
Abstract
Osteomyelitis is a devastating disease caused by microbial infection of bone. While the frequency of infection following elective orthopedic surgery is low, rates of reinfection are disturbingly high. Staphylococcus aureus is responsible for the majority of chronic osteomyelitis cases and is often considered to be incurable due to bacterial persistence deep within bone. Unfortunately, there is no consensus on clinical classifications of osteomyelitis and the ensuing treatment algorithm. Given the high patient morbidity, mortality, and economic burden caused by osteomyelitis, it is important to elucidate mechanisms of bone infection to inform novel strategies for prevention and curative treatment. Recent discoveries in this field have identified three distinct reservoirs of bacterial biofilm including: Staphylococcal abscess communities in the local soft tissue and bone marrow, glycocalyx formation on implant hardware and necrotic tissue, and colonization of the osteocyte-lacuno canalicular network (OLCN) of cortical bone. In contrast, S. aureus intracellular persistence in bone cells has not been substantiated in vivo, which challenges this mode of chronic osteomyelitis. There have also been major advances in our understanding of the immune proteome against S. aureus, from clinical studies of serum antibodies and media enriched for newly synthesized antibodies (MENSA), which may provide new opportunities for osteomyelitis diagnosis, prognosis, and vaccine development. Finally, novel therapies such as antimicrobial implant coatings and antibiotic impregnated 3D-printed scaffolds represent promising strategies for preventing and managing this devastating disease. Here, we review these recent advances and highlight translational opportunities towards a cure.
Similar content being viewed by others

Introduction
Osteomyelitis, defined as inflammation of the bone often caused by bacterial infection, is one of the oldest diseases in history.1 Infection of bone can be caused by endogenous seeding, known as hematogenous osteomyelitis,2 or by exogenous seeding, via contamination of a fracture site or surgical hardware during implantation.
With over 1.5 million total hip and total knee replacement (TKR) procedures performed each year,3,4 bone infection remains the most severe and devastating risk associated with orthopedic implants. It has been understood for decades that the addition of a foreign material to a biological environment provides a haven for bacterial attachment and colonization.5,6,7,8 Additionally, movement-induced wear on orthopedic prostheses causes the release of debris, resulting in local inflammation, and creating a favorable site for the development of infection.9
While advances in prophylaxis and aseptic surgical technique have decreased the incidence of orthopedic infection following hip or knee arthroplasty, rigorous intervention studies (e.g. outcomes from the Surgical Care Improvement Project (SCIP)10) have demonstrated that infection rates for elective surgery cannot be reduced below 1%–2%.10,11,12,13 Additionally, rates of recurrent or persistent infection following a two-stage revision surgery are still as high as 33%.13,14,15 Despite infection treatment strategies such as surgical site debridement, complete hardware exchange, and aggressive long-term antimicrobial therapy, infections continue to recur. In total, the cost for treatment of implant-associated osteomyelitis is projected to exceed $1.62 billion by 2020.16 These data are consistent with the conclusions from the 2018 International Consensus Meeting on Musculoskeletal Infection, which found that the incidences of infection for all orthopedic subspecialties range from 0.1% to 30%, at a cost of $17 000–$150 000 per patient.13
An astounding 75% of osteomyelitis cases are caused by pathogens of the Staphylococcus genus.17,18 Specifically, Staphylococcus aureus is the most common pathogen isolated from implant-associated ostemyelitis17,19,20 and over 50% of cases are caused by hard-to-treat methicillin-resistant S. aureus (MRSA) strains.21 For these reasons, S. aureus will be the primary focus of this review. Other osteomyelitis-causing pathogens include Enterococcus, Pseudomonas, and Streptococcus species.17
