GCTs, but detectable expression of RB1 on protein level only in teratomas, frequent deletions of RB1 and low expression of P16INK4A in GCNIS, seminomas and embryonal carcinomas (ECs) and overexpression of CyclinD2 (CCND2), as well as expression of CDK4 in GCNIS and invasive GCTs.14,15,16,17,18,19
In this study, we investigated the therapeutic potential of CDK4/6 inhibition in cisplatin-resistant and -sensitive paediatric and adult GCTs.
Methods
Cell culture
In this study, the following GCT cell lines were used: TCam-2 (seminoma), 2102EP, NCCIT, NT2/D1 (all EC), JAR, JEG3 and BeWo (all choriocarcinoma). In the cisplatin-resistant GCT sublines 2102EP-R, NCCIT-R and NT2/D1-R, cisplatin resistance was provoked by cultivating these EC cell lines for a prolonged time with increasing sub-lethal cisplatin concentrations as published.20,21 All GCT cell lines, MPAF fibroblasts and FS1 Sertoli cells were cultivated as described previously.22,23 The cell lines HaCat, HEK293 and VHF fibroblasts were cultivated in Dulbecco’s modified Eagle’s medium (10% foetal calf serum (FCS), 1% penicillin/streptomycin) and THP-1 cells in RPMI medium (10% FCS, 1% penicillin/streptomycin, 200 mM L-Glutamine) at 37 °C and 5% CO2. Short tandem repeat profiles of all cell lines are checked on a regular basis and are available upon request. For CDK4/6 inhibition, PaRi were dissolved in distilled water (pH 3) or dimethyl sulfoxide, respectively.
Measurement of cell viability and Caspase-3/7 activity
The triphenyl tetrazolium chloride (XTT) assays were performed as described previously.23 In summary, 4000 cells were plated onto 96-well plates 24 h before PaRi or solvent application. In a total of 96 h, every day viability was screened by adding 50 μl XTT (1 mg/ml) plus 0.5 μl phenazine methosulfate (1.25 mM) (both from Sigma-Aldrich, Taufkirchen, Germany) and measuring absorbance 4 h later in a ultraviolet–visible spectrometer (450 vs. 650 nm, BMG Labtech, Ortenberg, Germany). Each time point/concentration was measured in quadruplicates. LD50 (lethal dose, 50%) doses were calculated using the GraphPad Prism software version 6. Relative Caspase-3/7 activity was adjusted to the relative cell viability as measured by Caspase-Glo-3/7 Assay and CellTiter-Glo Luminescent Cell Viability Assay (both Promega, Madison, WI, USA), respectively, and performed according to the manufacturer’s protocols.
Flow cytometry (apoptotic and mitotic cells, cell cycle phase profiles)
Flow cytometry analyses of apoptosis rates and cell cycle distribution was performed by Annexin V/propidium iodide (PI) or PI staining, respectively. Briefly, for PI flow cytometry, cells were washed once with phosphate-buffered saline (PBS), fixed with Nicoletti buffer (0.1% Triton-X100, 0.1% sodium-citrate dehydrate dissolved in PBS), and stained with 50 μg/ml PI (Sigma-Aldrich) for 10 min at room temperature (RT). For apoptosis measurement, cells were washed once with Annexin V-binding buffer (10 mM HEPES pH 7.4, 150 mM sodium chloride, 5 mM potassium chloride, 5 mM magnesium chloride, 1.6 mM calcium chloride dissolved in H2O) before incubation with 5 μl Annexin V (ImmunoTools, Friesoythe, Germany) and 50 μg/ml PI (Sigma-Aldrich). For phospho-H3 (pH3) antibody staining, cells were permeabilised with 0.5% Triton X-100 before an 1 h incubation with a pH3 antibody at RT. Secondary antibodies and 50 μg/ml PI were added subsequently before flow cytometric measurement. For antibody details see Table 1. All flow cytometry analyses were performed using the MACSQuant Analyser (Miltenyi Biotech, Bergisch Gladbach, Germany). A total of 50,000 cells was measured for each sample/experiment.
RNA and protein isolation
RNA was isolated (including DNase I digestion) using the RNAeasy Mini Kit (Qiagen, Hilden, Germany) according to the manual.
Proteins were extracted by RIPA buffer containing 10% protease and phosphatase inhibitors (Sigma-Aldrich). Protein concentrations were assessed by the BCA Protein Assay Reagent Kit (Thermo Fisher Scientific, Dreieich, Germany).
Western blot
Twenty micrograms of whole protein lysates were used for western blotting. Staining with Ponceau S solution (Sigma-Aldrich); 0.5% in 5% acetic acid) confirmed a correct membrane transfer. Membranes were blocked in 5% non-fat milk or 5% bovine serum albumin (BSA) in PBS + 1% Tween-20 (PBST) for 1 h and then incubated with primary antibodies overnight at 4 °C. Secondary horseradish peroxidase (HRP)-conjugated antibodies were incubated for 1 h at RT. The ChemiDoc Imaging System (Bio-Rad, Düsseldorf, Germany) was used for imaging. For antibody details see Table 1.
Immunofluorescence staining
Cells were seeded in a 96-well plate and fixed with 4% formaldehyde (10 min) before permeabilisation with 0.2% Triton X-100 in PBS (5 min) with subsequent blocking in 1% BSA, 5% goat serum, 0.3 M glycine and 0.1% Tween-20 in PBS (1 h). Ki-67 antibody was incubated overnight at 4 °C before the addition of secondary antibody (1 h) and counterstaining with 0.5 μg 4′,6-diamidino-2-phenylindole (5 min). For antibody details see Table 1.
Immunohistochemistry
Immunohistochemical reactions were performed on 4-μm formalin-fixed and paraffin-embedded tissue sections as published previously.24 Antigen retrieval was carried out at 98 °C in ethylenediaminetetraacetic acid buffer (pH 9; 20 min). Primary antibodies were incubated for 30 min at RT. Afterwards, sections were incubated with an HRP-labelled secondary antibody at RT for 25 min. The substrate ‘DAB + Chromogen’ system was applied to visualise the target antigen (Dako, Hamburg, Germany). For antibody details see Table 1.
Quantitative reverse transcriptase-polymerase chain reaction
1 µg of RNA was in vitro transcribed into complementary DNA (cDNA) using 1 µl dNTP-Mix (10 mM), 1 µl Oligo(dT)18 Primer (0.5 µg/µl), 4 µl RT Buffer (5×), 1 µl Maxima H Minus Reverse Transcriptase (200 U/µl) and 0.5 µl RiboLock RNase Inhibitor (40 U/µl) (all Thermo Fisher Scientific) on a S1000 cycler (Bio-Rad) at 50 °C for 30 min. Further, quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) runs were performed on a 384-well C1000 cycler (both Bio-Rad). In general, all samples were analysed in technical triplicates using 7.34 ng of cDNA for each replicate and the SYBR-green-based Luna Universal qPCR Master Mix (New England Biolabs, Frankfurt a. M., Germany). At the end of each run, melting-curve analyses were performed. Oligonucleotide sequences are given in Table 2.
