
Abstract
Background
Patients harbouring the UGT1A1*28/*28 genotype are at risk of severe toxicity with the standard irinotecan dose. However, this dose is considerably lower than the dose that can be tolerated by UGT1A1*1/*1 and *1/*28 patients. This randomised phase II trial evaluated the efficacy and safety of the FOLFIRI regimen with high-dose irinotecan (HD-FOLFIRI) in metastatic colorectal cancer patients.
Methods
Eighty-two patients with the UGT1A1*1/*1 or the *1/*28 genotype were randomised to receive HD-FOLFIRI versus FOLFIRI. Patients with the UGT1A1*28/*28 genotype were excluded. In the experimental group, the irinotecan dose was 300 mg/m2 for UGT1A1*1/*1 and 260 mg/m2 for *1/*28 patients. In the control group, the dose was 180 mg/m2. We analysed the overall response rate (ORR), toxicity, and survival.
Results
The ORR was significantly higher in the HD-FOLFIRI group (67.5 versus 43.6%; p = 0.001 OR: 1.73 [95% CI:1.03–2.93]). Neutropenia (17.7%), diarrhoea (5.1%), and asthenia (5.1%) were the most common grade 3–4 toxicity. No differences were observed in severe toxicity (22.5% versus 20.5%), dose reduction (22.5% versus 28.2%), or prophylactic G-CSF (17.5% versus 12.8%). No difference in survival was found.
Conclusions
Patients with the UGT1A1*1/*1 and *1/*28 genotypes can receive high doses of irinotecan to achieve a more favourable ORR without significant adverse events.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is the second most frequent neoplasm in industrialised countries. Worldwide, it is the fourth deadliest type of cancer. At initial diagnosis, around 30% of patients have inoperable or metastatic disease, and >50% of patients will receive chemotherapy at some stage of the disease.1,2 Current cytotoxic agents are fluoropyrimidine-based regimens in combination with oxaliplatin or irinotecan.3
Irinotecan (CPT11) inhibits topoisomerase I, an enzyme needed to separate the DNA double helix during replication and transcription.4 This inhibition leads to cell death and is the basis of its antineoplastic effect. Irinotecan is converted to an active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38) by a carboxylesterase, and finally metabolised through the action of uridine diphosphate glucuronosyltransferase (UGT) enzymes. The predominant enzyme is UGT1A1, the enzyme that conjugates bilirubin.5 In the promoter region of the UGT1A1 gene, an extra TA dinucleotide characterises the genotype associated with Gilbert’s syndrome (chronic non-conjugated hyperbilirubinemia due to reduced UGT1A1 activity). The presence of this polymorphism (TA7) results in the UGT1A1*28 variant allele instead of the dominant allele of 6 TA repeats (UGT1A1*1). Patients who are homozygous for this variant (UGT1A1*28/*28) have less enzymatic activity and are predisposed to develop myelosuppression and severe diarrhoea when treated with irinotecan.6,7,8,9,10,11,12 Premature drug suspension and dose reduction as well as administration delays due to toxicity can decrease antitumour activity. Therefore, serious toxicity rates that decrease survival can be anticipated through genetic analysis of patients prior to treatment, thereby optimising and personalising irinotecan dosing. The United States of America Food and Drug Administration recognises the importance of UGT1A1 pharmacogenetics in predicting toxicity to irinotecan and describes the association between reduced enzymatic activity of the UGT1A1*28 allele and neutropenia after drug administration in their summary of product characteristics. The recommendation is to reduce the dose of irinotecan in homozygous patients.13 Despite the good will of this recommendation, however, the exact dose reduction needed to limit drug toxicity is not specified for *28/*28 patients.
