
Abstract
Background
Indoleamine 2,3-dioxygenase (IDO), the first step in the kynurenine pathway (KP), is upregulated in some cancers and represents an attractive therapeutic target given its role in tumour immune evasion. However, the recent failure of an IDO inhibitor in a late phase trial raises questions about this strategy.
Methods
Matched renal cell carcinoma (RCC) and normal kidney tissues were subject to proteomic profiling. Tissue immunohistochemistry and gene expression data were used to validate findings. Phenotypic effects of loss/gain of expression were examined in vitro.
Results
Quinolate phosphoribosyltransferase (QPRT), the final and rate-limiting enzyme in the KP, was identified as being downregulated in RCC. Loss of QPRT expression led to increased potential for anchorage-independent growth. Gene expression, mass spectrometry (clear cell and chromophobe RCC) and tissue immunohistochemistry (clear cell, papillary and chromophobe), confirmed loss or decreased expression of QPRT and showed downregulation of other KP enzymes, including kynurenine 3-monoxygenase (KMO) and 3-hydroxyanthranilate-3,4-dioxygenase (HAAO), with a concomitant maintenance or upregulation of nicotinamide phosphoribosyltransferase (NAMPT), the key enzyme in the NAD+ salvage pathway.
Conclusions
Widespread dysregulation of the KP is common in RCC and is likely to contribute to tumour immune evasion, carrying implications for effective therapeutic targeting of this critical pathway
Similar content being viewed by others


Background
Renal cancer is one of the ten most common adult cancers, accounting for over 100,000 deaths worldwide each year.1 It is also a cancer with one of the highest projected increases in incidence over the next two decades.2 Almost 90% of these cancers arise within the renal parenchyma and are termed renal cell carcinomas (RCCs). The most common (75%) histological subtype is clear cell RCC (conventional) (ccRCC), which are characterised by loss of the VHL tumour-suppressor gene, followed by papillary (10–15%) and chromophobe (5%) RCC. Each is considered to arise from distinct parts of the human nephron, are genetically distinct3 and vary considerably in their clinical behaviour.
Treatment options for patients with RCC have burgeoned in recent years, but despite this, it is a cancer that remains incurable for most patients with advanced disease. As in a number of other tumour types, immunotherapy, in the form of checkpoint inhibitors (CPIs), has come to the forefront of patient treatment, both in the first- and second-line settings. However, while some patients are observed to have deep and durable responses to these agents, many patients fail to respond.4 It appears therefore that, alone, CPIs are not always sufficient to overcome immune evasion and immune tolerance by tumours. Hence, increasing attention is being focussed on combining these drugs with other immune-modifying targeted agents.5
Alterations in cellular metabolism are a hallmark of cancer,6 most notably perhaps the ‘Warburg effect’, that describes the increased rate of glycolysis with reduced oxidative phosphorylation characteristic of tumours.7 We were among the first to demonstrate this comprehensively in renal cancers, using a proteomic approach, showing an upregulation in the majority of proteins in the glycolytic pathway and a parallel downregulation of mitochondrial enzymes in comparison to normal renal tissues,8 highlighting novel opportunities for therapeutic targeting.9
More recently, alteration in the metabolism of the essential amino acid tryptophan in cancer, through the kynurenine pathway, has come to wide attention as a mechanism by which tumours may escape immune control and promote disease progression. The enzymes indoleamine 2,3-dioxygenase (IDO1, IDO2) and tryptophan 2,3-dioxygenase (TDO) initiate the first steps in the kynurenine pathway, converting tryptophan to kynurenine, with the TDO-dependent pathway in the liver normally accounting for the majority of tryptophan metabolism and IDO-mediated metabolism predominantly occurring secondary to inflammation and cytokine-induced upregulation.10 In normal physiology, IDO plays an important role in tolerance to non-self-antigens, for example foetal antigens, where such immune non-responsiveness may be important.11,12 Upregulation of IDO leads to tryptophan depletion and kynurenine accumulation, which appear to work in concert to mediate immunosuppression, via T cell anergy and apoptosis and suppressed T cell differentiation.13 The harnessing of this phenomenon by tumours has led to the development of inhibitors of IDO1 that have progressed to clinical trials in combination with CPI. Despite much promise, initial results have, however, been disappointing and the future of these agents currently remains uncertain.14,15 This may be because of patient selection and lack of suitable profiling of immuno-regulating metabolism, emphasising the need for a deeper understanding of these pathways.
Here, using a proteomic-based approach, we show that the kynurenine pathway is more broadly disrupted than has been previously considered, extending beyond IDO1, that this is a common event in RCC and is not just restricted to the clear cell histological subtype and suggests possible redundancy in the pathway within the tumour setting. Our findings are of significance in terms of highlighting various aspects of this pathway for potential therapeutic targeting, patient stratification and may have implications for other cancers.
Methods
Reagents
Reagents were purchased as follows: general chemicals (Sigma, Poole, UK and VWR, Poole, UK), goat serum and human serum albumin, mouse monoclonal anti-β-actin antibody clone AC15 (Sigma), Hybond™C super NC membrane, Pharmalyte pH 3–10, IPG strips, dry strip cover fluid, bromophenol blue and PlusOne Silver Stain (GE Healthcare,Little Chalfont, UK), CHAPS (Calbiochem, San Diego, USA), LMP agarose, Minimum Essential Medium (MEM)-Alpha medium, L-glutamine, trypsin with EDTA, G418, HBSS and Antibody Diluent (Invitrogen Life Technologies, Paisley, UK), foetal calf serum (FCS; Harlan-Seralab, Sussex, UK); PBS (Oxoid, Basingstoke, UK), acrylamide (National Diagnostics, Hull, UK), OWL Silver Stain (OWL Separation Systems, Portsmouth, USA), trypsin sequencing grade (Promega, Southampton, UK), ACN (Rathburn, Walkerburn, UK), Complete™ mini protease inhibitor cocktail tablets (Roche, Lewes, UK), Envision™1 systems (Dako, Ely, UK), SuperSignal® West Dura Extended Duration Substrate (Pierce, Tattenhall, UK), Access Revelation solution (Menarini Diagnostics, Berkshire, UK), Bloxall, Impress Rabbit horseradish peroxidase (HRP)‐conjugated secondary antibodies, Impact DAB and horse serum (Vector Laboratories, Peterborough, UK), BCA protein assay (Thermo Scientific, Warrington, UK) mouse monoclonal anti-VHL clone Ig32 (BD Biosciences, Wokingham, UK), affinity-purified rabbit antibodies to kynurenine pathway components KMO (cat. nos. HPA056942 and HPA031115), KYNU (cat. no.HPA031686), NAMPT (cat. no. HPA047776) and IDO1 (cat. no. HPA023072) (Atlas Antibodies, Sweden), oligonucleotides (Eurogentec).
