
Abstract
The tissue-resident skeletal stem cells (SSCs), which are self-renewal and multipotent, continuously provide cells (including chondrocytes, bone cells, marrow adipocytes, and stromal cells) for the development and homeostasis of the skeletal system. In recent decade, utilizing fluorescence-activated cell sorting, lineage tracing, and single-cell sequencing, studies have identified various types of SSCs, plotted the lineage commitment trajectory, and partially revealed their properties under physiological and pathological conditions. In this review, we retrospect to SSCs identification and functional studies. We discuss the principles and approaches to identify bona fide SSCs, highlighting pioneering findings that plot the lineage atlas of SSCs. The roles of SSCs and progenitors in long bone, craniofacial tissues, and periosteum are systematically discussed. We further focus on disputes and challenges in SSC research.
Similar content being viewed by others

Introduction
The skeleton is comprised of mineralized matrix that protects organ and facilitates body movement. It is a reservoir for calcium and phosphate, regulating the systematic mineral ion metabolism.1,2 The bone marrow harbors niches for hematopoiesis.3 Bone also regulates systemic metabolism and closely interacts with other organs such as brain, kidney, intestine, and liver.4,5,6,7,8,9,10 Over the decades, the skeletal stem cells (SSCs) have gained great attention based on the recognition that SSCs are situated at the apex of the lineage differentiation trajectory and continuously provide cells for bone development, homeostasis, and injury repair.11,12,13,14 Understanding the properties and lineage commitment of SSCs help revealing the nature of organogenesis and aiding in the treatment of disorders such as congenital anomalies (e.g., dwarfism, cleidocranial dysplasia), age-related diseases (e.g., osteoporosis, osteoarthritis), tumor (e.g., chondromas, osteochondromas) and rare genetic diseases (e.g., fibrodysplasia ossificans progressiva, progressive osseous heteroplasia).15,16,17,18,19,20,21,22,23 However, the search for bona fide stem cells has been a long-striving goal in the fields of bone research. The intricate developmental phases, heterogenous cell types, and mineralized structure of the skeletal system make this work extremely challenging.24,25
Definition and characterization
The concept of SSCs or mesenchymal stem cells (MSCs) originates from the groundbreaking discovery that heterotopic transplantation of total bone marrow cell suspensions or boneless marrow fragments formed ossicles and reconstructed hematopoietic and reticular structures.26,27 This capacity was later ascribed to a population of self-renewable and multipotent stromal cells, which were defined as “mesenchymal stem cells”.28,29,30 The International Society of Cellular Therapies has proposed a minimal criteria to define MSCs, wherein the cells with plastic-adherent and tri-lineage differentiation ability (osteoblasts, chondrocytes, adipocytes), as well as with the expression of subsets of immunophenotypic makers (CD73, CD90, and CD105, but not CD34, CD45, CD14/CD11b, CD79a/CD19, and HLA-DR) were regarded as putative MSCs.31 With time, the MSC concept was extended to other tissues, such as fat, muscle, and dental pulp.32,33,34 It seems that researchers could always culture out a plate of “stem cells” from any mesenchymal tissue.35 However, this definition and characterization of MSCs was later considered inadequate, as it indeed yielded highly a heterogenous and unidentified population.11,12 Ectopic transplantation of these “stem cells” often failed to form a chondro-osteo structure or support a hematopoietic environment in vivo.12 The ambiguity of MSCs further jeopardizes the clinical translation as stem cell therapy failed to reach a predictable outcome.36
To address these issues, Paolo Bianco proposed the term “skeletal stem cells” to indicate the skeletal tissues-resident, self-renewable and multipotent cells that generate cartilage, bone, hematopoiesis-supporting stroma and marrow adipocytes.12,37 Importantly, it was advocated that, along with colony formation units (CFUs) and tri-lineage differentiation test in vitro, the self-renewal and multipotent properties should be tested with rigorous in vivo assays. Serial transplantation study is a golden standard to verify bona fide SSCs.11 Cells are sorted with presumptive stem cell surface markers and transplanted ectopically, and are then evaluated for the formation of the bone and bone marrow components. Presumptive SSCs again are sorted from the newly formed bone and then secondarily transplanted, to test the ability to form complete skeletal components.11,38,39,40 Likewise, the multipotency of presumptive SSCs are analyzed in situ, by co-labeling with different lineage markers. EdU (5-ethynyl-2′-deoxyuridine)/BrdU (5-bromo-2′-deoxyuridine) retention test further helps to verify the slow-cycling cells, generally regarded as stem cells.41,42 Overall, SSCs and MSCs indicate different cell populations and their meanings are not interchangeable. To some extent, SSCs can be regarded as more homogeneous subpopulations of MSCs. The use of “SSCs” is recommended to define the self-renewable and multipotent cells that are restricted to skeletal system and display explicit surface markers.
Identification and isolation
Two approaches are now widely used to identify SSCs, fluorescence-activated cell sorting (FACS) and transplantation assay, and Cre-loxP system-driven lineage tracing strategy.43,44 The identification and isolation of different lineages by FACS and transplantation assay is routinely used in hematopoietic domain, which facilitates the classification of differentiation trajectory and precise elucidation of homogenous cells subsets without contamination.45 However, as stem cells and progenitors are differentially labeled, often with several markers, visualization and functional study in situ are much limited. Besides, it is reported that in native hematopoietic environment, the maintenance of blood production during adulthood is contributed mainly by long-lived lineage-restricted progenitors rather than hematopoietic stem cells which are classically defined by transplantation assay into lethally irradiated mice.46
Lineage tracing, on the other hand, enables the spatial and temporal observation of SSCs and progenies in an unperturbed manner.43 Diverse site-specific recombinases are developed for lineage tracing, such as Cre-loxP, Dre-rox, and Flp-frt.47,48,49,50 Cre-loxP technology-based mouse models are most frequently constructed in the bone field. Briefly, Cre recombinase expressed under the control of different transgenes removes the loxP site and mediates the expression of reporter genes.50 Using the tamoxifen-induced CreER expression system, we are able to study the stem cells in a restricted time window.51 Besides, reporters directly driven by promoters of transgenes are widely used in combination with Cre-loxP technology, such as Col1(2.3 kb)-GFP.52 Dre-rox system is compatible with Cre-loxP system, and orthogonal recombinase system is developed that facilitates the label of intersect or subtractive parts of cell populations.53,54
Different reporter mice are available, such as R26R-LacZ (β-galactosidase), R26R-EYFP (yellow fluorescent protein), R26R-tdTomato, double-fluorescent mT/mG, and four fluorescent R26R-Confetti/Brainbow2.1 (Table 1).55,56,57,58,59 Especially, the mT/mG mice express membrane-targeted tandem dimer Tomato (mT) and membrane-targeted green fluorescent protein (mG) before and after Cre-mediated excision respectively.57 The R26R-Confetti mice stochastically express one of the YFP, tdTomato, GFP, and CFP (cyan fluorescent protein) after Cre-mediated excision.58 A single SSC and its progeny express the same fluorescent protein, so we can evaluate the clonogenic ability using R26R-Confetti mice.58 Nonetheless, the non-specificity of transgenes is an issue in lineage tracing.48 Occasionally, unwanted cell types are simultaneously labeled.60 Poor recombination is observed in some Cre/CreER mice as well.61 Osterix (Osx)-Cre mouse, a widely used transgenic model, unexpectedly displayed bone developmental defects.62 Besides, cells labeled by site-specific recombinase include SSCs and descendants, so it is almost impossible to sort out a homogenous stem cell population with a single transgene. Therefore, a thorough characterization of SSCs relies on both cell sorting and lineage tracing.
Current understanding in SSC research
Several goals are to be achieved in SSC research. The first is to plot the entire differentiation trajectory of SSCs based on authentic surface markers. The second is to answer how dysfunction of SSCs leads to pathogenesis. The third is to explore major factors that determine the fate commitment of SSCs.
Several studies have rigorously identified and tested the bona fide SSCs using either a combination of immunophenotypic markers or a specific promotor/enhancer that drives transgene expression.38,39,41,64,65,66,67,68,69,70 The lineage differentiation trajectory of several types of SSCs have been plotted, though their overlap and difference require clarification.38,66,71,72,73,74 The opinion that a single SSC lineage gives rise to all bone components is an oversimplification. Indeed, accumulating evidence indicates that more than one types of SSCs contribute to skeletal development and homeostasis at different stages (we will discuss about it in “Disputes and challenges of SSCs”). For example, during embryonic development, different waves of progenitors emerge to support rapid bone growth. The type II collagen (Col2)-Cre-labeled perichondrial and growth plate progenitors and Osx-Cre-labeled progenitors in periosteum and primary spongiosa actively participate in bone growth and bone marrow formation.67,69,75,76 With growth, these early progenitors are replaced by long-lived SSCs such as leptin-receptor-expressing (LepR+) perivascular cells.64,77
Studies have revealed several postnatal stem cell niches that support SSCs self-renewal, including the perivascular niches, growth plate niches, and periosteal niches.42,60,71,78 Despite divergent microenvironment that regulates SSCs behavior, SSCs spontaneously differentiate into osteoblasts.79 In fact, the intrinsic ability of SSCs to form bone rather than adipocytes is established epigenetically, in which the chromatin accessibility is predetermined for osteogenesis. The adipogenic process instead requires a substantial remodeling of the chromatin landscape where enhancers are activated de novo.79 It is currently unclear why SSCs possess different ability to generate chondrocytes, adipocytes and stromal cells. One possible explanation is the niches. For example, growth plate SSCs sequentially differentiate into chondrocytes and then osteoblasts, but perisinusoidal SSCs differentiate into either osteoblasts or adipocytes in a mutually exclusive way.41,80 The cells at growth plate niches are predominantly regulated by factors like fibroblast growth factors (FGF), Wnt, parathyroid hormone-related protein (PTHrP), and Indian hedgehog (Ihh), whereas SSCs residing in the bone marrow niches actively interact with endothelial and hematopoietic cells, and therefore possess a different transcriptional landscape and differentiation trajectory.73,81,82,83,84,85,86
Skeletal stem cells in long bones
Markers of SSCs
Various surface markers for SSCs and lineage-restricted progenitors have been identified. Here we summarize these key findings of both human and mice based on FACS (Tables 2 and 3) and lineage tracing (Table 4). Initially identified in human bone marrow and dental pulp, STRO-1+ perivascular stem cells were positive for α-smooth muscle actin (αSMA) and CD146.87 The pioneering study to identify a functional human SSC group to support hematopoietic microenvironment is by Bianco Laboratory.39 They identified a group of subendothelial/adventitial reticular CD45−CD146+ SSCs with in vivo self-renewal and clonogenic properties. All CFU-fibroblasts (CFU-Fs) selected from unfractionated bone marrow showed high level of CD146 expression. CD45−CD146+ SSCs transplanted subcutaneously repetitively gave rise to bone and hematopoiesis-associated stromal cells. This study also provides a standard template to functionally characterize SSCs in vitro and in vivo.39
Further study identified a small fraction of CD146+ cells that were PDGFRα+CD51+ in the bone marrow, which exhibited even higher clonogenic capacity. These PDGFRα+CD51+ cells were mainly present in fetal bone marrow and robustly expressed NESTIN and hematopoietic stem cell (HSC) maintenance genes.88 Moreover, a group of lin−/CD45−/CD271+ SSCs were identified.89 lin−/CD45−/CD271+ cells gave rise to bone and hematopoietic stroma when transplanted, regardless of CD146 expression. Interestingly, lin−/CD45−/CD271+/CD146+ SSCs were perivascular, while lin−/CD45−/CD271+/CD146−/low SSCs were endosteal.89
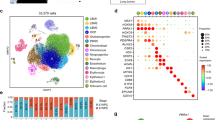
In 2018, Chan et al. plotted lineage differentiation trajectory of human SSCs. Combining single-cell RNA sequencing (scRNA-seq) with FACS, they identified CD45−CD235a−Tie2−CD31−PDPN+CD146−CD73+CD164+ cells as the bona fide human SSCs, which were able to form ectopic ossicles with hematopoiesis-supporting marrow stroma even after serial renal capsule transplantation.66 Interestingly, these SSCs can be isolated not only from human fetal and adult bone, but also from bone morphogenetic protein 2 (BMP2)-treated adipose stroma and induced pluripotent stem cells (iPSCs). Comparison between CD146+ SSCs and PDPN+CD146−CD73+CD164+ SSCs showed a higher colony-forming ability of the latter pupulation.66