In a previous study (EC07/90232), we classified patients with metastatic CRC (mCRC) treated with first-line chemotherapy (FOLFIRI regimen) according to their UGT1A1 genotype. The goal was to predict, diminish, and/or avoid toxicity and improve the therapeutic effect of chemotherapy. This optimisation was based on administering different doses depending on the genotype of the UGT1A1 gene. The results showed that the recommended dose of irinotecan within the FOLFIRI regimen (180 mg/m2) was considerably lower than the dose tolerated by UGT1A1*1/*1 and *1/*28 patients.14 These findings validated the results reported by an Italian group who performed a clinical phase I irinotecan dose-escalation trial according to the UGT1A1 genotype in mCRC patients treated with FOLFIRI.15 Based on these two studies, it has been proposed to individualise irinotecan dosing according to the UGT1A1 status so as to optimise treatment efficacy and tolerability. However, whether genotype-driven dosing will lead to differences in outcome has yet to be tested prospectively. For these reasons, the main objectives of the present randomised phase II trial were to evaluate the efficacy and safety of the FOLFIRI regimen with high-dose irinotecan (HD-FOLFIRI) in mCRC patients with a favourable UGT1A1 genotype (homozygous wild type *1/*1 and heterozygous *1/*28), while excluding patients genetically at risk for toxicity (*28/*28).
Methods
Study design
This randomised, multicentre, open-label, non-blinded phase II study was conducted in three hospitals in Spain: Hospital de la Santa Creu i Sant Pau, Barcelona; Hospital Universitario Mutua Terrassa, Terrassa; and Hospital de Mataró, Mataró, Barcelona. The protocol was approved by the institutional review board at each participating centre. All patients signed a written informed consent form before entering the study. The clinicaltrials.gov identifier was NCT01639326.
Patient eligibility
Patients with histologically confirmed diagnosis of mCRC and measurable disease defined via RECIST (Response Evaluation Criteria in Solid Tumour; version 1.1) were enrolled. Eligibility criteria were: UGT1A1*1/*1 or *1/*28 genotypes, no prior chemotherapy for metastatic disease, age ≥18–< 76 years, The Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, absolute neutrophil count ≥1500/μl, platelets ≥100,000/μl, creatinine clearance <1.5× the upper limit of normal (ULN), alanine transaminase and aspartate aminotransferase <2.5× the ULN ( < 5× the ULN in the presence of liver metastases), and total serum bilirubin ≤1.5 mg/dl. Patients with the UGT1A1*28/*28 genotype or carriers of other UGT1A1 alleles (*6, *36 (TA5), *37 (TA8)) were not eligible.
Randomisation and drug administration
Between June 2012 and October 2016, patients were randomised 1:1 to receive HD-FOLFIRI (experimental group) versus FOLFIRI (control group) every 2 weeks. Treatment allocation was conducted using block randomisation and stratified by centre. The Statistical Package for Social Sciences, Version 19.0 (SPSS Inc., Chicago, IL, USA) was used. Irinotecan doses for UGT1A1*1/*1 and *1/*28 patients in the experimental group were 300 and 260 mg/m2, respectively. These doses were chosen based on our previous phase I trial, in which a dose ≥260 mg/m2 was an independent predictor of a better response in mCRC patients with the *1/*1 or *1/*28 UGT1A1 genotypes.14 The standard irinotecan dose of 180 mg/m2 was administered in the control group. Irinotecan was administered as an intravenous infusion over 90 min on days 1 and 15, with leucovorin 400 mg/m2 administered concomitantly. 5-Fluorouracil was administered as a 400 mg/m2 bolus immediately after the irinotecan infusion, followed by 2400 mg/m2 over a 46-h continuous infusion on days 1 and 15. A cycle was 28 days. Before irinotecan was started, patients were pretreated with atropine 0.5 mg, dexamethasone 20 mg, and granisetron 1 mg. Diarrhoea was promptly treated at onset with oral intake of loperamide 4 mg, and oral intake of 2 mg for any further episodes. Granulocyte-colony stimulating factors (G-CSF) were allowed in patients who had grade ≥3 neutropenia during previous cycles. Treatment was continued until disease progression, unacceptable toxicity, withdrawal of consent, or investigator’s decision, whichever was earlier. To avoid potential bias regarding the primary objectives of the study, biological agents were not allowed.
