
Abstract
The continuing efforts to exploit the death receptor agonists, such as the tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), for cancer therapy, have largely been impaired by the anti-apoptotic and pro-survival signalling pathways leading to drug resistance. Cell migration, invasion, differentiation, immune evasion and anoikis resistance are plastic processes sharing features of the epithelial-to-mesenchymal transition (EMT) that have been shown to give cancer cells the ability to escape cell death upon cytotoxic treatments. EMT has recently been suggested to drive a heterogeneous cellular environment that appears favourable for tumour progression. Recent studies have highlighted a link between EMT and cell sensitivity to TRAIL, whereas others have highlighted their effects on the induction of EMT. This review aims to explore the molecular mechanisms by which death signals can elicit an increase in response heterogeneity in the metastasis context, and to evaluate the impact of these processes on cell responses to cancer therapeutics.
Similar content being viewed by others


The epithelial-to-mesenchymal transition (EMT) features
EMT is a physiological process that occurs during embryogenesis (type 1 EMT), wound healing phases (type 2 EMT) and metastasis (type 3 EMT). In these distinct situations where it undertakes development,1 cellular homoeostasis and repair,2 EMT is characterised by the loss of some epithelial features and, in parallel, by the gain of new mesenchymal properties such as the acquisition of invasive capacities and resistance to apoptosis. The newly acquired phenotype has stem cell capacities that confer pluripotency and plasticity to cells, but also a different sensitivity to both endogenous and environmental signals.3 It is a critical process for tumour initiation and progression.4
During EMT, E-cadherin, the main cadherin responsible for epithelial cell adherent junctions, but also a regulator of actin cytoskeleton homoeostasis and organisation,5,6 is downregulated primarily via transforming growth factor (TGF)-β/SMAD signalling, leading to a loss of cell–cell adhesion.7 To date, cancer cells with a low level of E-cadherin are considered invasive (or aggressive), whereas those with a high level are associated with stress resistance and survival.8 However, E-cadherin expression has recently been shown to be crucial for metastasis by preventing reactive oxygen species (ROS)-dependent cell death and by allowing cancer cell dissemination,9 a finding in accordance with clinical evidence showing metastatic E-cadherin+ tumours.10,11
Pathways other than TGF-β/SMAD also play a central role in relaying the EMT signal. This is the case for receptor tyrosine kinases (RTKs), Notch, Hedgehog and the canonical and the non-canonical Wnt pathways.12 They all activate EMT-inducing transcription factors (EMT-TFs). Among them, the Snail family (Snail and Slug), ZEB-1/2, TWIST1/2, TCF3, FOXC2, PRRX1, YAP/TAZ and SOX4/9 target E-cadherin repression or co-operate with core EMT-TFs.13 Because EMT-TFs are differentially expressed depending on the cancer stage, as observed, for example, in endometrioid endometrial carcinoma,14 the spatiotemporal expression of the EMT-TF TWIST1 can be a mechanism for the hierarchical role of EMT-TFs observed during cancer progression,15 whereas the miR-34/SNAIL and the miR-200/ZEB axis not only regulates E-cadherin expression, but also the hybrid phenotype via a double-negative feedback.16,17,18,19
Evidence concerning the acquisition of stem cell characteristics associated with the induction of partial EMT, also known as a hybrid E/M state, has been described and is associated with an increase in tumour-propagating cell (TPC) frequency.20 By showing that the earliest EMT state can exhibit a high TPC frequency, the authors demonstrated that intermediate states can also provide stem cell properties, leading to drug resistance and cancer progression, and this mechanism does not require the establishment of a full EMT as it had been assumed previously. Indeed, a report that EMT was not required for lung metastasis was based on the observation that metastasis in secondary sites mostly exhibited an epithelial phenotype.21 However, the actual tools and methods used to claim such a controversial conclusion have been questioned and found to be insufficient to rule out the EMT process during cancer progression.22 Therefore, the hybrid E/M system is still the proposed mechanism by which EMT drives metastatic dissemination.23,24,25
Cell death resistance has been shown in partial EMT states,26 and the hybrid E/M phenotype has been described in tumours as a source of cancer cell response heterogeneity with differences in sensitivity to apoptotic stimulus such as anoikis and anticancer drugs.27,28 The main signalling pathways involved in the development and regenerating processes are also involved in this mechanism in neoplastic tissue. Among them, the Notch/Jagged pathway stabilises the hybrid E/M phenotype and is necessary to expand the fraction of cancer stem cells (CSCs). This has been shown in a triple-negative breast cancer model under the influence of interleukin (IL)-6, a pro-inflammatory cytokine able to activate the EMT programme.29 Still in the mammary gland, the EMT programme also increases stem-like features through the control of the Hedgehog signalling pathway,30 whereas Wnt pathways regulate the stem cell programme of hybrid E/M phenotypes that accounts for increasing drug resistance.31,32,33
CSCs have tumorigenic potential that depends on the EMT state.33 EMT also confers stem cell properties to cancer cells by inducing non-genetic and heritable epigenetic changes;34 however, these newly acquired properties are known to be reversible through the induction of a mesenchymal-to-epithelial programme called MET,35 with the EMT/MET switch contributing to cell phenotypic plasticity.36,37 Because a large number of phenotypic states exist between partial and the well-differentiated states of EMT, this phenotypic diversity thus increases intra-tumoural heterogeneity38 and is a potential source of drug resistance observed in patients.39
EMT and cell survival
Evidence that EMT not only provides the mechanistic basis of metastasis, but also of resistance to apoptosis, has been demonstrated in different model tissues, such as breast,40 lung,21 prostate41 or pancreas,42 with a process potentially dependent on miR-200,43,44 TWIST and Snail1 expression.45,46 The decreased expression of key proteins, such as cadherins and integrins, is accompanied by the loss of cell adhesion with the extracellular matrix (ECM) and with neighbouring cells during the EMT process. These lead to the activation of intracellular pro-survival signals known as ‘non-canonical pathways’ and is mainly mediated by the phosphoinositide 3-kinase (PI3K)/Akt (also known as protein kinase B (PKB)) pathway.
The PI3K/Akt signalling pathway plays a pivotal role in controlling cancer cell survival. More specifically, it allows the activation of the mitogen-activated protein kinase (MAPK) pathway responsible for the activation of downstream p90RSK, thus inhibiting the pro-apoptotic protein Bad.47 In prostate cancer, PI3K/Akt signalling is activated downstream by the involvement of Notch and activation of the Hedgehog pathway. While Hedgehog increases the expression of the anti-apoptotic Bcl-2, Notch mediates pro-survival mechanisms under the control of Akt, thus leading to docetaxol resistance.48 Akt activation also drives nuclear factor (NF)-κB activation, which, in turn, controls the expression of the anti-apoptotic proteins FLICE-inhibitory protein (FLIP) and X-linked inhibitor of apoptosis protein (XIAP).49
The main EMT-TFs responsible for migration, invasion or dedifferentiation also play a role in cell survival by modulating the expression of pro- and anti-apoptotic proteins. For example, Twist increases Bcl-2, leading to apoptotic resistance,50 whereas SNAI1 interacts with poly(ADP-ribose) polymerase 1 (PARP1).51,52 Other apoptotic regulators are involved in EMT-dependent survival mechanisms. Among them, TGF-β, a tumour repressor with a dual role in cancer that depends on the environmental conditions such as matrix rigidity53 or cancer progression stage,54 has been shown to induce apoptosis and to interplay with the PI3K/Akt pathway.55
In conclusion, many environmental factors derived from the ECM, cancer-associated fibroblasts (CAFs), immune cells and vessels are responsible for the increase in EMT-TFs described above and are involved in multidrug resistance (MDR) phenomena. They not only regulate the expression of pro- or anti-apoptotic proteins, but also those of ABC transporter genes.56 Moreover, signals that induce EMT such as TGF-β could modulate the response of cancer cells to anticancer drugs (as with endogenous antimitotic signals) by cytokinesis failure, a heritable mechanism that leads to genomic instability.57 The dual role of TGF-β is applied in this context and can lead to opposite effects, depending on the cancer mutations and the model studied. For example, in Ras-mutant pancreatic cancer cells, sensitivity to apoptosis is then controlled by the TGF-β-induced EMT. In other words, TGF-β induces EMT and subsequent apoptosis confers a tumour-suppressive property to the EMT programme.58
Death receptor-mediated features of EMT
Activation of transmembrane receptors of the tumour necrosis factor superfamily (TNF-SF), such as Fas/CD95, TNF receptors 1 and 2 (TNF-R1/R2) and TNF-related apoptosis-inducing ligand (TRAIL) receptors 1 and 2 (TRAIL-R1/2, DR4/DR5), by their respective ligands Fas ligand (Fas-L), TNF-α and APO-2L/TRAIL (Table 1), can lead to the induction of cell death.59,60 The binding of the ligand to its receptor allows the formation of a death-inducing signalling complex (DISC), including caspase-8,61 which can transduce a pro-apoptotic signal via the caspase cascade, leading to cell death.62
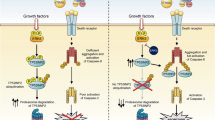
In addition to apoptosis, a range of cell responses are induced upon binding to death receptors. Among them, differentiation was shown to be regulated by TRAIL-induced caspase activation in intestinal cells,63 osteoclasts64 and keratinocytes.65 A close relationship exists between differentiation steps during the early stage of development and cancer progression, leading to metastasis, and involves common molecular factors and pathways,66 such as death receptor activation or dysregulation. Indeed, metastasis and invasion are processes associated with TRAIL treatment and are shown to be dependent on the NF-κB pathway.67 In human cholangiocarcinoma cell lines, TRAIL promotes cell migration and invasion under the control of the NF-κB-dependent pathway.68 As cancer progression can be initiated through the induction of EMT, involving different cell differentiation steps, it is important to better understand the molecular mechanisms leading to the acquisition of heterogenous EMT features upon death receptor engagement, which have a further impact on the cell response (Fig. 1).

Binding of death ligands can activate pathways, including caspase-8-dependent apoptosis and survival. Cancer cells that survive treatment can give rise to different responses, such as proliferation, senescence or differentiation. Epithelial-to-mesenchymal transition (EMT) is one of the cell biological processes that contributes to cellular plasticity, allowing cancer cells to switch from an epithelial state to a mesenchymal one. Cells lose their adhesion capacities, acquire stem cell characteristics (CSCs) and can migrate until invading secondary sites via the lymphatic system and the blood circulation. EMT also provides resistance to anoikis, an apoptotic process following loss of cell contacts with ECM and decreases the immunogenic response. Together, these events can participate in the increased resistance of circulating tumour cells (CTCs) alone or in clusters (CTMs), allowing cancer progression and metastasis. Finally, EMT increases response heterogeneity by enhancing cell diversity within the tumour, which can further increase clonal heterogeneity and cancer cell resistance to chemotherapies.
