
Abstract
Background
Pheochromocytoma (PCC) and paraganglioma (PGL) are uncommon neoplasms with high morbidity in advanced stages. Effective systemic treatments are limited.
Methods
A multisite phase 2 trial evaluated sunitinib in patients with progressive PCC/PGL. Patients received 50 mg orally for 4–6 weeks.
Results
Between May 2009 and May 2016, 25 patients were enroled. The median age was 50 years and 56% were male. Three patients (12%) received prior chemotherapy and 16 (64%) prior surgery. The DCR was 83% (95% CI: 61–95%) and median PFS 13.4 (95% CI: 5.3–24.6) months. Of 23 evaluable patients, 3 (13%) with germline mutations (SDHA, SDHB, RET) achieved a PR. The patient with mutated RET and MEN2A remains on treatment after 64 cycles. The median time on treatment was 12.4 (1–88.0) months. Grade 3 or 4 toxicities were as expected and manageable; fatigue (16%) and thrombocytopenia (16%) were most common. One patient with grade 3 hypertension and 2 with grade 3 cardiac events discontinued treatment.
Conclusion
Although the primary endpoint of disease control was met, the overall response rate of sunitinib was low in unselected patients with progressive PCC/PGL. Patients with germline variants in RET or in the subunits of SDH may derive greatest benefit.
Similar content being viewed by others

Background
Pheochromocytomas (PCCs) and extra-adrenal paragangliomas (PGL) are uncommon neuroendocrine neoplasms arising from the adrenal medulla and sympathetic/parasympathetic paraganglia respectively. Local treatments including surgical resection, radiation and radiolabelled agents are important therapeutic options when possible. Up to 20% of PCC/PGL can present with bulky inoperable disease, progressive symptoms or the presence of metastases, indicating high morbidity and a 5-year survival of less than 50%.1
In over 30% of cases of PCC/PGL, a predisposing germline mutation can be identified, the most common are inactivating mutations in subunits of the succinate dehydrogenenase (SDH) complex predominantly SDHB and SDHD.2,3 Other hereditary susceptibility genes include RET and von Hippel-Lindau (VHL) as part of the syndromes of multiple endocrine neoplasia 2 (MEN2) and VHL syndrome respectively; the tumour suppressor gene neurofibromatosis 1 (NF1),4 MYC associated factor X (MAX) and transmembrane protein 127 (TMEM127) and a number of additional genes.3 Recent molecular characterisation and mRNA expression profile clustering have identified germline VHL and SDH subunit mutations specific to a pseudohypoxia subgroup of PCC/PGL, or cluster 1 as previously published.5 Both genes are important in the regulation of hypoxia inducible factor (HIF) and angiogenesis.6,7 Subsets of sporadic PCC/PGL are also thought to exhibit high expression of angiogenic factors.8 Accordingly, case reports and small series have suggested a role for anti-angiogenic agents in treating this group of endocrine tumours.9,10,11
Systemic options for advanced PCC/PGL are limited. No prospective trials have been published to guide treatment decisions. Variations of the cytotoxic regimen of cyclophosphamide, vincristine and dacarbazine (CVD), and temozolomide regimens have been reported in small retrospective series and case studies, with variable response rates and limited quality data.12,13,14,15,16,17 In addition chemotherapy-related toxicities are common, including myelosuppression, neuropathy and gastrointestinal toxicity.12 Better treatment options are urgently needed. Sunitinib is an oral tyrosine kinase inhibitor targeting vascular endothelial factor receptor 1 and 2, PDGF-B receptor, RET and other tyrosine kinases including FGFR that is overexpressed in PCC/PGL.18 We conducted a multicentre open label single arm phase 2 study of sunitinib in patients with locally advanced unresectable or metastatic PCC/PGL who had demonstrated progression or who had disease-related symptoms.