For mouse skeletal stem cells, Matsuzaki et al. identified a group of nonhematopoietic PDGFRα+Sca1+ SSCs from adult mouse bone marrow.65 These cells resided in a perivascular space and were highly enriched with CFU-Fs. A single PDGFRα+Sca1+ cell was sufficient to give rise to mesenchymal and endothelial lineages. When freshly isolated and transplanted into lethally irradiated mice with HSCs, PDGFRα+Sca1+ SSCs were capable of self-renewal and differentiating into osteoblasts, adipocytes, and HSC-supporting stromal cells. However, cultured PDGFRα+Sca1+ cells failed to engraft into the recipient mice.65
Chan et al. performed clonal assay using Actin-CreERT driven rainbow reporter mouse and identified growth plate as a clonal region.38,91 A majority of growth plate cells were CD45−Ter119−Tie2−AlphaV+. Further screening identified eight subpopulations of CD45−Ter119−Tie2−AlphaV+ cells that gave rise to distinct components of bone. CD45−Ter119−Tie2−AlphaV+Thy−6C3−CD105−CD200+ (mouse skeletal stem cells, mSSCs) subpopulation was capable of self-renewal and generated other seven subpopulations through sequence of stages when transplanted beneath the renal capsule.38 A population of CD45−Ter119−CD31−CD166−CD146−Sca1+ (Sca1+) SSCs were also identified.74 Sca1+ SSCs generated CD45−Ter119−CD31−CD166−CD146+ (CD146+) intermediate progenitors and CD45−Ter119−CD31−CD166+CD146+ (CD166+) mature osteoprogenitors. Sca1+ SSCs also gave rise to C-X-C motif chemokine ligand 12-expressing (Cxcl12+) stromal cells and homed to bone marrow to support hematopoiesis when intravenously infused into sub-lethally irradiated mice.74
Utilizing Cre-loxP technology and fluorescent reporter mouse models, an array of SSCs and lineage-restricted progenitors are identified (Fig. 1). Here we briefly summarize the transgenes that specifically label these functional cells. Perivascular is common origin for mesenchymal stem cells, where several types of SSCs and progenitors were identified, including PDGFRα+, Nestin-GFP+ (Nes-GFP), LepR+, αSMA+, Ebf3+, and Cxcl12+ cells.61,64,68,78,92,93,94,95 Nes-GFP+ cells were perivascular and co-localized with HSCs, accounting for all CFU-Fs activity. The Nes-GFP+ “mesenspheres” could expand through serial heterotopic transplantation.78,96 Further study revealed some Nes-GFP+ cells were endothelial in embryonic perichondrium, and only the nonendothelial Nes-GFP+ cells generated osteoblast lineage. Meanwhile, cells targeted by Nes-CreER were predominantly endothelial in developing and adult bone marrow and the labeled cells make little contribution to early bone development, indicating the heterogeneity of Nes+ population.68 LepR+ SSCs mainly originated from fetal Col2+ precursors and gradually gave rise to bone cells and adipocytes in adult bone marrow.64,77

Schematics of skeletal stem cells and progenitors in mouse long bone. At fetal stage, limb bud mesenchymal cells express Prx1 and osteochondral progenitors are Sox9+. When fetal growth plate forms, Col2, Nes-GFP, and Osx mark progenitors at perichondrium. Neonatal long bone harbors various types of SSCs and progenitors, including Sox9+/Col2+/Acan+/Col10+ cells at growth plate, Gli1+/Osx+ cells at primary spongiosa, Ctsk+ cells at periosteum and Ranvier’s groove. Postnatal long bone is occupied with long-term SSCs and progenitors, including Grem1+/FoxA2+/PTHrP+/Sox9+/Col2+/Acan+/Col10+ cells at epiphysis and growth plate cartilage, Gli1+/Osx+ cells at primary spongiosa, PDGFRβ+/PαS+/Nes-GFP+/LepR+/Cxcl12+ cells in bone marrow, and αSMA+Mx1+/Ctsk+ cells at periosteum. Adult bone marrow is highly complex and contains endosteal, perisinusoidal, and periarteriolar niches, which harbor tissue-resident SSCs and progenitors essential for hematopoietic lineages
Growth plate and metaphyseal also harbor SSCs and progenitors, including Gremlin1+ (Grem1+), Sox9+, Aggrecan+ (Acan+), Col2+, FoxA2+, PTHrP+, Gli1+ and Osx+ cells.41,67,69,97,98 For instance, Grem1 marked a population of osteochondralreticular (OCR) stem cells in the metaphysis of long bone, which were more clonogenic than Nes-GFP+ SSCs.97 Embryonic perichondrium and postnatal periosteum are niches for SSCs.67,68,69,71,99 During early embryonic development, Sox9+, Col2+, Nes-GFP+, and Osx+ progenitors gave rise to bone marrow osteoblasts and stroma.67,68,69 Postnatal periosteum harbored cathepsin K-expressing (Ctsk+) SSCs and Mx1+αSMA+ SSCs which contributed to cortical bone formation and fracture repair (To be discussed in the session “Skeletal stem cells in periosteum”).71,99
Emergence of SSCs in limb bud
Long bones are lateral plate mesoderm derived and the development starts with the emergence of limb buds at embryonic day (E) 9.5 (forelimb) or E10.5 (hindlimb).25 Morphogens from the flank, apical ectodermal ridge (AER) and zone of polarizing activity (ZPA) guide the pattern formation and establish the identity of emerging skeletal elements.25,100,101,102,103 Since E11.5, the forelimb mesenchyme condensation starts. The cartilage primordium is initially formed where the mesenchymal cells differentiate into chondrocytes at the core of condensation and into perichondrial cells immediately outside the condensation under the guidance of the master transcriptional factor SOX9.104 The round immature chondrocytes at the central portion of the cartilage primordia then stop proliferating and undergo morphological change into hypertrophic chondrocytes, whose volume increases around 20-fold.105,106 The distally located round chondrocytes however continuously proliferate and give rise to flattened stacked chondrocytes, which together with hypertrophic chondrocytes form the fetal growth plate.
Prx1-Cre marked all the mesenchymal cells in the developing limb bud, which gave rise to chondrocytes, perichondrial cells, periosteal cells, osteoblasts, and stromal cells, but not muscle cells.107 Similarly, Sox9-Cre labeled a more restricted population of the limb mesenchyme from E10.0, and Sox9+ progenitors gave rise to chondrocytes and osteoblasts.108 However, it is unclear whether a particular group of limb mesenchymal progenitors generate Sox9+ osteochondral progenitors.
SSCs in endochondral bone formation
The hypertrophic chondrocytes of the fetal growth plate secrete Indian hedgehog (Ihh), which induces the expression of parathyroid hormone-related peptide (PTHrP) in the periarticular perichondrium that signals to PTHrP receptor in the pre-hypertrophic chondrocytes and delays Ihh production, and therefore a PTHrP-Ihh feedback is formed to maintain the growth plate structure.81,83 Besides this, Ihh directly targets to the perichondrial progenitors and commits their fate into Osx+ osteoblast precursors.68,109 Hypertrophic chondrocytes also secrete vascular endothelial growth factor (VEGF) and attract vascular invasion to form the primary ossification center.110
Col2-Cre marked perichondrial cells from E12.5, and Col2+ perichondrial cells gave rise to all the Col1a1-GFP osteoblasts at E14.5 and occupied the primary ossification center at E15.5.67,68 Absence of Runx2 is dispensable for the appearance of Col2+ perichondrial cells.67 Osx-cre marked perichondrial cells in a more restricted domain than Col2-Cre at E12.5. At E14.5, Osx+ cells resided in the inner perichondrium broader than Col1a1+ cells.67 Importantly, the perichondrial Osx+ pre-osteoblasts but not the Col1a1+ mature osteoblasts, translocated into the bone marrow along with the blood vessels in a pericyte-like fashion during development.111 These Osx+ progenitors transiently gave rise to trabecular osteoblasts, osteocytes and stromal cells, and disappeared along with longitudinal growth.69
A group of Nes-GFP+ cells were found in the perichondrium at E12.5, but these cells were endothelial. From E13.5, a group of nonendothelial CD31−Nes-GFP+ cells appeared in the innermost of the perichondrium and increased with endochondral bone formation. The emergence of CD31−Nes-GFP+ progenitors required Ihh and Runx2, and a portion of CD31−Nes-GFP + cells expressed Osx.68 Similar to Osx+ pre-osteoblasts, CD31−Nes-GFP+ cells invaded into the primary ossification center along the blood vessels. However, as Nes-GFP labeled both the endothelial and nonendothelial cells, Nes-CreER preferentially labeled the endothelial CD31+Nes-GFP+ cells.68 Thus, this marker seems not suitable for the tracing of osteoblast lineages. Embryonic Gli1+ progenitors also resided at perichondrium, articular cartilage surface, and Ranvier’s groove at E13.5, and contributed to primary ossification center at E18.5.98 To conclude, perichondrial cells participate in the formation of primary ossification center. It is likely that Col2+ cells give rise to CD31−Nes-GFP+ and Osx+ cells, which partially overlap.
Besides the contribution of perichondrial cells to the nascent bone marrow, growth plate hypertrophic chondrocytes transdifferentiate into osteoblast lineages and stromal cells.112,113,114 Col2-Cre not only labeled perichondrial cells but also growth plate chondrocytes since E12.5.67 A single tamoxifen injection at E13.5 to pulse chase the fate of Col2-CreER labeled cells further proved that Col2+ cells contributed to primary ossification center formation at E16.5 and postnatal day 0 (P0). Col2+ cells and descendants occupied both the primary ossification center and secondary ossification center (SOC) at P21.67 In fact, the rapid longitudinal bone growth during the fetal-neonatal period is supported by constant consumption of Col2+ chondroprogenitors, which generated short and multiclonal columns. The chondroprogenitors obtained the ability of self-renewal and formed large monoclonal columns only after the formation of SOC, along with the rise of a stem cell niche at the resting zone of the epiphyseal cartilage.42
As bone grows and vascular invasion progresses, it is possible that SSCs are established in the marrow cavity. Studies have uncovered different groups of SSCs located perivascular and support hematopoiesis postnatally.39,60,78,94,115,116 Though HSCs home to bone marrow around birth, whether the hematopoiesis-supporting cells are pre-established during fetal bone development is unclear.117 One interesting finding is the C-KIT+ mesenchymal cells.118 C-KIT ligand/C-KIT signaling is known to maintain HSCs in bone marrow. Postnatal C-KIT+ cells did not mark osteoblasts or adipocytes, but fetal C-KIT+ progenitors gave rise to bone and bone marrow stroma. The chondrocytes, pre-osteoblasts and bone marrow stromal cells at E16.5 were labeled when KitMerCreMer;R26R-tdTomato mice received tamoxifen at E12.5 and E14.5.118 However, it is unclear whether the bone marrow C-KIT+ cells are derived from perichondrial or growth plate chondrocytes, or re-established in bone marrow.
SSCs in postnatal long bone growth
From neonatal to early adolescence, the growth plate chondroprogenitors are the major source of osteoblasts and stromal cells. Tracing the fate of early postnatal Col2-expressing cells by a single tamoxifen injection to Col2-CreER;R26R-tdTomato mice at P3 showed that Col2+ cells gave rise to osteoblasts and stromal cells in the metaphysis and epiphysis, but not diaphysis at 1 month of age. These Col2CreER-P3 cells continued to become osteoblasts, stromal cells and adipocytes for 1 year.67 Besides Col2, genes involved in cartilage development including Sox9 and Acan, are able to mark growth plate chondroprogenitors. Both Sox9-CreER and Acan-CreER marked comparable populations of osteoblasts and stromal cells at least up to 6 months when receiving a single dose of tamoxifen at P3.67
More solid evidence that the growth plate harbors SSCs comes from the study of the PTHrP+ chondrocytes.41 A group of PTHrP-mCherry+ chondrocytes that were less proliferative appeared at the resting zone of growth plate at P3 and rapidly increased in number between P6 and P9. Lineage tracing using Pthrp-CreER;R26R-tdTomato mice and Pthrp-CreER;R26R-Confetti mice proved that a single PTHrP+ chondrocyte gave rise a longitudinal column. Long-term tracing showed that a fraction of PTHrP+ chondrocytes differentiated into osteoblasts and stromal cells, but not adipocytes in bone marrow. Besides, flow cytometry analysis showed PTHrP-mCherry+ resting chondrocytes exhibited immunophenotype that partially overlapped with the CD51+CD90−CD105−CD200+ mSSCs. The appearance of SOC was crucial for the self-renewal and clonality of PTHrP+ chondrocytes, as P12 PTHrP+ cells, but not P9 PTHrP+ chondrocytes, survived rounds of passage in vitro.41 This is in accordance with the finding that growth plate SSC niches emerged with the formation of SOC.42 Recently, a group of FoxA2+ SSCs residing at the top compartment of the resting zone and immediately adjacent to SOC were identified.119 FoxA2+ SSCs were distinct population from PTHrP+ cells and possessed higher clonogenicity and longevity. At early postnatal stage, FoxA2+ cells generated SOC bone cells and growth plate chondrocytes. After P28, FoxA2+ cells only contributed to chondrogenic lineages. The major function of FoxA2+ SSCs was to maintain growth plate turnover and regeneration after injury.