Established cell line VHL transfectants
Cell line pairs generated from the VHL-defective human RCC cell lines UMRC2, RCC4 and 786-0 by stable transfection with either empty vector or a wild-type (WT) VHL expression construct have been described previously.16,17,18 786-0, stably transfected with full-length VHL (786+VHL) or control vector (786+pRC) were obtained as gifts from W.G. Kaelin.16 Cells were maintained in MEM-α medium supplemented with 10% v/v FCS and 1% v/v L-glutamine, as previously described.19 All cell lines were screened for mycoplasma contamination.
Quinolate phosphoribosyltransferase (QPRT) expression and QPRT knockdown stable transfectants
QPRT gene expression constructs containing empty vector (pFB-HYG) or QPRT (pFB-HYG-QPRT) were created. The QPRT insert was made by PCR of human cDNA (forward primer: GTCAGTCGACCACCATGGACGCTGAAGGCC and reverse primer GACTCGAGCTAGTGGATTTTGGGCACTGGAGC) followed by digestion with Sal1 and Xho1 and ligation into the multiple cloning site of pFB. Correct sequence was confirmed by sequencing. These constructs were amplified in XL1 Blue competent Escherichia coli (Stratagene), then introduced into 293-PhoenixA cells using SiPORT transfection agent (Ambion). Supernatant containing viral particles was harvested on days 3 and 4, and polybrene was added to 8 μg/ml. Four ml of medium was used to infect each T75 flask of a VHL-defective 786-0 cell line lacking endogenous VHL followed by selection with hygromycin (0.5 mg/ml) and were designated 786+pFB and 786+QPRT, respectively. Cells were maintained in MEM-α medium supplemented with 10% FCS, 1% v/v L-glutamine, G418 (1 mg/ml) and hygromycin selection (0.5 mg/ml).
QPRT short hairpin RNA (shRNA) constructs containing a non-specific shRNA (pRetroSuper-shRNA-scramble) or shRNA targeting QPRT (pRetroSuper-shRNA-QPRT) were created using the following oligonucleotides (capitals indicate nucleotides corresponding to QPRT sequence or control, lower case indicates loop and linker sequences):
shQPRT:
Forward gatccccGCCCTTGATTTCTCCCTCAttcaagagaTGAGGGAGAAATCAAGGGCtttttggaaa
Reverse agcttttccaaaaaGCCCTTGATTTCTCCCTCAtctcttgaaTGAGGGAGAAATCAAGGGCggg
Scramble:
Forward
gatccccCTTCAGCCGTTACGCTCGGttcaagagaCCGAGCGTAACGGCTGAAGtttttggaaa
Reverse
agcttttccaaaaaCTTCAGCCGTTACGCTCGGtctcttgaaCCGAGCGTAACGGCTGAAGggg
Oligonucleotide pairs were annealed by heating to 100 °C for 2 min and cooling slowly to room temperature and ligated into HindIII/BglII-digested pRetroSuper-puro (a gift from Darren Tomlinson). Constructs were amplified in XL1 Blue competent E. coli (Stratagene), then introduced into 293-PhoenixA cells using SiPORT transfection agent (Ambion). Supernatant containing viral particles was harvested on days 3 and 4, and polybrene was added to 8 μg/ml. Four ml of the medium was used to infect each T75 flask of 786+VHL cells, followed by selection with puromycin (2 μg/ml). Cell lines were maintained in MEM-α medium supplemented with 10% FCS, 1% v/v L-glutamine, G418 (1 mg/ml) and puromycin (2 µg/ml).
Two-dimensional (2D) gel electrophoresis
For global protein profiling of 786-0 cells +/−VHL, protein extracts (80 µg protein for analytical gels and 1 mg for preparative gels) were analysed by 2D polyacrylamide gel electrophoresis (2D-PAGE) over a pH range of 4–7 using a combined IPGPhor and Multiphor approach.20 Protein samples were loaded onto IPG strips by overnight in-gel rehydration and focussing carried out for a total of 65 kVh. Strips were equilibrated in running buffer, placed onto polyacrylamide gels (10% resolving gel with 4% stacking gel) and electrophoresed overnight (12.5 °C, 18 mA/gel). Gels were stained using OWL silver stain and scanned using a Personal Densitometer SI (GE Healthcare), and images from triplicate gels were analysed using the Melanie 3 software. Preparative gels were stained with PlusOne™ Silver Stain using a modified staining protocol,21 and selected spots were excised and digested with trypsin.22 Peptides were analysed by Nano-LC (Ultimate, LC Packings (Dionex), Camberley, UK) followed by automated data-dependent mass spectrometry (MS)/MS using a Q-TOF mass spectrometer (Micromass, Manchester, UK). Protein identities were determined by searching the NCBI database using MS-TAG or MS-Pattern (prospector.ucsf.edu).
Renal tissue samples
Frozen renal tissue samples from 42 previously untreated patients who had undergone nephrectomy for sporadic ccRCC from December 2001 to December 2006 were obtained from the Leeds Multidisciplinary Research Tissue Bank (REC Ref 15/YH/0080). Tissue collection and processing was as previously described.19 Ten pairs of matched tumour/normal tissue were used for Western blot analysis. For the initial immunohistochemical (IHC) studies of QPRT, frozen tissue sections from 13 tumours and matched normal kidney samples were examined. Further examination of additional proteins and across other RCC subtypes was achieved through a tissue microarray (TMA), containing formalin-fixed paraffin-embedded (FFPE) tissue cores from a further 20 patients, reviewed and selected by an experienced pathologist, from 5 normal renal cortex, 5 normal renal medulla, 11 ccRCC, 6 papillary RCC and 3 chromophobe RCC samples, each arrayed in duplicate. In addition, previously generated liquid chromatography (LC)-MS/MS proteomic data sets analysing 13 matched tumour/normal pairs of ccRCC tissues and 7 matched pairs of chromophobe RCC tissues were also interrogated for the purposes of this study. This data set forms part of a larger proteogenomic study (manuscript in preparation). All included tumours were reviewed by an expert pathologist to confirm at least 70% viable tumour cells. Details of patients/tumours across the various substudies are presented in Supplementary Table 1.