Methods
Patients
Patients included in the study were ≥18 years of age, Eastern Cooperative Oncology Group (ECOG) performance status 0–2, with a histologically or cytologically confirmed diagnosis of PCC/PGL that was considered non-resectable or metastatic. To be enroled patients needed measurable disease and demonstrated radiological progression by RECIST1.1 criteria and/or biochemical progression. The protocol was later amended to allow newly presenting patients with tumour-related symptoms without radiological progression. Radiological progression was defined by the presence of new lesions or a 20% increase in the sum of the longest diameter of target lesions, comparing two computed-tomography (CT) scans performed within 13 months of each other. This time frame was included given that this patient population is scanned infrequently. These pre-treatment measurements were recorded to ascertain rate of growth over time, prior to sunitinib and comparing to on-treatment measurements. Biochemical progression was defined as a change ≥50% in urinary catecholamines and/or measured urinary metanephrines over 5–7 months. Adequate organ function was required, and patients were excluded for prior anti-angiogenic treatment, uncontrolled hypothyroidism, HIV infection, venous thromboembolism within 3 months or any major vascular event. Notably hypertension must have been controlled prior to enrolment (systolic ≤ 150 mmHg and/or diastolic ≤ 90 mmHg). CYP3A4 inhibitors and inducers were prohibited 7 and 12 days respectively prior to study commencement and throughout the trial. Patients with QTc prolongation (≥500 ms) or those receiving pro-arrhythmics were also excluded. Germline testing was not a study requirement however in those patients who did undergo germline evaluation, testing was performed as per institutional guidelines.
Study design
Procedures
In this single-arm multi-institutional phase 2 study, all patients received starting dose of sunitinib 50 mg orally, daily for 4 weeks, followed by 2 weeks observation; each cycle was 6 weeks in duration and was maintained even when doses were missed during the 4-week treatment period. Within 7 days of enrolment and prior to each cycle, all patients were required to have had a physical examination, blood chemistry including TSH and T4, and urinary analysis for protein excretion. Every second cycle, analysis of 24-h urinary collection for metanephrines and catecholamines, serum chromogranin A, together with a CT chest abdomen and pelvis were performed.
At week 2 and week 4 of treatment, patients had blood pressure and heart rate checks. Guidelines for the management of hypertension including the appropriate antihypertensives to be prescribed and required dose reductions were available within the protocol. In general blood pressure was to be maintained at less than 150/90 mmHg. Dose modifications were made for other grade 3 or 4 adverse events. Patients discontinued treatment for unacceptable toxicity, disease progression, investigator discretion or consent withdrawal.
Outcomes and statistical analyses
The primary endpoint of this study was disease control rate (DCR) defined as a partial response (PR), complete response (CR), or stable disease maintained for ≥12 weeks from the initiation of treatment. Tumour response to sunitinib was assessed as per RECIST 1.1 using CT-imaging. Clinically apparent lesions needed to be >10 mm by calliper measurement to be included as targets. Secondary endpoints included biochemical response as defined by >20% reduction in one of the following; 24-h urinary catecholamines (norepinephrine, epinephrine, dopamine) or total metanephrines, which was sustained for ≥12 weeks; progression free survival (PFS), overall survival (OS), overall response rate (ORR) and symptom control. Adverse events were assessed according to the revised NCI Common Terminology Criteria for Adverse Events (CTCAE), version 3.0 during treatment and through 28 days post discontinuation of sunitinib.
Data from all participating study sites were combined for the analyses. Summary data are reported for baseline demographics, efficacy and safety data. Categorical data are presented as frequencies and percentages and continuous data as the median and range unless stated otherwise. A multi-stage design as described by Fleming19 was employed, assuming an unacceptable response rate (defined by DCR) of 5% and a promising rate of 20% with type I and type II errors of 10% each. 14 response evaluable patients were entered in the first stage to accrue to the second stage if ≥1 response as defined by DCR was seen. The primary endpoint was met if ≥4 responses (as defined by the DCR) were found after a total enrolment of 28 patients. The percentage change in volume over time was plotted using a spider plot to assess antitumor activity with pre-treatment measurements also included. The Kaplan Meier method was used to assess PFS and OS. PFS was defined as the time from registration to progression or death from any cause and OS, the time from registration to death from any cause. Data lock occurred on the 31 January 2018.