Besides the growth plate chondroprogenitors that generate bone components, several types of SSCs or lineage-restricted progenitors emerge around the highly vascularized marrow space just beneath the growth plate and function as an important source for osteoblasts and marrow stromal cells.68,69,97,98 Neonatal Osx+ cells were located at primary spongiosa and around the cortical bone. A single tamoxifen injection to Osx-CreERT2;R26R-tdTomato mice at P5 labeled Tomato+ cells that gave rise to osteoblasts and osteocytes after 3 weeks. Surprisingly, Tomato+ cells continuously gave rise to long-lived marrow stromal cells at least for 32 weeks.69
Postnatal Gli1+ cells were observed at the chondro-osseous junction beneath the growth plate, which gave rise to Col1a1-GFP osteoblasts.98 Gli1+ cells highly expressed Pdgfra and Runx2, but not Lepr and Osx at 1 month of age. At 6 months, half amount of the Gli1+ cells were Lepr+, indicating postnatal Gli1+ cells gave rise to Lepr+ cells with age. Both hedgehog and Wnt signaling were required for the osteoblastic fate commitment of Gli1+ cells. However, Gli1+ were mainly enriched in young postnatal mice and dramatically diminished with age.98
Gremlin1 also labeled a group of postnatal non-perivascular osteochondroreticular (OCR) stem cells that located at the primitive mesenchyme and primary spongiosa at P1.97 Grem1+ cells gave rise to chondrocytes, osteoblasts, marrow stromal cells but not adipocytes. Four weeks after a single injection of tamoxifen to Grem1-CreERT;R26-tdTomato;col2.3-GFP mice, red fluorescence was observed in a majority of bone and chondrocytes within the metaphysis and epiphysis, but not in diaphysis. DTA-driven Grem1+ cells ablation significantly diminished postnatal bone formation in the femoral epiphysis. Compared with Nes-GFP+ cells, Grem1+ cells exhibited a higher CFU capacity. Transcriptional profiling of Grem1+ cells indicated higher osteochondrogenic potential and inhibition of adipogenesis.97
Aforementioned SSCs and progenitors are marked by promoters of the transgenes that are actively involved in skeletal development. This unavoidably poses a question that whether a panel of transgenes can mark SSCs once the genes play an essential role in bone development. One intriguing example is the Hox11 paralogous genes that specifically participate in zeugopod pattern formation.120,121 By lineage fate-mapping of Hoxa11-EGFP and Hoxa11-CreERT2-expressing cells, members in Wellik Laboratory demonstrated that Hox11 lineage-positive cells were sufficient to give rise to all the skeletal components regionally restricted to zeugopod throughout life, highlighting SSCs arise from the earliest stages of bone development and can be labeled by a region-specific marker.120,121 However, Dlx5-CreER only transiently labeled a group of cells and made little contribution to bone components, despite an essential role of Dlx5 in development.41 Therefore, it remains to be answered why only some promoters label the authentic SSC populations.
Skeletal stem cells in long bone maintenance
The fate transition of short-term progenitors to long-lived SSCs
The contribution of chondrocytes to bone marrow osteoblasts gradually decreases with the completion of long bone growth. Adult bone marrow SSCs become the major source of osteoblasts instead. Aforementioned studies have shown that the short-term perinatal Col2+ chondrocytes and Osx+ cells gave rise to long-lived bone marrow SSCs, indicating a reestablishment of cellular identity from osteochondral progenitors.
Using dual-recombinase fate-mapping system, Shu et al. captured the transition of chondrocytes to adolescent bone marrow SSCs.77 They first constructed Col2-Cre;LeprdreER;R26LSL-ZsGreen;R26RSR-tdTomato mice, in which the Col2-Cre-derived cells expressed ZsGreen and Lepr-Dre-derived cells expressed Tomato. After tamoxifen induction, a great portion of ZsGreen+ cells and Tomato+ cells overlapped, indicating the genetic relation of Col2+ cells and Lepr+ cells. A mutually exclusive tracing system (Col2dre;Leprcre;IR mice) was then developed, in which the first recombination prevented the other. The frequency of Lepr-Cre-marked ZsGreen+ cells was greatly reduced, accounting for only 11% in single-recombinase Leprcre;IR mice, rigorously demonstrating Col2 lineage sat upstream of Lepr lineage. Interestingly, the osteoblasts formed by Col2+ cells and Lepr+ cells exhibited a spatially separate distribution, where the Lepr-derived osteoblasts first emerged in diaphysis and progressively spread to metaphysis. The molecular mechanism regulating this transition remains to be answered. Nonetheless, growth plate Col2+Col10a1+ chondrocytes only gave rise to ~58% of Lepr+ cells. About 12% of the Lepr+ cells were derived from Col2+Col10a1− periosteal cells and the origin of the remaining 30% is unknown.77 One study compared the functional difference of growth plate (Col10a1+) and non-growth plate (Col10a1−) derived Lepr+ CFU-Fs. They exhibited similar osteogenic and adipogenic capacities in vitro. Surprisingly, when transplanted underneath renal capsule, the Col10a1+ CFU-Fs formed a complete ossicle with bone marrow whereas Col10a1− CFU-Fs only formed bone.122
Bone marrow LepR+ cells only comprised 0.3% of bone marrow cells, but accounted for 94% of CFU-Fs.64 LepR+ cells arose postnatally, residing around sinusoids and arterioles. LepR+ cells expressed MSC markers Prx1, PDGFRα, CD51 and Nes-GFPlow, and were highly enriched for hematopoiesis-supporting factor Scf-GFP and Cxcl12-DsRed. Lineage tracing showed LepR+ cells gave rise to a majority of osteoblasts and adipocytes in the bone marrow from 8 weeks of age. LepR+ cells remained quiescent physiologically, but were rapidly activated in response to irradiation and bone fracture. Transplantation of LepR+ cells into the bone marrow of sub-lethally irradiated mice supported bone, cartilage and adipocytes formation.64
A major difference between Col2+/Acan+ osteoprogenitors and Lepr+ SSCs lies in their function in supporting bone longitudinal growth and appositional remodeling, respectively.77 Loss of Runx2 in perinatal Acan+ cells retarded bone growth while in LepR+ cells reduced cortical bone thickness. Mechanical stimulus such as running only improved the osteoblastic differentiation of perinatal Acan+ cells but not adult LepR+ cells. Importantly, tamoxifen-induced expression of LepR-CreER cells at P1-P3 only gave rise to ~28% of all bone marrow LepR+ cells after 2 months, and these perinatal LepR+ cells made little contribution to bone formation, which makes LepR-Cre/LepR-CreER an ideal system to study postnatal adult bone marrow skeletal stem cells.77
Cxcl12-CreER labeled a dormant subset of Cxcl12-GFPhigh perisinusoidal stromal cells that resided in the central marrow and ubiquitously expressed Lepr. In response to cortical bone injury or marrow ablation, Cxcl12-CreER-expressing cells transformed into skeletal stem-like cells via Wnt signaling, and contributed to bone regeneration. This transition indicates the cellular plasticity of Cxcl12-CreER-expressing cells.123
Bone marrow SSCs and niches
Apart from the contribution to osteo-lineages, one major function of adult bone marrow SSCs is to support hematopoiesis.60,94,115,124,125 Perivascular stromal cells, including endothelial cells, LepR+ cells, CXCL12-abundant reticular (CAR) cells and Nes-GFP+ cells, are implicated in hematopoietic stem and progenitor cell maintenance by secreting CXCL12 or stem cell factor (SCF).78,94,126
Nestin was first identified to label bone marrow SSCs, but different Nestin transgenes, including Nes-GFP, Nes-Cherry, Nes-Cre, and Nes-CreER, seemed to label different groups of perivascular stromal and endothelial cells.60,78 It is now confirmed that Nes-GFP+ cells labeled perivascular SSCs and constituted HSC niche component. HSCs transplanted into lethally irradiated mice homed to Nes-GFP+ cells and ablation of Nes+ cells impaired HSC content.78 However, other studies reported that SCF or CXCL12 deletion in Nes-Cre/Nes-CreER-expressing cells hardly affect HSCs frequency and function.60
LepR+ cells largely overlapped with CAR cells and constituted the HSCs niche. Knockout of Scf from LepR+ cells but not Col2.3-Cre-expressing osteoblasts depleted quiescent HSCs from bone marrow.60 Ebf3-Cre also labeled CAR/LepR+ cells and Ebf3+ cells were self-renewable. Interestingly, Ebf3 deletion in LepR-Cre-expressing or Prx1-Cre-expressing cells led to osteosclerosis and HSCs depletion, indicating the important role of Ebf3 in maintaining stemness and HSCs niches.93
Endosteal niches harbored osteoprogenitors instead. It is noteworthy to mention that different from fetal bone where the perichondrial Osx+ osteoblast precursors gave rise to mature osteoblasts and osteocytes, adult osteocalcin+ osteoblasts and Osx+ pre-osteoblasts turned over rapidly, and were continuously replenished by SSCs or lineage-committed progenitors.127 The myxovirus resistance-1 (Mx1)-Cre labeled osteo-lineage-committed progenitors at endosteal bone that efficiently gave rise to osteoblasts but not adipocytes or chondrocytes in vivo, though labeled cells are capable of multi-lineage differentiation in vitro.127 Interestingly, by conditional deleting CXCL12 in different niches, two companion work demonstrated that osteo-lineage-committed cells supported lymphoid progenitors.94,115 In support of these findings, a recent study identified a subpopulation of periarteriolar LepR+osteolectin+ osteogenic progenitors. These cells were fast-dividing and short-lived, which was different from the quiescent nature of self-renewable LepR+ cells. LepR+osteolectin+ cells were responsive to mechanical stimulus and supported lymphopoiesis.128 Moreover, this study indicated that not all LepR+ perivascular cells are long-lived SSCs. Some subgroups of LepR+ population are indeed committed progenitors.
Bone marrow adipocytes precursors
Bone marrow adipocytes share a mesenchymal origin with osteoblasts.129,130 The fate decision of SSCs is tilted toward adipocytes during ageing or under pathological conditions such as myeloablation, high-fat diet and caloric restriction.22,86,129,131 As discussed above, many transgenes (e.g. Prx1-Cre, Osx-Cre, LepR-Cre) are able to mark marrow adipocytes.64,69,132 Importantly, Adiponectin-CreER (Adipoq-CreER) was reported to mark a subpopulation (~5%) of LepR-Cre-expressing cells that preferentially differentiated into adipocytes.133
Using flow cytometry and transplantation assays, Ambrosi et al. plotted the marrow adipocytic lineage differentiation trajectory.72 A population of multipotent perivascular CD45−CD31−Sca1+CD24+ progenitors gave rise to lineage-committed CD45−CD31−Sca1+CD24− adipogenic progenitor cells (APCs), which generated a more mature CD45−CD31−Sca1−Zfp423+ pre-adipocytes (preAd). Meanwhile, a group of CD45−CD31−Sca1−PDGFRα+ osteochondrogenic progenitors could be differentiated from CD45−CD31−Sca1+CD24+ progenitors in parallel.72
Likewise, a population of bone marrow adipogenic lineage precursors (MALPs) that situated downstream of SSCs and upstream of lipid-laden adipocytes were identified by Qin Lab.134,135 They profiled the Col2-Cre;R26R-tdTomato-expressing cells using scRNA-seq, and identified a cluster of MALPs that were efficiently labeled by Adipoq-Cre/Adipoq-CreER as stromal cells and pericytes since newborn. These cells were non-proliferative, non-lipid-laden (Perilipin−) and protruded to form a three-dimension network. MALPs could differentiate into lipid-laden Adipoq+Perilipin+ mature adipocytes. Ablation of MALPs with diphtheria toxin impaired blood vessels but increased bone formation in Adipoq-Cre;DTR mice, without significant influence on hematopoiesis.134 Long et al. reported the Col10a1-Cre-derived cells in the bone marrow were not pericytes, though they were associated with blood vessels.122 As growth plate Col10a1+ cells are immediately downstream of Col2a1+ cells, why do Adipoq-Cre-marked populations function as pericytes needs further clarification. More importantly, a recent study constructed the Adipoq-Cre;mTmG mice and traced the fate of Adipoq+ cells from 1 month to 9 months. They surprisingly identified that Adipoq+ cells contributed to bone formation with age. In 1-month-old mice, GFP-expressing cells were restricted to marrow stroma. In 9-month-old mice, the ratio of GFP-expressing osteocytes to all osteocytes almost reached 40%. This study indicated that at least a subset of Adipoq-Cre-expressing cells were bipotent.136
Skeletal stem cells in craniofacial bones
Origin of craniofacial SSCs
The craniofacial bones originate from the paraxial head mesoderm and the cranial neural crest cells (CNCCs).138,139 The anterior skull bones are derived from the CNCCs, whereas the posterior part originates from the paraxial head mesoderm.138 CNCCs are highly plastic compared to the trunk neural crest cells.140 CNCCs give rise to a majority of mesenchymal progenitors of facial cartilage and bone elements. Twist1 gene is activated upon delamination in the cranial compartment and directs CNCCs toward a mesenchymal fate. Intriguingly, sustained overexpression of Twist1 was sufficient to reverse the developmental program of the trunk crest to a mesenchymal route.140 Schwann cell precursors (SCPs) are direct progeny of neural crest and generate osteochondral progenitors in facial region.141 The contribution of individual CNCC to facial components and the spatial organization of the ectomesenchyme has been revealed.142 During early outgrowth and shaping of the facial regions, organization is mainly driven by oriented cells division, allocation and relocation but minimal individual cell migration, which indicates a conserved program of facial outgrowth.142
SSCs in alveolar bone