Death receptors co-operate with cell membrane components: TRAIL and loss of cell adhesion
TRAIL has been reported to induce the loss of adhesion followed by drug resistance in several cellular contexts. First, through the apoptotic pathways, TRAIL activates the cleavage of substrates involved in cell differentiation and remodelling.69 Then, by interacting with cadherins at the plasma membrane (Table 1), DR4/5 stabilisation and activation can be impaired, leading to changes in TRAIL sensitivity, as discussed below.
Occludin and claudin, two transmembrane proteins forming tight junctions (TJs), play a barrier role not only in controlling fluid transport but also in proliferation and differentiation of the epithelium. As an epithelial marker, occludin is downregulated during EMT.70 Epigenetic silencing of occludin leads to metastasis of cancer cells via modulation of unique sets of apoptosis-associated genes.71 Moreover, its knockdown decreases TRAIL-induced cell death, thus conferring an important role of occludin in apoptosis.72 Indeed, occludin (and to a lesser extent claudin) interacts physically with Fas associated via death domain (FADD) and DISC when tight junctions are disrupted, a phenomenon allowing cell defence (an antibacterial strategy) by activating the extrinsic cell death signal.73,74
Studies have reported that distribution of TRAIL receptors in lipid rafts can be linked to cell sensitivity to TRAIL ligand,75,76,77,78 but how they interact with membrane components remains poorly understood. First, DR4/5 can couple with E-cadherin.79 Secondly, E-cadherin/α-catenin linkage with dynamic cytoskeleton is essential for the efficient assembly of active death receptor complexes.80 Consequently, this receptor clustering allows formation of active TRAIL signalling complexes and sensitises some cancer cells to death induced by TRAIL. Because EMT leads to the dysregulation and disassembly of this E-cadherin–TRAIL complex, cancer cells with a mesenchymal phenotype increase their protection against TRAIL-induced apoptosis.81 However, in patients with early-detected colorectal cancer, DR4 and DR5 can be expressed in parallel with E-cadherin, but their co-localisation at the membrane is not systematic.82 Moreover, the potential interactions between the death receptors (including the decoy receptors known as death receptor competitors that lack the intracellular death domain responsible for the propagation of TRAIL-induced apoptotic signal) and cadherins remains unanswered, despite the potential mechanistic impact these cell processes hold.83
A study focusing on the natural anti-metastatic agent antrocin has shown that it could act as an EMT inhibitor, restoring E-cadherin protein levels in parallel with the increase in DR5 expression,84 whereas another study has shown that DR5 knockdown could increase E-cadherin expression and diminish migration in breast cancer, which further suggests a specific regulatory step.85 However, how DR4/DR5 and E-cadherin expression is simultaneously regulated is still not well understood. Nevertheless, the Hedgehog pathway and the modulation of some microRNAs (miRNAs) may be involved in this regulatory process. Indeed, TRAIL-induced apoptosis resistance in chronic conditions has been shown to be under the control of miR-21, miR-30c and miR-100 in lung cancer.86 The overexpression of these miRNAs inhibits the expression of caspases-3/8 and the EMT marker E-cadherin. They also activate the NF-κB pathway that regulates via a feedback loop the expression of the miRNAs involved. Among them, miR-21 seems to play a pivotal role in modulating the expression of both E-cadherin and DR4/DR5 as observed in a glioblastoma model.87 In this study, the Hedgehog inhibitor NPV-LDE-225 blocked the EMT process and allowed an increase in the efficiency of TRAIL-induced apoptosis by increasing DR4/DR5 and E-cadherin expression. Finally, it decreases not only miR-21 levels, but also stem-cell markers such as NANOG, OCT4, SOX2 and c-Myc, making both miR-21 and the Hedgehog signalling pathway possible master regulators of this mechanism. In addition, the natural Hedgehog inhibitor cordycepin can also induce apoptosis in breast cancer models, with the upregulation of DR4/DR5 and E-cadherin.88
More recently, the co-operation of cadherin/DR was studied in head and neck cancers (HNSCCs): N-cadherin, the major mesenchymal marker, has been shown to enhance cell growth by inhibiting apoptosis.89 N-cadherin overexpression was associated not only with an increase in DcR2 but also with a decrease in DR5, whereas its knockdown led to the opposite results, suggesting the existence of a signalling network between cadherins and death receptors. Moreover, N-cadherin was observed to interact with DcR2 in these same models, a process allowing cell survival via cleavage of caspases by activating the MAPK/extracellular signal-regulated kinase (ERK) pathway. Because the E-/N-cadherin switch is the hallmark of EMT90,91 and because cell sensitivity to TRAIL is changing with cell status (from epithelial to mesenchymal, and this could represent, in turn, a TRAIL-sensitivity marker. Further investigations are needed to clarify this possible regulation even if limited data exist in the literature supporting this regulatory process. Indeed, we know that both DR5 and DcR2 expression are under the control of P53, and a negative-feedback loop has been proposed between DcR2 and P53.92 Overexpression of P53 can lead to an increase in DcR2 which, in turn, can attenuate DR5-mediated apoptosis.93
While E-cadherin co-operates with DR5 at the membrane level of epithelial cancer cells and N-cadherin with DcR2 in mesenchymal cells, another member of the cadherin family has been described to physically interact with TRAIL receptors, namely FAT1. FAT1 is a cadherin-like protein with tumour-suppressor functions, which plays a central role in developmental processes and cell communication.94,95 This adhesive molecule is highly expressed in several foetal epithelia, but its mutation leads to an aberrant activation of the WNT signalling pathway resulting in tumorigenesis.96 In glioblastoma cells, FAT1 acts as a negative regulator of DR4/5. After knockdown of FAT1, cancer cells became more sensitive to TRAIL-induced apoptosis, a process very similar to those mentioned above: by interacting with FAT1, DRs finally prevent DISC activation.97
Death receptors co-operate physically with other membrane proteins not specifically involved in EMT, but also involved in cancer progression, leading to metastasis. For example, carcinoembryonic antigen (CEA, CD66e), mainly found in colorectal cancer, is a cell-surface glycoprotein that is increased along with DR5 when cells are in suspension. Interestingly, it binds and inhibits DR5, resulting in the decreased activity of caspase-8. An increase in cell survival (in vitro) and colonisation of secondary tissues (in vivo) were also observed. Together, these events stimulate cancer progression and metastasis.98
EMT regulates TRAIL sensitivity
Targeting mesenchymal cancer cells by displaying stem cell characteristics with TRAIL has been proposed to reduce resistance in different cancers, such as squamous and adenocarcinoma lung cancer.99 This association is emerging in other pathologies such as biliary atresia, a common viral-dependent cholangiopathy where EMT has been shown to block biliary innate immune response via TRAIL-mediated apoptosis100,101 or in hepatitis B virus (HBV) infection, where HBV may activate in certain conditions an EMT-like state that is ER-stress dependent.102
Interestingly, some homologies have been observed between differential sensitivity to TRAIL and the EMT process. TRAIL resistance of non-genetic origins from variable activation and expression levels of pro- and anti-apoptotic proteins103,104,–105 has been shown to be transient and sustainable.106 Similar observations have been made in the EMT context. Indeed, during cell division, variable partitioning of macromolecules in daughter cells was proposed to increase EMT heterogeneity,107 illustrating that non-genetic mechanisms play an important role in cellular heterogeneity and plasticity, leading to different cell states. Because cancer cells can switch from an epithelial state to a mesenchymal one in order to adapt to the tumour microenvironment and to progress to metastasis, the intermediate states known as hybrid E/M linked to differences in sensitivity to chemotherapeutic agents108 are now emerging as promising targets against cancer progression.109
One of the first observations was related to nitric oxide (NO) donors such as DETANONOate. This chemical can sensitise cancer cells to TRAIL-induced apoptosis through different mechanisms. First, it contributes to increase the expression of Raf kinase inhibitor protein (RKIP), a metastatic tumour suppressor. Then, it inhibits both the NF-κB pathway responsible for cell resistance to chemotherapies and the YY1 transcription factor, which is, in turn, responsible for the regulation of Fas and DR5 (the main receptor for TRAIL). Finally, NO donors contribute to the inhibition of the Snail transcription factor, an E-cadherin repressor, thus repressing the EMT process. In brief, by dysregulating the NF-κB/Snail/YY1/RKIP/phosphatase and tensin homologue (PTEN) axis, NO donors prevent metastatic potential and resistance to apoptosis.110,111 Similar observations have been found in urothelial cancer cell lines where mesenchymal cells showed higher resistance to TRAIL treatments than epithelial cells. Indeed, the latter have a lower level of XIAP and Bcl-2 proteins that account in part for the anti-apoptotic effects. These data appear to be an additional point in favour of the importance of targeting EMT markers and/or processes as a strategy against cancer progression.112
A compelling observation suggesting a link between EMT and resistance to TRAIL-induced apoptosis is the deregulation of transcription factors such as Snail and Slug.45,113 Both are not only involved in the downregulation of adherent proteins known as epithelial markers such as E-cadherin, claudins or occludins, but also in the inhibition of pro-apoptotic proteins such as Bcl-2, Bid, Puma and caspase-9. Moreover, the upregulation of Snail and Slug leads to the increase in P53 protein levels that mediate resistance through anoikis.114 Thus, reverting EMT appears to be a strategy to sensitise cancer cells to TRAIL therapy. Srivastava et al. used a benzamide histone deacetylase inhibitor (MS-275 also called entinostat) to target histone deacetylase (HDAC) 1/3, leading to an increase in the apoptosis-inducing potential of TRAIL in different cancer cell lines in vitro.113 This treatment enabled to sensitise TRAIL-resistant cancer cells, a phenomenon also observed in vivo (breast cancer xenografts in nude mice) where MS-275 inhibits EMT, decreases NF-κB pathway activation and finally increased DR4/DR5 receptor and pro-apoptotic protein expression. In pancreatic CSCs, the same team demonstrated that a GLI transcription factor inhibitor (GANT-61), which targets the Hedgehog pathway, allowed EMT inhibition in parallel with an increase in DR4 and DR5 expression.115
Another mechanism proposed previously is the dysregulation of miRNAs, especially Mir-9, which has been found to be downregulated in many cancers.116 This miRNA can modulate the expression of interferon (IFN)-induced genes and MHC class I molecules. Among these IFN-induced genes, TRAIL has been shown to be one of them. Indeed, an increase in Mir-9 is associated with overexpression of TRAIL.117 TRAIL overexpression was also found in MCF-7 cancer cells that have acquired resistance to metformin treatment. By inducing autophagy in certain cancer cells, TRAIL can protect cells by blunting the cytotoxicity of the treatment, thus contributing to TRAIL resistance.118 Mir-9 is also known to interact with the TGF-β signalling pathway during EMT;119 however, information is still lacking about TRAIL sensitivity. It has only been reported that TGFβ-induced EMT plays a critical role during irradiation of the breast cancer cell line HMLE, leading to radioresistance of the stem-like breast cancer cells generated. Indeed, in this study, mesenchymal CD24−/low/CD44+ CSCs were shown to exert apoptosis resistance through differential activation of death receptors such as TRAIL and in parallel via the increased expression of the anti-apoptotic marker survivin.120 The changes observed in TRAIL gene expression are likely to be associated with an EMT signature in such cases. Furthermore, another miRNA candidate has been proposed to play such an important role in TRAIL-induced apoptosis resistance. For example, by downregulating the PI3K/Akt regulator PTEN, miR221 induces EMT and invasiveness of breast cancer cells.121