Affymetrix/Illumina HT-12v4 expression arrays and Illumina 450k DNA methylation array
The Affymetrix expression array analysis of GCT tissues (GCNIS, n = 3; seminomas, n = 4; ECs, n = 3; teratomas, n = 3; normal testis tissue (NTT), n = 4) was performed previously and re-analysed in the context of this study.25 Illumina microarray expression data of parental GCT cell lines (TCam-2, n = 5; 2102EP, n = 5, NCCIT, n = 4; JAR, n = 2; FS1, n = 4; MPAF, n = 4) was extracted from previous studies available via Gene Expression Omnibus (GEO, ncbi.nlm.nih.gov/geo/) (GSE71239, GSE71269, GSE79065, GSE60698).23,26,27,28,29 The Illumina 450k DNA methylation array was generated in a previous study and re-analysed in the context of this study (GSE76709).30
RNA-sequencing
RNA samples used for transcriptome analyses were quantified (Qubit RNA HS Assay, Thermo Fisher Scientific) and quality was measured by capillary electrophoresis using the Fragment Analyser and the ‘Total RNA Standard Sensitivity Assay’ (Agilent Technologies Inc., Santa Clara, USA). The library preparation was performed according to the manufacturer’s protocol (VAHTS Stranded mRNA-Seq Library Prep Kit). Three hundred nanograms of total RNA was used for mRNA capturing, fragmentation, synthesis of cDNA, adapter ligation and library amplification. Bead purified libraries were normalised and finally sequenced on the HiSeq 3000/4000 System (Illumina Inc., San Diego, USA) with a read setup of 1 × 150 base pairs (bp). The bcl2fastq tool was used to convert the bcl files to fastq files as well for adapter trimming and demultiplexing. Data analyses on fastq files were conducted with CLC Genomics Workbench (version 12.0.2, Qiagen). The reads of all probes were adapter (Illumina TruSeq) and quality trimmed (using the default parameters: bases below Q13 were trimmed from the end of the reads, ambiguous nucleotides max. 2). Mapping was done against the Homo sapiens (hg38) (25 May 2017) genome sequence. Statistical differential expression tests were determined using the ‘Differential Expression in Two Groups’ tool (version 1.02). The resulting p values were corrected for multiple testing by false discovery rate and Bonferroni correction. A p value of ≤0.05 was considered significant.
Online analyses tools
Venn diagrams were generated using Venny 2.1 (https://bioinfogp.cnb.csic.es/tools/venny/).31 The STRING algorithm was used to predict interactive networks from RNA-seq data (https://string-db.org).32 Functional annotation analyses were performed by DAVID (https://david.ncifcrf.gov/home.jsp).33,34 In functional annotation analyses of commonly deregulated genes in GCT cells, only categories (UP_Keywords) with at least five members and p values ≤0.05 were considered significant. For functional annotation analysis of genes deregulated in each GCT cell line, only categories (UP_Keywords) with at least ten members and a p value ≤0.05 was considered significant. Only genes related to an official human gene symbol were included. ‘The Cancer Genome Atlas’ (TCGA) datasets were analysed for isoform/gene expression, DNA methylation and copy number alterations (CNAs) using the UCSC Xena browser (https://xena.ucsc.edu) and the cBioPortal (https://www.cbioportal.org).35,36,37
Results
In this study, we analysed the potential of CDK4 and CDK6 inhibitors palbociclib (PF-00080665, Pfizer Ltd.) and ribociclib (GST0000015996, Novartis Pharma AG) as therapeutic options for cisplatin-resistant and -sensitive GCTs. First, by re-evaluating microarray data of GCT tissues and cell lines, as well as by western blot analyses of GCT cell lines, we screened for expression of CDK4/CDK4 and CDK6/CDK6 on RNA and protein level, respectively (Fig. 1a, b). CDK4/CDK4 was highly expressed in GCT tissues (GCNIS, seminomas, ECs, teratomas) and cell lines (TCam-2, seminoma; 2102EP, NCCIT, both EC; JAR, choriocarcinomas) (Fig. 1a, b). Additionally, CDK4 expression was detectable in NTT, Sertoli cells (FS1) and fibroblasts (MPAF) (Fig. 1a). In contrast, CDK6/CDK6 expression was absent or very weak in all tissues and cell lines analysed (Fig. 1a, b). Interestingly, in paediatric type I GCTs, expression of CDK6 was higher than CDK4 (Fig. 1a, inlay in upper panel). We also confirmed CDK4 expression on protein level by immunohistochemistry of formalin-fixed-paraffin-embedded GCT tissues and found mainly cytoplasmatic, but also nuclear staining in seminomas (n = 8), ECs (n = 7), yolk-sac tumours (n = 4) and teratomas (n = 3) (Fig. 1c). Using the UCSC Xena browser, we screened the TCGA dataset of testicular GCT tissues for expression of CDK4/6 and their different isoforms in GCT tissues.36 The isoforms CDK4-201 (ENST00000257904.11) and CDK6-201 (ENST00000265734.8) seemed to be the predominantly expressed isoforms in GCT (purple) and normal testis tissue (green) (Supplementary Fig. S1A, B). We stratified the TCGA dataset of 156 samples into a seminoma expression signature (SOX17, PRAME, PRDM1 positive; SOX2, DNMT3B, GAL negative) and an EC expression signature (SOX2, DNMT3B, GAL positive; SOX17, PRAME, PRDM1 negative) (Supplementary Fig. S1C). Additionally, we included AFP and β-hCG (CGB), demonstrating that AFP positivity is associated with the EC signature (indicative of yolk-sac tumour components), while β-hCG positivity can be found in both expression signatures (indicative of choriocarcinoma component in EC signature and choriocarcinoma/trophoblast component in seminoma signature) (Supplementary Fig. S1C). CDK4 was strongly expressed in both seminoma and EC signatures, while CDK6 expression was less intense compared to CDK4. Interestingly, in contrast to CDK4, CDK6 positivity was clearly associated with a non-seminomatous signature (Supplementary Fig. S1C).

a Analysis of CDK4/6 expression in GCT tissues (type II GCTs, upper panel, Affymetrix microarray; type I GCTs, inlay in upper panel, qRT-PCR) and cell lines (middle panel: Illumina microarray; lower panel: RNA-seq data, RPKM = reads per kilobase million). As controls, normal testis tissue (NTT), the Sertoli cell line FS1 and fibroblasts (MPAF) were included. Standard deviation is given above bars. b Western blot analysis of CDK4, CDK6, RB1 and phospho-RB1 (pRB1) protein levels in GCT cell lines and controls (fibroblasts, Sertoli cells). HepG2 and HeLa cells served as positive controls for CDK4 and CDK6. GAPDH was used as housekeeper and for normalisation. c Immunohistochemical staining of CDK4 in GCT tissues (seminoma, EC, yolk-sac tumour and teratoma). Scale bar: 500 μm.