Study objectives
The primary objectives were: (1) to determine the effect of higher doses of irinotecan on the efficacy of FOLFIRI as assessed by overall response rate (ORR); and (2) to evaluate safety according to the NCI Common Terminology Criteria for Adverse Events (version 3.0). Objective tumour response was evaluated by the investigator at each centre using the modified RECIST (version 1.1); no independent review was performed. Tumour response was evaluated every 10 weeks (±2 weeks) according to the standard of care at each centre. Secondary objectives were progression-free survival (PFS) and overall survival (OS). PFS was defined from the time of drug administration to the occurrence of progressive disease or death, whichever occurred first. OS was defined from the time of drug administration to the date of death. Patients not meeting the criteria by the cutoff date were censored at the last contact date.
Genotyping assays
Genomic DNA was extracted from peripheral leukocytes by the salting-out procedure.16 The TA index of the UGT1A1 promoter was genotyped by fragment sizing. Polymerase chain reaction was performed in a total volume of 25 µl containing template DNA (80 ng/µl), according to Monaghan et al.17 The primers used were a forward primer that was modified by the addition of a 50 fluorescent-labelled FAM and an unlabelled reverse primer (UGT-FAM_F; 50-GTCACGTGACACAGTCAAAC-30, UGT_R 50-TTTGCTCCTGCCAGAGGTT-30). The PCR product (TA*1, 98 bp; TA*28, 100 bp), the internal size standard, and Hi-Di formamide (GeneScan 500, Applied Biosystems, Foster City, CA, USA) were mixed. The samples were then run in the ABI Prism 3100 Genetic Analyzer (Applied Biosystems). Fragment sizes were determined by comparison with the internal standard GeneScan 500 using the local Southern algorithm and analysed by the GeneMapper software version 3.5 (Applied Biosystems). Homozygous-dominant and heterozygous- and homozygous-recessive sequenced samples were included on every run as a quality control. Genotypes were assigned based on the number of TA repeats in each allele (i.e., TA*1/TA*1, TA*1/TA*28, and TA*28/TA*28).
KRAS and NRAS mutations in exons 2, 3, and 4 and BRAF V600E mutation were assessed on tumour DNA. Genomic DNA was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) as specified by the manufacturer’s instructions. Mutational analysis was performed using standard PCR conditions and primers for exons 2, 3, and 4 of KRAS and NRAS genes and for exon 15 of BRAF gene. The thermal cycling conditions were an initial 12 min at 94°C, followed by 40 cycles of 45 s at 94°C, 45 s at primer annealing temperature of 55°C, 10 min at 72°C, and a final extension of 10 min at 72°C. Each sample underwent capillary electrophoresis on an ABI 3500 Genetic Analyzer (Applied Biosystems).
Statistical analysis
All data were analysed using the Statistical Package for Social Sciences, Version 19.0, software (SPSS Inc., Chicago, IL, USA). ORR (complete+partial response) was compared between groups using chi-square or Fisher’s exact test for Count Data. Based on the report from Van Cutsem et al., the ORR was reported as 38.7% for the FOLFIRI regimen.18 An estimated difference to the order of 30% was assumed between the control and the experimental groups. The target sample size was therefore determined to be 96 patients, distributed between the two groups. The effects of the irinotecan dose on PFS and OS were estimated using the Kaplan–Meier estimator, and differences were tested using the log-rank test. For all comparisons, a two-sided p value <0.05 was considered significant, so the level was set at 5% (α = 0.05) and the β level was set at 20% (β = 0.20).
Results
Patient population
Between June 2012 and October 2016, the UGT1A1 genotype was analysed in mCRC patients (Fig. 1). Owing to 4 years of slower-than anticipated accrual, the trial was terminated early. Eighty-two patients harbouring UGT1A1*1/*1 or *1/*28 genotypes were enrolled from the three centres, with 41 patients randomly assigned to HD-FOLFIRI (experimental group) and 41 patients to FOLFIRI (control group). Three patients were not included in the final study because they did not receive the pre-planned dose for the endpoint analyses. Therefore, 79 patients were evaluated for toxicity and for response to treatment. Patient characteristics are summarised in Table 1.