Distinctive from long bone, our understanding of niches and stem cells in alveolar bone is lacking. We have plotted a single-cell atlas of mouse mandibular alveolar bone and identified a microenvironment with a higher portion of mature immune cells compared with long bone marrow. A high level of Oncostatin M (Osm) was found in alveolar bone marrow monocytes/macrophages, which promoted osteogenic differentiation and inhibited adipogenic differentiation of alveolar MSCs.143 Further, we have identified a tissue-resident LepR+ subpopulation in alveolar bone of the adult mice, which contributed to socket healing of tooth extraction. The biological behavior of LepR+ cells were regulated by PTH/PTH1R signaling.116
SM22α-lineage niche cells have been recently found to regulate regeneration of alveolar bone.144 They were quiescent physiologically and expanded following bone injury. Moreover, SM22α-lineage niche cells did not act as stem cells and instead regulated alveolar bone regeneration via PDGFRβ-triggered hydrogen sulfide (H2S) production.144
Cranial suture stem cells
Adult Gli1+ suture stem cells (SuSCs) were responsible for maintenance and injury repair of cranial bones.145 Ablation of Gli1+ SuSCs caused craniosynostosis and growth retardation of mouse skull.145 The bone regeneration was faster when injury site was located closer to the suture, supporting that Gli1+ SuSCs are the source for the cranial bone regeneration.146 Craniosynostosis of Twist1+/− mice model showed reduced number of Gli1+ SuSCs, suggesting that diminished SuSCs may account for suture closure.145 A translational research showed Gli1+ SuSCs-based functional cranial suture regeneration corrected skull deformity along with cognitive defect of Twist1+/− mice model. The regenerated suture created a niche for endogenous SuSCs migration to support cranial bone homeostasis and injury repair.147
Distraction osteogenesis is adopted to correct underdevelopment of the skull and maxilla. In mouse suture expansion model, Gli1+ SuSCs contributed to bone remodeling upon tensile force loading. Reduction of Wnt signaling decreased such contribution.148
Axin2-expressing population within cranial suture were long-term self-renewal and clonogenic. Axin2+ SuSCs contributed to injury repair of cranial bones in a cell autonomous manner.149 CD51+CD200+ cells were confirmed as resident SSCs in cranial sutures.150 Reduced CD51+CD200+ population was found in posterior frontal suture closure and craniosynostosis. Wnt activation increased CD51+CD200+ cells and rescued craniosynostosis by preventing suture closure.150
Prx1-expressing cells were identified in the cranial suture mesenchyme at P12 of Prx1-CreER;EGFP mouse model.151 Prx1+ cells were identified in the posterior frontal sutures, coronal sutures, sagittal sutures, and lambdoid sutures by intravital microscopy.152 Prx1+ cells progressively reduced from 8-week-old mice to 32-week-old mice. Ablation of postnatal Prx1+ lineage using DTA showed that they were required for cranial bone defect regeneration, but not for calvarial growth.152
Stem cells in periodontal ligament
Postnatal MSCs of human periodontal ligament was firstly isolated in 2004.153 CD45−Ter119−Tie2−CD51+Thy−6C3−CD105−CD200+ mSSCs were recently sorted out from mouse periodontal ligament (mPDL) tissue.154 mPDL-derived SSCs exhibited clonogenic, cementogenic and odontogenic capacity.154
Gli1+ cells were multipotent stem cells within mouse molar PDL and contributed to turnover of periodontal supporting tissue.155 PDL Gli1+ cells resided around the neurovascular bundle especially in apical region of the mouse molar. PDL Gli1+ cells were responsive to Wnt signaling and removal of occlusal force inhibited Gli1+ cells activation by upregulating sclerostin.155 Mechanoresponsive property of Gli1+ cells was supported by Yap, a classical mechano-transduction factor.156 Using Gli1LacZ/+ and Gli1-CreERT2;R26RtdTomato mouse models, study showed cellular cementoblasts did not express Gli1 (β-galactosidase-), and Gli1+ cells contributed to cementum formation (Tomato+). Ablation of Gli1+ cells using DTA reduced cementogenesis. Reduction of Wnt/β-catenin signaling in Gli1+ cells decreased cementum formation postnatally.157 PDL is responsive to Wnt signaling.158 Axin2+ cells were identified as PDL stem cells supporting the homeostasis of the periodontium. Orthodontic tension force increased the number of PDL Axin2+ cells.159 In response to tooth extraction, Axin2+ cells contributed to bone healing, and healing accelerated when WNT3A protein was applied.160 Axin2+ lineage also contributed to cementum formation at postnatal rapid growth period.161 Comparatively, K14+ epithelial cells were initially active at early development and reduced in number at postnatal period from P28 to P56. Ablation of Axin2+ lineage caused cementum hypoplasia. Activation of Wnt/β-catenin signaling in Axin2+ lineage promoted cellular cementogenesis.161 CD90+ cells also gave rise to cementoblasts during postnatal development. During adult homeostasis, however, only Axin2+ contributed to cementogenesis, whereas CD90+ cells were reactivated in mice with periodontitis.162
Postnatal Prx1+ cells were found within the PDL of mouse incisor and ablation of Prx1+ lineage at P3 led to PDL enlargement at P21.163 Prx1+ cells contributed to regeneration of non-critical size periodontal defect. Prx1+ cells were also identified within human PDL.163 Deletion of Pth1r using Prx1-Cre arrested the eruption of the mandibular incisor.164 Lineage tracing of postnatally Prx1-Cre;R26R-tdTomato mouse showed Prx1+ cells were mainly restricted to the first molars, in which PDL Prx1+ cells contributed to almost all types of mesenchymal cells. PDL reconstruction of the transplanted molar required Prx1+ cells from the alveolar bone of the recipient mice.165
We recently constructed a LepR-CreER;tdTomato mouse model and identified a subset of LepR+ cells within the PDL region. Such LepR+ subsets were specifically located adjacent to blood vessels of the PDL and contributed to root furcation bone healing when PDL injury was induced (unpublished data). We also found a Ctsk+ subpopulation within the PDL using Ctsk-Cre;tdTomato mice. Ctsk+ cells were present throughout the entire PDL including furcation area and apical area. Ctsk+ cells also contributed to cementum and alveolar bone development, which share a developmental origin with PDL (unpublished data).
Stem cells in Schneiderian membrane
The contribution of the sinus membrane to bone regeneration is widely concerned. Postoperative new bone formation was reported after maxillary sinus augmentation, suggesting pluripotent stem cells exist in the sinus membrane.166 In an experiment by Helms Lab, the posterior paranasal sinus in a preclinical mouse model served as a recipient site for autografts or bone graft substitutes, and autograft indicated faster new bone formation rate. Internal periosteum-derived Wnt-responsive progenitors of the maxillary bone was found to contribute to new bone formation.167
A subset of Krt14+Ctsk+ cells have been identified as osteoprogenitors, which contribute to homeostasis and injury-induced osteogenesis of maxillary sinus floor. Such subset exhibited both epithelial and mesenchymal properties, and specifically played a role in bone regeneration after maxillary sinus floor lifting. Lineage tracing with dual recombinases showed that descendants of Krt14+Ctsk+ progenitors, which are Krt14−Ctsk+, underwent robust osteogenesis. Similarly, a Krt14+Ctsk+ subset was found in human Schneiderian membrane as well (Fig. 2).168

Schematics of craniofacial progenitors for bone formation. Craniofacial region harbors various cell sources. From the latest literatures and our research, the indicated cell sources play crucial roles in craniofacial bone formation and healing
Stem cells in other craniofacial tissues
A stem cell niche inhabits in tooth.169 Stem cells from human exfoliated deciduous teeth were identified.170 Stem cells in dental pulp and from oral mucosa were characterized and reported.171,172 Pericyte-derived and nonpericyte-derived MSCs altogether contributed to tooth growth and repair.173 A small population of Gli1+ slow-cycling MSCs supported by neurovascular bundle was identified to contribute to mouse incisor continuous growth.174 Proteolipid protein (PLP) was also reported to label slow-cycling cells in mouse incisor and contributed to a proportion of odontoblasts, suggesting the glial origin of dental pulp of mouse incisor.175
Stem cell-based temporomandibular joint regeneration is extensively studied.176,177,178 Potential of various human stem cells to regenerate TMJ has been explored and in vivo studies are expected.176 Notably, fibrocartilage stem cells (FCSCs) at the superficial zone of the condylar cartilage were identified with αSMA-CreERT2 and they generated and maintained the cartilage structure. A single FCSC was sufficient to form cartilage anlage, and recapitulated endochondral ossification to form bone and support hematopoiesis in vivo.179 Moreover, FCSCs could be isolated from human TMJ cartilage, suggesting the translational potential for condylar cartilage treatment of TMJ disorders.180,181
Skeletal stem cells in periosteum
Periosteal SSCs in long bone
Periosteum is derived from outer layer of embryonic perichondrium, and harbors SSC niches that support cortical bone formation and architecture, and bone fracture repair postnatally (Fig. 3).71,99,182,183,184,185 Periosteal cells displayed an embryonic Prx1-Cre-expressing mesenchymal origin and were not derived from vascular invasion.186 Ctsk-Cre marked periosteal heterogenous mesenchymal cells despites its ability to label endosteal osteoclasts.71,187 FACS analysis divided CTSK-mGFP cells into three groups, the CD200+CD105− periosteal stem cells (PSCs), CD200−CD105− periosteal progenitor 1 (PP1) cells, and CD105+CD200variable periosteal progenitor 2 (PP2) cells. PSCs first appeared at perichondrium of E14.5 femur and displayed self-renewal ability and clonal multipotency. PSCs generated the whole spectrum of CTSK-mGFP cells after serial transplantation assays in mammary fat pad. Therefore, PSCs are bona fide SSCs. Interestingly, PSCs did not support hematopoiesis and directly differentiated into osteoblasts via intramembranous ossification at physiological condition or when transplanted under the kidney capsule. However, PSCs remained plastic and contributed to fracture callus formation via both intramembranous and endochondral bone formation. Blocking the osteogenic ability of Ctsk-Cre-expressing cells via knockout of Osx dramatically impaired the cortical bone formation and fracture repair.71

Skeletal stem cells in periosteum. Periosteum of long bones, calvaria, and mandible harbors SSCs that participate in bone development and injury repair
Ctsk-Cre-mediated deletion of liver kinase b1 (Lkb1) led to osteosarcoma via activation of mammalian target of rapamycin complex 1 (mTORC1).188 Besides, Ctsk-Cre also labeled progenitors at the perichondrial groove of Ranvier, a recognized niche for stem cells.189,190 Knockout of Ptpn11, genes encoding tyrosine phosphatase SHP2, in Ctsk-Cre-expressing cells caused metachondromatosis due to overactivation of hedgehog signaling at the perichondrial groove of Ranvier.189 Thus, these studies indicate that postnatal periosteal/perichondrial Ctsk-Cre-expressing cells are an origin of tumor when dysregulated. Moreover, Ctsk-Cre labeled PSCs played an indispensable role in maintaining postnatal growth plate structure and prolonged longitudinal bone growth by secreting IHH. Interestingly, IHH production from developing growth plate progressively reduced with age, whereas postnatal PSCs-derived IHH supported the proliferation of growth plate resting zone SSCs, indicating a crosstalk between periosteal and growth plate stem cells.191
αSMA was identified to mark periosteal progenitors that differentiated into osteoblasts and chondrocytes during fracture healing.95 Ortinau et al. further identified a group of Mx1+αSMA+ periosteal cells. Around 80% of Mx1+αSMA+ cells expressed SSC immunophenotypic markers (CD105+CD140a+) whereas only ~10% of Mx1−αSMA+ cells were CD105+CD140a+. Mx1+αSMA+ periosteal cells were long-lived population and continuously gave rise to osteoblasts. Moreover, Mx1+αSMA+ periosteal cells expressed the C–C Motif Chemokine Ligand 5 (CCL5) receptor CCR3 and CCR5, and migrated in a CCL5-dependent manner to bone injury site.99 Nevertheless, the relationship between CTSK-mGFP PSCs and Mx1+αSMA+ SSCs is currently unknown.
Periosteal SSCs in calvarial bone
In a mouse model of calvarial injury, Mx1+αSMA+ cells migrated to and resided in the center of the injury site, which differentiated into Mx1+αSMA− bone cells. Administration of CCL5 promoted migration of Mx1+αSMA+ periosteal cells to the injury site for calvaria repair (Fig. 3).99
Periosteal SSCs in mandible
By mapping the transcriptional landscape of the human mandible periosteum, a Ctsk+ subset was identified. Further identification presented Ctsk+Ly6a+ subset as periosteal progenitors that were activated during injury repair (Fig. 3).192 The periosteum SSCs are regulated by schwann cells in a paracrine manner.193 Myelin PLP+ schwann cells played a role in tissue repair.194,195,196 By constructing an inferior alveolar nerve (IAN) denervation mouse model, study showed adult schwann cell paucity reduced contribution of SSCs to mandibular healing.193
Mandible periosteal SSCs are mechanoresponsive. Members in Longaker Lab analyzed the chromatin and transcriptional profile of SSCs during mandible distraction osteogenesis, and found an activation of CNCC-like program in SSCs in focal adhesion kinase (FAK)-dependent manner. This study indicated that during mouse mandible regeneration, SSCs could reverse to developmental stage and regained a CNCC-like identity.197
Single-cell landscape of skeletal components
Single-cell transcriptomics have revealed a more complex landscape of the skeletal components.198,199 The differentiation trajectory of progenitors during bone development and homeostasis was plotted, unveiling the heterogeneity of SSCs.