We asked, if DNA methylation might influence CDK4/6 expression in GCTs (Supplementary Fig. S2A). In CDK4-positive and -negative GCT tissues from the TCGA cohort, we found mainly hypomethylated CpG dinucleotides around the transcription start site (TSS) and the gene body of CDK4 (Supplementary Fig. S2A). In CDK6-positive and -negative GCT tissues, the TSS presented as hypomethylated, while we found a region ranging from the middle of the gene body to the 3′ end of the gene, where high DNA methylation levels correlated to high CDK6 expression (and vice versa) (Supplementary Fig. S2A, black box). Up to now, the consequence of this finding remains elusive. These methylation dynamics/profiles could also be found in GCT cell lines (Supplementary Fig. S2B). In conclusion, although the CDK4 gene locus seems rather hypomethylated, while the CDK6 gene body seems hypermethylated, no correlation between CDK4/6 expression and DNA methylation became obvious. At the most, one might speculate that the high CDK6 gene body methylation might limit expression of CDK6 and might be responsible for the low CDK6 expression in GCTs compared to CDK4 expression.
In GCT cell lines, we also screened for the expression of the CDK4 targets RB1 and phospho-RB1 (pRB1) (Fig. 1b). On protein level, RB1 and pRB1 were highly detectable in GCT cell lines, while the control cells showed only moderate RB1 and weak pRB1 levels (Fig. 1b).
We screened three studies of GCT tissues for common CNAs (amplifications and deletions) in seminomas and non-seminomas (TCGA Pan-Cancer Atlas, Firehose Legacy35,36) (Supplementary data S1A). We found 11 common CNAs in seminoma and non-seminoma tissues, 19 in seminomas only and 24 in non-seminomas only (Supplementary data S1A). Among them, we found the cell cycle- and CDK4/6-associated genes CCND2 (seminomas 9.8%, non-seminomas 16.8%), CDKN1B (P27; seminomas 15.3%, non-seminomas 16.4%) and MDM2 (seminomas 4%, non-seminomas 3.6%) commonly amplified in seminomas and non-seminomas (Supplementary data S1A). Interestingly, CDK6 was amplified in 2.6% of seminomas, while CCND3 (CyclinD3; 1.7%) and CHEK1 (6.8%) were amplified or deleted in non-seminomas, respectively (Supplementary data S1A). Thus, GCT patients harbouring mutations in these cell cycle- and CDK4/6-related genes might respond differently to CDK4/6 inhibition than non-mutated patients.
Next, we analysed the effect of PaRi on the viability of GCT cell lines by XTT assays. All GCT cell lines analysed displayed a strong reduction in viability 24–96 h after a single PaRi application (Fig. 2 and Supplementary Fig. S3). In detail, the GCT cell lines presented as more sensitive to palbociclib (LD50 range: 2.76–24.85 μM) than ribociclib (LD50 range: 14.58–43.65 μM), and among the different GCT entities, choriocarcinoma cell lines presented as least sensitive to PaRi (palbociclib average LD50: 19.67 μM; ribociclib average LD50: 40.02 μM) (Fig. 2). Interestingly, cisplatin-resistant EC subclones (−R) showed the highest sensitivity towards PaRi compared to their parental cells and all other tested cells (Fig. 2). As controls, fibroblasts (VHF2, MPAF), Sertoli cells (FS1), monocytes (THP-1), keratinocytes (HaCat) and kidney cells (HEK293) were included. On the average, the control cells (palbociclib average LD50: 22.62 μM; ribociclib average LD50: 44.96 μM) presented as less sensitive towards PaRi than the GCT cells (palbociclib average LD50: 12.39 μM; ribociclib average LD50: 34.05 μM) (Fig. 2).

GraphPad Prism-based calculation of LD50 doses in GCT cell lines treated with PaRi based on XTT data (Supplementary Fig. S3). As controls, fibroblasts (VHF2, MPAF), the Sertoli cell line FS1, the monocyte cell line THP-1, the keratinocyte cell line HaCat and the kidney cell line HEK293 were included. Additionally, the breast cancer cell line MCF7 was included.
We asked, if a combinatorial application of PaRi and cisplatin might enhance the therapeutic efficacy compared to a cisplatin-based monotherapy. Thus, we treated cisplatin-resistant and -sensitive GCT cell lines 2102EP(-R) and NT2/D1(-R) with cisplatin (5 μM/2.5 μM) alone or in combination with 5 μM palbociclib or 15 μM ribociclib (Fig. 3). Compared to the cisplatin or PaRi monotherapy, the combinatorial treatment reduced viability considerably stronger.

XTT data of cisplatin-resistant and -sensitive GCT cell lines 2102EP(-R) and NT27D1(-R) treated once with palbociclib (5 μM), ribociclib (15 μM) or cisplatin (5/2.5 μM) alone and in combination. Changes in viability (compared to solvent-treated controls) were measured over 96 h.
We also measured the effects of PaRi on the cell cycle and apoptosis rates by flow cytometry (Fig. 4a, b). As CDK4/6 inhibitors, PaRi should induce a semi-senescent state by blocking the cell cycle transition from G1/S phase. Interestingly, 16 h after PaRi (10 μM /25μM) treatment most GCT cell lines (TCam-2, NCCIT(-R), JAR, JEG3, BeWo) accumulated in G1/G0 phase of the cell cycle, but two EC cell lines including their cisplatin-resistant subclones accumulated in G2/M phase (2102EP(-R), NT2/D1(-R)) (Fig. 4a). The effects on the cell cycle were highly similar between palbo- and ribociclib. In control cells, the cell cycle phase distribution was affected considerably weaker (Fig. 4a). A Ki-67 immunofluorescent staining confirmed reduced proliferation rates after PaRi treatment of TCam-2, 2012EP and JAR cells (Fig. S4). To test, if the G2/M phase arrested cell lines were blocked at the end of G2 phase or enter mitosis under PaRi treatment, we counted phosphorylated histone H3-positive (pH3) cells as a marker of cells in M phase (Fig. 4b). Only NT2/D1 cells showed an increase in pH3 positive cells, while all other GCT cell lines showed a reduced number in pH3 positive cells.

a, b Flow cytometry-based analysis of cell cycle phase distribution (a) and apoptosis rates (b) in GCT cell lines and controls (fibroblasts and Sertoli cells) 16 h after PaRi treatment (10 μM/25 μM). c Phospho-histone H3-positive (pH3+) cells were counted by flow cytometry in solvent and PaRi-treated GCT cells. Differences are given in percentage. d Western blot analysis of PARP (total and cleaved) protein levels in GCT cell lines and MPAF fibroblasts treated for 16 h with PaRi (10 μM/25 μM). Only a long exposure of MPAF enables detection of total PARP. β-Actin was used as housekeeper and for normalisation. e Changes in Caspase-3/7 activity (as fold change) in GCT cell lines after PaRi treatment compared to solvent-treated controls.