Lu et al. proposed a mechanism of EMT-dependent inhibition of apoptosis where loss of E-cadherin (which binds selectively to DR4 and DR5 but not to Fas owing to the DISC formation and caspase-8 activation) drives cancer-cell resistance to TRAIL treatment.79 Another study reported that EMT reversal by ML327, an isoxazole-based small-molecule probe that represses E-cadherin levels and partially reverses the EMT phenotype, is accompanied by an enhanced response to TRAIL in carcinoma cells and this was in an E-cadherin-independent manner.122
Involvement of the mitochondrial pathway in models such as melanoma is also critical in TRAIL sensitivity,123 but its relationship with EMT remains less well described. In lung cancer, when the EMT marker MUC1 (responsible for pro-oncogenic signal transduction) is silenced, TRAIL treatment becomes more efficient. This increased sensitivity is possibly due to the MUC1–BAX association, leading to prevention of mitochondrial permeabilisation in response to apoptotic stimuli.124
Depending on the EMT status and on the expression levels of pro- and anti-apoptotic proteins under the control of the EMT-TFs, cancer cells will respond to anticancer therapies differently, with greater sensitivity in epithelial cells.125
TRAIL and resistance to anoikis in the metastatic context
The term ‘anoikis', from the Greek anoikos ‘without a home’, was proposed in the 1990s by Frisch and Francis126 to describe an apoptosis phenomenon following loss of cell-to-ECM interactions. The authors explained that anoikis occurs to abrogate an escape mechanism, meaning the possibility for a cell to reattach in an inappropriate tissue. This mechanism allows the limitation of oncogenic transformation without disrupting plasticity and cell migration necessary during development, repair and cell tissue homoeostasis. Anoikis and its resistance also increases the diversity of phenotypes.127 Thus, resistance to anoikis became a hallmark of malignant cells with their ability to grow under anchorage-independent conditions.128
In epithelial cells, anchorage to ECM represents an environmental signal that is mediated by integrins. Indeed, integrins β1 and β3 subunits, when in contact with ECM components, such as collagens, phosphorylate focal adhesion kinase (FAK), which, in turn phosphorylates Akt, leading to inhibition of pro-apoptotic proteins such as Bad. Consequently, the lack of ligation of integrins β1 and β3 subunits induces a decrease in both FAK protein and activity, but also those of the proto-oncogene tyrosine protein kinase Src or integrin-linked kinase (ILK), leading to the inhibition of the pro-survival Akt pathway.128,129
Evidence for a function of death receptors in anoikis has been described previously.130 When MDCK and HaCat cells lose their interactions with ECM, a caspase-8-dependent apoptotic cascade is triggered. This increasing caspase-8 activity after cell detachment occurs through FADD recruitment without DR4/DR5 activation, a process observed independently of the binding of death ligands.131 The authors also observed that Bcl-2 and Bcl-XL inhibit caspase-8-induced anoikis probably via a mitochondrial positive-feedback pathway by caspase-3. These data were further supported by another study showing that extrinsic apoptosis leading to anoikis was also triggered by caspase-8 in keratinocytes.129 This work revealed the positive feedback described above as a complementary interaction between the two apoptotic pathways. A negative post-transcriptional regulation of DR5 via miR126-3p was also proposed to explain the decrease in extrinsic apoptotic pathway signalling without affecting death receptor mRNA levels,132 but how TRAIL is associated with anoikis resistance during cancer progression remains unanswered.
Although DRs drive anoikis in normal cells, they fail to induce such a process in malignant cells, probably via a FLIP-dependent process.133 In breast cancer, cell anchorage suppresses TRAIL gene expression, whereas detachment increases its level. The autocrine role of endogenous TRAIL was then suspected to be associated with anoikis through activation of DR5 (and to a less extent DR4). Because the detached cells were found to be more sensitive to TRAIL, circulating tumour cells (CTCs) were considered as a potential target for TRAIL therapy.134 In fact, DR4/DR5 signalling allows caspase activation, leading to cleavage of Akt proteins and to their decreased expression levels. Because the Akt pathway plays a central role in mediating survival signalling, cell detachment via loss of integrin interactions with ECM is the first step in the inhibition of this anti-apoptotic signalling.135 In CRC cells, DR5 increases in cell suspension. The use of antagonists or DR5 knockdown is sufficient to inhibit anoikis, whereas no effects were observed concerning DR4. Exogenous TRAIL failed to increase anoikis as observed in a breast cancer model, and finally the proposed mechanism hypothesises that DR5 is activated by cross-linked soluble and membrane-bound TRAIL ligand.128
The mechanisms of anoikis resistance are numerous and they depend on the mutation status of the cancer cell model studied. Although one can suspect that a constitutive activation of pro-survival pathways could inhibit the apoptotic processes engaged after the loss of anchorage, thanks to acquired mutations, non-genetic heterogeneity associated with differences in protein expression levels can also largely impact the cell-fate decision. In the specific case of TRAIL-induced anoikis resistance, several mechanisms have been reported over the last two decades. For example, a decrease in DR4/DR5 expression has been described to explain such a resistance. In hepatoma cells, a low level of DR4/DR5 expression was associated with resistance of the TRAIL-induced apoptotic cascade even if upregulation of TRAIL mRNA was observed.136 Yet, no modulation in DR4/DR5 expression was observed between attached and detached human colon epithelial cells where TRAIL resistance was shown. Only increases in FAK and ILK activities and, secondly, the activation of the downstream Akt pathway, protect colon cells from TRAIL-induced apoptosis.137 Similar conclusions were reported in an ovarian model138 and in HL-60 cells.139 Interestingly, FAK not only stimulates the Akt pathway activation, but also interacts with caspase-8 in an adhesion-dependent manner, thus blocking the apoptotic extrinsic pathway in this condition;140,141 however, how TRAIL interacts with the integrin/FAK/Akt pathway remains unclear. More recently, TRAIL was described as a mediator of FAK signalling in the regulation of entosis (an invasion process involving two cells, where one is merging via the cytoplasm with the other) and necrosis in primary human mammary epithelial cells.127 Indeed, during detachment-induced cell death, even if TRAIL is rapidly increasing and this is for a long time (from 3 h to 72 h), FAK successfully inhibits TRAIL and protects cells during all of the processes.
Generally, the mechanisms of anoikis resistance linked to TRAIL treatment are shared with common apoptotic resistance mechanisms, especially those that interact with the extrinsic pathway. Indeed, a decrease in caspase-8 expression and its activity is associated with TRAIL resistance.142 Modulation of c-FLIP protein levels, the main endogenous pro-caspase-8 inhibitor143 and also an increase in the IAP protein family144 are other targets and regulators of this TRAIL-dependent resistance.
TRAIL regulates the PD-L1-dependent immunogenic response
In lung cancer or melanoma, programmed cell death protein-1 (PD-1)/programmed death-ligand 1 (PD-L1) expression and activation is an indicator of poor prognosis for patients,145,146 but their inhibition has become a strategy to stimulate the immune response and increase cell death.147,148 There is a growing body of evidence suggesting intricate regulation processes between TRAIL and PD-L1 expression. In 2010, Tu et al. analysed the effect of the hepatitis C virus core protein (HCVc) on human liver and especially on innate immune Kupffer cells (KCs). They found that it was able to induce the upregulation of PD-L1 under interleukins (IL-1β–IL10) and TNF-α secretion, along with the inhibition of the cell-surface expression of the cytotoxic molecule TRAIL, a process dependent on the activation of the PI3K/Akt pathway.149 Moreover, in chronic lymphocytic leukaemia (CLL), the therapeutic agent trabectedin induces apoptosis of both human primary leukaemic cells, selected myeloid and lymphoid immunosuppressive cells mainly through the TRAIL/TNF pathway. In parallel, trabectedin also blocks the PD-1/PD-L1 axis by targeting PD-L1+ CLL cells, PD-L1+ monocytes/macrophages and PD-1+ T cells.150 Complementary data were reported in murine melanoma151 and in hepatocellular carcinoma cells.152 Even if this association is not completely understood, we now know that IFN-γ, a cytokine responsible for the increase in expression of PD-L1, can also sensitise cancer cells to TRAIL-mediated apoptosis through downregulation of c-FLIP.153,154 Based on the relationship between immune cells of the tumour microenvironment and cancer cells, a very attractive approach has been proposed using a bifunctional fusion protein, designated anti-PD-L1:TRAIL,155 that successfully targets both immune cells (myeloid effector cells and T-cell activity) and cancer cells sensitised by this method.156
EMT plays a central role in immunogenicity. It has been shown to promote metastasis via immunosuppression,157,158 but evidence that PD-L1 overexpression correlates with the induction of EMT has been demonstrated in non-small-cell lung carcinoma (NSCLC) and more recently in breast cancer via a ZEB-1/miR-200 mechanism.159,160 Upstream of this signalling cascade, glycogen synthase kinase (GSK)-3β/β-catenin controls the ZEB-1/miR-200 axis and allows β-catenin nuclear translocation under the negative control of SDH5, a succinate dehydrogenase component of the tricarboxylic acid cycle.161,162 In NSCLCs, EMT specifically regulates PD-L1 expression with the need of epigenetic reprogramming, thus leading to immune escape.163 This mechanism requires both demethylation of the PD-L1 promoter due to TGF-β action and activation of NF-κB via TNF-α stimulation, but is not accompanied by an increase in DR4/DR5 or TRAIL expression,164 suggesting that an inversely proportional relationship between the expression of PD-L1 and the increase in resistance to TRAIL dependent on the decrease in DR expression would occur under the control of EMT. This proposed mechanism has also been observed in glioblastoma (GBM) where cannabidiol (CBD) upregulated the gene and protein expression of DR5/TRAIL-R2 and sensitises GBM cells to TRAIL-induced apoptosis. The authors observed that, as expected, CBD caused a notable decrease in GBM surface levels of PD-L1.165
Different regulation pathways have been proposed to explain the simultaneous expression of TRAIL receptors and PD-L1. In tumour IFN-driven resistance, stimulation of cancer cells by IFN-γ leads to the nuclear translocation of signal transducer and activator of transcription 1 (STAT1). The activation of the IFN-γ/STAT1 axis is then responsible for the increase in PD-L1 and in parallel the decrease in TRAIL-R2.166,167 Blockade of the IFN-γ receptor in this same resistant model leads to the increase in TRAIL-R2 and allows natural killer (NK) cells to stimulate extrinsic apoptosis in the cancer cells. Another regulation highlighting the role of miRNA-429 in PD-L1 expression and TRAIL sensitivity has been recently described. Indeed, miR-429 is a member of the miR-200 family that can inhibit ZEB-1/2 or PTEN/Akt upregulation making this miRNA an EMT regulator.168 In gastric cancer, PD-L1 is positively correlated with TRAIL resistance where miR-429 is downregulated.169 The authors observed that miR-429 targets the 3′ untranslated region (UTR) of PD-L1. They proposed a mechanism where PD-L1 interacts with phosphorylated epidermal growth factor receptor (EGFR), leading to the activation of the pro-survival Akt pathway, thus blocking the TRAIL-dependent apoptotic process.