Study oversight
The study was approved by the appropriate ethics committee or institutional review board at each study site. The study was conducted in accordance with Guidelines for Good Clinical Practice. All patients provided written informed prior to enrolment.
Results
Patient characteristics
In total, 25 of the planned 28 patients were enroled between May 2009 and May 2016 from three centres within Canada and one in the Netherlands. The study was closed on the 18th January 2017 due to slow accrual, demographics and clinical characteristics are shown in Table 1. Fourteen patients had PCC (56%) and 11 PGL (44%). Twenty-three (92%) had metastatic disease and 2 patients had locally advanced disease. The majority (n = 20, 80%) of patients were enroled based on radiologic and/or biochemical progression. Five patients (20%) presenting with a recent diagnosis of advanced PCC/PGL were enroled based on rapid symptomatic progression. One patient (SNP-010) had evidence of progressive metastatic disease to bone only together with biochemical progression and although did not have measurable disease by RECIST at baseline, was allowed to enrol given burden of pain related symptoms.
Genetic testing results were available in 60% (15/25) of patients. Of these 15 patients, 9 (60%) had germline mutations identified, including 5 SDHB mutations, 1 SDHA mutation, 1 SDHC mutation, 1 RET mutation in a patient with MEN2A and 1 MAX mutation. The median time from first diagnosis to enrolment was 33.6 months (3– >132). Surgical resection of primary or metastatic sites had been previously performed in 16 (64%) patients. Three patients received prior chemotherapy for advanced disease, 1 patient received cisplatin/vinorelbine, 1 patient CVD and 1 patient had received two prior regimens including carboplatin/etoposide, and temozolomide/capecitabine.
Treatment outcomes
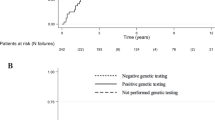
The median duration on treatment was 12.4 (1–88.0) months at data cut-off (Table 2). The median dose intensity was 40 mg (25 mg−50 mg) with 14 patients (56%) requiring dose reductions. Twenty-three patients were evaluable for response. Three patients (13%) achieved a partial response and 16 (70%) had stable disease ≥12 weeks as best response. The objective response rate (RECIST) was 13% and the DCR was therefore 83% (95% CI: 56–93%), meeting our primary endpoint. At 24 weeks 61% had evidence of disease control. The median PFS was 13.4 (95% CI: 5.3–24.6) months; 21 of 25 patients had a PFS event (1 death and 20 patients had disease progression, four patients are censored including two who remain on study and two who were lost to follow-up, Fig. 1). The median overall survival has not yet been reached; 9 (36%) patients have died (Fig. 2).

Kaplan Meier plot of progression-free survival through to 31 January 2018

Kaplan Meier plot of overall survival through to 31 January 2018
In the 3 patients achieving a PR, all three were found to have germline mutations. A patient with PCC who had a RET mutation (SNP-001) had a total reduction in tumour volume of 64% as best response and remains on treatment after 64 cycles (7 years) (Fig. 3). This patient also had significant biochemical responses (Fig. 4 and Supplementary Fig. 1–3). The second patient (SNP-004) with PCC who had a germline SDHB mutation had a 47% reduction in tumour volume as best response after 23 cycles but stopped treatment due to progression in non-target lesions together with evidence of clinical progression. Both of these patients reported improvements in pain with SNP-001 reporting a complete resolution in pain related to disease burden. The third patient (SNP-107, PGL) with a germline SDHA mutation who after 7 cycles had a 56% reduction in target tumour volume, subsequently developed new lesions after 10 cycles of sunitinib.