Immunocytochemistry, immunohistochemistry and western blotting for QPRT
Initial analysis of cell lines for QPRT was undertaken using affinity-purified rabbit antiserum to QPRT.23 For all other studies, custom rabbit antiserum to QPRT was raised (Eurogentec, Belgium) by immunising rabbits with peptides (CDLVLLDNFKPEELHP or CVAGTRKTTPGFRLVE).
Multi-well slides of 786-0+/−VHL cell lines were fixed in acetone for 2 min, air dried, washed briefly in TBS-T, endogenous peroxidase blocked using 0.6% v/v hydrogen peroxide in methanol for 5 min and washed again. After overnight incubation at 4 °C in rabbit antiserum to QPRT diluted 1:20,000 in TBS/0.1% w/v HSA with 0.1% w/v sodium azide, slides were washed in TBS-T and labelled using the rabbit EnVision+ detection system with DAB substrate according to the manufacturer’s instructions. Slides were counterstained with Mayer’s haematoxylin and mounted using DePeX mounting medium. Negative control sections were probed with an irrelevant antibody. Immunohistochemistry on sections of frozen tumour and normal tissue (5 µm OCT-embedded) was similarly performed.22
Western blotting of protein lysates of cell lines or matched tumour/normal tissue was performed as previously described24 with samples separated by 10% sodium dodecyl sulfate (SDS)-PAGE and transferred to Hybond-C Super NC membrane in Towbin’s buffer. After blocking with TBS-T/10% w/v dried skimmed milk, blots were probed with antibodies to QPRT (1:20,000), VHL (1 µg/mL) and β-actin (5 ng/ml; protein-loading control). After washing, blots were incubated with anti-rabbit or anti-mouse HRP-conjugated Envision+ reagent, then washed again and exposed to film. In all cases, western blots were normalised using densitometric scanning of parallel Coomassie blue-stained gels for total protein load, given the limitations of housekeeping genes.25 Additional blotting against beta actin was variably employed.
Measurement of quinolinic acid (QUIN) in renal tissue samples
Sections from matched pairs of frozen tumour/normal tissue were cut into 1 M HCL and stored at −80 °C. QUIN levels were measured in Schwarz laboratory by gas chromatography MS, performed as previously described.26
Effects of QPRT on cell proliferation
786+pFB and 786+QPRT cells in an exponential phase of growth were harvested and plated in 96-well plates (1 × 104 cells/well) and cultured for 24, 48 and 72 h at 37 °C with 5% CO2/95% air. Viable cells were quantified using WST-1 reagent according to the manufacturer’s protocol, and absorbance was measured at 450 and 650 nm. Wells were seeded in triplicate, and three independent experiments were run.
Effects of QPRT on anchorage-independent colony-formation assay
To investigate anchorage-independent growth of 786-0 cell lines stably transduced with constructs of interest, WT VHL (786+VHL)+shRNA scramble control, WT VHL (786+VHL)+shRNA QPRT, VHL negative+empty vector (786+pFB) and VHL negative+QPRT (786+QPRT) were cultured at 4 × 104 cells/well as previously described.27 Viable colonies were stained with 8 mM p-iodonitrotetrazolium violet, and colonies with a diameter of >1 mm were then counted within 10 random fields of view using an eyepiece graticule (1 cm2 area, made up of 10 ×10 mm squares) on a bright field microscope. The mean number of colonies per 10 cm2 from four independent experiments was determined. Statistical significance was assessed by Student’s T test. The NIH3T3 cell lines containing either a control or H1047R vector were employed as negative and positive controls for the assay, respectively.
Interrogation of LC-MS/MS RCC proteomic data sets for kynurenine pathway changes
As part of a large proteogenomic study of RCC (manuscript in preparation), LC-MS/MS proteomic data sets were created containing 13 matched tumour/normal pairs of ccRCC and 7 matched tumour/normal pairs of chromophobe RCC following expert pathological review of the selected blocks. We were able to interrogate this data for evidence of protein expression of enzymes from the kynurenine pathway. For each sample, 30-µm sections equivalent to 3 cm2 surface area of tissue were lysed in excess lysis solution (250 µl of 3% SDS in 50 mM Tris-HCl, pH 7.6), and DNA was sheared with brief sonication. Samples were then heated at 95 °C for 10 min, centrifuged at 13,000 × g for 8 min, supernatant removed, and protein concentration was measured by BCA assay. Dithiothreitol was added to samples at a final concentration of 30 mM, and samples were heated at 95 °C for 5 min. Seventy μg of protein was processed by the STrap protocol as previously described.28 Label-free MS and data analysis were conducted essentially as previously described29 but using an EASY-nLC 1000 UHPLC system connected to a capillary emitter column (75 μm inner diameter, packed with 3 μ Pursuit C18 media) hyphenated to an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Data were processed against the Uniprot human protein database using the Maxquant 1.3.0.5 software. Maximum false discovery rates were set to 0.01. PEP and Q-values calculate the probability of false identification for the proteins described in this study as being extremely low (p < 0.001).
IHC analysis of the kynurenine pathway
TMA FFPE sections (4 μm) were mounted onto Plus Frost slides, and dewaxing and epitope recovery was carried out by heating in a Cookworks pressure cooker for 5 min on high followed by 25 min on low temperature in Access Revelation solution. Endogenous peroxidase activity was blocked in Bloxall and 2.5% v/v normal horse serum used as a protein block. Antibodies were optimally diluted in Antibody Diluent and detected using rabbit HRP‐conjugated secondary antibodies followed by Impact DAB substrate for 5 min at room temperature, then counterstained with Mayer’s haematoxylin for 30 s dehydrated, cleared in xylene and mounted in DPX.
Results
QPRT is downregulated in ccRCC
From our 2D-PAGE-based comparison of whole-cell lysates of 786-0−/+VHL cells, we identified QPRT as being undetectable in −VHL cells and expressed at relatively high levels following re-introduction of WT VHL (Fig. 1a and Supplementary Table 2). This was confirmed using immunocytochemistry and western blotting of the 786-0+/−VHL cell lines (Fig. 1b, c). However, this apparent VHL-dependent expression of QPRT was not observed in UMRC2 and RCC4+/−VHL cell line pairs (Fig. 1d), which all retained expression. Western blotting of frozen tissue lysates from ten ccRCC/normal kidney matched pairs with tumour VHL mutation status defined in all but one pair (Fig. 1e) showed significant loss or downregulation of QPRT expression in ccRCC tissues compared to their normal counterparts in nine cases, although no relationship with VHL mutation status or mutation type was apparent. The loss of QPRT in clear cell tumours was confirmed in 12/13 patients by IHC (Fig. 1f).