Long bone at single-cell resolution
Human embryonic SSCs ontogeny was dissected by scRNA-seq.90 Cells in limb buds at 5 weeks post conception (WPC) were divided into 10 subsets, of which 4 subsets were of mesenchymal origin. They were limb bud mesenchymal subsets 1–3 (LBM1–3) that were PRRX1high and differentially expressed PDGFRA, and osteochondrogenic progenitors (OCPs) that were PRRX1+, SOX9low and PDGFRAhigh. LBM1 and LBM2 were distally positioned, preferentially expressing 5′ HOX genes, while LBM3 and OCPs were at proximal end, preferentially expressing 3′ HOX genes. LBM2 was highly proliferative, enriched with metabolic process and closely associated with AER, probably annotating the progress zone. Nevertheless, a comparative analysis showed a much lower portion of AER cluster and lack of LBM2 in mouse hindlimb buds at E11.5, indicating an accelerated maturation of mouse limb bud. Embryonic long bone was already formed at 8 WPC, and the osteochondral lineage was divided into 7 subsets, including three OCP subsets. They were the limb bud-derived mesenchymal cells (LBDMCs), BMSCs and embryonic skeletal stem/progenitor cells (eSSPCs). LBDMCs highly expressed TWIST2 and sat upstream of BMSCs (CXCL12high and PDGFRAhigh) and eSSPCs, a novel perichondrial subset that was highly enriched with FOXP1/2/4 regulons. The PDGFRAlow/–PDPN+CADM1+ cells could enrich the self-renewable eSSPCs and gave rise to osteochondral lineages in vivo, but did not reconstitute hematopoietic environment. Besides, the eSSPCs population could be identified in mouse E15.5 long bones.90
Kelly et al. plotted the single-cell landscape of mouse embryonic hindlimb development.200 Whole hindlimb tissue was dissociated for scRNA analysis at four time points (E11.5, E13.5, E15.5. E18.5). Seven clusters were identified across all time points, representing cartilage, bone/tendon, skin, muscle, blood, vasculature and cell cycle. An unsupervised algorithm predicting the developmental trajectories showed a branch of musculoskeletal precursors arose at E11.5 and the vasculature branch arose at last.200
Single-cell transcriptomics helped to prove the hypothesis that non-mitotic hypertrophic chondrocytes dedifferentiate into skeletal stem and progenitor cells, which then generate osteoblasts and adipocytes in the bone marrow.122 Col10a1-Cre;R26R-tdTomato-marked hypertrophic chondrocytes and progenies at E16.5 and 2 months of age were sorted for scRNA-seq and in vivo validation. At E16.5 when primary ossification center starts to form, scRNA-seq identified an intermediate cluster that linked hypertrophic chondrocytes to osteoblasts, and exhibited SSCs-like properties. Similarly, 3 clusters of marrow-associated cells derived from hypertrophic chondrocytes at 2 months of age exhibited a more primitive state compared with osteoblasts, and highly expressed SSC markers Lepr, Gremlin1, Pdfgra, Cxcl12 and Pdgfrb. Nearly 25%–45% of marrow SSCs were derived from hypertrophic chondrocytes, and perichondrium/periosteum might constitute the other portion of SSCs in bone marrow.122
In align with this endochondral ossification process, a group of mesenchyme-derived fatty acid binding protein 5 (FABP5)-expressing septoclasts that aided in matrix degradation and chondrocyte phagocytosis were reported by Adams Laboratory.201 FABP5+ cells were located at the chondro-osseous border and expressed PDGFRα and PDGFRβ. Single-cell analysis of the PDGFRα-GFP cells identified a small portion of cells that were FABP5+ and Mmp9+. This FABP5+ cluster emerged from and was closely associated with the PDGFRβ+ metaphyseal MSCs populations.201
Postnatal bone marrow consists of highly complicated environment with mesenchymal and hematopoietic cells forming different niches that guarantee the physiological function of hematopoiesis and bone remodeling. Wolock et al. isolated bone marrow nonhematopoietic and non-endosteal stromal cells from adult mice for scRNA-seq, and confirmed that MSCs committed fate into either adipocytes or osteoblasts/chondrocytes. Interestingly, cultured stromal cells exhibited a different landscape with freshly isolated stromal cells, with a larger proportion of cultured cells resembling osteoblast progenitors.202
A more detailed clustering of BMSCs from P21 bone marrow was exploited by Sivaraj et al.137 Seven subclusters were identified, including diaphyseal MSC 1 and 2 (dpMSC1, dpMSC2), metaphyseal MSC (mpMSC), proliferating BMSC (P-BMSCs), osteoprogenitor cells 1 and 2 (OPC1, OPC2), and osteoblasts (OBs). Trajectory analysis indicated mpMSCs were at the center and gave rise to other cell types. However, Lepr expression was detected only in cluster dpMSC1 and dpMSC2, but not in mpMSC. Diaphyseal LepR+ cells were located around sinusoids and highly express Kitl and Cxcl12. A lineage tracing using LepR-Cre;R26R-tdTomato mice showed that metaphyseal tdTomato+ cells did not express dpMSC immunophenotypic markers ESM1 and PDGFRβ, indicating the heterogeneity of LepR+ populations.137 In an integrated analysis of bone marrow environment, LepR+ cells were clustered into four subpopulations.203 Cluster P1 (Mgphigh) and P2 (Lplhigh) highly expressed adipogenic markers, and were closely associated with sinusoidal capillaries. In contrast, cluster P3 (Wif1high) and P4 (Spp1highIbsphigh) were osteolineage-primed, and were located to the trabecular portion. Intriguingly, the P1 and P2 subsets were highly enriched for CFUs, and accounted for the majority of CFU-Fs of total LepR+ cells.203
In particular, a study combining scRNA-seq with spatial transcriptomics revealed a highly heterogenous landscape of the bone marrow stromal cells.80 In specific, CAR cells located at sinusoidal (Adipo-CAR, Cxcl12+Alpl-) and arteriolar/non-vascular (Osteo-CAR, Cxcl12+Alpl+) niches displayed distinct transcriptional profile favoring adipogenesis and osteogenesis respectively. Adipo-CAR cells exhibited a transcriptional pattern highly similar to LepR+ cells.80 However, previous work by Morrison laboratory showed LepR+ cells as main source for both bone and adipocytes in adult bone marrow, with the lepR+osteolectin+ subsets and Adipoq-CreER-expressing subsets giving rise to bone cells and adipocytes respectively.64,128,133 Thus, the relationship of LepR+, LepR+osteolectin+, Adipo-CAR and Osteo-CAR cells are to be elucidated.
Scadden Lab proposed taxonomy for bone marrow stroma that included 17 clusters.204 They consisted of MSCs (Lepr+, Cxcl12+), two osteo-lineage cell (OLC) subsets (Bglap+), four chondrocyte subsets (Acan+, Col2a1+), five fibroblast subsets (S100a4+), three bone marrow-derived endothelial cells (BMEC) subsets (Cdh5+), pericytes (Acta2+) and two clusters that uniformly expressed markers of chondrocytes, osteoblasts and fibroblasts. Here the Lepr+ MSCs highly expressed lepr, adipoq, Cxcl12, Kitl and angiopoietin-1, and were therefore annotated with pre-adipocytic features. Four subsets were further subclustered in Lepr-MSCs. Interestingly, one subset expressed a higher level of OLC-specific genes Osx and Alpl, indicating a continuous transition from Lepr-MSCs to OLCs. Besides, the two OLC clusters were originated from two distinct differentiation trajectories and exhibited different hematopoietic potential. The four chondrocyte subsets in the diffusion map, on the other hand, displayed a classical endochondral bone formation process.204
Besides the taxonomy of bone marrow stroma from transcriptional level, Scadden Lab further clustered the stroma subpopulations using mass cytometry (CyTOF)-based single-cell protein analysis. A total of 28 subsets were defined, among which 14 subsets expressed hematopoietic cytokines. Irradiation eradicated most of the populations, including LepR+ and Nes+ subsets, while CD73+ subset is found highly resilient to irradiation.205 Zhong et al. compared the bone marrow mesenchymal lineages from 3-month and 16-month-old Col2-Cre;R26R-tdTomato mice. A smaller number of early mesenchymal progenitors and a shift toward adipogenic transcriptome were observed in 16-months mice.134 Together, these studies demonstrate the extreme complexity of the bone marrow stromal components and provide valuable sources that could be exploited in the further work.
Craniofacial bone at single-cell resolution
scRNA-seq is extensively applicated in craniofacial research.143,206,207 Calvaria cells collected from P4 pups were subjected to sequencing.208 Transcriptomes of the freshly isolated calvaria osteoblasts and cultured osteoblasts varied. Similar transcriptome was indicated between freshly isolated calvaria osteoblasts and long bone osteoblasts.208 Cells of coronal sutures from E15.5 and E17.5 were collected for scRNA-seq.209 Pre-osteoblasts between suture fronts and periosteum were distinct. A subpopulation above the cranial suture shared ligament characters and persisted into mice adulthood. A chondrogenic-like subpopulation was identified in the dura.209 Holmes et al. also isolated coronal suture cells of E16.5 and E18.5 for scRNA-seq.210 Seven populations were identified and the expression of Hedgehog Interacting Protein (Hhip) was found to mark mesenchymal population. Hhip-expressing cells at neonatal stage contributed to bone growth of calvaria.210 A single-cell atlas of human calvaria at 8 WPC was plotted. The data indicated that the proportions of osteoprogenitors and perichondrial mesenchymal stromal cells were much higher in calvarial bones compared to long bones. Twelve subsets were identified, of which the neural crest-derived cells (NCDC) cluster was PDGFRAlow, PDPN+, and CADM1+, resided in the outer layer of the sagittal suture and mimicked the long bone eSSPC phenotype.90
Disputes and challenges of SSCs
Skeletal stem cells of different origins
A general agreement in SSC field is the diverse origins of SSCs. Unlike hematopoietic system that a group of clearly defined stem and progenitor cells generate the whole lineages arranged by different immunophenotypic markers, the skeletal elements are established and replenished with different sources.117 The human SSCs (hSSCs) and mouse SSCs (mSSCs) lineage trees plotted by Longaker Laboratory are currently the most comprehensive and plausible paradigm that can be referred in skeletal stem cell research.38,66 Nevertheless, some disputes regarding the mSSCs lineage map should be mentioned.
One is that in their experiments skeletal tissue was harvested from P3 mice when SOC and hematopoietic niches are not formed yet.211 One study detected the mSSCs in adult mice (8–16 weeks) and observed a contamination of Col2.3-GFP+ osteoblasts in endosteal mSSCs (~40% GFP+ cells in CD45−CD51+Thy−6C3−CD105−CD200+ cells) and progeny BCSP (~20% GFP+ cells in CD45−CD51+Thy−6C3−CD105+ cells) subpopulations, as well as in periosteal SSC and BCSP. Indeed, over 90% of Col2.3-GFP osteoblasts expressed CD200.212 Another study showed a rapid decrease of mSSCs and BCSP population with age. At E13.5, ~ 4 × 104 SSCs were identified from 105 cells, but the number plumped to less than 100 SSCs per 105 cells at 8 weeks.73 Another issue is the location of mSSCs and BCSP, which were preferentially located at metaphysis and epiphysis. Few cells were detected in diaphysis and mSSCs failed to generate adipocytes even under strong external adipogenic induction in vitro.73 Therefore, mSSCs might produce a majority of bone components at embryonic and juvenile stage, but do not support hematopoiesis and bone remodeling at adult stage.
The perivascular CD45−CD31−Sca1+CD24+ progenitors in adult bone identified by Ambrosi et al. demonstrated another SSC source.72 This group of progenitors mainly resided in epiphysis and could be isolated from diaphysis as well. They exhibited a hematopoietic-supportive and adipogenic transcriptome, and were capable of generating bone, cartilage, stroma, and adipocytes when transplanted under renal capsule. scRNA-seq showed that mSSCs (CD45−CD51+Thy−6C3−CD105−CD200+ cells) and perivascular CD45−CD31−Sca1+CD24+ SSCs were separate clusters, supporting the notion of diverse origins of SSCs.73 Interestingly, a rare endothelial group of LNGFR+ cells in human fetal and regenerative bone marrow went through an endothelial-to-mesenchymal transition under the IL-33 signaling.213 Besides, Schwann cell precursors contributed to long bone development by transdifferentiating into mesenchymal cells.141 Neural crest-derived Nes+ cells differentiated into hematopoiesis-supporting SSCs as well.214
Relation of SSCs labeled by reporter mouse models
There are around 20 types of transgene labels that are reported to identify SSCs and early progenitors. More than 30 types of Cre/CreER-driven reporter mice are constructed to characterize the spatiotemporal feature of SSCs. The information these studies provide is sometimes contradictory and partial overlap of different groups of SSCs are frequently observed. However, several important indications can be addressed from these overwhelming results.