PaRi considerably induced apoptosis in GCT cell lines (Fig. 4c), except for NCCIT(-R). In the control cells, nearly no apoptosis induction could be observed (Fig. 4c). We confirmed induction of apoptosis in GCT cell lines after PaRi treatment by western blot detection of cleaved PARP (Fig. 4d). In MPAF fibroblasts, no PARP cleavage could be detected, even after a prolonged exposure of the membrane (Fig. 4d). Additionally, after PaRi application we found increased activity of Caspase-3/7 in GCT cell lines (Fig. 4e). The activity levels resembled the levels of apoptosis induction found in the Annexin V/PI analysis (Fig. 4c vs. e).
Accumulation of GCT cells in either in the G1/G0 or G2/M phase of the cell cycle and the induction of apoptosis pointed at an alternative mode of action of PaRi apart from the canonical CDK4–RB1 axis, prompting us to decipher the molecular mode of action of PaRi in GCT cells in more detail. We performed RNA-sequencing (RNA-seq) analyses of GCT cells (TCam-2, 2102EP, NCCIT, JAR, JEG3 and fibroblasts (MPAF)) treated for 16 h with 10 μM palbociclib to measure transcriptome-wide changes in gene expression. High quality (RNA quality number = 10) of all sequenced RNAs was verified by Agilent Fragment Analyser (Supplementary Fig. S5A). A principal component analysis demonstrated that palbociclib led to the strongest differences in the transcriptome in the choriocarcinoma cell lines JEG3 and JAR and the EC cell lines 2102EP and NCCIT followed by TCam-2, while MPAF fibroblasts displayed the weakest effects (Supplementary Fig. S5B).
We identified all transcripts deregulated (p values ≤ 0.05, fold change ≥ 1.5) in GCT cell lines after palbociclib treatment compared to solvent-treated cells (Supplementary data S1B). Among them, 39 transcripts were deregulated in all GCT cell lines tested (Fig. 5a, Venn diagram; Supplementary data S1C).31 We excluded all transcripts that could not be matched to an official gene symbol and were differentially expressed between the GCT cell lines, to end up with 23 commonly deregulated genes, of which 15 were upregulated and 8 downregulated (Fig. 5a). Ten of these genes were also deregulated in MPAF fibroblasts (Fig. 5a, red labelled genes, fold change ≥1.5). We used the STRING algorithm to identify putative interactions (Fig. 5b).32 Basically, three interactive networks were predicted: (1) consisting of VLDLR, FOS, VEGFA and DDIT4, (2) consisting of SLC7A11 and SLC1A4 and (3) consisting of POLE2, KIF20A, AURKB, UHRF1 and CDCA3. Interestingly, genes also deregulated in MPAF fibroblasts (underlined in red) were mainly found in network (3). Based on a DAVID gene ontology (GO) analysis, the commonly deregulated genes in GCT cells could be associated with (ranked by frequency) cellular growth processes (genes in network (3)), such as mitosis, cell cycle and proliferation (light blue), protein regulatory processes (light green), regulation of transcription (red), drug response (black), developmental processes (yellow), cell surface (pink) and disease (grey) (Fig. 5c and Supplementary data S1D).33,34 Thus, downregulation of the network (3) genes (SPC25, KIF20A, AURKB, UHRF1 and CDCA3) is a common palbociclib response in GCT cell lines and fibroblasts affecting the cell cycle. We confirmed RNA-seq data by qRT-PCR analysis of FOS, DDIT4, VEGFA, UHFR1 and AURKB in palbociclib-treated GCT cell lines and fibroblasts (Fig. 5d). Additionally, we confirmed downregulation of AURKB in palbociclib-treated, cisplatin-resistant and -sensitive GCT cells by western blotting (Fig. 5e). Importantly, we demonstrated that these genes/proteins become deregulated in a similar way after ribociclib treatment of GCT cells and MPAF, arguing further for a common effect of CDK4/6 inhibition (Fig. 5d, e).

a RNA-seq identified commonly deregulated transcripts in GCT cell lines 16 h after 10 μM palbociclib treatment. In MPAF control cells, 10 of the 23 transcripts were also deregulated (≥fold change 1.5) after palbociclib application (red labelled genes). b STRING-based interaction prediction of the commonly deregulated genes. Genes also deregulated in fibroblasts are labelled in red. c DAVID-based GO analysis of the commonly deregulated genes. d Validation of RNA-seq data by qRT-PCR in GCT cell lines and MPAF fibroblast treated with PaRi (10 μM/25 μM) for 16 h. GAPDH was used as housekeeper. Data are given as fold changes (PaRi treated vs. solvent control). Standard deviation is given above bars. e Validation of AURKB downregulation on protein level by western blotting. GCT cells were treated with PaRi (10 μM/25 μM) for 16 h. β-ACTIN was used as housekeeper. Densitometric evaluation of western blot data is given on the right side.
Next, we analysed the individual effects of palbociclib on each GCT entity (seminoma, ECs, choriocarcinomas) and MPAF fibroblasts by a DAVID GO analysis (Supplementary Figs. S6 and S7A). Again, it became obvious that mostly genes associated with the cell cycle were deregulated (Supplementary Figs. S6 and S7A; labelled in light blue), which is in line with the expected molecular effect caused by the palbociclib-mediated CDK4/6 inhibition (pseudo-senescent cell state; G1/S-phase arrest). To narrow down the cell type-/tumour entity-specific effects in more detail, we performed a STRING analysis of all genes related to the GO term ‘cell cycle’ and ended up with networks, presumably representing the key factors of each entity that regulate/influence the cell cycle in response to CDK4/6 inhibition (Supplementary Fig. S6, Supplementary data S1E).
Independent of our set thresholds, we also screened for expression of cell cycle-related factors like cyclins, CDKs, CDK inhibitors and necessary co-factors (Supplementary data S1F). Here, we observed a common trend in the upregulation of CCND1/2/3, CDK6/7 and E2F5, while CCNA2, CCNB1/2, CDK1, CDKN2D/3, CDC2/3/4/7/20/25A/25B/25C and E2F1/2 were downregulated (Supplementary Fig. S7B). Mostly, in MPAF fibroblasts the same trends in deregulation were observed (except for CCND2/D3 and CDK6) (Supplementary Fig. S7B). Thus, PaRi induce quite similar deregulations in canonical cell cycle regulators in GCT and fibroblast cells. We confirmed downregulation of CCNA2 and CCNB1 in PaRi-treated, cisplatin-resistant and -sensitive GCT cell lines by western blotting (Supplementary Fig. S7C). In contrast to upregulation of RNA level, CCND1/D2 were not altered on protein levels in ribociclib-treated GCT cell lines (Supplementary Fig. S7C).