a 2D-PAGE comparing 786-0 cell lines, +/−wild-type VHL. The protein spot identified as QPRT is arrowed. b Immunocytochemistry of 786-0+/− cell lines for QPRT. c–e Western blotting for QPRT in c 786-0+/−VHL cell lines; d 786-0, RCC4 and UMRC2 cell lines +/−wild-type VHL; e Paired normal/ccRCC tumour tissue lysates. VHL mutation status is indicated (one tumour was of unknown VHL status: FS frame shift, N nonsense, M mis-sense). Promoter methylation status was analysed in one of the two tumours containing no VHL mutation and confirmed as negative f Immunohistochemistry for QPRT of a representative example of normal and ccRCC tissue (×40 magnification). g Quinolinic acid content of paired normal/ccRCC tumour tissue samples determined by mass spectrometry.
Taken together, these results indicate that loss of QPRT is a common event in ccRCC although regulation by VHL in vitro is cell line dependent. QPRT catalyses the conversion of QUIN, produced within the kynurenine pathway, to nicotinamide adenine dinucleotide (NAD+). The reaction catalysed by QPRT is a rate-limiting step in this pathway, therefore loss of QPRT may lead to an increase in the level of QUIN. In support of this hypothesis, we found increased levels of QUIN in ccRCC tissues relative to patient-matched normal kidney cortex (Fig. 1g).
Knockdown of QPRT in 786-0 cells increases cellular anchorage-independent growth in vitro
786-0 cells (VHL-negative) transfected with a QPRT expression construct (786+QPRT) showed stable overexpression of QPRT (Fig. 2a). Conversely, transfection of 786+VHL (VHL-expressing) cells with an shRNA construct targeting QPRT (786+VHL+shQPRT) cells significantly reduced QPRT protein expression (Fig. 2b). QPRT expression in cells transfected with empty vector was unaffected.

a Western blot showing QPRT expression in control and QPRT-transfected VHL negative 786-0 cell lines. b QPRT expression in control and anti-QPRT shRNA transfected, VHL-expressing 786-0 cell lines. A single band at the expected molecular weight was observed. c, d Relative cell number of 786-0 cell lines plus or minus QPRT, as measured using WST1 assay. e, f Impact of QPRT transfection or knockdown on relative colony number in soft agar colony-formation assay and representative images. Colony number per 10 cm2 are provided as a mean of the sum over four independent experiments and then standardised as a percentage to either 786+pRC or 786+VHL controls. Significant differences are indicated (asterisk (*); 786+QPRT versus 786+pFB, p = 0.017, 786+VHL+shQPRT versus either control, p = 0.001).
Using this model, no significant effect of QPRT loss/gain on cell proliferation was observed (Fig. 2c, d). However, in a soft agar colony-formation assay (Fig. 2e, f), whereas all 786-0 cells, irrespective of whether +/−VHL, formed small colonies (diameter of >1 mm), 786+QPRT cells showed significantly reduced colony formation as compared to 786+pRC controls (p = 0.017) (Fig. 2e) and 786+VHL+shQPRT cells showed a significantly (p = 0.001) higher frequency of colonies relative to both the 786+VHL+scramble and 786+VHL cells (Fig. 2f). Colony size was also affected by QPRT expression status. Among QPRT-positive cell lines, only 10% of colonies were estimated to exceed 3 mm in diameter, versus approximately 50% of colonies among QPRT-negative cells lines.
Dysregulation of the kynurenine pathway occurs at multiple points and is common to both ccRCC and chromophobe RCC
Interrogation of existing LC-MS/MS proteomic data sets across other proteins within the kynurenine pathway confirmed the decrease in QPRT in ccRCC, identifying a total of 6 unique peptides (Supplementary Table 3), which together represent 18.5% of the entire protein sequence. A consistent and significant decrease in the number of QPRT peptides identified, and in normalised (LFQ) peptide intensities (representing relative quantification), was seen in ccRCC tissues compared with normal kidney tissues, and similar findings were observed for other enzymes of the kynurenine pathway, namely 3-hydroxyanthranilite 3,4-dioxygenase (HAAO) and kynurenine 3-monoxygenase (KMO) (Fig. 3a). Conversely, nicotinamide phosphoribosyl transferase (NAMPT; which is a key enzyme in the production of NAD+ via the alternative salvage pathway) was upregulated in most tumour samples (Fig. 3a). IDO was not detected in any samples. In the data set for the seven matched pairs of chromophobe RCC versus normal kidney, very similar results as for ccRCC were obtained, with expression of QPRT, KMO and HAAO being below the level of detection in tumours (with one exception). NAMPT again showed increased expression in the tumours and was undetectable in all but one of the normal kidney tissue samples (Fig. 3b).

Mass spectrometric relative concentrations (LFQ intensity) of proteins in the kynurenine pathway for paired normal kidney/tumour tissue samples (black and grey bars, respectively) for a ccRCC and b chromophobe RCC. Numbers along x-axis refer to assigned tissue sample number. Differences between groups by Wilcoxon matched-pairs sign rank test: Clear cell RCC: QPRT (p < 0.001), KMO (p < 0.001), HAAO (p = 0.01), and NAMPT (p < 0.01); Chromophobe RCC: QPRT (p < 0.016), KMO (p < 0.016), HAAO (p < 0.016), and NAMPT (p < 0.016).
Four other kynurenine pathway proteins were also detected in our proteomic data sets. Kynurenine formamidase (AFMID), kynureninase (KYNU) and kynurenine aminotransferase 1 and 3 (KYAT1, KYAT3) were all observed in the chromophobe data set, while AFMID and KYAT3 were also detected in the ccRCC data. All four proteins showed a pattern of presence in normal tissue and loss in tumour tissue. However, the number of peptides detected in each sample was low (1–3), and the intensities were near to the limit of detection. For this reason, while these data suggest a pattern of altered expression for these four proteins, it should not yet be considered as conclusive.