S. aureus is an extremely versatile opportunistic pathogen that can infect nearly every organ system in the human body causing life-threatening disease,22 while maintaining the ability to asymptomatically colonize 20%–60% of individuals.23 The invasive success of S. aureus infection can be attributed to its arsenal of virulence factors and resistance mechanisms including secreted toxins,24 adherence as a means of immune evasion,25 biofilm formation,26,27 the creation of slow growing small colony variant (SCV) subpopulations,28,29 and the development of antimicrobial resistance.30 As a result of these highly evolved pathogenic mechanisms of persistence, clinical S. aureus osteomyelitis recurrence after decades of quiescence remains an important problem.31,32,33
It has been over 200 years since Sir Benjamin Brodie described the bacterial abscess in bone that bears his name,34 and 40 years since William Costerton’s biofilm hypothesis explained the pathogenic mode of existence by which sessile bacteria adhere to implants and necrotic tissue during chronic infection.35 Based on these fundamental concepts of bone infection, a standard of care treatment for implant-associated osteomyelitis, most notably prosthetic joint infection (PJI), was established in the 1970s and involves: (1) removal of the infected implant, (2) extensive surgical debridement of adjacent bone and soft tissues, and (3) filling of the bone void with antibiotic-loaded acrylic cement. In a seminal, retrospective analysis of 825 one-stage reimplantations using this approach for infected total hip arthroplasties, Buchholz et al. documented in 1984 that S. aureus was the most commonly encountered organism, and that the 5-year success (survival) rate was only 77%.36 Remarkably, the results from the 2018 International Consensus Meeting on Musculoskeletal Infections reported no changes in PJI infection rates, the primary pathogen, treatment algorithm, and poor outcomes, since this original standard of care was established half a century ago.8,13,37 However, there have been recent basic and translational science advances in our understanding of microbial pathogenesis, antibiotic resistance, and the osteoimmunology of bone infection that warrant reevaluation of clinical management for bone infection. Thus, the goal of this review is to highlight these potential breakthroughs, which challenge the scientific premise of established paradigms, including “acute and chronic” osteomyelitis, intracellular infection of bone cells, and the efficacy of antibiotic-laden bone cement. Additionally, by reviewing emerging concepts in bone infection, with specific focus on S. aureus pathogenesis in chronic osteomyelitis, we aim to discuss novel diagnostics, immunizations, and therapies that could be transformative for this harmful condition.
Treatment of peri-PJI
Although the rate of infection in primary hip and knee arthroplasties is low, rates of reinfection are reported as high as 40%.13,15 These infections frequently result in implant failure due to devastating effects on bone and soft tissue, requiring hardware removal for successful treatment. Because of the absence of an effective therapy for implant-associated osteomyelitis and lack of consistent guidelines for treatment, there is great debate over the appropriate procedure for managing total joint replacement (TJR) infections.
Principles of curative surgery in implant-associated osteomyelitis involve a variety of factors including infection severity, the infecting pathogen, and the condition of the patient.38 In the case of “acute” or low-level infection without complicating factors, such as significant comorbidity or loosening of the prosthesis, a surgeon may choose to treat with a regimen of debridement, antibiotics, irrigation, and retention (DAIR).39,40,41 Historically, DAIR has shown success rates as low as 14%,42 and as high as 100%.43 More recent studies have found DAIR success rates in appropriate patients to be ~70%–75%.40,44,45,46,47 Not surprisingly, retrospective studies of PJI revealed that patient health and the species of infecting organism significantly affected the success of DAIR treatment.48 DAIR failures and implant-associated bone infections that are not in this “acute” category must undergo hardware removal-revision surgery. Unfortunately, specific guidelines for the choice of treatment with hardware retention are widely variable and lacking scientific evidence,49,50,51,52 making the important decision to retain hardware a contentious and highly subjective choice.