Of note, PaRi treatment led to an increased CDK4 protein level in most GCT cell lines, but not in MPAF fibroblasts, pointing at a putative PaRi counteracting mechanism in GCT cell lines (Supplementary Fig. S7D).
From the cell cycle analysis, we observed that 2102EP(-R) and NT2/D1(-R) accumulated in the G2/M phase instead of the G1/G0 phase of the cell cycle (Fig. 4a). To identify factors that might dictate this decision, we analysed all genes related to the GO term ‘cell cycle’ and are exclusively expressed in 2102EP cells by the STRING algorithm (Supplementary Fig. S8A, Supplementary data S1G). Within this group, we found genes involved in the G2/M transition in the cell cycle (Supplementary Fig. S8A). Two genes (NAE1, HMGA2) were linked to all mentioned GO categories, six genes (CDC25C, CDC7, CNTRL, HAUS1, HAUS2, HAUS6) were linked to four out of five detected categories, two genes (CDK3, CHEK1) were linked to three out of five categories and five genes (TERF1, BORA, PPM1D, MASTL, PLK3) were related to two out of five categories (Supplementary Fig. S8A). Of these 15 genes, 13 were downregulated and two upregulated (CDK3, PLK3) in expression (Supplementary Fig. S8B). Interestingly, CDK3 is able to promote G1/S checkpoint transition in an RB1-independent manner.38
Taken together, some EC cells are able to bypass the PaRi-induced G1/S arrest and reach the G2/M phase of the cell cycle, but downregulation of a plethora of the G2/M phase-regulating genes impedes completion of cell cycle and leads to induction of apoptosis.
Discussion
In this study, we demonstrated the utility of CDK4/6 inhibitors palbociclib and ribociclib in treating cisplatin-resistant and -sensitive GCTs. GCTs presented as more sensitive towards PaRi than analysed control cells (including MCF7 breast cancer cells), opening a putative therapeutic window for patients suffering from GCT. Although, it has to be mentioned that results gathered in this study by comparing GCT cell lines to one breast cancer cell lines is not enough to draw reliable conclusions on the differences between GCTs and breast cancers regarding PaRi treatment. In the future, comparing large tumour cohorts of each entity would be necessary.
Among the different GCT entities, choriocarcinoma cells presented as least sensitive and EC cells as most sensitive. Interestingly, Schmidt et al.18 demonstrated that among seminomas and non-seminomas, ECs show highest levels of CDK4 expression, thus in ECs sensitivity to CDK4/6 inhibition correlates to high levels of CDK4 expression.18 Additionally, in our study cisplatin-resistant EC subclones showed a further increased sensitivity compared to parental cells. This suggests that mechanisms causing cisplatin resistance might in turn cause sensitisation towards inhibition of cell cycle regulators, that is, CDK4 or application of PaRi is able to revert the cisplatin-resistant phenotype. In line, combination of cisplatin with PaRi might be a novel therapeutic option for treatment of GCT patients, since combining both drugs considerably increased therapeutic efficacy. It remains elusive if combining PaRi with cisplatin leads to additive or synergistic effects.
So far, only one early clinical study (phase 2, 29 patients) tested the potential of the CDK4/6 inhibitor palbociclib in treating incurable, refractory, RB1-expressing GCTs.39 In this study, especially mature teratomas (including cases with malignant transformation) had a favourable 24-week progression-free survival rate.39 In a follow-up study, Narayan et al.40 performed a retrospective analysis with long-term follow-up data of the patient cohort with unresectable mature teratoma treated with palbociclib and demonstrated that in 12 patients palbociclib might lead to a clinically meaningful delay in disease-related clinical events. Further, a randomised, blinded and placebo-controlled clinical study (NCT02300987) analysed the suitability of ribociclib to treat relapsed, refractory, incurable teratoma with recent progression. In this study, only 10 participants were recruited and thus numbers are quite limited (results pending). Nevertheless, CDK4/6 inhibition seems to be promising in treating especially mature teratomas, including teratomas with malignant transformation. The beneficial effect of palbociclib on the progression-free survival of non-teratoma GCT patients seems to be considerably weaker compared to patients with teratoma.39 Nevertheless, in the study of Vaughn et al.39, only a limited number of patients as well as only RB1-expressing GCT were included and only palbociclib was tested. Based on the result of our study, in GCTs PaRi do not only act via the canonical CDK4–RB1 axis and provoke effects beyond a cell cycle block, that is, apoptosis. Thus, it would be interesting and worth to (a) screen a larger cohort of type I and type II GCT patients with (b) no restrictions to RB1 status and to (c) include ribociclib and abemaciclib (Lilly) as alternatives to palbociclib. Additionally, we found that PaRi treatment led to an upregulation of CDK4 in GCT cell lines, which we postulated might counteract the effects of PaRi and might cause resistance. It would be interesting to screen the patients treated by Vaughn et al.39 for upregulation of CDK4 under therapy. If upregulation of CDK4 would be restricted to non-teratomatous GCT patients, the reduced activity of palbociclib in these patients could be explained.
In type II adult GCT cells CDK4 was highly expressed, while CDK6 expression was very low, confirming results of Schmidt et al.,18 who also detected high CDK4 in 41% of GCTs (most strongly in ECs) and reduced CDK6 expression in 64% of seminomas and 23% of non-seminomas. Thus, in adult type II GCTs CDK4 seems to be the primary target of PaRi. In contrast, in all type I paediatric GCTs analysed, CDK6 was expressed considerably higher than CDK4. In conclusion, although differing in expression of PaRi target molecules, PaRi might be a therapeutic option for both type I and type II GCTs.
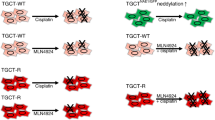
In type II GCT cell lines, PaRi induced a pseudo-senescent state, mostly leading to accumulation of cells in the G1/G0 phase of the cell cycle (Fig. 6a). Nevertheless, especially EC cells seem capable of escaping the G1/G0 arrest and continuing the cell cycle (Fig. 6b, c). Our results suggested that EC cell lines either accumulate at the end of the G2 phase or are able to enter mitosis, but without completing this process (Fig. 6b, c).

In GCTs, PaRi affects the cell cycle in three ways: a by inducing G1/G0 arrest via the CDK4–pRB1 axis, b by bypassing the G1/S checkpoint by upregulation of CDK3 and inducing an arrest at the end of the G2 phase or c by downregulating mitosis regulators leading to termination of mitosis. All described effects are not tolerated by GCT cells, eventually resulting in apoptosis.
Furthermore, PaRi induced apoptosis in cisplatin-resistant and parental GCT cell lines, but not in the tested control cells. Induction of apoptosis after PaRi has also been described in bladder cancer cells and T cell acute lymphoblastic leukaemia, while in pancreatic ductal adenocarcinoma cell lines, CDK4/6 inhibition alone did not induce apoptosis.41,42,43 Thus, induction of apoptosis in response to CDK4/6 inhibition seems to be tumour type dependent, but is in general of benefit in terms of a therapeutic application.