In addition, dysregulation of expression of QPRT, KMO, HAAO and NAMPT, largely mirroring changes observed at the protein level, was confirmed transcriptomically through examination of our previously generated RNA-seq data among 45 matched tumour (ccRCC)/normal tissue pairs (Fig. 4).30 Equivalent data for papillary and chromophobe RCCs are shown in Supplementary Figs, 1 and 2, respectively, based on data from The Cancer Genome Atlas Research Network.31

Data derived from transcriptomic (RNA-seq) analysis of 45 paired normal kidney/tumour tissue samples.30 Numbers along x-axis refer to assigned tissue sample number. FPKM fragments per kilo bases of exons per million mapped reads. Differences between groups by Wilcoxon matched-pairs sign rank test: QPRT (p < 0.001), KMO (p = 0.004), HAAO (p = 0.001), and NAMPT (p < 0.001).
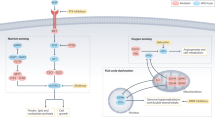
The observed changes and how these impact on the kynurenine pathway are summarised in Fig. 5.

The various enzymes and intermediates generated through this de novo synthesis of NAD+ from tryptophan are shown together with the salvage pathway route for generation of NAD+ from nicotinamide, catalysed by NAMPT. Enzymes for which we have strong evidence of downregulation in RCC tissues are marked with a solid grey arrow. Enzymes or substrates observed to be upregulated are marked with a hatched arrow. Enzymes with evidence to suggest they may be down regulated are marked by an open grey arrow.
IHC TMA analysis of the kynurenine pathway by TMA
In normal renal cortex, weak-to-moderate granular staining of proximal tubules (predominantly cytoplasmic) was observed for IDO1 and moderate/strong expression of KYNU, QPRT and KMO, with absent-to-moderate staining for NAMPT. In the case of KMO, staining was noticeably localised to basolateral aspect of the tubules. Glomerular reactivity was also seen for NAMPT and KYNU. In the medulla, tubules were largely negative, and two cases showed weak expression of IDO1, NAMPT and QPRT, although one of these showed distinct populations of tubules with moderate QPRT staining (Fig. 6).

Examples of the immunohistochemical staining patterns observed for IDO1, KMO, KYNU, QPRT and NAMPT in the normal renal cortex and medulla and different subtypes of RCC (clear cell, papillary and chromophobe) using a TMA. Scale bar (displayed in first core) = 60 μm.
In ccRCC cases, expression of IDO1, KYNU, QPRT and KMO was greatly reduced compared with normal kidney cortex and, in some cases, was absent (Fig. 6, Supplementary Table 4). IDO1 was not detected in tumour cells but only in occasional inflammatory cells or in endothelial cells, and KMO1 was also seen in some endothelial cells. For QPRT, five cases were completely negative, four cases showed only focal positivity and two weak-to-moderate staining. Conversely, NAMPT expression was absent from clear cell tumour cells in two cases but positive in five (from weak to strong) with focal positivity seen in a further four cases. Positive staining was also seen with occasional inflammatory cells and endothelial cells. Of note, the strongest staining for any of the enzymes amongst the ccRCC cases was observed in the rhabdoid cells contained in tumour 4728. A similar pattern of generally reduced staining compared with normal kidney was also seen with papillary and chromophobe cases and generally weak-to-moderate NAMPT. The findings were consistent with our MS results and demonstrate that disruption of the kynurenine pathway is a feature of RCC across histological subtypes.
Discussion
This study provides the first comprehensive demonstration of a wide and coordinate dysregulation of the kynurenine pathway in RCC and that this is a common, unifying event, highlighting its importance in the pathogenesis of these cancers and potentially providing insights of relevance to therapeutic targeting.
The kynurenine pathway is the main route for degradation of the essential amino acid tryptophan and de novo synthesis of NAD+, generating numerous other active intermediate metabolites including kynurenine, kynurenic acid, anthranilic acid, picolinic acid and QUIN. Dysregulation of the pathway was initially highlighted in neuropsychiatric disorders but has now been implicated more widely10 and increasingly in tumorigenesis and immune evasion. Although in the liver, where the majority of tryptophan degradation occurs, constitutively expressed TDO2 is the initial rate-limiting enzyme in the pathway, in most other tissues IDO1 is the first and rate-limiting enzyme. Inducible by inflammatory cytokines, IDO/IDO1 has been reported to be expressed in many cancers and hypothesised to play a role in tryptophan degradation and accumulation of active metabolites in the kynurenine pathway, both of which result in T cell/immune suppression leading to the idea of IDO1 inhibitors as useful anticancer therapeutics to overcome immune resistance, for example in combination with vaccine strategies.13,32 However, IDO1 expression is highly dependent on tumour type, and many cancers, including renal, melanoma and thyroid, have absent or low expression of IDO1 in tumour cells in most cases.32 This has been confirmed subsequently with the demonstration of IDO1 expression in endothelial cells and macrophages in RCC tissues33,34 and a recent extensive study across many cancers where, although 80% of renal carcinomas were positive, IDO1 was absent from tumour cells and present predominantly in endothelial cells or in some lymphocyte-rich stroma.35 This in agreement with our IHC results and with the relatively low expression being undetectable by MS.
We initially observed that in a VHL+/− cell line pair, expression of QPRT increased following the introduction of VHL. QPRT is the final enzyme in the kynurenine pathway, converting QUIN to NAD+. However, it is apparent that this is not mainly VHL dependent and may be an indirect effect, since this was not seen in other VHL-transfectant cell line models and changes in QPRT expression were subsequently shown to occur in ccRCC independent of VHL mutation status and in chromophobe and papillary RCC tissues where VHL is not involved. We subsequently learnt that the 786-pRC cell line we employed also contains a p53 mutation (personal communication from WG Kaelin to R Craven), although we do not believe this to be implicated either, since p53 mutations are rarely seen in RCC.3 The underlying biology leading to such dysregulation, and whether common or divergent mechanisms are responsible, remains uncertain, but its consistency across more than one type of RCC suggests that it is a key and potentially early event.