Finally, in KRAS-mutated cancer cells, oncogenic RAS allows the stabilisation of PD-L1 mRNA, leading to its increase and escape from immunosurveillance. This phenomenon partly accounts for the chemotherapeutic resistance observed. Interestingly in pancreatic ductal adenocarcinoma (PDCA), cancer cells also express endogenous TRAIL with an autocrine function. Via DR5 activation, TRAIL stimulates the migration and invasion of KRAS-mutated cancer cells in a Rac1-dependent manner. Knowing that Rac1 is usually inhibited via Rho-associated protein kinase (ROCK) under the control of KRAS in normal conditions, the authors proposed a new strategy to target both KRAS and TRAIL to stimulate the immunogenic response and increase patient survival.170
Death receptor expression in circulating tumour cells (CTCs/CTMs)
CTCs are considered as putative precursors that might contribute, alone or in clusters, to cancer cell dissemination in the body, leading to metastasis.171,172 This cancer progression step is often called ‘the leukaemic phase’ of solid tumours as suggested by Mocellin et al.173 In patients’ blood, not only are CTCs collected but also apoptotic CTCs and CTC clusters described as circulating tumour microemboli (CTM) with higher metastatic potential. Together, they represent poor prognostic and pharmacodynamic biomarkers of solid tumours.174,175,176,–177 Remarkably, only a small proportion of CTCs can give rise to metastasis.178,179
Anoikis resistance appears to be critical for the aetiology of CTCs.180,181,–182 CTCs from prostate cancer cells lose their adhesive capacity through downregulation of E-cadherin, γ-catenin and β4 integrin with, in parallel, the gain of anti-apoptotic mechanisms increasing their resistance to cytotoxic stresses induced by immune cells.183 Among them, the authors observed a decrease in heat-shock protein 90β family member 1 (HSP90B1), a chaperone protein that not only enables escape from immune surveillance, but also increases Bcl-2 under the control of Akt pathway signalling activation. In another model, namely pancreatic cancer cells, Wnt2 was proposed as a candidate CTC gene. Wnt2 has been shown to be responsible for anoikis resistance through the activation of the non-canonical WNT/TAK1/IFN1 signalling pathway.184 Such examples are emerging in the literature, but all have a double signature in common: the decrease in epithelial markers and the gain of anti-apoptotic capacities as observed during EMT.
In CTCs from breast cancer, the molecular features of EMT were found inversely correlated with TRAIL plasma cytokine expression.185 Unfortunately, DR expression levels were not reported in this study. However, it seems that soluble TRAIL could have only weak apoptotic effects on CTCs independently of the DR concentrations as observed in a computational model.186 Different regulatory processes were proposed to understand DR modulations in CTCs, such as the c-Jun N-terminal kinase (JNK) pathway. In pancreatic CSCs, JNK inhibition allows the decrease in DcR1 via an IL-8-dependent autocrine process, while DR4/5 expression is increased, thereby sensitising cells to TRAIL treatment.187 Consequently, the authors observed diminished tumour burden and number of CTCs. Autophagic processes have also been shown to regulate sensitivity of CTCs to TRAIL,188 and to protect invasive cancer cells from anoikis.189,190 In a breast cancer cell line, DR4/5 expression is decreased in cell suspension in contrast to adherent cells, thus increasing TRAIL resistance. Mechanistically, DR4/5 undergo a rapid endocytosis, sequestration in the nucleus and degradation in the autophagosome.191
Given that EMT provides mesenchymal cells with the ability to resist to apoptosis, anoikis and some stem cell characteristics (regulated by different factors such as TGF-β, Wnt or Notch192), more evidence is needed to evaluate whether death receptor agonists could favour the emergence of CTCs through EMT mechanisms and further assess the sensitivity of CTCs to these drugs.
Conclusions and perspectives
Activation of death receptors allows pleiotropic effects whether related to cell death (apoptosis, necrosis, necroptosis, pyroptosis…) or to survival (differentiation, division, migration, entosis, EMT…). However, cell fate will ultimately depend on a wide range of environmental and cell contexts with both genetic and non-genetic variations. This response heterogeneity is at the origin of cell resistance, an adaptive mechanism that impairs cancer drug development and therapeutic strategies.193 In this review, we examined how EMT participates to increase this response heterogeneity which, in turn, enhances cancer cell survival. There are other possible mechanisms by which EMT could increase response heterogeneity through interactions with the tumour microenvironment. First, cancer cell growth is usually accompanied with a decrease in the availability of oxygen and other necessary elements within the tumour. This transient ischaemia stimulates the expression of the hypoxia-inducible factor family (HIF-1) that mediates the angiogenic response and controls different EMT-TFs (TCF3, ZEB-1/2 and TWIST1) responsible for E-cadherin downregulation.194,195 Secondly, carcinoma-associated fibroblasts (CAFs) are stroma cells that secrete soluble TGF-β, matrix metalloproteinases (MMPs), hepatocyte growth factor (HGF) and urokinase-type plasminogen activator (uPA). These CAFs are also recruited and activated from resident fibroblasts via the equivalent secretion of factors produced by cancer cells in EMT.196 Finally, inflammation stimulates and maintains EMT through production of cytokines (TGF-β, TNF-α, IL-1β, IL-6, IL-8, chemokine (C–X–C motif) ligand 1 (CXCL1) and CC chemokine ligand 18 (CCL18)) by infiltrating immune cells, including tumour-associated macrophages (TAMs) and lymphocytes.197,198 Because the EMT programme is regulated temporally and spatially (activation at the invasive front of the tumour), the differential communication between cancer cells and the microenvironment can further contribute to increase response heterogeneity to drug treatments.29,199
References
Nieto, M., Sargent, M., Wilkinson, D. & Cooke, J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science 264, 835–839 (1994).
Yan, C., Grimm, W. A., Garner, W. L., Qin, L., Travis, T., Tan, N. et al. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-α through bone morphogenic protein-2. Am. J. Pathol. 176, 2247–2258 (2010).
Gupta, P. B., Pastushenko, I., Skibinski, A., Blanpain, C. & Kuperwasser, C. Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 24, 65–78 (2019).
Pastushenko, I. & Blanpain, C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 29, 212–226 (2019).
Lombaerts, M., van Wezel, T., Philippo, K., Dierssen, J. W. F., Zimmerman, R. M. E., Oosting, J. et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br. J. Cancer 94, 661–671 (2006).
Shankar, J. & Nabi, I. R. Actin cytoskeleton regulation of epithelial mesenchymal transition in metastatic cancer cells. PLoS ONE 10, e0119954 (2015).
Xu, J., Lamouille, S. & Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172 (2009).
Gomis, R. R. Survival skills ensure that cancer spreads. Nature 573, 353–354 (2019).
Padmanaban, V., Krol, I., Suhail, Y., Szczerba, B. M., Aceto, N., Bader, J. S. et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573, 439–444 (2019).
Hugo, H. J., Gunasinghe, N. P. A. D., Hollier, B. G., Tanaka, T., Blick, T., Toh, A. et al. Epithelial requirement for in vitro proliferation and xenograft growth and metastasis of MDA-MB-468 human breast cancer cells: oncogenic rather than tumor-suppressive role of E-cadherin. Breast Cancer Res. 19, 86 (2017).
Na, T.-Y., Schecterson, L., Mendonsa, A. M. & Gumbiner, B. M. The functional activity of E-cadherin controls tumor cell metastasis at multiple steps. Proc. Natl Acad. Sci. USA 117, 5931–5937 (2020).
Mohammadinejad, R., Biagioni, A., Arunkumar, G., Shapiro, R., Chang, K.-C., Sedeeq, M. et al. EMT signaling: potential contribution of CRISPR/Cas gene editing. Cell. Mol. Life Sci. 77, 2701–2722 (2020).
Stemmler, M. P., Eccles, R. L., Brabletz, S. & Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 21, 102–112 (2019).
Montserrat, N., Mozos, A., Llobet, D., Dolcet, X., Pons, C., de Herreros, A. G. et al. Epithelial to mesenchymal transition in early stage endometrioid endometrial carcinoma. Hum. Pathol. 43, 632–643 (2012).
Tsai, J. H., Donaher, J. L., Murphy, D. A., Chau, S. & Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736 (2012).
Díaz-López, A., Díaz-Martín, J., Moreno-Bueno, G., Cuevas, E. P., Santos, V., Olmeda, D. et al. Zeb1 and Snail1 engage miR-200f transcriptional and epigenetic regulation during EMT: EMT players controlling epithelial plasticity. Int. J. Cancer 136, E62–E73 (2015).