One is the contribution of embryonic/early postnatal skeletal progenitors to postnatal SSCs. Current evidence supports that embryonic/early postnatal short-term progenitors in the perichondrium and growth plate invade into the bone marrow and re-establish into postnatal long-residing SSCs. For instance, cells marked by Osx-CreER at perinatal stage generated long-lived Cxcl12+ stromal cells in the adult bone marrow.69 The same is true for most populations, such as Col2-CreER, Sox9-CreER, Acan-CreER, Gli1-CreER, and PTHrP-CreER-marked cells perinatally.41,67,98 The de-differentiation of Col10a1+ hypertrophic chondrocytes into PDGFRαH2B-GFP-expressing SSCs were visualized.122 More importantly, by constructing a mutually exclusive tracing system, Zhou Laboratory unambiguously demonstrated that Col2+ progenitors gave rise to LepR+ SSCs.77
Second is the overlap of different populations marked by transgenes. At embryonic and perinatal stages, progenitors in the growth plate or immediately beneath it are simultaneously labeled by various transgenes. The postnatal marrow SSCs populations are also highly overlapped. Zhou et al. comprehensively characterized the CFU activity of various SSCs populations.64 These populations were differentially enriched for CFU-Fs, indicating some overlaps among them. Specifically, LepR+ cells denoted the PDGFRα+CD45−Ter119− cells, uniformly expressed Prx1, and enriched for almost all the CFU-Fs. LepR+ cells also highly express Scf-GFP and Cxcl12-DsRed and Nes-GFPlow, so it is thought that LepR+ cells denote CAR cells.
Third is that none of the transgenes faithfully label a homogenous SSC population. Col2-Cre labeled all the chondrocytes and progenies including osteoblasts, stromal cells and adipocytes.67 PTHrP-mCherry more specifically labeled SSCs and early progenitors, but their contribution to bone marrow was modest.41 Notably, Lepr expression was detected in osteo-lineage, endothelial cells, pericytes, and fibroblasts, besides in Lepr-SSCs.204 Therefore, the interpretation of phenotype by Lepr-Cre-driven gene alteration should be cautious. The employment of LepR-CreER system might be a more reliable option than continuous LepR-Cre that is active throughout development. Alternatively, the dual-recombinase-activated lineage tracing (DeaLT) system that eliminates the interference from nontarget cells can delineate the stem cell fate more precisely.54
Technique challenges in skeletal stem cell research
The mineralized structure makes skeletal research difficult. To isolate and culture SSCs, flushing of bone marrow, mechanical crush of the compact bone, enzymatically digestion, and cell sorting procedures are preferentially applied.44,215,216 Digestion releases the endosteal SSCs and yields a higher frequency of SSCs compared with flushing method. However, collagenase digestion is a stress that alters cellular transcriptome.202,217 It is also recommended to frequently change medium and adjust the trypsinization time to minimize hematopoietic contamination.215 Culture of the bone chips to allow fibroblast-like cells migration is also reported to yield stem cells.216 SSCs cultured as nonadherent mesenchymal spheres are reported as well.96 Low oxygen (2%) culture is also recommended.38 Nevertheless, different methods often yield different cell populations. Morphological difference can be easily discerned in cell culture. For example, the PDGFRα+Sca1+ cells were spindle-like while PDGFRα+Sca1− cells appeared more rounded.65 Cell culture impacts SSCs properties as well. PDGFRα+Sca1+ cells lost the ability to home to bone marrow niches once cultured in vitro, while freshly isolated ones successfully reconstructed the vascular niche for hematopoiesis in lethally irradiated recipient mice.65
The functional assessment of SSCs after isolation is another topic. Tri-lineage differentiation assays in vitro are routinely harnessed with well-established protocols.215,216 The osteogenic and adipogenic differentiation was induced with monolayer cell culture, whereas chondrogenic differentiation was preferentially performed with cell pellet. In vivo assays were achieved by transplantation, in which SSCs were loaded into scaffold such as Matrigel and embedded under the skin, the mammary fat pad or kidney capsule of the immunodeficient mice.38,41,44,71 Weeks after transplantation the tissue was subject to histological analysis for the formation of bone, cartilage, stroma and adipose tissue. Bone marrow transplantation is an optional choice and is technically challenging because the mice have to survive irradiation first and then intravenous injection of SSCs is performed.65,127,218 Donor cells labeled with fluorescence and the reporter recipient mice are recommended as bone marrow comprises of huge amounts of cells difficult to discern. Real-time in vivo imaging is harnessed as well and SSCs are usually transplanted at calvarial site for visualization of cell migration and injury repair.99
The construction of Cre-loxP system for lineage tracing is challenging, as it requires both specific and efficient recombination. The mouse models constructed by Nestin transgenes are highly variable that Nes-Cre and Nes-GFP labeled different populations.60,68 PDGFRα-CreER, on the other hand, recombinased poorly in bone marrow PDGFRα+ cells, which were enriched for almost all the CFU-Fs.61,64
The sensitivity of single-cell sequencing should be rigorously interrogated as well. Members in Warman Laboratory treated mice with vehicle or sclerostin-neutralizing antibody, but failed to identify significant changes of bone anabolism-associated transcripts in osteoblasts.208 This study indicates the underpower of scRNA-seq at some settings. More confusingly, unexpected results are obtained from lineage tracing and scRNA-seq experiments even with robust tools. Unwanted cell types are unavoidably labeled or sorted, and hematopoietic lineages often contaminate the mesenchymal cells. Recent scRNA-seq results highlight the contamination issue.123,134 In a single-cell sequencing experiment performed by Ono group, the GFPhigh population gated from Cxcl12GFP/+;Cxcl12-creER;R26RtdTomato bone marrow were subjected to sequencing, but the results yield a substantial fraction (~26.2%) of Cxcl12-GFPneg myeloid cells, lymphocytes, and erythroid cells.123 Similar contamination was observed in Col2-Cre;R26R-tdTomato mice. When the top 1% tdTomato+ endosteal cells were sorted for scRNA-seq, a large number of non-mesenchymal cells were observed. Surprisingly, these cells were validated for detectable Tdt and Col2a1 expression.134 The reason and solution to such massive contamination remains to be answered.
The visualization of skeletal components remains an issue for decades.219 Long-term decalcification diminishes the epitopes on cells. Undecalcified frozen hard-tissue section using Kawamot’s Film method is now routinely harnessed in our lab for von-kossa staining and immunofluorescence.220,221 We also use the method developed by Adams Laboratory to prepare samples for three-dimension visualization.222,223 Tissue section with 20–100 μm thickness obtained with this method are subjected for immunostaining and confocal visualization. Besides, tissue-clearance technique is applied in our lab.224,225 For example, we are able to visualize the bone-implant interfaces after tissue-clearance with two-photon excitation microscope, which is almost unapplicable with other methods.
Conclusions
This review summarizes the key findings of skeletal stem cells in the recent decade. The application of lineage tracing and high-throughput sequencing have greatly broadened and deepened our understanding of the skeletal development, homeostasis, and injury repair. Several aspects of skeletal stem cells are warranted for future exploration. New sample preparation methods and lineage tracing models that achieve efficient and reliable evaluation of the behavior of skeletal cells are to be explored. The crosstalk within bone marrow niches, and the mechanism that determines fate determination of SSCs remain to be elucidated. The cells and regulatory mechanism in craniofacial system exhibit some unique properties compared with long bones, and are to be uncovered as well.
References
Chande, S. & Bergwitz, C. Role of phosphate sensing in bone and mineral metabolism. Nat. Rev. Endocrinol. 14, 637–655 (2018).
Farr, J. N. & Khosla, S. Skeletal changes through the lifespan—from growth to senescence. Nat. Rev. Endocrinol. 11, 513–521 (2015).
Morrison, S. J. & Scadden, D. T. The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334 (2014).
van Gastel, N. & Carmeliet, G. Metabolic regulation of skeletal cell fate and function in physiology and disease. Nat. Metab. 3, 11–20 (2021).
Karsenty, G. & Khosla, S. The crosstalk between bone remodeling and energy metabolism: a translational perspective. Cell Metab. 34, 805–817 (2022).
Lu, K. et al. Defects in a liver-bone axis contribute to hepatic osteodystrophy disease progression. Cell Metab. 34, 441–457.e7 (2022).
Berger, J. M. et al. Mediation of the acute stress response by the skeleton. Cell Metab. 30, 890–902.e8 (2019).
Whyte, M. P. Hypophosphatasia — aetiology, nosology, pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 12, 233–246 (2016).
Sugisawa, E. et al. RNA sensing by gut Piezo1 is essential for systemic serotonin synthesis. Cell 182, 609–624.e21 (2020).
Han, Y., You, X., Xing, W., Zhang, Z. & Zou, W. Paracrine and endocrine actions of bone—the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 6, 16 (2018).
Bianco, P. et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat. Med. 19, 35–42 (2013).
Bianco, P. & Robey, P. G. Skeletal stem cells. Development 142, 1023–1027 (2015).
Marecic, O. et al. Identification and characterization of an injury-induced skeletal progenitor. Proc. Natl. Acad. Sci. USA 112, 9920–9925 (2015).
Ono, N. & Kronenberg, H. M. Bone repair and stem cells. Curr. Opin. Genet. Dev. 40, 103–107 (2016).
Klingseisen, A. & Jackson, A. P. Mechanisms and pathways of growth failure in primordial dwarfism. Genes Dev. 25, 2011–2024 (2011).
Shiang, R. et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 78, 335–342 (1994).
Singer, F. R. Paget’s disease of bone—genetic and environmental factors. Nat. Rev. Endocrinol. 11, 662–671 (2015).
Hendrickx, G., Boudin, E. & Van Hul, W. A look behind the scenes: the risk and pathogenesis of primary osteoporosis. Nat. Rev. Rheumatol. 11, 462–474 (2015).
Bovée, J. V. M. G., Hogendoorn, P. C. W., Wunder, J. S. & Alman, B. A. Cartilage tumours and bone development: molecular pathology and possible therapeutic targets. Nat. Rev. Cancer 10, 481–488 (2010).
Pignolo, R. J., Ramaswamy, G., Fong, J. T., Shore, E. M. & Kaplan, F. S. Progressive osseous heteroplasia: diagnosis, treatment, and prognosis. Appl. Clin. Genet. 8, 37–48 (2015).
Farrow, E., Nicot, R., Wiss, A., Laborde, A. & Ferri, J. Cleidocranial dysplasia: a review of clinical, radiological, genetic implications and a guidelines proposal. J. Craniofac. Surg. 29, 382–389 (2018).
Deng, P. et al. Loss of KDM4B exacerbates bone-fat imbalance and mesenchymal stromal cell exhaustion in skeletal aging. Cell Stem Cell 28, 1057–1073.e7 (2021).
Wang, Y. et al. Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat. Commun. 11, 5596 (2020).
Chai, Y. & Maxson, R. E. Recent advances in craniofacial morphogenesis. Dev. Dyn. 235, 2353–2375 (2006).
Zeller, R., López-Ríos, J. & Zuniga, A. Vertebrate limb bud development: moving towards integrative analysis of organogenesis. Nat. Rev. Genet. 10, 845–858 (2009).
Friedenstein, A. J., Piatetzky-Shapiro, I. I. & Petrakova, K. V. Osteogenesis in transplants of bone marrow cells. J. Embryol. Exp. Morphol. 16, 381–390 (1966).
Tavassoli, M. & Crosby, W. H. Transplantation of marrow to extramedullary sites. Science 161, 54–56 (1968).
Castro-Malaspina, H. et al. Characterization of human bone marrow fibroblast colony-forming cells (CFU-F) and their progeny. Blood 56, 289–301 (1980).
Owen, M. & Friedenstein, A. J. Stromal stem cells: marrow-derived osteogenic precursors. Ciba Found. Symp. 136, 42–60 (1988).
Caplan, A. I. Mesenchymal stem cells. J. Orthop. Res. 9, 641–650 (1991).
Dominici, M. et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317 (2006).
Prieto González, E. A. Heterogeneity in adipose stem cells. Adv. Exp. Med. Biol. 1123, 119–150 (2019).
De Micheli, A. J. et al. Single-cell analysis of the muscle stem cell hierarchy identifies heterotypic communication signals involved in skeletal muscle regeneration. Cell Rep. 30, 3583–3595.e5 (2020).
Campanella, V. Dental stem cells: current research and future applications. Eur. J. Paediatr. Dent. 19, 257 (2018).
Sacchetti, B. et al. No Identical ‘mesenchymal stem cells’ at different times and sites: human committed progenitors of distinct origin and differentiation potential are incorporated as adventitial cells in microvessels. Stem Cell Rep. 6, 897–913 (2016).
Galipeau, J. & Sensébé, L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell 22, 824–833 (2018).
Bianco, P., Robey, P. G. & Simmons, P. J. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell 2, 313–319 (2008).
Chan, C. K. F. et al. Identification and specification of the mouse skeletal stem cell. Cell 160, 285–298 (2015).
Sacchetti, B. et al. Self-Renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131, 324–336 (2007).
Ambrosi, T. H., Longaker, M. T. & Chan, C. K. F. A revised perspective of skeletal stem cell biology. Front. Cell Dev. Biol. 7, 189 (2019).
Mizuhashi, K. et al. Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature 563, 254–258 (2018).
Newton, P. T. et al. A radical switch in clonality reveals a stem cell niche in the epiphyseal growth plate. Nature 567, 234–238 (2019).
Kretzschmar, K. & Watt, F. M. Lineage tracing. Cell 148, 33–45 (2012).
Gulati, G. S. et al. Isolation and functional assessment of mouse skeletal stem cell lineage. Nat. Protoc. 13, 1294–1309 (2018).
Frisch, B. J. Hematopoietic stem cell cultures and assays. Methods Mol. Biol. 2230, 467–477 (2021).