On a molecular level, PaRi considerably affected cell cycle-related genes in GCT cells, but interestingly factors that mediate this response were different between the GCT entities. Although regulators of different cell cycle phases become deregulated upon PaRi application, GCTs mostly accumulate already in the G1/G0 phase. This G1/0 phase accumulation seemed to be the result of downregulation of early cell cycle phase regulators E2F/1, CDCA3, KIF20A, SPC25, UHRF1, CDKN2D and CDKN3 (Fig. 6a).
Interestingly, CDK3 is able to promote the G1/S-phase transition in a RB1-independent manner38 (Fig. 6b). Thus, in EC cells, upregulation of CDK3 might enable progression beyond the G1/S-phase checkpoint, independent of the palbociclib-inhibited CDK4–RB1 axis (Fig. 6b). Then, EC cells either arrest at the end of the G2 phase or enter mitosis without completing this process (Fig. 6b, c). PLK3 regulates the M phase of the cell cycle by interacting with CDC25C (becomes inactivated by PLK3) and CHEK1. After palbociclib treatment, PLK3 was upregulated in 2102EP cells, while CDC25C and CHEK1 were downregulated (Supplementary Fig. S8A, B). Thus, upregulation of PLK3 might lead to downregulation of CDC25C and CHEK1. Additionally, CHEK1 is deleted in 6.8% of non-seminomas, suggesting that deletion of CHEK1 might be of benefit for efficacy of a CDK4/6 inhibitor treatment in non-seminomas (Supplementary data S1A). Furthermore, downregulation of the human augmin complex genes HAUS1/2/6, which play an important role in mitotic spindle and centrosome integrity, was observed after PaRi application (Supplementary Fig. S8A, B). These effects combined might prevent the EC cells from completing mitosis (Fig. 6b, c). Independent of the cell cycle state, the cells arrested in apoptosis was induced in GCT cell lines after PaRi application, suggesting that even cells that enter mitosis do not complete this process. We conclude that GCT cells are not able to tolerate the pseudo-senescent state (either at G1/S checkpoint or when accumulating at the end of G2 phase) or a misregulated mitosis and undergo apoptosis soon after (Fig. 6a–c). Thus, we hypothesise that induction of apoptosis is not a directly provoked effect by PaRi, but a consequence of the cells’ inability to tolerate the prolonged alterations in the cell cycle.
We observed that CDK4 is upregulated on protein level after PaRi treatment. We propose that GCT cells try to counteract inhibition of CDK4 by upregulating its expression, leading to the production of new (and more) CDK4. Thus, GCT cells might become resistant to PaRi treatment over time, but we found no upregulation of the CDK4 downstream target CCND1/CCND2. Upregulation of CCND in response to CDK4/6 inhibition has been shown in several publications and was linked to acquired resistance by bypassing cytostasis via a CyclinD1-CDK2-mediated S-phase entry.12,44,45 GCT cell lines seem to bypass this mechanism as GCT cell lines predominately accumulated in the G1/G0 phase of the cell cycle after PaRi treatment and neither upregulated CyclinD1/2 on protein level nor significantly upregulated CDK2 on RNA level (Supplementary Fig. S7C, Supplementary data S1B).
In our analysis of CNAs, we found that CCND2 is commonly amplified in seminomas and non-seminomas (Supplementary data S1A). Maybe, those patients are more prone to become resistant to CDK4/6 inhibition than non-CCND1-amplified patients. Advances in PROteolysis TArgeting Chimeras (PROTACs) suppressing CDK4/6 were recently introduced in breast cancer and glioblastoma cell lines, where CDK4 and CDK6 protein expressions were reduced considerably.46 Thus, a PROTAC targeting CDK4/6 could be used as an alternative to PaRi, if resistance to these drugs might be an issue.
Conclusions
In conclusion, palbociclib and ribociclib present a new putative therapeutic option for cisplatin-resistant and -sensitive paediatric and adult GCTs. Nevertheless, it has to be shown in the future whether mechanisms and drug kinetics/dynamics found and measured in this study on artificial cell lines can be mirrored to the in vivo situation. Therefore, large type I and II GCT cohorts have to be analysed in detail. Additionally, further experiments will have to reveal whether GCTs are able to acquire resistance towards PaRi and if a combinatorial treatment of cisplatin and PaRi can be of benefit for the patients. Up to now, it remains elusive if CDK4/6 inhibition might revert a cisplatin-resistant phenotype or if mechanisms causing cisplatin resistance in parallel sensitise GCT cells towards drugs that interfere with the cell cycle.
References
Oosterhuis, J. W. & Looijenga, L. H. J. Testicular germ-cell tumours in a broader perspective. Nat. Rev. Cancer 5, 210–222 (2005).
Cheng, L., Albers, P., Berney, D. M., Feldman, D. R., Daugaard, G., Gilligan, T. et al. Testicular cancer. Nat. Rev. Dis. Prim. 4, 29 (2018).
Berney, D. M., Looijenga, L. H. J., Idrees, M., Oosterhuis, J. W., Rajpert-De Meyts, E., Ulbright, T. M. et al. Germ cell neoplasia in situ (GCNIS): evolution of the current nomenclature for testicular pre-invasive germ cell malignancy. Histopathology 69, 7–10 (2016).
Moch, H., Cubilla, A. L., Humphrey, P. A., Reuter, V. E. & Ulbright, T. M. The 2016 WHO Classification of Tumours of the urinary system and male genital organs—part a: renal, penile, and testicular tumours. Eur. Urol. 70, 93–105 (2016).
Park, J. S., Kim, J., Elghiaty, A. & Ham, W. S. Recent global trends in testicular cancer incidence and mortality. Medicine (United States). 97, e12390 (2018).
Oing, C., Seidel, C., Bokemeyer, C., Oing, C., Seidel, C. & Bokemeyer, C. Expert review of anticancer therapy therapeutic approaches for refractory germ cell cancer therapeutic approaches for refractory germ cell cancer. Expert Rev. Anticancer Ther. 18, 389–397 (2018).
Oing, C., Giannatempo, P., Honecker, F., Oechsle, K., Bokemeyer, C., Beyer, J. Palliative treatment of germ cell cancer. Cancer Treat. Rev. 71, 102–107 (2018).
Oing, C. & Lorch, A. The role of salvage high-dose chemotherapy in relapsed male germ cell tumors. Oncol. Res. Treat. 41, 365–369 (2018).
Hamilton, E. & Infante, J. R. Targeting CDK4/6 in patients with cancer. Cancer Treatment Rev. 45, 129–138 (2016).
Schettini, F., De Santo, I., Rea, C. G., De Placido, P., Formisano, L., Giuliano, M. et al. CDK 4/6 inhibitors as single agent in advanced solid tumors. Front. Oncol. 8, 608 (2018).