Grelet, S., McShane, A., Geslain, R. & Howe, P. H. Pleiotropic roles of non-coding RNAs in TGF-β-mediated epithelial-mesenchymal transition and their functions in tumor progression. Cancers 9, 75 (2017).
Lu, M., Jolly, M. K., Levine, H., Onuchic, J. N. & Ben-Jacob, E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl Acad. Sci. USA 110, 18144–18149 (2013).
Wellner, U., Schubert, J., Burk, U. C., Schmalhofer, O., Zhu, F., Sonntag, A. et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 11, 1487–1495 (2009).
Pastushenko, I., Brisebarre, A., Sifrim, A., Fioramonti, M., Revenco, T., Boumahdi, S. et al. Identification of the tumour transition states occurring during EMT. Nature 556, 463–468 (2018).
Fischer, K. R., Durrans, A., Lee, S., Sheng, J., Li, F., Wong, S. T. C. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476 (2015).
Ye, X., Brabletz, T., Kang, Y., Longmore, G. D., Nieto, M. A., Stanger, B. Z. et al. Upholding a role for EMT in breast cancer metastasis. Nature 547, E1–E3 (2017).
Yu, M., Bardia, A., Wittner, B. S., Stott, S. L., Smas, M. E., Ting, D. T. et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584 (2013).
Tran, H. D., Luitel, K., Kim, M., Zhang, K., Longmore, G. D. & Tran, D. D. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 74, 6330–6340 (2014).
Beerling, E., Seinstra, D., de Wit, E., Kester, L., van der Velden, D., Maynard, C. et al. Plasticity between epithelial and mesenchymal states unlinks EMT from metastasis-enhancing stem cell capacity. Cell Rep. 14, 2281–2288 (2016).
Jolly, M. K., Somarelli, J. A., Sheth, M., Biddle, A., Tripathi, S. C., Armstrong, A. J. et al. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 194, 161–184 (2019).
Gupta, S. & Maitra, A. EMT: matter of life or death? Cell 164, 840–842 (2016).
Tan, T. Z., Miow, Q. H., Miki, Y., Noda, T., Mori, S., Huang, R. Y. et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 6, 1279–1293 (2014).
Bocci, F., Gearhart-Serna, L., Boareto, M., Ribeiro, M., Ben-Jacob, E., Devi, G. R. et al. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl Acad. Sci. USA 116, 148–157 (2019).
Guen, V. J., Chavarria, T. E., Kröger, C., Ye, X., Weinberg, R. A. & Lees, J. A. EMT programs promote basal mammary stem cell and tumor-initiating cell stemness by inducing primary ciliogenesis and Hedgehog signaling. Proc. Natl Acad. Sci. USA 114, E10532–E10539 (2017).
Bierie, B., Pierce, S. E., Kroeger, C., Stover, D. G., Pattabiraman, D. R., Thiru, P. et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl Acad. Sci. USA 114, E2337–E2346 (2017).
Grosse-Wilde, A., Fouquier d’Hérouël, A., McIntosh, E., Ertaylan, G., Skupin, A., Kuestner, R. E. et al. Stemness of the hybrid epithelial/mesenchymal state in breast cancer and its association with poor survival. PLoS ONE 10, e0126522 (2015).
Kröger, C., Afeyan, A., Mraz, J., Eaton, E. N., Reinhardt, F., Khodor, Y. L. et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl Acad. Sci. USA 116, 7353–7362 (2019).
Tellez, C. S., Juri, D. E., Do, K., Bernauer, A. M., Thomas, C. L., Damiani, L. A. et al. EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res. 71, 3087–3097 (2011).
Dongre, A. & Weinberg, R. A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 20, 69–84 (2019).
Biddle, A., Gammon, L., Liang, X., Costea, D. E. & Mackenzie, I. C. Phenotypic plasticity determines cancer stem cell therapeutic resistance in oral squamous cell carcinoma. EBioMedicine 4, 138–145 (2016).
Brabletz, T. To differentiate or not-routes towards metastasis. Nat. Rev. Cancer 12, 425–436 (2012).
Jolly, M. K. & Celià-Terrassa, T. Dynamics of phenotypic heterogeneity associated with EMT and stemness during cancer progression. J. Clin. Med. 8, 1542 (2019).
Navas, T., Kinders, R. J., Lawrence, S. M., Ferry-Galow, K. V., Borgel, S., Hollingshead, M. G. et al. Clinical evolution of epithelial-mesenchymal transition in human carcinomas. Cancer Res. 80, 304–318 (2019).
Xu, X., Zhang, L., He, X., Zhang, P., Sun, C., Xu, X. et al. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem. Biophys. Res. Commun. 502, 160–165 (2018).
Pu, H., Horbinski, C., Hensley, P. J., Matuszak, E. A., Atkinson, T. & Kyprianou, N. PARP-1 regulates epithelial-mesenchymal transition (EMT) in prostate tumorigenesis. Carcinogenesis 35, 2592–2601 (2014).
Zheng, X., Carstens, J. L., Kim, J., Scheible, M., Kaye, J., Sugimoto, H. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530 (2015).
Puhr, M., Hoefer, J., Schäfer, G., Erb, H. H. H., Oh, S. J., Klocker, H. et al. Epithelial-to-mesenchymal transition leads to docetaxel resistance in prostate cancer and is mediated by reduced expression of miR-200c and miR-205. Am. J. Pathol. 181, 2188–2201 (2012).
Radisky, D. C. miR-200c at the nexus of epithelial-mesenchymal transition, resistance to apoptosis, and the breast cancer stem cell phenotype. Breast Cancer Res. 110, bcr2885 (2011).
Kurrey, N. K., Jalgaonkar, S. P., Joglekar, A. V., Ghanate, A. D., Chaskar, P. D., Doiphode, R. Y. et al. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells Dayt. Ohio 27, 2059–2068 (2009).
Smit, M. A., Geiger, T. R., Song, J.-Y., Gitelman, I. & Peeper, D. S. A twist-snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Mol. Cell. Biol. 29, 3722–3737 (2009).
Tan, Y., Ruan, H., Demeter, M. R. & Comb, M. J. p90(RSK) blocks bad-mediated cell death via a protein kinase C-dependent pathway. J. Biol. Chem. 274, 34859–34867 (1999).
Domingo-Domenech, J., Vidal, S. J., Rodriguez-Bravo, V., Castillo-Martin, M., Quinn, S. A., Rodriguez-Barrueco, R. et al. Suppression of Acquired docetaxel resistance in prostate cancer through depletion of Notch- and Hedgehog-dependent tumor-initiating cells. Cancer Cell 22, 373–388 (2012).
Mitsiades, C. S., Mitsiades, N., Poulaki, V., Schlossman, R., Akiyama, M., Chauhan, D. et al. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: therapeutic implications. Oncogene 21, 5673–5683 (2002).
Cao, Z., Livas, T. & Kyprianou, N. Anoikis and EMT: lethal ‘liaisons’ during cancer progression. Crit. Rev. Oncog. 21, 155–168 (2016).
Rodríguez, M. I., González-Flores, A., Dantzer, F., Collard, J., de Herreros, A. G. & Oliver, F. J. Poly(ADP-ribose)-dependent regulation of Snail1 protein stability. Oncogene 30, 4365–4372 (2011).
Stanisavljevic, J., Porta-de-la-Riva, M., Batlle, R., de Herreros, A. G. & Baulida, J. The p65 subunit of NF-κB and PARP1 assist Snail1 in activating fibronectin transcription. J. Cell Sci. 124, 4161–4171 (2011).
Leight, J. L., Wozniak, M. A., Chen, S., Lynch, M. L. & Chen, C. S. Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial-mesenchymal transition. Mol. Biol. Cell 23, 781–791 (2012).
Song, J. EMT or apoptosis: a decision for TGF-beta. Cell Res. 17, 289–290 (2007).
Zhang, L., Zhou, F. & ten Dijke, P. Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 38, 612–620 (2013).
Erin, N., Grahovac, J., Brozovic, A. & Efferth, T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist. Updat. 53, 100715 (2020).
Comaills, V., Kabeche, L., Morris, R., Buisson, R., Yu, M., Madden, M. W. et al. Genomic instability is induced by persistent proliferation of cells undergoing epithelial-to-mesenchymal transition. Cell Rep. 17, 2632–2647 (2016).
David, C. J., Huang, Y.-H., Chen, M., Su, J., Zou, Y., Bardeesy, N. et al. TGF-β tumor suppression through a lethal EMT. Cell 164, 1015–1030 (2016).
Holbrook, J., Lara-Reyna, S., Jarosz-Griffiths, H. & McDermott, M. F. Tumour necrosis factor signalling in health and disease. F1000Research 8, 111 (2019).
Rossin, A., Miloro, G. & Hueber, A.-O. TRAIL and FasL functions in cancer and autoimmune diseases: towards an increasing complexity. Cancers 11, 639 (2019).
Seyrek, K., Ivanisenko, N. V., Richter, M., Hillert, L. K., König, C. & Lavrik, I. N. Controlling cell death through post-translational modifications of DED proteins. Trends Cell Biol. 30, 354–369 (2020).
Kretz, A.-L., Trauzold, A., Hillenbrand, A., Knippschild, U., Henne-Bruns, D., von Karstedt, S. et al. TRAILblazing strategies for cancer treatment. Cancers 11, 456 (2019).
Rimondi, E., Secchiero, P., Quaroni, A., Zerbinati, C., Capitani, S. & Zauli, G. Involvement of TRAIL/TRAIL-receptors in human intestinal cell differentiation. J. Cell. Physiol. 206, 647–654 (2006).
Yen, M.-L., Tsai, H.-F., Wu, Y.-Y., Hwa, H.-L., Lee, B.-H. & Hsu, P.-N. TNF-related apoptosis-inducing ligand (TRAIL) induces osteoclast differentiation from monocyte/macrophage lineage precursor cells. Mol. Immunol. 45, 2205–2213 (2008).
Wu, N.-L., Lee, T.-A., Tsai, T.-L. & Lin, W.-W. TRAIL-induced keratinocyte differentiation requires caspase activation and p63 expression. J. Investig. Dermatol. 131, 874–883 (2011).
Yang, L., Shi, P., Zhao, G., Xu, J., Peng, W., Zhang, J. et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 5, 8 (2020).
Malhi, H. & Gores, G. J. TRAIL resistance results in cancer progression: a TRAIL to perdition? Oncogene 25, 7333–7335 (2006).