Spider plot showing responses to treatment over time and pre-trial tumour growth

Waterfall plot showing % change in 24-h urinary metanephrine results. Those with no baseline values or follow-up results are indicated. The median (range) cycle to nadir for patients with abnormal baseline results with at least 1 follow-up result (n = 12) 3 (2, 45)
Sixteen patients achieved stable disease ≥ 12 weeks as best response by RECIST 1.1 criteria. Of these patients 6 had reductions of tumour volume greater than 10% (range: 12–27%). Three of these patients had germline mutations in SDH. In the 6 remaining patients with identified germline mutations in predisposition genes, 4 patients (3 SDHB and 1 SDHC) had prolonged stability by RECIST 1.1 lasting beyond 16 cycles with one patient remaining on treatment after 33 cycles (SNP-010). This patient who did not have measurable disease at baseline reported significant reductions in pain. One additional patient with an SDHB mutation had a short period of disease stability, stopping treatment for progression after four cycles. One further patient with a MAX mutation progressed on treatment. Disease stability was seen in ten further patients.
Biochemical and symptom changes
Timed urinary catecholamine collection over 24 h included measurements of norepinephrine, epinephrine, dopamine and total metanephrines. Three patients had normal results and 6 had no follow-up results recorded; of 16 evaluable. 11 had evidence of a biochemical response. Fractionated urinary metanephrines were captured in some patients but were not measured routinely at opening of the trial. Reference ranges varied between institutions. Elevated levels of total 24-h urinary metanephrines were evident in 15 of 21 patients with available baseline values (71%). Of these 15, 12 had subsequent values recorded, of which seven (58%) (3 PGL, 4 PCC) had a greater than 20% decline in values more lasting more than 12 weeks (Fig. 4). One of these 7 patient (SNP-001) also had a RECIST response. The two other patients with partial responses had normal baseline metanephrines. The three patients without subsequent values had received only 1–2 cycles of treatment with evidence of clinical or radiologic progression.
Baseline levels of norepinephrine were available in 20 patients (80%); of these 16 patients had elevated levels. Twelve of 16 patients (75%) had follow-up results, of these 7 patients had a greater than 20% biochemical reduction. The median cycle to best response was achieved at cycle 3 (range: 3–30, Supplementary Fig. 1). Epinephrine baseline results were available in 19 (76%) patients of which 6 had abnormal values. One patient had no follow-up results and 4 of 5 patients had evidence of a biochemical response, the median cycle to response was 4 (Supplementary Fig. 2). Dopamine urinary results were abnormal in 8 of 20 patients with available baseline results. Three of 5 patients with follow-up measurements had a biochemical response (Supplementary Fig. 3). Follow-up values were predominantly not available because of study cessation or progressive clinical decline.
In the five patients enroled based on symptoms only, the median number of cycles received was 4 (2–10). Three of these patients had an ECOG of 2 at diagnosis without improvement on study; two reported reductions in pain scores.
Toxicity
Reported AEs are shown in Table 3. The AEs were as expected for sunitinib and manageable. The most common grade 1–2 AEs were fatigue (68%), nausea and/or vomiting (68%) and palmar-plantar dyserthrodysesthesia (64%). Grade 3 events occurred in 14 (56%) patients and grade 4 in 3 patients (12%). Five (20%) patients discontinued sunitinib because of adverse events, including 1 patient with grade 2 nausea/vomiting, 1 patient with grade 2 elevation in creatinine and 1 patient with grade 3 hypertension. Two cardiac events occurred leading to discontinuation; 1 patient developed a restrictive cardiomyopathy related to study drug and one patient also had a myocardial infarction following episodic hypertensive episodes. There were no grade 5 events.
Discussion
Pheochromocytoma (PCC) and paraganglionoma (PGL) are rare neuroendocrine neoplasms with limited systemic options in the setting of progressive inoperable or metastatic disease. Comprehensive molecular profiling together with increased germline detection has allowed for a more complete understanding of this heterogeneous group of tumours, providing potential biomarker-derived subgroups for drug targets.5
This phase 2 trial met its primary endpoint; however objective response rates has of sunitinib in an unselected group of patients with progressive and/or symptomatic disease were low. Durable responses, however were seen in one patient with a RET mutation and two with SDH mutations, suggesting potential biomarkers of response to this agent. This study represents the largest prospective analysis of a multi tyrosine kinase inhibitor in advanced PCC/PGL. The disease control rate was 83% and median PFS 13.4 months at data-cut-off. In addition, biochemical responses were evident in 11/16 (69%) evaluable patients.