CONSORT diagram
Efficacy
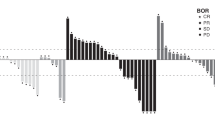
Table 2 shows the results of the tumour response per-protocol analysis. The ORR of patients treated with HD-FOLFIRI was significantly higher than in patients treated with FOLFIRI (67.5% versus 43.6%; p = 0.001 odds ratio (OR): 1.73 [95% confidence interval (CI): 1.03–2.93]). In the intention to treat analysis, the ORR was 65.9% in the experimental group versus 43.9% in the control group (p = 0.046 OR: 1.64 [95% CI: 0.99–2.72]). There were no interactions between ORR and clinical characteristics (sex, age, ECOG, tumour location, or synchronous disease). Metastatic surgical resection was performed in 15 patients (22.5% in HD-FOLFIRI and 15.4% in FOLFIRI) and was associated with ORR (29.5% versus 5.7%; p = 0.007).
There were no interactions between ORR and UGT1A1 or RAS status (Table 2). However, when BRAF mutated tumours were considered, the ORR was 41.7% in the HD-FOLFIRI group versus no objective response in the control group (p = 0.003).
Survival
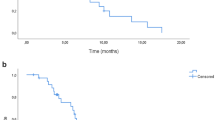
The median follow-up period was 18 months (range, 1.8–45 months). The cutoff date for PFS and OS was December 2016. At the time of analysis, 89% of the patients had progressed (90% in the experimental group and 87% in the control group). The median PFS and OS were 8.6 and 26 months in the experimental group (HD-FOLFIRI) and 8.2 and 17.6 months in the control group (FOLFIRI) (p = 0.46 hazard ratio (HR) 0.84 [95% CI: 0.52–1.35] and p = 0.74 HR 0.90 [95% CI: 0.49–1.67], respectively) (Supplementary Figure 1).
PFS was significantly associated with ECOG performance status (9.9 months in ECOG 0 versus 7.2 months in ECOG 1) and metastatic resection (15.5 months in patients who underwent surgery versus 7.8 months in the remaining cases). In terms of OS, patients with metastatic surgery achieved a better outcome than patients who did not undergone surgery (median not reached versus 18.4 months).
No statistically significant difference in PFS or OS was found between the experimental and control groups according to RAS or BRAF status. In RAS/BRAF wild-type tumours, the median PFS and OS were 9.8 versus 8.6 months and 29 versus 17 months, respectively. In RAS-mutated tumours, the median PFS and OS were 8.5 versus 8.1 months and 14 versus 17.6 months, respectively. In BRAF-mutated tumours, the median PFS and OS were 8.3 versus 7.6 months and 22 versus 11.7 months, respectively.
Table 3 shows the results of the multivariate survival analysis. Multivariate analysis showed a significant association between metastatic resection with both PFS and OS. When patients undergoing metastasectomy were excluded, BRAF- and RAS-mutated tumours had a shorter PFS and OS than wild-type tumours (Table 4).
Toxicity
Table 5 summarises the toxicity data available for the 79 patients who received treatment. The non-haematological toxicities that were frequently reported were asthenia and adverse gastrointestinal effects, such as diarrhoea and nausea/vomiting. With regard to haematological toxicities, the most frequent were anaemia, neutropenia, and leukopenia. Most of the toxicities were grade 1 or 2. No significant differences in grade 3–4 toxicities were observed between patients treated with HD-FOLFIRI or standard irinotecan dose (FOLFIRI): diarrhoea (2.5% versus 7.7%), asthenia (5% versus 5.1%), leukopenia (7.5% versus 2.6%), and neutropenia (15% versus 20.5%). Febrile neutropenia was observed in 2 patients in each arm (5% versus 5.1%). No differences were observed in serious adverse events (22.5% versus 20.5%), dose reduction (22.5% versus 28.2%), or prophylactic use of G-CSF (17.5% versus 12.8%) irrespectively of irinotecan dose.