Sun, J. et al. Clonal dynamics of native haematopoiesis. Nature 514, 322–327 (2014).
Han, X. et al. A suite of new Dre recombinase drivers markedly expands the ability to perform intersectional genetic targeting. Cell Stem Cell 28, 1160–1176.e7 (2021).
He, L. et al. Enhancing the precision of genetic lineage tracing using dual recombinases. Nat. Med. 23, 1488–1498 (2017).
Theodosiou, N. A. & Xu, T. Use of FLP/FRT system to study Drosophila development. Methods 14, 355–365 (1998).
McLellan, M. A., Rosenthal, N. A. & Pinto, A. R. Cre-loxP-mediated recombination: general principles and experimental considerations. Curr. Protoc. Mouse Biol. 7, 1–12 (2017).
Feil, S., Valtcheva, N. & Feil, R. Inducible Cre mice. Methods Mol. Biol. 530, 343–363 (2009).
Kalajzic, Z. et al. Directing the expression of a green fluorescent protein transgene in differentiated osteoblasts: comparison between rat type I collagen and rat osteocalcin promoters. Bone 31, 654–660 (2002).
Zhao, H. & Zhou, B. Dual genetic approaches for deciphering cell fate plasticity in vivo: more than double. Curr. Opin. Cell Biol. 61, 101–109 (2019).
He, L. et al. Genetic lineage tracing of resident stem cells by DeaLT. Nat. Protoc. 13, 2217–2246 (2018).
Soriano, P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71 (1999).
Srinivas, S. et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 (2001).
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. & Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010).
Abe, T. & Fujimori, T. Reporter mouse lines for fluorescence imaging. Dev. Growth Differ. 55, 390–405 (2013).
Ding, L., Saunders, T. L., Enikolopov, G. & Morrison, S. J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462 (2012).
Rivers, L. E. et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat. Neurosci. 11, 1392–1401 (2008).
Huang, W. & Olsen, B. R. Skeletal defects in Osterix-Cre transgenic mice. Transgenic Res. 24, 167–172 (2015).
Livet, J. et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 450, 56–62 (2007).
Zhou, B. O., Yue, R., Murphy, M. M., Peyer, J. G. & Morrison, S. J. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 15, 154–168 (2014).
Morikawa, S. et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J. Exp. Med. 206, 2483–2496 (2009).
Chan, C. K. F. et al. Identification of the human skeletal stem cell. Cell 175, 43–56.e21 (2018).
Ono, N., Ono, W., Nagasawa, T. & Kronenberg, H. M. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat. Cell Biol. 16, 1157–1167 (2014).
Ono, N. et al. Vasculature-associated cells expressing nestin in developing bones encompass early cells in the osteoblast and endothelial lineage. Dev. Cell 29, 330–339 (2014).
Mizoguchi, T. et al. Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Dev. Cell 29, 340–349 (2014).
Nusspaumer, G. et al. Ontogenic identification and analysis of mesenchymal stromal cell populations during mouse limb and long bone development. Stem Cell Rep. 9, 1124–1138 (2017).
Debnath, S. et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 562, 133–139 (2018).
Ambrosi, T. H. et al. Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell 20, 771–784.e6 (2017).
Ambrosi, T. H. et al. Distinct skeletal stem cell types orchestrate long bone skeletogenesis. eLife 10, e66063 (2021).
Hu, X. et al. Identification of a common mesenchymal stromal progenitor for the adult haematopoietic niche. Nat. Commun. 7, 13095 (2016).
Nakashima, K. et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17–29 (2002).
Liu, Y. et al. Osterix-cre labeled progenitor cells contribute to the formation and maintenance of the bone marrow stroma. PLoS One 8, e71318 (2013).
Shu, H. S. et al. Tracing the skeletal progenitor transition during postnatal bone formation. Cell Stem Cell 28, 2122–2136.e3 (2021).
Méndez-Ferrer, S. et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829–834 (2010).
Rauch, A. et al. Osteogenesis depends on commissioning of a network of stem cell transcription factors that act as repressors of adipogenesis. Nat. Genet. 51, 716–727 (2019).
Baccin, C. et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 22, 38–48 (2020).
Lanske, B. et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 273, 663–666 (1996).
Hallett, S. A. et al. Chondrocytes in the resting zone of the growth plate are maintained in a Wnt-inhibitory environment. eLife 10, e64513 (2021).
Vortkamp, A. et al. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 273, 613–622 (1996).
Ornitz, D. M. & Marie, P. J. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 29, 1463–1486 (2015).
Salazar, V. S., Gamer, L. W. & Rosen, V. BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 12, 203–221 (2016).
Song, L. et al. Loss of wnt/β-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J. Bone Miner. Res. 27, 2344–2358 (2012).
Shi, S. & Gronthos, S. Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J. Bone Miner. Res. 18, 696–704 (2003).
Pinho, S. et al. PDGFRα and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 210, 1351–1367 (2013).
Tormin, A. et al. CD146 expression on primary nonhematopoietic bone marrow stem cells is correlated with in situ localization. Blood 117, 5067–5077 (2011).
He, J. et al. Dissecting human embryonic skeletal stem cell ontogeny by single-cell transcriptomic and functional analyses. Cell Res. 31, 742–757 (2021).
Chan, C. K. F. et al. Clonal precursor of bone, cartilage, and hematopoietic niche stromal cells. Proc. Natl. Acad. Sci. USA 110, 12643–12648 (2013).
Crisan, M. et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3, 301–313 (2008).
Seike, M., Omatsu, Y., Watanabe, H., Kondoh, G. & Nagasawa, T. Stem cell niche-specific Ebf3 maintains the bone marrow cavity. Genes Dev. 32, 359–372 (2018).
Greenbaum, A. et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230 (2013).
Matthews, B. G. et al. Analysis of αSMA-labeled progenitor cell commitment identifies notch signaling as an important pathway in fracture healing. J. Bone Miner. Res. 29, 1283–1294 (2014).
Isern, J. et al. Self-renewing human bone marrow mesenspheres promote hematopoietic stem cell expansion. Cell Rep. 3, 1714–1724 (2013).
Worthley, D. L. et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160, 269–284 (2015).
Shi, Y. et al. Gli1 identifies osteogenic progenitors for bone formation and fracture repair. Nat. Commun. 8, 2043 (2017).
Ortinau, L. C. et al. Identification of functionally distinct Mx1+αSMA+ periosteal skeletal stem cells. Cell Stem Cell 25, 784–796.e5 (2019).
Petit, F., Sears, K. E. & Ahituv, N. Limb development: a paradigm of gene regulation. Nat. Rev. Genet. 18, 245–258 (2017).
Zhulyn, O. et al. A switch from low to high Shh activity regulates establishment of limb progenitors and signaling centers. Dev. Cell 29, 241–249 (2014).
Cooper, K. L. et al. Initiation of proximal-distal patterning in the vertebrate limb by signals and growth. Science 332, 1083–1086 (2011).
Roselló-Díez, A., Ros, M. A. & Torres, M. Diffusible signals, not autonomous mechanisms, determine the main proximodistal limb subdivision. Science 332, 1086–1088 (2011).
Kronenberg, H. M. Developmental regulation of the growth plate. Nature 423, 332–336 (2003).
Li, Y. et al. Dynamic imaging of the growth plate cartilage reveals multiple contributors to skeletal morphogenesis. Nat. Commun. 6, 6798 (2015).
Cooper, K. L. et al. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 495, 375–378 (2013).
Logan, M. et al. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80 (2002).
Akiyama‡, H. et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl. Acad. Sci. USA 102, 14665–14670 (2005).
Kronenberg, H. M. The role of the perichondrium in fetal bone development. Ann. N. Y. Acad. Sci. 1116, 59–64 (2007).
Gerber, H. P. et al. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 5, 623–628 (1999).
Maes, C. et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell 19, 329–344 (2010).
Zhou, X. et al. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 10, e1004820 (2014).
Yang, L., Tsang, K. Y., Tang, H. C., Chan, D. & Cheah, K. S. E. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 111, 12097–12102 (2014).
Yang, G. et al. Osteogenic fate of hypertrophic chondrocytes. Cell Res. 24, 1266–1269 (2014).
Ding, L. & Morrison, S. J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235 (2013).
Zhang, D. et al. LepR-expressing stem cells are essential for alveolar bone regeneration. J. Dent. Res. 99, 1279–1286 (2020).
Dzierzak, E. & Speck, N. A. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat. Immunol. 9, 129–136 (2008).
He, D. D. et al. C-KIT expression distinguishes fetal from postnatal skeletal progenitors. Stem Cell Rep. 14, 614–630 (2020).
Muruganandan, S. et al. A FoxA2+ long-term stem cell population is necessary for growth plate cartilage regeneration after injury. Nat. Commun. 13, 2515 (2022).
Pineault, K. M., Song, J. Y., Kozloff, K. M., Lucas, D. & Wellik, D. M. Hox11 expressing regional skeletal stem cells are progenitors for osteoblasts, chondrocytes and adipocytes throughout life. Nat. Commun. 10, 3168 (2019).
Rux, D. R. et al. Regionally restricted hox function in adult bone marrow multipotent mesenchymal stem/stromal cells. Dev. Cell 39, 653–666 (2016).
Long, J. T. et al. Hypertrophic chondrocytes serve as a reservoir for marrow-associated skeletal stem and progenitor cells, osteoblasts, and adipocytes during skeletal development. eLife 11, e76932 (2022).
Matsushita, Y. et al. A Wnt-mediated transformation of the bone marrow stromal cell identity orchestrates skeletal regeneration. Nat. Commun. 11, 332 (2020).
Kunisaki, Y. et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643 (2013).
Omatsu, Y. et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 33, 387–399 (2010).
Comazzetto, S. et al. Restricted hematopoietic progenitors and erythropoiesis require SCF from leptin receptor+ niche cells in the bone marrow. Cell Stem Cell 24, 477–486.e6 (2019).
Park, D. et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 10, 259–272 (2012).
Shen, B. et al. A mechanosensitive peri-arteriolar niche for osteogenesis and lymphopoiesis. Nature 591, 438–444 (2021).
Li, Q., Wu, Y. & Kang, N. Marrow adipose tissue: its origin, function, and regulation in bone remodeling and regeneration. Stem Cells Int. 2018, 1–11 (2018).
Wu, Y. et al. Mettl3-mediated m6A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat. Commun. 9, 4772 (2018).
Fan, Y. et al. Parathyroid hormone directs bone marrow mesenchymal cell fate. Cell Metab. 25, 661–672 (2017).
Yue, R., Zhou, B. O., Shimada, I. S., Zhao, Z. & Morrison, S. J. Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell 18, 782–796 (2016).
Zhou, B. O. et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat. Cell Biol. 19, 891–903 (2017).
Zhong, L. et al. Single cell transcriptomics identifies a unique adipose lineage cell population that regulates bone marrow environment. eLife 9, e54695 (2020).
Yu, W. et al. Bone marrow adipogenic lineage precursors promote osteoclastogenesis in bone remodeling and pathologic bone loss. J. Clin. Investig 131, 140214 (2021).
Palmisano, B. et al. GsαR201C and estrogen reveal different subsets of bone marrow adiponectin expressing osteogenic cells. Bone Res. 10, 50 (2022).
Sivaraj, K. K. et al. Regional specialization and fate specification of bone stromal cells in skeletal development. Cell Rep. 36, 109352 (2021).
Ferguson, J. W. & Atit, R. P. A tale of two cities: the genetic mechanisms governing calvarial bone development. Genesis 57, e23248 (2019).
Shah, H. N. et al. Craniofacial and long bone development in the context of distraction osteogenesis. Plast. Reconstr. Surg. 147, 54e–65e (2021).
Soldatov, R. et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science 364, eaas9536 (2019).
Xie, M. et al. Schwann cell precursors contribute to skeletal formation during embryonic development in mice and zebrafish. Proc. Natl. Acad. Sci. USA 116, 15068–15073 (2019).
Kaucka, M. et al. Analysis of neural crest-derived clones reveals novel aspects of facial development. Sci. Adv. 2, e1600060 (2016).
Lin, W. et al. Mapping the immune microenvironment for mandibular alveolar bone homeostasis at single-cell resolution. Bone Res. 9, 17 (2021).
Zhou, X. et al. SM22alpha-lineage niche cells regulate intramembranous bone regeneration via PDGFRbeta-triggered hydrogen sulfide production. Cell Rep. 39, 110750 (2022).
Zhao, H. et al. The suture provides a niche for mesenchymal stem cells of craniofacial bones. Nat. Cell Biol. 17, 386–396 (2015).
Park, S., Zhao, H., Urata, M. & Chai, Y. Sutures possess strong regenerative capacity for calvarial bone injury. Stem Cells Dev. 25, 1801–1807 (2016).
Yu, M. et al. Cranial suture regeneration mitigates skull and neurocognitive defects in craniosynostosis. Cell 184, 243–256.e18 (2021).
Jing, D. et al. Response of Gli1+ suture stem cells to mechanical force upon suture expansion. J. Bone Miner. Res. 37, 1307–1320 (2022).
Maruyama, T., Jeong, J., Sheu, T.J. & Hsu, W. Stem cells of the suture mesenchyme in craniofacial bone development, repair and regeneration. Nat. Commun. 7, 10526 (2016).