As demonstrated in the spider plot, there is a change in the growth kinetics of a number of these cases, suggesting a durable disease control benefit from sunitinib rather than selection of patients with indolent disease. SNP-004 and SNP-104 had evidence of 22 and 16% growth in tumour volume approximately 12 weeks prior to study entry and subsequently had best tumour responses of −47 and −26% respectively. SNP-001 had growth of 6% in the prior 12 weeks and has had tumour shrinkage > 55%, remaining on treatment. The subset (n = 5) enroled for tumour-related symptoms appeared to have the shortest exposure to sunitinib with fairly early discontinuation due to functional decline and progressive disease. Patients were not required to undergo genetic testing as part of this study however of the three patients with a partial response, germline mutations in SDHB, SDHA and RET were evident. In the patient with MEN2A syndrome, the response is ongoing for more than 7 years. Prolonged stability was also present in four other patients with germline SDH mutations. An additional 10 patients in whom genetic testing results were not available or negative had stable disease as best response.
The results of our prospective trial are corroborated in other series of multi-targeted tyrosine kinase inhibitors.11,20 In the combined retrospective analysis from MD Anderson and Institute Gustavvy-Roussy, 3 patients (18%) of 17 reported developed a partial response and an additional 5 patients had disease stability resulting in a DCR of 47%. Of the 8 patients achieving disease control, 6 had germline inactivating mutations in SDHB or VHL genes.11 In this study the PFS was 4.1 months, attributed to early cessation due to side effects. Evaluation of sunitinib in our clinical trial, where blood pressure was rigorously controlled has allowed for a more accurate determination of efficacy. Although grade 3 adverse events were present in over 50%, the sample size in this study is small and similar percentages have been reported.21
The SDH genes which include SDHA, SDHB, SDHC, SDHD encode the subunits of the succinate dehydrogenase complex (SDH) together with the assembly factor SDHAF2. Recent molecular characterisation of PCC/PGL identifies four major subgroups by unsupervised clustering of mRNA expression profiles. Germline SDH and VHL mutations are represented in the pseudohypoxia group or cluster 1 supporting prior studies.5,22,23,24 Disruption of the SDH or mitochondrial II complex results in the accumulation of succinate which in turn inhibits HIF-alpha prolyl hydroxylases activating HIF-1 alpha which in turn autoregulates the cycle suppressing SDHB levels.6,24 Stabilisation and activation of the transcription factor HIF-1 alpha regulates the hypoxic response and as a result upregulates multiple factors involved in angiogenesis, including VEGF and PDGF.25 Similarly in the presence of VHL mutations, HIF-1 alpha and HIF 2- alpha become constitutively active given the failure of proteasomal degradation and polyubiquitination.26 The response seen in our study in the pseudohypoxia group which is frequently enriched in angiogenic pathway activation, is akin to that seen in renal cell cancer where sunitinib has been a standard of care for many years.
Relevant to this trial, mutations in RET are categorised within a kinase signalling subtype also known as cluster 2.22,23,24 Notably the best responder in this study was a patient with a mutation in RET and MEN2A. Sunitinib is tyrosine kinase inhibitor that also targets RET.27 This suggests that sunitinib in addition to targeting pseudohypoxia subgroups may have additional activity in patients with RET mutations.
Algorithms for the treatment of advanced PCC/PGL are limited by a lack of prospective studies. Chemotherapy (CVD) in this population has been reported in a retrospective study to provide response rates of 55% however survival at 5 years was less than 50%.12 In the largest institutional experience with chemotherapy to date, 33% of patients had a response defined as tumour shrinkage or normalised blood pressure. Of the 54 patients in this study 24% had tumour shrinkage and 51% were alive at 5-years.15 Similarly in a retrospective study of temozolomide in metastatic PGL/PCC response rates were 33% and median PFS 13.4 months similar to the PFS in this study. These survival outcomes are similar to that reported in our study.