Ishimura, N., Isomoto, H., Bronk, S. F. & Gores, G. J. Trail induces cell migration and invasion in apoptosis-resistant cholangiocarcinoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G129–G136 (2006).
Julien, O. & Wells, J. A. Caspases and their substrates. Cell Death Differ. 24, 1380–1389 (2017).
Ikenouchi, J., Matsuda, M., Furuse, M. & Tsukita, S. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J. Cell Sci. 116, 1959–1967 (2003).
Osanai, M., Murata, M., Nishikiori, N., Chiba, H., Kojima, T. & Sawada, N. Epigenetic silencing of occludin promotes tumorigenic and metastatic properties of cancer cells via modulations of unique sets of apoptosis-associated genes. Cancer Res. 66, 9125–9133 (2006).
Rachow, S., Zorn-Kruppa, M., Ohnemus, U., Kirschner, N., Vidal-y-Sy, S., von den Driesch, P. et al. Occludin is involved in adhesion, apoptosis, differentiation and Ca2+-homeostasis of human keratinocytes: implications for tumorigenesis. PLoS ONE 8, e55116 (2013).
Beeman, N., Webb, P. G. & Baumgartner, H. K. Occludin is required for apoptosis when claudin-claudin interactions are disrupted. Cell Death Dis. 3, e273 (2012).
Beeman, N. E., Baumgartner, H. K., Webb, P. G., Schaack, J. B. & Neville, M. C. Disruption of occludin function in polarized epithelial cells activates the extrinsic pathway of apoptosis leading to cell extrusion without loss of transepithelial resistance. BMC Cell Biol. 10, 85 (2009).
Ouyang, W., Yang, C., Zhang, S., Liu, Y., Yang, B., Zhang, J. et al. Absence of death receptor translocation into lipid rafts in acquired TRAIL-resistant NSCLC cells. Int. J. Oncol. 42, 699–711 (2013).
Ouyang, W., Yang, C., Liu, Y., Xiong, J., Zhang, J., Zhong, Y. et al. Redistribution of DR4 and DR5 in lipid rafts accounts for the sensitivity to TRAIL in NSCLC cells. Int. J. Oncol. 39, 1577–1586 (2011).
Vanoosten, R. L., Moore, J. M., Ludwig, A. T. & Griffith, T. S. Depsipeptide (FR901228) enhances the cytotoxic activity of TRAIL by redistributing TRAIL receptor to membrane lipid rafts. Mol. Ther. 11, 542–552 (2005).
VanOosten, R. L., Moore, J. M., Karacay, B. & Griffith, T. S. Histone deacetylase inhibitors modulate renal cell carcinoma sensitivity to TRAIL/Apo-2L-induced apoptosis by enhancing TRAIL-R2 expression. Cancer Biol. Ther. 4, 1104–1112 (2005).
Lu, M., Marsters, S., Ye, X., Luis, E., Gonzalez, L. & Ashkenazi, A. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol. Cell 54, 987–998 (2014).
Ratheesh, A. & Yap, A. S. A bigger picture: classical cadherins and the dynamic actin cytoskeleton. Nat. Rev. Mol. Cell Biol. 13, 673–679 (2012).
Padmanabhan, C., Rellinger, E. J., Zhu, J., An, H., Woodbury, L. G., Chung, D. H. et al. cFLIP critically modulates apoptotic resistance in epithelial-to-mesenchymal transition. Oncotarget 8, 101072–101086 (2017).
Kriegl, L., Jung, A., Horst, D., Rizzani, A., Jackstadt, R., Hermeking, H. et al. Microsatellite instability, KRAS mutations and cellular distribution of TRAIL-receptors in early stage colorectal cancer. PLoS ONE 7, e51654 (2012).
Gallegos, L. L. & Brugge, J. S. Live free or die: cell-cell adhesion regulates sensitivity to trail-induced apoptosis. Dev. Cell 30, 3–4 (2014).
Chiu, K.-Y., Wu, C.-C., Chia, C.-H., Hsu, S.-L. & Tzeng, Y.-M. Inhibition of growth, migration and invasion of human bladder cancer cells by antrocin, a sesquiterpene lactone isolated from Antrodia cinnamomea, and its molecular mechanisms. Cancer Lett. 373, 174–184 (2016).
Fritsche, H., Heilmann, T., Tower, R. J., Hauser, C., von Au, A., El-Sheikh, D. et al. TRAIL-R2 promotes skeletal metastasis in a breast cancer xenograft mouse model. Oncotarget 6, 9502–9516 (2015).
Jeon, Y.-J., Middleton, J., Kim, T., Laganà, A., Piovan, C., Secchiero, P. et al. A set of NF-κB–regulated microRNAs induces acquired TRAIL resistance in Lung cancer. Proc. Natl Acad. Sci. USA 112, E3355–E3364 (2015).
Fu, J., Rodova, M., Nanta, R., Meeker, D., Van Veldhuizen, P. J., Srivastava, R. K. et al. NPV-LDE-225 (Erismodegib) inhibits epithelial mesenchymal transition and self-renewal of glioblastoma initiating cells by regulating miR-21, miR-128, and miR-200. Neuro-Oncol. 15, 691–706 (2013).
Liu, C., Qi, M., Li, L., Yuan, Y., Wu, X. & Fu, J. Natural cordycepin induces apoptosis and suppresses metastasis in breast cancer cells by inhibiting the Hedgehog pathway. Food Funct. 11, 2107–2116 (2020).
Nguyen, P. T., Nguyen, D., Chea, C., Miyauchi, M., Fujii, M. & Takata, T. Interaction between N-cadherin and decoy receptor-2 regulates apoptosis in head and neck cancer. Oncotarget 9, 31516–31530 (2018).
Araki, K., Shimura, T., Suzuki, H., Tsutsumi, S., Wada, W., Yajima, T. et al. E/N-cadherin switch mediates cancer progression via TGF-β-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br. J. Cancer 105, 1885–1893 (2011).
Gravdal, K., Halvorsen, O. J., Haukaas, S. A. & Akslen, L. A. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin. Cancer Res. 13, 7003–7011 (2007).
Sheikh, M. & Fornace, A. Death and decoy receptors and p53-mediated apoptosis. Leukemia 14, 1509–1513 (2000).
Meng, R. D., McDonald, E. R., Sheikh, M. S., Fornace, A. J. & El-Deiry, W. S. The TRAIL decoy receptor TRUNDD (DcR2, TRAIL-R4) is induced by adenovirus-p53 overexpression and can delay TRAIL-, p53-, and KILLER/DR5-dependent colon cancer apoptosis. Mol. Ther. 1, 130–144 (2000).
Blair, S. & McNeill, H. Big roles for fat cadherins. Curr. Opin. Cell Biol. 51, 73–80 (2018).
Fulford, A. D. & McNeill, H. Fat/Dachsous family cadherins in cell and tissue organisation. Curr. Opin. Cell Biol. 62, 96–103 (2020).
Morris, L. G. T., Kaufman, A. M., Gong, Y., Ramaswami, D., Walsh, L. A., Turcan, Ş. et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 45, 253–261 (2013).
Kranz, D. & Boutros, M. A synthetic lethal screen identifies FAT1 as an antagonist of caspase-8 in extrinsic apoptosis. EMBO J. 33, 181–197 (2014).
Samara, R. N., Laguinge, L. M. & Jessup, J. M. Carcinoembryonic antigen inhibits anoikis in colorectal carcinoma cells by interfering with TRAIL-R2 (DR5) signaling. Cancer Res. 67, 4774–4782 (2007).
Loebinger, M. R., Sage, E. K., Davies, D. & Janes, S. M. TRAIL-expressing mesenchymal stem cells kill the putative cancer stem cell population. Br. J. Cancer 103, 1692–1697 (2010).
Harada, K. Sclerosing and obstructive cholangiopathy in biliary atresia: mechanisms and association with biliary innate immunity. Pediatr. Surg. Int. 33, 1243–1248 (2017).
Harada, K. & Nakanuma, Y. Biliary innate immunity: function and modulation. Mediators Inflamm. 2010, 373878 (2010).
Khatun, M., Mondal, R. K., Pal, S., Baidya, A., Bishnu, D., Banerjee, P. et al. Distinctiveness in virological features and pathogenic potentials of subgenotypes D1, D2, D3 and D5 of Hepatitis B virus. Sci. Rep. 8, 8055 (2018).
Spencer, S. L., Gaudet, S., Albeck, J. G., Burke, J. M. & Sorger, P. K. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459, 428–432 (2009).
Roux, J., Hafner, M., Bandara, S., Sims, J. J., Hudson, H., Chai, D. et al. Fractional killing arises from cell-to-cell variability in overcoming a caspase activity threshold. Mol. Syst. Biol. 11, 803 (2015).
Meyer, M., Paquet, A., Arguel, M.-J., Peyre, L., Gomes-Pereira, L. C., Lebrigand, K. et al. Profiling the non-genetic origins of cancer drug resistance with a single-cell functional genomics approach using predictive cell dynamics. Cell Syst. 11, 367–374.e5 (2020).
Flusberg, D. A., Roux, J., Spencer, S. L. & Sorger, P. K. Cells surviving fractional killing by TRAIL exhibit transient but sustainable resistance and inflammatory phenotypes. Mol. Biol. Cell 24, 2186–2200 (2013).
Tripathi, S., Chakraborty, P., Levine, H. & Jolly, M. K. A mechanism for epithelial-mesenchymal heterogeneity in a population of cancer cells. PLOS Comput. Biol. 16, e1007619 (2020).
Jolly, M. K., Tripathi, S. C., Jia, D., Mooney, S. M., Celiktas, M., Hanash, S. M. et al. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 7, 27067–27084 (2016).
Hari, K., Sabuwala, B., Subramani, B. V., La Porta, C. A. M., Zapperi, S., Font-Clos, F. et al. Identifying inhibitors of epithelial–mesenchymal plasticity using a network topology-based approach. Npj Syst. Biol. Appl. 6, 15 (2020).
Bonavida, B., Baritaki, S., Huerta-Yepez, S., Vega, M. I., Chatterjee, D. & Yeung, K. Novel therapeutic applications of nitric oxide donors in cancer: roles in chemo- and immunosensitization to apoptosis and inhibition of metastases. Nitric Oxide 19, 152–157 (2008).
Bonavida, B. & Garban, H. Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics. Redox Biol. 6, 486–494 (2015).
McConkey, D. J., Choi, W., Marquis, L., Martin, F., Williams, M. B., Shah, J. et al. Role of epithelial-to-mesenchymal transition (EMT) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev. 28, 335–344 (2009).