As predicted, we found that that loss of QPRT was associated with increased QUIN in tumour tissue, and metabolomic studies of urine and tissue samples from RCC patients reported higher concentrations of quinolinate compared with healthy controls36 or normal tissue.37 QUIN has been reported to activate β-Catenin and increase proliferation in colon cancer cell lines,38 and in RCC cell lines, variable effects of quinolinate on cell viability or proliferation have been reported although very different quinolinate concentrations have been used across studies.36,39 We did not observe increased cell proliferation in our in vitro QPRT knockdown model. However, we did see a pronounced increase in anchorage-independent growth in response to loss of QPRT. It is possible that this is mediated by QUIN accumulation and exerting this effect through a potential autocrine loop involving N-methyl-d-aspartate receptors (NMDARs), since this receptor–ligand binding is important for its role in neurological disease40 and expression of NMDAR subunits has been demonstrated in the normal kidney cortex and medulla and across multiple cancer types, with receptor blockade reducing cancer cell proliferation and invasiveness in numerous cancers in vitro.41 QUIN is also known to be an immune modulator. For example, treatment with QUIN induced the selective apoptosis in vitro of murine thymocytes and of T helper type 1 (Th1) but not of Th2 cells, and mice treated with QUIN had significantly reduced levels of immature thymocytes in the thymus.42 In a microenvironment deficient in tryptophan, QUIN was found to inhibit proliferation of both lymphocytes and natural killer (NK) cells.43 Interestingly, accumulation of QUIN has been reported to occur in human gliomas but accompanied by increased QPRT expression and supporting NAD generation through this pathway rather than the NAMPT-mediated pathway.44 QPRT was shown to be induced by oxidative stress, temozolomide and irradiation and to be associated with poorer prognosis in recurrent tumours after radiochemotherapy, potentially through increasing resistance. These results suggest that targeting QPRT itself may be a potential therapeutic option and indeed data from a cell line model have implicated upregulation of QPRT as conferring resistance to NAMPT inhibitors.45
Examination of a parallel existing LC-MS/MS proteomic data set generated by our group as part of an ongoing proteogenomic study in RCC not only confirmed loss of QPRT in RCC but also showed loss of five other enzymes in the kynurenine pathway, namely HAAO, KMO, kynurenine formamidase and KYAT1 and 2. Remarkably little is known about these enzymes in cancer although upregulation of KMO has been described in hepatocellular carcinoma.46 This may be due at least in part to the lack of availability of good antibodies, at least until recently, which is why we generated our own QPRT antibody. Critically, one can expect that loss of KMO, alongside an increase in IDO expression, will lead to the accumulation in tumour tissue of kynurenine. In metabolomic studies of mouse RCC xenografts and human RCC tumours, significantly lower tryptophan and higher kynurenine levels and higher quinolinate and kynurenine levels, respectively, were seen in tumours compared with controls.37,47 A metabolomic study involving ccRCC along with chromophobe and papillary tissue samples also identified elevated kynurenine compared with controls but in ccRCC cases only.48 The immune-suppressive properties of kynurenine are well described, and it has been shown to inhibit T cell and NK cell proliferation and promote immune suppression via the aryl hydrocarbon receptor.32,43,49 Furthermore, it can promote cancer cell survival and motility.38,49 3-Hydroxyanthranilic acid, the substrate for HAAO, has also been shown to have multiple roles in promoting tumour immune evasion, by promoting apoptosis of Th1 and NK cells, promoting differentiation of regulatory T cells and inhibiting T cell proliferation.10,42
This novel observation, that multiple enzymes in the kynurenine pathway downstream of IDO are all downregulated and in a manner likely to promote tumorigenesis and immune evasion, is striking. The loss of QPRT, and hence the blockade of de novo biosynthesis of NAD+ may initially seem at odds with the requirement of cells, and especially cancer cells, for this molecule.50 However, NAD+ may also be made from nicotinamide via the salvage pathway of which the enzyme NAMPT is a key component, and this pathway is often preferred in cancer cells.51 Consistent with this, we observed upregulation of NAMPT occurring as a common event in RCC, confirming a recent IHC study.52 As such, NAMPT forms an attractive therapeutic target, and it is of note that KPT-9274, a NAMPT/PAK4 inhibitor, has recently been reported to have activity against renal cancer xenografts.53
The kynurenine pathway forms a particularly attractive target for therapy, since it seems to regulate tolerance to non-self-antigens, rather than to self-antigens,11,12 thus reducing the risk of immune-related adverse events often seen with immune-checkpoint inhibition. Whether inhibition of IDO1 alone is sufficient to overcome pathway dysregulation is uncertain, as exemplified by the recent negative results seen with epacadostat in patients with melanoma.14 Indeed, a planned Phase 3 trial of this agent in combination with pembrolizumab in patients with RCC has been halted based on these findings. While combined IDO1/TDO inhibitors are currently in early Phase trials (NCT03208959), our results suggest that, in RCC at least, the kynurenine pathway is much more widely dysregulated and may require further downstream modulation in addition to IDO inhibition.
Our study has its limitations and a number of questions remain unanswered that would need to be explored in future studies. Whether the phenotypic consequences of loss or gain of QPRT extend beyond effects on colony formation, for example, and whether such changes are consistently observed across RCC cell lines, remains uncertain. Furthermore, since the 786-0 cell line is known to contain a phosphatase and tensin homologue (PTEN) mutation (although rarely observed in ccRCC tissues),30 it would be of interest to examine how deficiency of PTEN and/or dysregulation of the phosphoinositide-3 kinase/AKT/mammalian target of rapamycin signalling pathway impacts QPRT expression. We have also not examined how dysregulation of the kynurenine pathway in RCC correlates with patient outcomes.
In conclusion, we have demonstrated a widespread and apparently coordinated dysregulation of the kynurenine pathway in RCC. These findings have implications for current strategies aimed at therapeutically targeting this critical pathway and highlight the potential for novel treatment strategies, such as inhibition of NAMPT.
References
Ferlay, J., Soerjomataram, I., Dikshit, R., Eser, S., Mathers, C., Rebelo, M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–E386 (2015).
Smittenaar, C. R., Petersen, K. A., Stewart, K. & Moitt, N. Cancer incidence and mortality projections in the UK until 2035. Br. J. Cancer 115, 1147–1155 (2016).
Ricketts, C. J., De Cubas, A. A., Fan, H., Smith, C. C., Lang, M., Reznik, E. et al. The Cancer Genome Atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 23, 3698 (2018).
Motzer, R. J., Escudier, B., McDermott, D. F., George, S., Hammers, H. J., Srinivas, S. et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 373, 1803–1813 (2015).
Stewart, G. D., De Santis, M., Escudier, B., Powles, T. & Sonpavde, G. Immunotherapy for renal cancer: sequencing and combinations. Eur. Urol. Focus 2, 582–588 (2016).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
Unwin, R. D., Craven, R. A., Harnden, P., Hanrahan, S., Totty, N., Knowles, M. et al. Proteomic changes in renal cancer and co-ordinate demonstration of both the glycolytic and mitochondrial aspects of the Warburg effect. Proteomics 3, 1620–1632 (2003).
Srinivasan, R., Ricketts, C. J., Sourbier, C. & Linehan, W. M. New strategies in renal cell carcinoma: targeting the genetic and metabolic basis of disease. Clin. Cancer Res. 21, 10–17 (2015).