The response rate to 131I-MIBG in patients with metastatic PCC/PGL demonstrating 123 I-MIBG avidity was 22% with an estimated 5-year survival of 64%.28 In that study two patients died from myelodysplastic syndrome and one as a result of ARDs, most patients received 1–2 infusions only.28 Notably SDHB- associated PCC/PGL were more likely to respond to 131I-MIBG. Although response rates to sunitinib in our study were less than those described above, the median number of cycles in patients with a germline mutation was 22 (1–64), more than 2.5 years with one patient on treatment more than 7 years with a significant reduction in tumour volume.
Definitive prognostic variables in PCC/PGL are limited and the true survival when progressive and/or metastatic disease is present has been difficult to determine due to study biases and the heterogeneity in definitions of metastatic disease.29 Despite this, tumour progression is the most frequent cause of death in patients with advanced PCC/PGL underscoring the need for systemic agents.30
This study has several limitations. For a variety of reasons, a proportion of patients did not have genetic testing and given the small numbers, this trial is not able to define prognostic or predictive biomarkers. Furthermore, genetic panel testing may have varied between institution and during the study period. Biochemical response assessment was limited because plasma metanephrine analysis was not performed throughout the study and only total but not fractionated urinary metanephrines were available.31 Given the multisite nature of the study, a number of patients did not have either baseline or follow-up samples which makes biochemical interpretation less than complete. Furthermore, plasma metanephrines/normetanephrine or their fractionated urinary counterparts are now standard practice in this population,31 but this was not part of the study protocol in 2009. Hypertension in this trial must have been adequately controlled prior to study entry, and as a result only two patients experienced grade 3/4 hypertension. Patient-reported outcomes were not integrated into the study which was initiated in May 2009. It should however be noted that 3 patients reported significant reductions in pain scores. Furthermore, the improvement in symptoms and health-related quality of life could have been under-reported. Although our study, like others, did not fully accrue,20 this trial represents the largest prospective study of a targeted approach in patients with progressive PCC/PGL. The low response rates suggest the need for a greater understanding of the heterogeneity of the disease and the molecular drivers. Dual inhibition or multi targeted agents may be required to overcome drug resistance and improve efficacy.32
In summary, sunitinib in this prospective trial had modest activity as a single agent in an unselected population and could be considered a systemic option. The FIRSTMAPPP trial which is randomising sunitinib at a dose of 37.5 mg to placebo will help establish the role of sunitinib in this setting. While those with germline mutations in the SDH subunit genes and pseudohypoxia signatures may derive great benefit, we also show that RET mutation may be a biomarker for response to this agent that can target other pathways implicated in the kinase signalling subtype. Larger collaborative efforts will be needed in the future to conduct prospective biomarker-driven trials in this rare, molecularly heterogeneous group of neuroendocrine neoplasms.
References
Ayala-Ramirez, M., Feng, L., Johnson, M. M., Ejaz, S., Habra, M. A., Rich, T. et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J. Clin. Endocrinol. Metab. 96, 717–725 (2011).
Ricketts, C. J., Forman, J. R., Rattenberry, E., Bradshaw, N., Lalloo, F., Izatt, L. et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum. Mutat. 31, 41–51 (2010).
Turchini, J., Cheung, V. K. Y., Tischler, A. S., De Krijger, R. R., Gill, A. J. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology 72, 97–105 (2018).
Amar, L., Bertherat, J., Baudin, E., Ajzenberg, C., Bressac-de Paillerets, B., Chabre, O. et al. Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. 23, 8812–8818 (2005).
Fishbein, L., Leshchiner, I., Walter, V., Danilova, L., Robertson, A. G., Johnson, A. R. et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 31, 181–193 (2017).