Srivastava, R. K., Kurzrock, R. & Shankar, S. MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits angiogenesis and metastasis, and reverses epithelial-mesenchymal transition in vivo. Mol. Cancer Ther. 9, 3254–3266 (2010).
Kruyt, F. A. E. & Schuringa, J. J. Apoptosis and cancer stem cells: Implications for apoptosis targeted therapy. Biochem. Pharmacol. 80, 423–430 (2010).
Fu, J., Rodova, M., Roy, S. K., Sharma, J., Singh, K. P., Srivastava, R. K. et al. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 330, 22–32 (2013).
Nowek, K., Wiemer, E. A. C. & Jongen-Lavrencic, M. The versatile nature of miR-9/9* in human cancer. Oncotarget 9, 20838–20854 (2018).
Gao, F., Zhao, Z.-L., Zhao, W.-T., Fan, Q.-R., Wang, S.-C., Li, J. et al. miR-9 modulates the expression of interferon-regulated genes and MHC class I molecules in human nasopharyngeal carcinoma cells. Biochem. Biophys. Res. Commun. 431, 610–616 (2013).
Oliveras-Ferraros, C., Vazquez-Martin, A., Cuyàs, E., Corominas-Faja, B., Rodríguez-Gallego, E., Fernández-Arroyo, S. et al. Acquired resistance to metformin in breast cancer cells triggers transcriptome reprogramming toward a degradome-related metastatic stem-like profile. Cell Cycle 13, 1132–1144 (2014).
Wang, H., Wu, Q., Zhang, Y., Zhang, H.-N., Wang, Y.-B. & Wang, W. TGF-β1-induced epithelial-mesenchymal transition in lung cancer cells involves upregulation of miR-9 and downregulation of its target, E-cadherin. Cell. Mol. Biol. Lett. 22, 22 (2017).
Konge, J., Leteurtre, F., Goislard, M., Biard, D., Morel-Altmeyer, S., Vaurijoux, A. et al. Breast cancer stem cell-like cells generated during TGFβ-induced EMT are radioresistant. Oncotarget 9, 23519–23531 (2018).
Wang, H., Xu, C., Kong, X., Li, X., Kong, X., Wang, Y. et al. Trail resistance induces epithelial-mesenchymal transition and enhances invasiveness by suppressing PTEN via miR-221 in breast cancer. PLoS ONE 9, e99067 (2014).
Rellinger, E. J., Padmanabhan, C., Qiao, J., Appert, A., Waterson, A. G., Lindsley, C. W. et al. ML327 induces apoptosis and sensitizes Ewing sarcoma cells to TNF-related apoptosis-inducing ligand. Biochem. Biophys. Res. Commun. 491, 463–468 (2017).
Quast, S.-A., Berger, A., Plötz, M. & Eberle, J. Sensitization of melanoma cells for TRAIL-induced apoptosis by activation of mitochondrial pathways via Bax. Eur. J. Cell Biol. 93, 42–48 (2014).
David, J. M., Hamilton, D. H. & Palena, C. MUC1 upregulation promotes immune resistance in tumor cells undergoing brachyury-mediated epithelial-mesenchymal transition. Oncoimmunology 5, e1117738 (2016).
Du, B. & Shim, J. S. Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules 21, 965 (2016).
Frisch, S. M. & Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 124, 619–626 (1994).
Ishikawa, F., Ushida, K., Mori, K. & Shibanuma, M. Loss of anchorage primarily induces non-apoptotic cell death in a human mammary epithelial cell line under atypical focal adhesion kinase signaling. Cell Death Dis. 6, e1619 (2015).
Laguinge, L. M., Samara, R. N., Wang, W., El-Deiry, W. S., Corner, G., Augenlicht, L. et al. DR5 receptor mediates anoikis in human colorectal carcinoma cell lines. Cancer Res. 68, 909–917 (2008).
Marconi, A., Atzei, P., Panza, C., Fila, C., Tiberio, R., Truzzi, F. et al. FLICE/caspase-8 activation triggers anoikis induced by beta1-integrin blockade in human keratinocytes. J. Cell Sci. 117, 5815–5823 (2004).
Frisch, S. M. Evidence for a function of death-receptor-related, death-domain-containing proteins in anoikis. Curr. Biol. 9, 1047–1049 (1999).
Rytömaa, M., Martins, L. M. & Downward, J. Involvement of FADD and caspase-8 signalling in detachment-induced apoptosis. Curr. Biol. 9, 1043–1046 (1999).
Escate, R., Padro, T. & Badimon, L. LDL accelerates monocyte to macrophage differentiation: effects on adhesion and anoikis. Atherosclerosis 246, 177–186 (2016).
Mawji, I. A., Simpson, C. D., Hurren, R., Gronda, M., Williams, M. A., Filmus, J. et al. Critical role for Fas-associated death domain-like interleukin-1-converting enzyme-like inhibitory protein in anoikis resistance and distant tumor formation. J. Natl Cancer Inst. 99, 811–822 (2007).
Goldberg, G. S., Jin, Z., Ichikawa, H., Naito, A., Ohki, M., El-Deiry, W. S. et al. Global effects of anchorage on gene expression during mammary carcinoma cell growth reveal role of tumor necrosis factor-related apoptosis-inducing ligand in anoikis. Cancer Res. 61, 1334–1337 (2001).
Bachelder, R. E., Wendt, M. A., Fujita, N., Tsuruo, T. & Mercurio, A. M. The cleavage of Akt/protein kinase B by death receptor signaling is an important event in detachment-induced apoptosis. J. Biol. Chem. 276, 34702–34707 (2001).
Cao, L., Han, L., Zhang, Z., Li, J., Qu, Z., Du, J. et al. Involvement of anoikis-resistance in the metastasis of hepatoma cells. Exp. Cell Res. 315, 1148–1156 (2009).
Kočí, L., Hýžd’alová, M., Vaculová, A., Hofmanová, J. & Kozubík, A. Detachment-mediated resistance to TRAIL-induced apoptosis is associated with stimulation of the PI3K/Akt pathway in fetal and adenocarcinoma epithelial colon cells. Cytokine 55, 34–39 (2011).
Lane, D., Goncharenko-Khaider, N., Rancourt, C. & Piché, A. Ovarian cancer ascites protects from TRAIL-induced cell death through alphavbeta5 integrin-mediated focal adhesion kinase and Akt activation. Oncogene 29, 3519–3531 (2010).
Tamagiku, Y., Sonoda, Y., Kunisawa, M., Ichikawa, D., Murakami, Y., Aizu-Yokota, E. et al. Down-regulation of procaspase-8 expression by focal adhesion kinase protects HL-60 cells from TRAIL-induced apoptosis. Biochem. Biophys. Res. Commun. 323, 445–452 (2004).
Barbero, S., Mielgo, A., Torres, V., Teitz, T., Shields, D. J., Mikolon, D. et al. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res. 69, 3755–3763 (2009).
Hess, F., Estrugo, D., Fischer, A., Belka, C. & Cordes, N. Integrin-linked kinase interacts with caspase-9 and -8 in an adhesion-dependent manner for promoting radiation-induced apoptosis in human leukemia cells. Oncogene 26, 1372–1384 (2007).
Capper, D., Gaiser, T., Hartmann, C., Habel, A., Mueller, W., Herold-Mende, C. et al. Stem-cell-like glioma cells are resistant to TRAIL/Apo2L and exhibit down-regulation of caspase-8 by promoter methylation. Acta Neuropathol. 117, 445–456 (2009).
Tazzari, P. L., Tabellini, G., Ricci, F., Papa, V., Bortul, R., Chiarini, F. et al. Synergistic proapoptotic activity of recombinant TRAIL plus the Akt inhibitor Perifosine in acute myelogenous leukemia cells. Cancer Res. 68, 9394–9403 (2008).
Peng, H., Huang, Y., Duan, Z., Erdmann, N., Xu, D., Herek, S. et al. Cellular IAP1 regulates TRAIL-induced apoptosis in human fetal cortical neural progenitor cells. J. Neurosci. Res. 82, 295–305 (2005).
Boffa, D. J., Graf, R. P., Salazar, M. C., Hoag, J., Lu, D., Krupa, R. et al. Cellular expression of PD-L1 in the peripheral blood of lung cancer patients is associated with worse survival. Cancer Epidemiol. Prev. Biomark. 26, 1139–1145 (2017).
Gibney, G. T., Weiner, L. M. & Atkins, M. B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 17, e542–e551 (2016).
Akinleye, A. & Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 12, 92 (2019).
Tumeh, P. C., Harview, C. L., Yearley, J. H., Shintaku, I. P., Taylor, E. J. M., Robert, L. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014).
Tu, Z., Pierce, R. H., Kurtis, J., Kuroki, Y., Crispe, I. N. & Orloff, M. S. Hepatitis C virus core protein subverts the antiviral activities of human Kupffer cells. Gastroenterology 138, 305–314 (2010).
Banerjee, P., Zhang, R., Ivan, C., Galletti, G., Clise-Dwyer, K., Barbaglio, F. et al. Trabectedin reveals a strategy of immunomodulation in chronic lymphocytic leukemia. Cancer Immunol. Res. 7, 2036–2051 (2019).
Bahrambeigi, V., Ahmadi, N., Moisyadi, S., Urschitz, J., Salehi, R. & Haghjooy Javanmard, S. PhiC31/PiggyBac modified stromal stem cells: effect of interferon γ and/or tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) on murine melanoma. Mol. Cancer 13, 255 (2014).
Hwang, S., Han, J., Baek, J.-S., Tak, E., Song, G.-W., Lee, S.-G. et al. Cytotoxicity of human hepatic intrasinusoidal CD56bright natural killer cells against hepatocellular carcinoma cells. Int. J. Mol. Sci. 20, 1564 (2019).
Langaas, V., Shahzidi, S., Johnsen, J. I., Smedsrød, B. & Sveinbjørnsson, B. Interferon-gamma modulates TRAIL-mediated apoptosis in human colon carcinoma cells. Anticancer Res. 21, 3733–3738 (2001).
Ruiz-Ruiz, C. & López-Rivas, A. Mitochondria-dependent and -independent mechanisms in tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis are both regulated by interferon-gamma in human breast tumour cells. Biochem. J. 365, 825–832 (2002).
Bremer, E. Targeting of the tumor necrosis factor receptor superfamily for cancer immunotherapy. ISRN Oncol. 2013, 371854 (2013).