Badawy, A. A. Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int. J. Tryptophan Res. 10, 1178646917691938 (2017).
Munn, D. H., Zhou, M., Attwood, J. T., Bondarev, I., Conway, S. J., Marshall, B. et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 281, 1191–1193 (1998).
Prendergast, G. C. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene 27, 3889–3900 (2008).
Platten, M., Wick, W. & Van den Eynde, B. J. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 72, 5435–5440 (2012).
Long, G. V., Dummer, R., Hamid, O., Gajewski, T. F., Caglevic, C., Dalle, S. et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 20, 1083–1097 (2019).
Jung, K. H., LoRusso, P., Burris, H., Gordon, M., Bang, Y. J., Hellmann, M. D. et al. Phase I Study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) administered with PD-L1 inhibitor (Atezolizumab) in advanced solid tumors. Clin. Cancer Res. 25, 3220–3228 (2019).
Iliopoulos, O., Kibel, A., Gray, S. & Kaelin, W. G. Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat. Med. 1, 822–826 (1995).
Zatyka, M., Morrissey, C., Kuzmin, I., Lerman, M. I., Latif, F., Richards, F. M. et al. Genetic and functional analysis of the von Hippel-Lindau (VHL) tumour suppressor gene promoter. J. Med. Genet. 39, 463–472 (2002).
Clifford, S. C., Cockman, M. E., Smallwood, A. C., Mole, D. R., Woodward, E. R., Maxwell, P. H. et al. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum. Mol. Genet. 10, 1029–1038 (2001).
Craven, R. A., Hanrahan, S., Totty, N., Harnden, P., Stanley, A. J., Maher, E. R. et al. Proteomic identification of a role for the von Hippel Lindau tumour suppressor in changes in the expression of mitochondrial proteins and septin 2 in renal cell carcinoma. Proteomics 6, 3880–3893 (2006).
Craven, R. A., Totty, N., Harnden, P., Selby, P. J. & Banks, R. E. Laser capture microdissection and two-dimensional polyacrylamide gel electrophoresis: evaluation of tissue preparation and sample limitations. Am. J. Pathol. 160, 815–822 (2002).
Yan, J. X., Wait, R., Berkelman, T., Harry, R. A., Westbrook, J. A., Wheeler, C. H. et al. A modified silver staining protocol for visualization of proteins compatible with matrix-assisted laser desorption/ionization and electrospray ionization-mass spectrometry. Electrophoresis 21, 3666–3672 (2000).
Craven, R. A., Stanley, A. J., Hanrahan, S., Dods, J., Unwin, R., Totty, N. et al. Proteomic analysis of primary cell lines identifies protein changes present in renal cell carcinoma. Proteomics 6, 2853–2864 (2006).
Okuno, E. & Schwarcz, R. Purification of quinolinic acid phosphoribosyltransferase from rat liver and brain. Biochim. Biophys. Acta 841, 112–119 (1985).
Aggelis, V., Craven, R. A., Peng, J., Harnden, P., Cairns, D. A., Maher, E. R. et al. Proteomic identification of differentially expressed plasma membrane proteins in renal cell carcinoma by stable isotope labelling of a von Hippel-Lindau transfectant cell line model. Proteomics 9, 2118–2130 (2009).
Ferguson, R. E., Carroll, H. P., Harris, A., Maher, E. R., Selby, P. J. & Banks, R. E. Housekeeping proteins: a preliminary study illustrating some limitations as useful references in protein expression studies. Proteomics 5, 566–571 (2005).
Guidetti, P., Luthi-Carter, R. E., Augood, S. J. & Schwarcz, R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol. Dis. 17, 455–461 (2004).
Ross, R. L., Askham, J. M. & Knowles, M. A. PIK3CA mutation spectrum in urothelial carcinoma reflects cell context-dependent signaling and phenotypic outputs. Oncogene 32, 768–776 (2013).
Zougman, A., Selby, P. J. & Banks, R. E. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 14, 1006–1000 (2014).
Zougman, A. & Banks, R. E. C-STrap sample preparation method—in-situ cysteinyl peptide capture for bottom-up proteomics analysis in the STrap format. PLoS ONE 10, e0138775 (2015).
Scelo, G., Riazalhosseini, Y., Greger, L., Letourneau, L., Gonzalez-Porta, M., Wozniak, M. B. et al. Variation in genomic landscape of clear cell renal cell carcinoma across Europe. Nat. Commun. 5, 5135 (2014).
TCGA Research Network. https://www.cancer.gov/tcga
Uyttenhove, C., Pilotte, L., Theate, I., Stroobant, V., Colau, D., Parmentier, N. et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 9, 1269–1274 (2003).
Riesenberg, R., Weiler, C., Spring, O., Eder, M., Buchner, A., Popp, T. et al. Expression of indoleamine 2,3-dioxygenase in tumor endothelial cells correlates with long-term survival of patients with renal cell carcinoma. Clin. Cancer Res. 13, 6993–7002 (2007).
Lucarelli, G., Rutigliano, M., Ferro, M., Giglio, A., Intini, A., Triggiano, F. et al. Activation of the kynurenine pathway predicts poor outcome in patients with clear cell renal cell carcinoma. Urol. Oncol. 35, 461.e15–461.e27 (2017).
Theate, I., van Baren, N., Pilotte, L., Moulin, P., Larrieu, P., Renauld, J. C. et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol. Res. 3, 161–172 (2015).
Kim, K., Taylor, S. L., Ganti, S., Guo, L., Osier, M. V. & Weiss, R. H. Urine metabolomic analysis identifies potential biomarkers and pathogenic pathways in kidney cancer. OMICS 15, 293–303 (2011).
Wettersten, H. I., Hakimi, A. A., Morin, D., Bianchi, C., Johnstone, M. E., Donohoe, D. R. et al. Grade-dependent metabolic reprogramming in kidney cancer revealed by combined proteomics and metabolomics analysis. Cancer Res. 75, 2541–2552 (2015).
Thaker, A. I., Rao, M. S., Bishnupuri, K. S., Kerr, T. A., Foster, L., Marinshaw, J. M. et al. IDO1 metabolites activate beta-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology 145, 416.e4–425.e4 (2013).
Trott, J. F., Kim, J., Abu Aboud, O., Wettersten, H., Stewart, B., Berryhill, G. et al. Inhibiting tryptophan metabolism enhances interferon therapy in kidney cancer. Oncotarget 7, 66540–66557 (2016).