Menon, S. et al. Skeletal stem and progenitor cells maintain cranial suture patency and prevent craniosynostosis. Nat. Commun. 12, 4640 (2021).
Ouyang, Z. et al. Prx1 and 3.2kb Col1a1 promoters target distinct bone cell populations in transgenic mice. Bone 58, 136–145 (2014).
Wilk, K. et al. Postnatal calvarial skeletal stem cells expressing PRX1 reside exclusively in the calvarial sutures and are required for bone regeneration. Stem Cell Rep. 8, 933–946 (2017).
Seo, B.-M. et al. Investigation of multipotent postnatal stem cells from human periodontal ligament. Lancet 364, 149–155 (2004).
Liang, J. F. et al. Identification of dental stem cells similar to skeletal stem cells. J. Dent. Res. 101, 1092–1100 (2022).
Men, Y. et al. Gli1+ periodontium stem cells are regulated by osteocytes and occlusal force. Dev. Cell 54, 639–654 (2020).
Liu, A. Q. et al. Mechanosensing by Gli1(+) cells contributes to the orthodontic force-induced bone remodelling. Cell Prolif. 53, e12810 (2020).
Xie, X., Xu, C., Zhao, H., Wang, J. & Feng, J. Q. A biphasic feature of Gli1(+)-mesenchymal progenitors during cementogenesis that is positively controlled by Wnt/β-catenin signaling. J. Dent. Res. 100, 1289–1298 (2021).
Lim, W. H. et al. Wnt signaling regulates homeostasis of the periodontal ligament. J. Periodontal Res 49, 751–759 (2014).
Wang, K. et al. Axin2+ PDL cells directly contribute to new alveolar bone formation in response to orthodontic tension force. J. Dent. Res. 101, 695–703 (2022).
Yuan, X. et al. A Wnt-responsive PDL population effectuates extraction socket healing. J. Dent. Res. 97, 803–809 (2018).
Xie, X. et al. Axin2(+)-mesenchymal PDL cells, instead of K14(+) epithelial cells, play a key role in rapid cementum growth. J. Dent. Res. 98, 1262–1270 (2019).
Zhao, J., Faure, L., Adameyko, I. & Sharpe, P. T. Stem cell contributions to cementoblast differentiation in healthy periodontal ligament and periodontitis. Stem Cells 39, 92–102 (2021).
Bassir, S. H. et al. Prx1 expressing cells are required for periodontal regeneration of the mouse incisor. Front. Physiol. 10, 591 (2019).
Cui, C. et al. Role of PTH1R signaling in Prx1+ mesenchymal progenitors during eruption. J. Dent. Res. 99, 1296–1305 (2020).
Gong, X. et al. Tracing PRX1(+) cells during molar formation and periodontal ligament reconstruction. Int J. Oral. Sci. 14, 5 (2022).
Dragonas, P., Katsaros, T., Schiavo, J., Galindo-Moreno, P. & Avila-Ortiz, G. Osteogenic capacity of the sinus membrane following maxillary sinus augmentation procedures: a systematic review. Int. J. Oral. Implantol. Berl. Ger. 13, 213–232 (2020).
Coyac, B. R., Wu, M., Bahat, D. J., Wolf, B. J. & Helms, J. A. Biology of sinus floor augmentation with an autograft versus a bone graft substitute in a preclinical in vivo experimental model. Clin. Oral. Implants Res. 32, 916–927 (2021).
Weng, Y. et al. A novel lineage of osteoprogenitor cells with dual epithelial and mesenchymal properties govern maxillofacial bone homeostasis and regeneration after MSFL. Cell Res. 32, 814–830 (2022).
Sharpe, P. T. Dental mesenchymal stem cells. Development 143, 2273–2280 (2016).
Miura, M. et al. SHED: stem cells from human exfoliated deciduous teeth. Proc. Natl. Acad. Sci. USA 100, 5807–5812 (2003).
Sloan, A. J. & Smith, A. J. Stem cells and the dental pulp: potential roles in dentine regeneration and repair. Oral. Dis. 13, 151–157 (2007).
Zhang, Q. Z., Nguyen, A. L., Yu, W. H. & Le, A. D. Human oral mucosa and gingiva: a unique reservoir for mesenchymal stem cells. J. Dent. Res. 91, 1011–1018 (2012).
Feng, J., Mantesso, A., De Bari, C., Nishiyama, A. & Sharpe, P. T. Dual origin of mesenchymal stem cells contributing to organ growth and repair. Proc. Natl. Acad. Sci. USA 108, 6503–6508 (2011).
Zhao, H. et al. Secretion of Shh by a neurovascular bundle niche supports mesenchymal stem cell homeostasis in the adult mouse incisor. Cell Stem Cell 23, 147 (2018).
Kaukua, N. et al. Glial origin of mesenchymal stem cells in a tooth model system. Nature 513, 551–554 (2014).
Gong, S., Emperumal, C. P., Al-Eryani, K. & Enciso, R. Regeneration of temporomandibular joint using in vitro human stem cells: a review. J. Tissue Eng. Regen. Med. 16, 591–604 (2022).
Matheus, H. R., Özdemir, Ş. D. & Guastaldi, F. P. S. Stem cell-based therapies for temporomandibular joint osteoarthritis and regeneration of cartilage/osteochondral defects: a systematic review of preclinical experiments. Osteoarthr. Cartil. 30, 1174–1185 (2022).
Van Bellinghen, X. et al. Temporomandibular joint regenerative medicine. Int. J. Mol. Sci. 19(2), 446 (2018).
Embree, M. C. et al. Exploiting endogenous fibrocartilage stem cells to regenerate cartilage and repair joint injury. Nat. Commun. 7, 13073 (2016).
Bi, R. et al. Identification of human temporomandibular joint fibrocartilage stem cells with distinct chondrogenic capacity. Osteoarthr. Cartil. 28, 842–852 (2020).
Fan, Y. et al. Fibrocartilage stem cells in the temporomandibular joint: insights from animal and human studies. Front. Cell Dev. Biol. 9, 665995 (2021).
Colnot, C. Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J. Bone Miner. Res. 24, 274–282 (2009).
Colnot, C., Zhang, X., & Knothe Tate, M. L. Current insights on the regenerative potential of the periosteum: molecular, cellular, and endogenous engineering approaches. J. Orthop. Res. 30, 1869–1878 (2012).
Xu, J. et al. PDGFRα reporter activity identifies periosteal progenitor cells critical for bone formation and fracture repair. Bone Res. 10, 7 (2022).
He, X. et al. Sox9 positive periosteal cells in fracture repair of the adult mammalian long bone. Bone 103, 12–19 (2017).
Kawanami, A., Matsushita, T., Chan, Y. Y. & Murakami, S. Mice expressing GFP and CreER in osteochondro progenitor cells in the periosteum. Biochem. Biophys. Res. Commun. 386, 477–482 (2009).
Lotinun, S. et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J. Clin. Investig. 123, 666–681 (2013).
Han, Y. et al. Lkb1 deletion in periosteal mesenchymal progenitors induces osteogenic tumors through mTORC1 activation. J. Clin. Investig. 129, 1895–1909 (2019).
Yang, W. et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 499, 491–495 (2013).
Karlsson, C., Thornemo, M., Henriksson, H. B. & Lindahl, A. Identification of a stem cell niche in the zone of Ranvier within the knee joint. J. Anat. 215, 355–363 (2009).
Tsukasaki, M. et al. Periosteal stem cells control growth plate stem cells during postnatal skeletal growth. Nat. Commun. 13, 4166 (2022).
Ding, Y., Mo, C., Geng, J., Li, J. & Sun, Y. Identification of periosteal osteogenic progenitors in Jawbone. J. Dent. Res. 101, 1101–1109 (2022).
Jones, R. E. et al. Skeletal stem cell-schwann cell circuitry in mandibular repair. Cell Rep. 28, 2757–2766 e5 (2019).
Carr, M. J. et al. Mesenchymal precursor cells in adult nerves contribute to mammalian tissue repair and regeneration. Cell Stem Cell 24, 240–256.e9 (2019).
Clements, M. P. et al. The wound microenvironment reprograms schwann cells to invasive mesenchymal-like cells to drive peripheral nerve regeneration. Neuron 96, 98–114 e7 (2017).
Johnston, A. P. et al. Dedifferentiated schwann cell precursors secreting paracrine factors are required for regeneration of the mammalian digit tip. Cell Stem Cell 19, 433–448 (2016).
Ransom, R. C. et al. Mechanoresponsive stem cells acquire neural crest fate in jaw regeneration. Nature 563, 514–521 (2018).
Greenblatt, M. B., Ono, N., Ayturk, U. M., Debnath, S. & Lalani, S. The unmixing problem: a guide to applying single‐cell RNA sequencing to bone. J. Bone Miner. Res. 34, 1207–1219 (2019).
Li, X. & Wang, C.-Y. From bulk, single-cell to spatial RNA sequencing. Int. J. Oral. Sci. 13, 36 (2021).
Kelly, N. H., Huynh, N. P. T. & Guilak, F. Single cell RNA-sequencing reveals cellular heterogeneity and trajectories of lineage specification during murine embryonic limb development. Matrix Biol. J. Int. Soc. Matrix Biol. 89, 1–10 (2020).
Sivaraj, K. K. et al. Mesenchymal stromal cell-derived septoclasts resorb cartilage during developmental ossification and fracture healing. Nat. Commun. 13, 571 (2022).
Wolock, S. L. et al. Mapping distinct bone marrow niche populations and their differentiation paths. Cell Rep. 28, 302–311.e5 (2019).
Tikhonova, A. N. et al. The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228 (2019).
Baryawno, N. et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell 177, 1915–1932.e16 (2019).
Severe, N. et al. Stress-induced changes in bone marrow stromal cell populations revealed through single-cell protein expression mapping. Cell Stem Cell 25, 570–583.e7 (2019).
Yianni, V. & Sharpe, P. T. Single cell RNA-seq: cell isolation and data analysis. Methods Mol. Methods Mol. Biol. 2403, 81–89 (2022).
Krivanek, J. et al. Dental cell type atlas reveals stem and differentiated cell types in mouse and human teeth. Nat. Commun. 11, 4816 (2020).
Ayturk, U. M. et al. Single-cell RNA sequencing of calvarial and long-bone endocortical cells. J. Bone Min. Res. 35, 1981–1991 (2020).
Farmer, D. T. et al. The developing mouse coronal suture at single-cell resolution. Nat. Commun. 12, 4797 (2021).
Holmes, G. et al. Single-cell analysis identifies a key role for Hhip in murine coronal suture development. Nat. Commun. 12, 7132 (2021).
Xie, M. & Chagin, A. S. The epiphyseal secondary ossification center: evolution, development and function. Bone 142, 115701 (2021).
Matthews, B. G. et al. Heterogeneity of murine periosteum progenitors involved in fracture healing. eLife 10, e58534 (2021).
Kenswil, K. J. G. et al. Endothelium-derived stromal cells contribute to hematopoietic bone marrow niche formation. Cell Stem Cell 28, 653–670.e11 (2021).
Isern, J. et al. The neural crest is a source of mesenchymal stem cells with specialized hematopoietic stem cell niche function. eLife 3, e03696 (2014).
Soleimani, M. & Nadri, S. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat. Protoc. 4, 102–106 (2009).
Zhu, H. et al. A protocol for isolation and culture of mesenchymal stem cells from mouse compact bone. Nat. Protoc. 5, 550–560 (2010).
van den Brink, S. C. et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936 (2017).
Park, E. et al. Bone marrow transplantation procedures in mice to study clonal hematopoiesis. J. Vis. Exp. 171, e61875 (2021).
Coutu, D. L., Kokkaliaris, K. D., Kunz, L. & Schroeder, T. Three-dimensional map of nonhematopoietic bone and bone-marrow cells and molecules. Nat. Biotechnol. 35, 1202–1210 (2017).
Kawamoto, T. & Kawamoto, K. Preparation of thin frozen sections from nonfixed and undecalcified hard tissues using Kawamot’s Film Method (2012). Vol. 1130. In Skeletal Development and Repair (ed. Hilton, M. J.) 149–164 (Humana Press, 2014).
Li, Q. et al. Ubiquitin-specific protease 34 inhibits osteoclast differentiation by regulating NF-κB signaling. J. Bone Miner. Res. 35, 1597–1608 (2020).
Kusumbe, A. P., Ramasamy, S. K., Starsichova, A. & Adams, R. H. Sample preparation for high-resolution 3D confocal imaging of mouse skeletal tissue. Nat. Protoc. 10, 1904–1914 (2015).
Langen, U. H. et al. Cell–matrix signals specify bone endothelial cells during developmental osteogenesis. Nat. Cell Biol. 19, 189–201 (2017).
Jing, D. et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Res. 28, 803–818 (2018).
Zhan, Y., Wu, H., Liu, L., Lin, J. & Zhang, S. Organic solvent-based tissue clearing techniques and their applications. J. Biophotonics 14, e202000413 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82125006). Figures were created with BioRender.com.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Q., Xu, R., Lei, K. et al. Insights into skeletal stem cells. Bone Res 10, 61 (2022). https://doi.org/10.1038/s41413-022-00235-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-022-00235-8
This article is cited by
-
Primary cilia support cartilage regeneration after injury
International Journal of Oral Science (2023)