As infection progresses, eradication of bacteria from implant hardware surfaces becomes the primary concern of surgeons because of the formation of robust biofilms. Therefore, a complete hardware exchange is recommended in cases of “chronic” or longer duration infection. Hardware exchange surgeries can be performed in 1 or 2 stages, depending on the use of local antibiotic delivery mechanisms, such as antibiotic-laden beads or bone cement that must be removed in a follow-up revision surgery. The advantage of single-stage vs. two-stage is currently unclear and seemingly largely dependent on the specific case.53,54
Initially, progressive osteomyelitis is evaluated by radiology to visualize bone lysis or erosion with associated soft tissue pathology (Fig. 1a). Single-stage revision is typically pursued if the microorganism is treatable, if the bone remains stable when the hardware is removed,55 or if the patient’s health suggests the need to limit surgical procedures. In a single-stage revision surgery, hardware removal and replacement are conducted in a single surgical procedure along with radical debridement of infected tissue and antibiotic treatment (Fig. 1b, c). When infected hardware is removed following revision surgery, extensive biofilm can be visualized on the surface of the implants immediately adjacent to bone (Fig. 1d, e). High magnification scanning electron micrographs show S. aureus embedded within an extracellular matrix of biofilm (Fig. 1e, g).

Removal of necrotic bone and biofilm contaminated components during revision surgery for MRSA-infected total joint replacements (TJR). a–c The indications for this single-stage revision for a MRSA-infected total hip replacement is shown. a Radiographic evidence of the septic TJR in the pre-op X-ray are periosteal reaction and a non-united femoral fracture (yellow arrows). b The open infected thigh requires removal of necrotic soft tissue and white (dead) bone, adjacent to live (red) bone that needs to be retained for successful limb salvage. Complete removal of the dead bone, cement, and necrotic tissues creates a healthier environment for the new prosthesis. c Post-op X-ray of the femoral defect with modular hip prosthesis. d–g Bacterial biofilm on explanted hardware components. Photographs of the surface of a femoral total knee replacement component before (d) and after (e) osmium tetroxide staining identifying bacterial biofilm on the bone cement. f SEM of the explanted hardware reveals biofilm bacteria (yellow arrow) on the surface of the implant (x10 000) and g bacteria attached to fibrin on the explanted hardware (x10 000)
On the other hand, in a two-stage revision procedure the hardware is removed. Antibiotic-laden bone cement (poly(methyl methacrylate (PMMA)) beads or spacers are implanted for dead space management and to locally deliver a high concentration of antibiotics, such as gentamicin or vancomycin56 to reduce detrimental systemic effects. While local delivery of antibiotics can be effective in eradicating infection, it may also introduce additional risks leading to bacterial resistance and recurrence of infection. It has been shown that antibiotic release efficiency from beads is low due to an early burst release, resulting in antibiotic concentrations below the minimum inhibitory concentrations.57 Administration of antibiotics in low concentrations can trigger the formation of antibiotic-resistant pathogens and SCVs, leading to recurrence of infection.29 Additionally, beads and bone cement spacers that are no longer eluting antibiotics provide additional surfaces for biofilm formation.58 Finally, the risk of reinfection increases with each additional revision surgery.
In all treatment scenarios, success is dependent on complete removal of infected and devitalized tissue and hardware alongside an array of possible patient risk factors. Unfortunately, there is no definitive way to ascertain if debridement is successful or not, as dormant and biofilm-dwelling bacteria often cannot be completely removed using this procedure. More importantly, specific guidelines for treating at risk populations are not uniform. As a result, the incidence of infection recurrence following PJI revision is extremely high following all treatments. In a study investigating outcomes of 16 622 TKR infections, 20.8% were treated with incision and drainage (I&D), 15.9% with I&D and liner exchange, 22.7% with single-stage revision, 39.7% with two-stage revision, and 0.98% with amputation.59 The results of this study showed that patients who underwent I&D had the highest rate of infection recurrence (28.2%), whereas patients who underwent two-stage revision had the lowest rate of recurrence (19%) at 1 year.59 Regardless of the treatment chosen, rates of reinfection are alarmingly high, suggesting the need for continued research on novel diagnostics and therapeutics to combat recurrent infection.