Selak, M. A., Armour, S. M., MacKenzie, E. D., Boulahbel, H., Watson, D. G., Mansfield, K. D. et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 7, 77–85 (2005).
Kim, W. Y., Kaelin, W. G. Role of VHL gene mutation in human cancer. J. Clin. Oncol. 22, 4991–5004 (2004).
Favier, J., Igaz, P., Burnichon, N., Amar, L., Libe, R., Badoual, C. et al. Rationale for anti-angiogenic therapy in pheochromocytoma and paraganglioma. Endocr. Pathol. 23, 34–42 (2012).
Joshua, A. M., Ezzat, S., Asa, S. L., Evans, A., Broom, R., Freeman, M. et al. Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J. Clin. Endocrinol. Metab. 94, 5–9 (2009).
Jimenez, C., Cabanillas, M. E., Santarpia, L., Jonasch, E., Kyle, K. L., Lano, E. A. et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease-related tumors. J. Clin. Endocrinol. Metab. 94, 386–391 (2009).
Ayala-Ramirez, M., Chougnet, C. N., Habra, M. A., Palmer, J. L., Leboulleux, S., Cabanillas, M. E. et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J. Clin. Endocrinol. Metab. 97, 4040–4050 (2012).
Huang, H., Abraham, J., Hung, E., Averbuch, S., Merino, M., Steinberg, S. M. et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer 113, 2020–2028 (2008).
Patel, S. R., Winchester, D. J., Benjamin, R. S. A 15-year experience with chemotherapy of patients with paraganglioma. Cancer 76, 1476–1480 (1995).
Tanabe, A., Naruse, M., Nomura, K., Tsuiki, M., Tsumagari, A., Ichihara, A. Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine in patients with malignant pheochromocytoma and paraganglioma. Horm. Cancer 4, 103–110 (2013).
Ayala-Ramirez, M., Feng, L., Habra, M. A., Rich, T., Dickson, P. V., Perrier, N. et al. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer 118, 2804–2812 (2012).
Niemeijer, N. D., Alblas, G., van Hulsteijn, L. T., Dekkers, O. M., Corssmit, E. P. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis. Clin. Endocrinol. 81, 642–651 (2014).
Hadoux, J., Favier, J., Scoazec, J. Y., Leboulleux, S., Al Ghuzlan, A., Caramella, C. et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int J. Cancer 135, 2711–2720 (2014).
Cassol, C. A., Winer, D., Liu, W., Guo, M., Ezzat, S., Asa, S. L. Tyrosine kinase receptors as molecular targets in pheochromocytomas and paragangliomas. Mod. Pathol. 27, 1050–1062 (2014).
Fleming, T. R. One-sample multiple testing procedure for phase II clinical trials. Biometrics 38, 143–151 (1982).
Jasim, S., Suman, V. J., Jimenez, C., Harris, P., Sideras, K., Burton, J. K. et al. Phase II trial of pazopanib in advanced/progressive malignant pheochromocytoma and paraganglioma. Endocrine 57, 220–225 (2017).
Mejean, A., Ravaud, A., Thezenas, S., Colas, S., Beauval, J. B., Bensalah, K. et al. Sunitinib alone or after nephrectomy in metastatic renal-cell carcinoma. N. Engl. J. Med. 379, 417–427 (2018).
Eisenhofer, G., Huynh, T. T., Pacak, K., Brouwers, F. M., Walther, M. M., Linehan, W. M. et al. Distinct gene expression profiles in norepinephrine and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr. Relat. Cancer 11, 897–911 (2004).
Lopez-Jimenez, E., Gomez-Lopez, G., Leandro-Garcia, L. J., Munoz, I., Schiavi, F., Montero-Conde, C. et al. Research resource: transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol. Endocrinol. 24, 2382–2391 (2010).
Dahia, P. L., Ross, K. N., Wright, M. E., Hayashida, C. Y., Santagata, S., Barontini, M. et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 1, 72–80 (2005).
Forsythe, J. A., Jiang, B. H., Iyer, N. V., Agani, F., Leung, S. W., Koos, R. D. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell Biol. 16, 4604–4613 (1996).