Laderian, B. & Fojo, T. CDK4/6 inhibition as a therapeutic strategy in breast cancer: palbociclib, ribociclib, and abemaciclib. Semin. Oncol. 44, 395–403 (2017).
Paternot, S., Colleoni, B., Bisteau, X. & Roger, P. P. The CDK4/CDK6 inhibitor PD0332991 paradoxically stabilizes activated cyclin D3-CDK4/6 complexes. Cell Cycle 13, 2879–2888 (2014).
Knudsen, E. S. & Witkiewicz, A. K. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer 3, 39–55 (2017).
Strohmeyer, T., Reissmann, P., Cordon-Cardo, C., Hartmann, M., Ackermann, R. & Slamon, D. Correlation between retinoblastoma gene expression and differentiation in human testicular tumors. Proc. Natl Acad. Sci. USA 88, 6662–6666 (1991).
Murty, V. V., Houldsworth, J., Baldwin, S., Reuter, V., Hunziker, W., Besmer, P. et al. Allelic deletions in the long arm of chromosome 12 identify sites of candidate tumor suppressor genes in male germ cell tumors. Proc. Natl Acad. Sci. USA 89, 11006–11010 (1992).
Houldsworth, J., Reuter, V., Bosl, G. J. & Chaganti, R. S. K. Aberrant expression of Cyclin D2 is an early event in human male germ cell tumorigenesis. Cell Growth Differ. 8, 293–299 (1997).
Chaganti, R. S. K. & Houldsworth, J. Genetics and biology of adult human male germ cell tumors. Cancer Res. 60, 1475–1482 (2000).
Schmidt, B. A., Rose, A., Steinhoff, C., Strohmeyer, T., Hartmann, M. & Ackermann, R. Up-regulation of cyclin-dependent kinase 4/cyclin D2 expression but down-regulation of cyclin-dependent kinase 2/cyclin E in testicular germ cell tumors. Cancer Res. 61, 4214–4221 (2001).
Bartkova, J., Lukas, C., Sørensen, C., Rajpert-De Meyts, E., Skakkebæk, N. E., Lukas, J. et al. Deregulation of the RB pathway in human testicular germ cell tumours. J. Pathol. 200, 149–56 (2003).
Oechsle, K., Honecker, F., Cheng, T., Mayer, F., Czaykowski, P., Winquist, E. et al. Preclinical and clinical activity of sunitinib in patients with cisplatin-refractory or multiply relapsed germ cell tumors: a Canadian Urologic Oncology Group/German Testicular Cancer Study Group cooperative study. Ann. Oncol. 22, 2654–2660 (2011).
Fenske, A. E., Glaesener, S., Bokemeyer, C., Thomale, J., Dahm-Daphi, J., Honecker, F. et al. Cisplatin resistance induced in germ cell tumour cells is due to reduced susceptibility towards cell death but not to altered DNA damage induction or repair. Cancer Lett. 324, 171–178 (2012).
Nettersheim, D., Heukamp, L. C., Fronhoffs, F., Grewe, M. J., Haas, N., Waha, A. et al. Analysis of TET Expression/Activity and 5mC oxidation during normal and malignant germ cell development. PLoS ONE 8, e82881 (2013).
Nettersheim, D., Jostes, S., Fabry, M., Honecker, F., Schumacher, V., Kirfel, J. et al. A signaling cascade including ARID1A, GADD45B and DUSP1 induces apoptosis and affects the cell cycle of germ cell cancers after romidepsin treatment. Oncotarget 7, 74931–74946 (2016).
Bremmer, F., Bohnenberger, H., Küffer, S., Oellerich, T., Serve, H., Urlaub, H. et al. Proteomic comparison of malignant human germ cell tumor cell lines. Dis. Markers 2019, 8298524 (2019).
Eckert, D., Nettersheim, D., Heukamp, L. C., Kitazawa, S., Biermann, K. & Schorle, H. TCam-2 but not JKT-1 cells resemble seminoma in cell culture. Cell Tissue Res. 82, 214–223 (2008).
Nettersheim, D., Jostes, S., Sharma, R., Schneider, S., Hofmann, A., Ferreira, H. J. et al. BMP inhibition in seminomas initiates acquisition of pluripotency via NODAL signaling resulting in reprogramming to an embryonal carcinoma. PLoS Genet. 11, e1005415 (2015).
Nettersheim, D., Heimsoeth, A., Jostes, S., Schneider, S., Fellermeyer, M., Hofmann, A. et al. SOX2 is essential for in vivo reprogramming of seminoma-like TCam-2 cells to an embryonal carcinoma-like fate. Oncotarget 7, 47095–47110 (2016).
Jostes, S., Nettersheim, D., Fellermeyer, M., Schneider, S., Hafezi, F., Honecker, F. et al. The bromodomain inhibitor JQ1 triggers growth arrest and apoptosis in testicular germ cell tumours in vitro and in vivo. J. Cell. Mol. Med. 21, 1300–1314 (2017).
Nettersheim, D., Arndt, I., Sharma, R., Riesenberg, S., Jostes, S., Schneider, S. et al. The cancer/testis-antigen PRAME supports the pluripotency network and represses somatic and germ cell differentiation programs in seminomas. Br. J. Cancer 115, 454–464 (2016).
Nettersheim, D., Berger, D., Jostes, S., Skowron, M. & Schorle, H. Deciphering the molecular effects of romidepsin on germ cell tumours: DHRS2 is involved in cell cycle arrest but not apoptosis or induction of romidepsin effectors. J. Cell. Mol. Med. 23, 670–679 (2019).
Oliveros, J. C. VENNY. An interactive tool for comparing lists with Venn Diagrams. http://bioinfogp.cnb.csic.es/tools/venny/index.html.bioinfogp.cnb.csic.es/tools/venny/index.html (2007).
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J. et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Bagrodia, A., Lee, B. H., Lee, W., Cha, E. K., Sfakianos, J. P., Iyer, G. et al. Genetic determinants of cisplatin resistance in patients with advanced germ cell tumors. J. Clin. Oncol. 34, 4000–4007 (2016).
Shen, H., Shih, J., Hollern, D. P., Wang, L., Bowlby, R., Tickoo, S. K. et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 23, 3392–3406 (2018).
Goldman, M., Craft, B., Hastie, M., Repečka, K., Kamath, A., McDade F. et al. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv (2019). https://doi.org/10.1101/326470.
Hofmann, F. & Livingston, D. M. Differential effects of cdk2 and cdk3 on the control of pRb and E2F function during G1 exit. Genes Dev. 10, 851–861 (1996).
Vaughn, D. J., Hwang, W. T., Lal, P., Rosen, M. A., Gallagher, M. & O’Dwyer, P. J. Phase 2 trial of the cyclin-dependent kinase 4/6 inhibitor palbociclib in patients with retinoblastoma protein-expressing germ cell tumors. Cancer 121, 1463–1468 (2015).