Hendriks, D., He, Y., Koopmans, I., Wiersma, V. R., van Ginkel, R. J., Samplonius, D. F. et al. Programmed death ligand 1 (PD-L1)-targeted TRAIL combines PD-L1-mediated checkpoint inhibition with TRAIL-mediated apoptosis induction. Oncoimmunology 5, e1202390 (2016).
Kudo-Saito, C., Shirako, H., Ohike, M., Tsukamoto, N. & Kawakami, Y. CCL2 is critical for immunosuppression to promote cancer metastasis. Clin. Exp. Metastasis 30, 393–405 (2013).
Kudo-Saito, C., Shirako, H., Takeuchi, T. & Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 15, 195–206 (2009).
Chen, L., Gibbons, D. L., Goswami, S., Cortez, M. A., Ahn, Y.-H., Byers, L. A. et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 5, 5241 (2014).
Noman, M. Z., Janji, B., Abdou, A., Hasmim, M., Terry, S., Tan, T. Z. et al. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 6, e1263412 (2017).
Spranger, S., Bao, R. & Gajewski, T. F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015).
Tuo, Z., Zong, Y., Li, J., Xiao, G., Zhang, F., Li, G. et al. PD-L1 regulation by SDH5 via β-catenin/ZEB1 signaling. Oncoimmunology 8, 1655361 (2019).
Evanno, E., Godet, J., Piccirilli, N., Guilhot, J., Milin, S., Gombert, J. M. et al. Tri-methylation of H3K79 is decreased in TGF-β1-induced epithelial-to-mesenchymal transition in lung cancer. Clin. Epigenetics 9, 80 (2017).
Asgarova, A., Asgarov, K., Godet, Y., Peixoto, P., Nadaradjane, A., Boyer-Guittaut, M. et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology 7, e1423170 (2018).
Ivanov, V. N., Wu, J., Wang, T. J. C. & Hei, T. K. Inhibition of ATM kinase upregulates levels of cell death induced by cannabidiol and γ-irradiation in human glioblastoma cells. Oncotarget 10, 825–846 (2019).
Benci, J. L., Xu, B., Qiu, Y., Wu, T. J., Dada, H., Twyman-Saint Victor, C. et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167, 1540–1554.e12 (2016).
Benci, J. L., Johnson, L. R., Choa, R., Xu, Y., Qiu, J., Zhou, Z. et al. Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell 178, 933–948.e14 (2019).
Guo, C. M., Liu, S. Q. & Sun, M. Z. miR-429 as biomarker for diagnosis, treatment and prognosis of cancers and its potential action mechanisms: a systematic literature review. Neoplasma 67, 215–228 (2020).
Lv, J., Guo, T., Qu, X., Che, X., Li, C., Wang, S. et al. PD-L1 under regulation of miR-429 influences the sensitivity of gastric cancer cells to TRAIL by binding of EGFR. Front. Oncol. 10, 1067 (2020).
von Karstedt, S. & Walczak, H. An unexpected turn of fortune: targeting TRAIL-Rs in KRAS-driven cancer. Cell Death Discov. 6, 14 (2020).
Aceto, N., Bardia, A., Miyamoto, D. T., Donaldson, M. C., Wittner, B. S., Spencer, J. A. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122 (2014).
Gkountela, S., Castro-Giner, F., Szczerba, B. M., Vetter, M., Landin, J., Scherrer, R. et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 176, 98–112.e14 (2019).
Mocellin, S., Keilholz, U., Rossi, C. R. & Nitti, D. Circulating tumor cells: the ‘leukemic phase’ of solid cancers. Trends Mol. Med. 12, 130–139 (2006).
Hou, J.-M., Krebs, M. G., Lancashire, L., Sloane, R., Backen, A., Swain, R. K. et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J. Clin. Oncol. 30, 525–532 (2012).
Liu, Y., Ling, Y., Qi, Q., Lan, F., Zhu, M., Zhang, Y. et al. Prognostic value of circulating tumor cells in advanced gastric cancer patients receiving chemotherapy. Mol. Clin. Oncol. 6, 235–242 (2017).
Marín-Aguilera, M., Reig, Ò., Lozano, J. J., Jiménez, N., García-Recio, S., Erill, N. et al. Molecular profiling of peripheral blood is associated with circulating tumor cells content and poor survival in metastatic castration-resistant prostate cancer. Oncotarget 6, 10604–10616 (2015).
Martini, V., Timme-Bronsert, S., Fichtner-Feigl, S., Hoeppner, J. & Kulemann, B. Circulating tumor cells in pancreatic cancer: current perspectives. Cancers 11, 1659 (2019).
Micalizzi, D. S., Maheswaran, S. & Haber, D. A. A conduit to metastasis: circulating tumor cell biology. Genes Dev. 31, 1827–1840 (2017).
Weiss, L., Mayhew, E., Rapp, D. G. & Holmes, J. C. Metastatic inefficiency in mice bearing B16 melanomas. Br. J. Cancer 45, 44–53 (1982).
Braunholz, D., Saki, M., Niehr, F., Öztürk, M., Borràs Puértolas, B., Konschak, R. et al. Spheroid culture of head and neck cancer cells reveals an important role of EGFR signalling in anchorage independent survival. PLoS ONE 11, e0163149 (2016).
Pantel, K. & Speicher, M. R. The biology of circulating tumor cells. Oncogene 35, 1216–1224 (2016).
Yao, X., Choudhury, A. D., Yamanaka, Y. J., Adalsteinsson, V. A., Gierahn, T. M., Williamson, C. A. et al. Functional analysis of single cells identifies a rare subset of circulating tumor cells with malignant traits. Integr. Biol. 6, 388–398 (2014).
Howard, E. W., Leung, S. C. L., Yuen, H. F., Chua, C. W., Lee, D. T., Chan, K. W. et al. Decreased adhesiveness, resistance to anoikis and suppression of GRP94 are integral to the survival of circulating tumor cells in prostate cancer. Clin. Exp. Metastasis 25, 497–508 (2008).
Yu, M., Ting, D. T., Stott, S. L., Wittner, B. S., Ozsolak, F., Paul, S. et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature 487, 510–513 (2012).
Mego, M., Cholujova, D., Minarik, G., Sedlackova, T., Gronesova, P., Karaba, M. et al. CXCR4-SDF-1 interaction potentially mediates trafficking of circulating tumor cells in primary breast cancer. BMC Cancer 16, 127 (2016).
Lederman, E. E., Hope, J. M. & King, M. R. Mass action kinetic model of apoptosis by TRAIL-functionalized leukocytes. Front. Oncol. 8, 410 (2018).
Recio-Boiles, A., Ilmer, M., Rhea, P. R., Kettlun, C., Heinemann, M. L., Ruetering, J. et al. JNK pathway inhibition selectively primes pancreatic cancer stem cells to TRAIL-induced apoptosis without affecting the physiology of normal tissue resident stem cells. Oncotarget 7, 9890–9906 (2016).
Twomey, J. D. & Zhang, B. Circulating tumor cells develop resistance to TRAIL-induced apoptosis through autophagic removal of death receptor 5: evidence from an in vitro model. Cancers 11, 94 (2019).
Avivar-Valderas, A., Bobrovnikova-Marjon, E., Alan Diehl, J., Bardeesy, N., Debnath, J. & Aguirre-Ghiso, J. A. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 32, 4932–4940 (2013).
Fung, C., Lock, R., Gao, S., Salas, E. & Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 19, 797–806 (2008).
Di, X., Zhang, G., Zhang, Y., Takeda, K., Rivera Rosado, L. A. & Zhang, B. Accumulation of autophagosomes in breast cancer cells induces TRAIL resistance through downregulation of surface expression of death receptors 4 and 5. Oncotarget 4, 1349–1364 (2013).
Mani, S. A., Guo, W., Liao, M.-J., Eaton, E. N., Ayyanan, A., Zhou, A. Y. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008).
Boumahdi, S. & de Sauvage, F. J. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 19, 39–56 (2020).
De Francesco, E. M., Maggiolini, M. & Musti, A. M. Crosstalk between Notch, HIF-1α and GPER in breast cancer EMT. Int. J. Mol. Sci. 19, 2011 (2018).
Higgins, D. F., Kimura, K., Bernhardt, W. M., Shrimanker, N., Akai, Y., Hohenstein, B. et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 117, 3810–3820 (2007).
Fiori, M. E., Di Franco, S., Villanova, L., Bianca, P., Stassi, G. & De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 18, 70 (2019).
Karlsson, M. C., Gonzalez, S. F., Welin, J. & Fuxe, J. Epithelial-mesenchymal transition in cancer metastasis through the lymphatic system. Mol. Oncol. 11, 781–791 (2017).
Suarez-Carmona, M., Lesage, J., Cataldo, D. & Gilles, C. EMT and inflammation: inseparable actors of cancer progression. Mol. Oncol. 11, 805–823 (2017).
Williams, E. D., Gao, D., Redfern, A. & Thompson, E. W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 19, 716–732 (2019).
Author information
Authors and Affiliations
Contributions
Writing—original draft: L.P., M.M. and J.R.; writing—review and editing: L.P. and J.R.; resources: P.H. and J.R.; funding acquisition: J.R.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Data availability
Not applicable.
Competing interests
The authors declare no competing interests.
Funding information
This work was funded by a Marie Curie International Incoming Fellowship within the 7th European Community Framework Programme (proposal SysBioDRez, no. 626190, call reference: FP7- PEOPLE-2013-IIF), an INCa Plan Cancer Biologie des Systèmes, ITMO Cancer (proposal IMoDRez, N°18CB001-00), a young investigator award (Emergence Jeunes Chercheurs) from Canceropôle Provence-Alpes-Côte d’Azur, the French National Cancer Institute (INCa) and the Provence-Alpes-Côte d’Azur Region, to J.R.
Additional information
Note This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peyre, L., Meyer, M., Hofman, P. et al. TRAIL receptor-induced features of epithelial-to-mesenchymal transition increase tumour phenotypic heterogeneity: potential cell survival mechanisms. Br J Cancer 124, 91–101 (2021). https://doi.org/10.1038/s41416-020-01177-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-020-01177-w
This article is cited by
-
CDH1 overexpression sensitizes TRAIL resistant breast cancer cells towards rhTRAIL induced apoptosis
Molecular Biology Reports (2023)
-
The Prognosis of Cancer Depends on the Interplay of Autophagy, Apoptosis, and Anoikis within the Tumor Microenvironment
Cell Biochemistry and Biophysics (2023)
-
Metastasis: complexity thwarts precision targeting
British Journal of Cancer (2021)