Gossage, L., Eisen, T. Alterations in VHL as potential biomarkers in renal-cell carcinoma. Nat. Rev. Clin. Oncol. 7, 277–288 (2010).
Roskoski, R. Jr., Sadeghi-Nejad, A. Role of RET protein-tyrosine kinase inhibitors in the treatment RET-driven thyroid and lung cancers. Pharm. Res. 128, 1–17 (2018).
Gonias, S., Goldsby, R., Matthay, K. K., Hawkins, R., Price, D., Huberty, J. et al. Phase II study of high-dose [131I]metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J. Clin. Oncol. 27, 4162–4168 (2009).
Hamidi, O., Young, W. F. Jr., Gruber, L., Smestad, J., Yan, Q., Ponce, O. J. et al. Outcomes of patients with metastatic phaeochromocytoma and paraganglioma: a systematic review and meta-analysis. Clin. Endocrinol. 87, 440–450 (2017).
Baudin, E., Habra, M. A., Deschamps, F., Cote, G., Dumont, F., Cabanillas, M. et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur. J. Endocrinol. 171, R111–R122 (2014).
Lenders, J. W., Duh, Q. Y., Eisenhofer, G., Gimenez-Roqueplo, A. P., Grebe, S. K., Murad, M. H. et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 99, 1915–1942 (2014).
Nolting, S., Grossman, A. B. Signaling pathways in pheochromocytomas and paragangliomas: prospects for future therapies. Endocr. Pathol. 23, 21–33 (2012).
Author information
Authors and Affiliations
Contributions
G.O.K.: Patient materials, data collection, manuscript writing, statistical analysis SE. Patient materials, data collection, manuscript writing, statistical analysis A.J.: Study design, protocol development, patient materials, manuscript writing I.B.: patient materials, manuscript writing R.L.A.: Study design, protocol development, patient materials, manuscript writing H.O.: patient materials, manuscript writing M.K.: patient materials, manuscript writing D.R.: patient materials, manuscript writing S.C.: patient materials, manuscript writing L.W.: statistical analyses, manuscript writing K.B.: data collection, manuscript writing A.H.: Patient materials, manuscript writing S.A.: Patient materials, protocol development, manuscript writing J.K.: Study design, protocol development, patient materials, manuscript writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent
This study was approved by the University Health Network (UHN) Research Ethics Board (REB) and submitted at each participating site. REB protocol number 08-0108. All patients provided informed consent as per study protocol. This consent included patient consent to collect clinical, demographic, genetic and outcome data for study publication. This study was conducted in accordance with the Declaration of Helsinki.
Data availability
This study is available withinthis publication and in the supplementary material. Further patient data can be requested from the corresponding author.
Note
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Funding
This study was funded as part of an Investigator Initiated Trial from Pfizer Canada. Dr Jennifer Knox, the Principal Investigator was the study sponsor.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
O’Kane, G.M., Ezzat, S., Joshua, A.M. et al. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: the SNIPP trial. Br J Cancer 120, 1113–1119 (2019). https://doi.org/10.1038/s41416-019-0474-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-019-0474-x
This article is cited by
-
Management of phaeochromocytoma and paraganglioma in patients with germline SDHB pathogenic variants: an international expert Consensus statement
Nature Reviews Endocrinology (2024)
-
Cabozantinib for different endocrine tumours: killing two birds with one stone. A systematic review of the literature
Endocrine (2023)
-
Locally invasive recurrence or metastasis of pheochromocytoma into the liver?—clinicopathological challenges
World Journal of Surgical Oncology (2022)
-
New Directions in Treatment of Metastatic or Advanced Pheochromocytomas and Sympathetic Paragangliomas: an American, Contemporary, Pragmatic Approach
Current Oncology Reports (2022)
-
Favorable outcome in advanced pheochromocytoma and paraganglioma after hypofractionated intensity modulated radiotherapy
Journal of Endocrinological Investigation (2022)