Discussion
In this study, we continued our line of research by conducting a randomised phase II trial to evaluate the efficacy and safety of the FOLFIRI regimen with HD-FOLFIRI as first-line therapy in patients with mCRC. Our findings confirmed that HD-FOLFIRI increased ORR without adding toxicity in patients with a favourable UGT1A1 genotype (*1/*1 or *1/*28).
Previous prospective studies have shown that the recommended dose of 180 mg/m2 for irinotecan in the FOLFIRI regimen is considerably lower than the dose that can be tolerated by non-UGT1A1*28/*28 mCRC patients. In a Caucasian population study, Toffoli et al.15 established that 370 mg/m2 in *1/*1 genotype and 310 mg/m2 in *1/*28 genotype can be safely administered every 2 weeks in patients undergoing first-line treatment for mCRC treated with FOLFIRI. Similar maximum tolerated doses (MTD) (390 mg/m2 in *1/*1 and 340 mg/m2 *1/*28 patients) were validated the following year in a study conducted by our group.14 In an Asian population, in addition to UGT1A1*28, the variant UGT1A1*6 has been associated with significantly increased related toxicities. In a phase I study, Korean patients were genotyped for UGT1A1 and stratified according to the number of defective alleles (DA): *28 and/or *6. The recommended doses were 300 (0 DA), 270 (1 DA), and 150 (2 DA) mg/m2.19
The effect of adding a biologic drug to genotype-guide dosing of FOLFIRI has recently been explored. Two initial retrospective studies in Asian patients were performed by the same group of investigators. They showed that patients with mCRC with pre-therapeutic UGT1A1 genotyping and subsequent irinotecan dose escalation can achieve a more favourable response and outcome without a significant increase in toxicity while using the FOLFIRI-plus-bevacizumab regimen.20,21
More recently, Toffoli et al. published a dose-finding study in first-line mCRC patients treated with FOLFIRI plus bevacizumab to establish the MTD of irinotecan in *1/*1 and *1/*28 patients.22 Again, the MTD of irinotecan was 310 mg/m2 for UGT1A1*1/*1 patients and 260 mg/m2 for *1/*28 patients. The most common dose-limiting toxicities were neutropenia (46%) and diarrhoea (38%), but 65% of the patients treated at the MTD did not require a reduction of irinotecan. These authors also demonstrated that bevacizumab did not alter the pharmacokinetics of irinotecan.
High-dose FOLFIRI (irinotecan 260 mg/m2 for UGT1A1*1/*1 and *1/*28 genotypes and 220 mg/m2 for UGT1A1*28/*28 genotypes) combined with cetuximab has been explored in a multicentre phase II study (ERBIFORT) in patients with potentially resectable liver metastases of CRC.23 This regimen yielded high response rates and enabled complete resection of hepatic metastases in most patients. Thanks to the irinotecan dose adaptation according to UGT1A1 pharmacogenomics status, this treatment schedule was less toxic yet as effective as the intense combination of FOLFIRINOX plus cetuximab reported by Assenat et al. in a phase II trial.24
It is now widely accepted that the dosing recommendations obtained from traditional, non-genotype-directed clinical trials should be revised in light of validated genetic markers of toxicity risk. The lack of patient stratification based on genotype might result in significant underdosing of patient subgroups. However, phase II–III clinical trials are required to determine whether these irinotecan genotype-guided doses—with or without a biological agent—imply a higher antitumour efficacy.
To our knowledge, the present work is the first randomised phase II trial to show higher antitumour efficacy when a genotype-guided dose of irinotecan is considered. It should be noted that this scheme not only increases the response rate and metastasis surgery but also improves efficacy in patients with worse prognosis, such as mutated BRAF. However, the limitations of our study should be considered. In addition to those previously mentioned concerning the early termination due to low accrual, there was an imbalance between experimental and control arms regarding the UGT1A1*1/*1 and *1/*28 genotypes and tumour sidedness, which could have favoured the experimental arm. Therefore, UGT1A1 genotype and primary tumour location should have been included as stratification factors to avoid bias that could have influenced the results in survival or disease control rates. Moreover, lower doses than in our previous phase I study were used and the FOLFIRI schedule is now given together with biologics in the first-line setting for most mCRC patients, which limits the incorporation of high doses of irinotecan regimens in clinical practice. Regarding toxicity aspects, current evidence highlights the association between dihydropyrimidine dehydrogenase deficiency and an increased risk of fluoropyrimidine-related toxicity.25 The additive value of combined UGT1A1/DPYD genotype analysis could improve the safety of irinotecan plus fluoropyrimidine combinations and should be considered in further studies using these drugs.