Heyes, M. P., Saito, K., Crowley, J. S., Davis, L. E., Demitrack, M. A., Der, M. et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 115(Pt 5), 1249–1273 (1992).
Hogan-Cann, A. D. & Anderson, C. M. Physiological roles of non-neuronal NMDA receptors. Trends Pharmacol. Sci. 37, 750–767 (2016).
Fallarino, F., Grohmann, U., Vacca, C., Orabona, C., Spreca, A., Fioretti, M. C. et al. T cell apoptosis by kynurenines. Adv. Exp. Med. Biol. 527, 183–190 (2003).
Frumento, G., Rotondo, R., Tonetti, M., Damonte, G., Benatti, U. & Ferrara, G. B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 196, 459–468 (2002).
Sahm, F., Oezen, I., Opitz, C. A., Radlwimmer, B., von Deimling, A., Ahrendt, T. et al. The endogenous tryptophan metabolite and NAD+ precursor quinolinic acid confers resistance of gliomas to oxidative stress. Cancer Res. 73, 3225–3234 (2013).
Guo, J., Lam, L. T., Longenecker, K. L., Bui, M. H., Idler, K. B., Glaser, K. B. et al. Identification of novel resistance mechanisms to NAMPT inhibition via the de novo NAD(+) biosynthesis pathway and NAMPT mutation. Biochem. Biophys. Res. Commun. 491, 681–686 (2017).
Jin, H., Zhang, Y., You, H., Tao, X., Wang, C., Jin, G. et al. Prognostic significance of kynurenine 3-monooxygenase and effects on proliferation, migration, and invasion of human hepatocellular carcinoma. Sci. Rep. 5, 10466 (2015).
Ganti, S., Taylor, S. L., Abu Aboud, O., Yang, J., Evans, C., Osier, M. V. et al. Kidney tumor biomarkers revealed by simultaneous multiple matrix metabolomics analysis. Cancer Res. 72, 3471–3479 (2012).
Schaeffeler, E., Buttner, F., Reustle, A., Klumpp, V., Winter, S., Rausch, S. et al. Metabolic and lipidomic reprogramming in renal cell carcinoma subtypes reflects regions of tumor origin. Eur. Urol. Focus 5, 608–618 (2019).
Opitz, C. A., Litzenburger, U. M., Sahm, F., Ott, M., Tritschler, I., Trump, S. et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 (2011).
Chiarugi, A., Dolle, C., Felici, R. & Ziegler, M. The NAD metabolome-a key determinant of cancer cell biology. Nat. Rev. Cancer 12, 741–752 (2012).
Kennedy, B. E., Sharif, T., Martell, E., Dai, C., Kim, Y., Lee, P. W. et al. NAD(+) salvage pathway in cancer metabolism and therapy. Pharmacol. Res. 114, 274–283 (2016).
Shackelford, R. E., Abdulsattar, J., Wei, E. X., Cotelingam, J., Coppola, D. & Herrera, G. A. Increased nicotinamide phosphoribosyltransferase and cystathionine-beta-synthase in renal oncocytomas, renal urothelial carcinoma, and renal clear cell carcinoma. Anticancer Res. 37, 3423–3427 (2017).
Abu Aboud, O., Chen, C. H., Senapedis, W., Baloglu, E., Argueta, C. & Weiss, R. H. Dual and specific inhibition of NAMPT and PAK4 By KPT-9274 decreases kidney cancer growth. Mol. Cancer Ther. 15, 2119–2129 (2016).
Acknowledgements
We are grateful to the patients for donating samples and the staff of the Leeds Biobanking and Sample Processing Lab and Leeds Multidisciplinary RTB for samples and Anke Bruning-Richardson for technical support with the TMA. We thank the staff of the oncology and urology departments of Leeds Teaching Hospitals NHS Trust who assisted in patient recruitment, Professor W Kaelin for the 786-VHL cell lines, Robert Schwarz for providing antibody to QPRT, Darren Tomlinson for providing the parental QPRT vector, Anthea Stanley for laboratory support and Nick Totty and Sarah Hanrahan for the sequencing of the 2D-PAGE proteins.
Author information
Authors and Affiliations
Contributions
R.E.B., P.S.J. and N.S.V. supervised the study with A.H. and E.R.M. also contributing to study design; R.A.C. and R.E.B. conducted the initial 2D-PAGE work; J. Brown facilitated access to tissue specimens and clinical data; K.R.D., S.T., R.S. and A.Z. conducted the mass spectrometric studies; K.R.D., M.K. and J. Burns conducted and oversaw phenotypic studies; T.F. performed the assay for tissue QUIN concentrations; M.S. and H.S. performed the IHC; S.B. and M.M. provided expert pathological review; L.F., A.B., Y.R., M.L. and G.S. provided transcriptomic data sets; N.H. analysed the data and, together with R.A.C., N.S.V. and R.E.B., drafted the manuscript; all authors provided comments on the manuscript and had final approval of the submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Leeds East Research Ethics Committee (ethical approval 15/YH/0080) and performed in accordance with the Declaration of Helsinki. All samples were obtained with the patients’ informed consent.
Consent for publication
No individually identifiable data are presented.
Data availability
RNA-seq data, generated through our ICGC CAGEKID study, has been deposited in a public repository as described elsewhere: Scelo et al.30 Proteomic data sets are available on request.
Competing interests
A.H. is Editor-in-Chief of British Journal of Cancer. R.S.’s current role is Publishing Editor for British Journal of Cancer.
Funding information
This work was supported by funding from the University of Leeds and Cancer Research UK. E.R.M. is funded by the European Research Council (Advanced Researcher Award), NIHR (Senior Investigator Award and Cambridge NIHR Biomedical Research Centre) and Cancer Research UK Cambridge Cancer Centre. The views expressed are those of the authors and not necessarily those of the NHS or Department of Health. The University of Cambridge has received salary support in respect of E.R.M. from the NHS in the East of England through the Clinical Academic Reserve.
Additional information
Note This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hornigold, N., Dunn, K.R., Craven, R.A. et al. Dysregulation at multiple points of the kynurenine pathway is a ubiquitous feature of renal cancer: implications for tumour immune evasion. Br J Cancer 123, 137–147 (2020). https://doi.org/10.1038/s41416-020-0874-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-020-0874-y
This article is cited by
-
Prediction of metabolites associated with somatic mutations in cancers by using genome-scale metabolic models and mutation data
Genome Biology (2024)
-
Kynurenine pathway in kidney diseases
Pharmacological Reports (2022)