Narayan, V., Hwang, W. T., Lal, P., Rosen, M. A., Gallagher, M., O’Dwyer, P. J. et al. Cyclin-dependent kinase 4/6 inhibition for the treatment of unresectable mature teratoma: long-term follow-up of a phase II study. Clin. Genitourin. Cancer 14, 504–510 (2016).
Choi, Y. J., Li, X., Hydbring, P., Sanda, T., Stefano, J., Christie, A. L. et al. The requirement for Cyclin D function in tumor maintenance. Cancer Cell 22, 438–451 (2012).
Franco, J., Balaji, U., Freinkman, E., Witkiewicz, A. K. & Knudsen, E. S. Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep. 14, 979–990 (2016).
Zhang, G., Ma, F., Li, L., Li, J., Li, P., Zeng, S. et al. Palbociclib triggers apoptosis in bladder cancer cells by Cdk2-induced Rad9-mediated reorganization of the Bak.Bcl-xl complex. Biochem. Pharmacol. 163, 133–141 (2019).
Dean, J. L., Thangavel, C., McClendon, A. K., Reed, C. A. & Knudsen, E. S. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 29, 4018–4032 (2010).
Herrera-Abreu, M. T., Palafox, M., Asghar, U., Rivas, M. A., Cutts, R. J., Garcia-Murillas, I. et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 76, 2301–2313 (2016).
Zhao, B. & Burgess, K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem. Commun. 55, 2704–2707 (2019).
Acknowledgements
We thank Anna Pehlke for the excellent technical assistance. We also thank Pfizer Ltd and Novartis Pharma AG for providing palbociclib and ribociclib, respectively (MTA to D.N.: WI238366, PLSMTA18FEB46).
Author information
Authors and Affiliations
Contributions
Conceptualisation: D.N. Data curation: M.A.S., M.V., A.W., T.K.B., F.B. and P.P. Formal analysis: M.A.S., M.V., A.W., T.K.B., F.B., P.P. and D.N. Investigation: M.A.S., M.V., A.W., T.K.B., F.B. and P.P. Methodology: D.N., P.P. and K.K. Project administration: D.N. and P.A. Resources: D.N., P.A., S.S. and G.C. Software: P.P., K.K. and M.A.S. Supervision: D.N. Validation: M.A.S., M.V., A.W. and T.K.B. Visualisation: D.N., M.A.S. and P.P. Writing— original draft: D.N. and M.A.S. Writing—review and editing: D.N., P.A., S.S. and G.C.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ethics committee of the Heinrich-Heine University Düsseldorf (EC-HHU-D) raised no concerns about using all analysed cell lines for in vitro experiments and drug screening (ethics votes 2018-178 and 2019-412). All type I GCT samples were cryopreserved and anonymised during the MAKEI 96-study and provided by the MAKEI 96-biobank with no concerns raised by EC-HHU-D about analysing these samples (ethic votes 837 and 2019-822). The cell lines TCam-2 was kindly provided by Sohei Kitazawa via Hubert Schorle (Department of Developmental Pathology, Institute of Pathology, University of Bonn Medical School, Bonn, Germany). 2102EP, NCCIT, NT2/D1 and the cisplatin-resistant sublines 2102EP-R, NCCIT-R and NT2/D1-R were kindly provided by Friedemann Honecker (ZeTuP Silberturm, St. Gallen, Switzerland) and Christoph Oing (Department of Oncology, Haematology and Bone Marrow Transplantation with Division of Pneumology, University Medical Centre Hamburg-Eppendorf, Hamburg, Germany). The cell lines JAR, JEG3 and BeWo were purchased from ATCC. MPAF and VHF2 fibroblasts were kindly provided by Michael Peitz (Core Facility Cell Programming, Life & Brain GmbH, Bonn, Germany) and Wolfgang Schulz (Department of Urology, Urological Research Lab, University Hospital Düsseldorf, Düsseldorf, Germany), respectively. FS1 Sertoli cells were kindly provided Valerie Schumacher (Harvard Medical School, Department of Paediatrics, Boston, MA, USA). The cell lines HaCat and HEK293 were provided by Wolfgang Schulz (Department of Urology, Urological Research Lab, University Hospital Düsseldorf, Düsseldorf, Germany).
Data availability
All data published in this study is available upon request to the corresponding author. Illumina gene expression microarray data are available via GEO (ncbi.nlm.nih.gov/geo/) (GSE71239, GSE71269, GSE79065, GSE60698). The Illumina 450k DNA methylation array data is also available via GEO (GSE76709). The Cancer Genome Atlas’ (TCGA) datasets can be accessed via the UCSC Xena browser (https://xena.ucsc.edu) and cBioPortal (https://www.cbioportal.org).
Competing interests
Pfizer Ltd and Novartis Pharma AG neither influenced interpretation of the data or writing of the manuscript nor financially supported this study, except providing the CDK4/6 inhibitors. The authors declare no competing interests.
Funding information
D.N. (CIO D), P.A. (CIO D), G.C. (CIO B) and S.S. (CIO B) cooperated in this study as part of the lighthouse project ‘Germ Cell Tumours’ of the ‘Centrum für Integrierte Onkologie – Aachen, Bonn, Cologne, Düsseldorf’ (CIO ABCD). D.N. P.P. and K.K. cooperated in this study as part of the ‘Biologisch-Medizinisches Forschungszentrum’ (BMFZ) of the Heinrich-Heine University Düsseldorf. D.N., P.A. and F.B. cooperated in this study as members of the ‘German Testicular Cancer Study Group’ (GTCSG) of the ‘Deutsche Krebsgesellschaft’. F.B. is supported by the Wilhelm Sander-Stiftung (grant numbers 2016.041.1 and 2016.041.2). Open access funding provided by Projekt DEAL.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Skowron, M.A., Vermeulen, M., Winkelhausen, A. et al. CDK4/6 inhibition presents as a therapeutic option for paediatric and adult germ cell tumours and induces cell cycle arrest and apoptosis via canonical and non-canonical mechanisms. Br J Cancer 123, 378–391 (2020). https://doi.org/10.1038/s41416-020-0891-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-020-0891-x
This article is cited by
-
Apoptosis and eryptosis: similarities and differences
Apoptosis (2024)
-
Molecular and histopathological characterization of seminoma patients with highly elevated human chorionic gonadotropin levels in the serum
Virchows Archiv (2023)
-
Therapeutical interference with the epigenetic landscape of germ cell tumors: a comparative drug study and new mechanistical insights
Clinical Epigenetics (2022)
-
Cisplatinrefraktäre Keimzelltumoren – molekulare Grundlagen und klinische Konzepte
Der Onkologe (2021)
-
The component of the m6A writer complex VIRMA is implicated in aggressive tumor phenotype, DNA damage response and cisplatin resistance in germ cell tumors
Journal of Experimental & Clinical Cancer Research (2021)