To conclude, the findings from our study confirm the safety of chemotherapy with HD-FOLFIRI and indicate that this strategy, although does not show an improvement in survival, improves ORR. A randomised phase II–III study is needed to validate irinotecan intensification according to UGT1A1 pharmacogenetic status combined with a biological drug adapted to RAS status.
References
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J. Cancer 136, E359–E386 (2015).
Scnall, S. & MacDonald, J. S. Gastrointestinal cancers: Colorectal carcinoma, In Manual of Oncologic Therapeutics 3rd edn. (eds McDonald, J. S., Haller, D. G. & Mayer, R. J.) (Lippincott Company, Michigan, 2004).
Tournigand, C. et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J. Clin. Oncol. 22, 229–237 (2004).
Kunimoto, T. et al. Antitumor activity of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxy-camptothec in, a novel water-soluble derivative of camptothecin, against murine tumors. Cancer Res. 47, 5944–5947 (1987).
Iyer, L. et al. Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J. Clin. Invest. 101, 847–854 (1998).
Marcuello, E. et al. UGT1A1 gene variations and irinotecan treatment in patients with metastatic colorectal cancer. Br. J. Cancer 91, 678–682 (2004).
Ando, Y. et al. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res. 60, 6921–6926 (2000).
Innocenti, F. et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J. Clin. Oncol. 22, 1382–1388 (2004).
Rouits, E. et al. Relevance of different UGT1A1 polymorphisms in irinotecan-induced toxicity: a molecular and clinical study of 75 patients. Clin. Cancer Res. 10, 5151–5159 (2004).
Toffoli, G. et al. The role of UGT1A1*28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J. Clin. Oncol. 24, 3061–3068 (2006).
Hoskins, J. M., Goldberg, R. M., Qu, P., Ibrahim, J. G. & McLeod, H. L. UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J. Natl. Cancer Inst. 99, 1290–1295 (2007).
Kim, T. W. & Innocenti, F. Insights, challenges, and future directions in irinogenetics. Ther. Drug Monit. 29, 265–270 (2007).
United States Food and Drug Administration: Camptosar label. http://www.fda.gov/cder/foi/label/2005/020571s024,027,028lbl.pdf
Marcuello, E. et al. A genotype-directed phase I-IV dose-finding study of irinotecan in combination with fluorouracil/leucovorin as first-line treatment in advanced colorectal cancer. Br. J. Cancer 105, 53–57 (2011).
Toffoli, G. et al. Genotype-driven phase I study of irinotecan administered in combination with fluorouracil/leucovorin in patients with metastatic colorectal cancer. J. Clin. Oncol. 28, 866–871 (2010).
Miller, S. A., Dykes, D. D. & Polesky, H. F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16, 1215 (1998).
Monaghan, G., Ryan, M., Seddon, R., Hume, R. & Burchell, B. Genetic variation in bilirubin UPD-glucuronosyltransferase gene promoter and Gilbert’s syndrome. Lancet 347, 578–581 (1996).
Van Cutsem, E. et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 360, 1408–1417 (2009).
Kim, K. P. et al. A phase I study of UGT1A1 *28/*6 genotype-directed dosing of irinotecan (CPT-11) in Korean patients with metastatic colorectal cancer receiving FOLFIRI. Oncology 88, 164–172 (2015).
Lu, C. Y. et al. Prognostic advantage of irinotecan dose escalation according to uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) genotyping in patients with metastatic colorectal cancer treated with bevacizumab combined with 5-fluorouracil/leucovorin with irinotecan in a first-line setting. Transl. Res. 164, 169–176 (2014).
Lu, C. Y. et al. Clinical implication of UGT1A1 promoter polymorphism for irinotecan dose escalation in metastatic colorectal cancer patients treated with bevacizumab combined with FOLFIRI in the first-line setting. Transl. Oncol. 8, 474–479 (2015).
Toffoli, G. et al. Genotype-guided dosing study of FOLFIRI plus bevacizumab in patients with metastatic colorectal cancer. Clin. Cancer Res. 23, 918–924 (2017).
Phelip, J. M. et al. High resectability rate of initially unresectable colorectal liver metastases after UGT1A1-adapted high-dose irinotecan combined with LV5FU2 and cetuximab: a multicenter phase II study (ERBIFORT). Ann. Surg. Oncol. 23, 2161–2166 (2016).
Assenat, E. et al. Cetuximab plus FOLFIRINOX (ERBIRINOX) as first-line treatment for unresectable metastatic colorectal cancer: a phase II trial. Oncologist 16, 1557–1564 (2011).
Henricks, L. M., Opdam, F. L., Beijnen, J. H., Cats, A. & Schellens, J. H. M. DPYD genotype-guided dose individualization to improve patient safety of fluoropyrimidine therapy: call for a drug label update. Ann. Oncol. 28, 2915–2922 (2017).
Acknowledgements
In addition to the authors listed on the title page, the following investigators and Institutions contributed to this study: Marta Martín-Richard, Hospital de la Santa Creu i Sant Pau; and Montserrat Zanui, Rosa Querol, and Pilar Lianes, Hospital de Mataró, Barcelona, Spain. We are indebted to the patients who participated in the study. We also extend our sincere thanks to Montserrat Baiget for her invaluable support for this project. This work has been financed by the Ministry of Health, Social Services and Equality (Spain) with the co-finantiation of FEDER funds (EC11-336).
Author contributions
D.P., M.T., and A.S. designed the study. D.P., M.T., J.F.-P., A.S., A.C.V., L.C., and I.S. included and followed patients. D.P. provided clinical data. P.R. and J.S. performed sequencing experiments. D.P. performed statistical analyses. D.P. and P.R. designed the figures. A.B. supervised the study. D.P., M.T., A.S., P.R., and J.S. wrote the manuscript. All coauthors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All subjects gave written consent. The study protocol was approved by the hospital’s ethics committee. The study was performed according to the guidelines of the Declaration of Helsinki.
Consent for publication
All subjects gave written consent for publication:
Competing interests
D.P. has declared scientific advisory role for Amgen, Sanofi, Merck Serono, and Servier. M.T. has declared scientific advisory role for Amgen, Sanofi, and F. Hoffmann-La Roche Ltd. A.B. has declared scientific advisory role and has participated on the speaker’s bureau for Novartis, Pfizer, F. Hoffmann-La Roche Ltd, MSD, Pierre Fabre, Lilly. and Pallex. A.B. has received institutional grant support from F. Hoffmann-La Roche Ltd, Pfizer, Lilly. and Bristol Myers Squibb. The other authors declare no competing interests.
Note
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Additional information
Note: This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Electronic supplementary material
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Páez, D., Tobeña, M., Fernández-Plana, J. et al. Pharmacogenetic clinical randomised phase II trial to evaluate the efficacy and safety of FOLFIRI with high-dose irinotecan (HD-FOLFIRI) in metastatic colorectal cancer patients according to their UGT1A 1 genotype. Br J Cancer 120, 190–195 (2019). https://doi.org/10.1038/s41416-018-0348-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-018-0348-7
This article is cited by
-
A phase-II study based on dose adjustment according to UGT1A1 polymorphism: is irinotecan underdosed in first-line FOLFIRI regimen for mCRC?
Cancer Chemotherapy and Pharmacology (2024)
-
Germinal Immunogenetics predict treatment outcome for PD-1/PD-L1 checkpoint inhibitors
Investigational New Drugs (2020)