New definitions of “biofilm” in chronically infected bone
Due to the absence of an effective treatment for progressive osteomyelitis, there is a profound need for research to elucidate the mechanism of recurrent S. aureus osteomyelitis. To this end, recent breakthroughs have identified three pathogenically distinct reservoirs of biofilm bacteria in osteomyelitis (Fig. 2), each of which must be treated effectively or the bone infection will likely persist or recur.60

Three distinct reservoirs of bacteria in chronic osteomyelitis. Chronic implant-associated osteomyelitis was established in mice with S. aureus as previously described,84,93,117,267 and the bacterial burden: (1) in Staphylococcus abscess communities (SACs) assessed by histology (a–e), (2) on the implant assessed by SEM (f–j), and (3) in cortical bone assessed by TEM (k–m) is shown. Micrographs of orange G/alcian blue-stained histology of tibiae 7 days (a) and 14 days (c) post-infection are shown highlighting SACs (arrows) in the bone marrow and adjacent soft tissues. The boxed regions in (a), (c) are shown in Brown and Brenn-stained parallel section (b, d) to highlight the Gram+ bacteria (dark blue) surrounded by dead and dying neutrophils following NETosis (red cells), which are surrounded by a ring of macrophages (white layer). Chronic infection is clearly established by day 14, as evidenced by the complete replacement of hematopoietic bone marrow (BM) with inflammatory tissue, and the presence of M2 macrophages (brown cells) surrounding the SAC, as seen by immunostaining with antibody against arginase-1 (e). Biofilm formation on the implant commences with planktonic bacterial adhesion (f), as illustrated in this case of in vitro S. aureus attachment onto a stainless-steel wire incubated in a flow chamber system (×10 000). Following transtibial implantation, the planktonic bacteria rapidly transition to biofilm (g), seen as uniform glycocalyx coating the stainless-steel pin 14 days post-op (×200). High power images of the biofilm on the implant reveal cocci adhering to fibrin strands (h, ×2 500), and clusters of S. aureus forming bacterial pods (i, ×5 000). By day 14 post-infection, bacterial emigration from the pod is complete, as evidenced by the empty lacunae (j, ×30 000). S. aureus colonization of cortical bone commences with eradication of bone lining cells to expose canaliculi (blue arrows) leading to an embedded osteocyte (OC) (k, ×6 000). Subsequently, S. aureus invasion and propagation through osteocyte lacuno-canalicular network (OLCN) renders the biofilm bacteria (*) inaccessible to activated neutrophils outside the bone (blue arrows) (l, ×1 800). The uninhibited bacteria demineralize and consume the cortical bone to expands a canaliculus, and propagate into neighboring canaliculi (yellow arrow), to reach a distant osteocyte (red arrow). m High power TEM (×12 000) of the osteocyte in (l) killed by S. aureus bacterial occupation of its lacunar space
Staphylococcal Abscess communities (SACs)
SACs are most commonly observed in skin,61 but can be formed in virtually every tissue type following dissemination.62 Typically described as a host-induced mechanism for infection control, abscess formation is actually a dynamic process controlled by both host and pathogen signals. While innate immune cells actively sequester infected tissue and associated pathogens within an abscess, in doing so, they confer protection to pathogens at the core of the abscess.63 The formation of an abscess creates a physical barrier that prevents immune cell penetration and facilitates long-term bacterial persistence. As a result, abscesses rarely resolve without exogenous antimicrobial treatment.64
Peri-prosthetic abscess formation begins with neutrophil recruitment to the site of an acute infection, stimulated by the local release of cytokines and chemokines by host cells. Activated neutrophils proceed to combat extracellular bacteria by phagocytosis, degranulation, and generation of neutrophil extracellular traps (NETosis).65 Concurrently, S. aureus is capable of attacking neutrophils and other phagocytes by releasing cytolytic toxins, which create pores in host cell membranes.66 S. aureus also resists host cell attack via an array of immunosuppressive mechanisms,67,68 most notably chemotaxis inhibitory protein of staphylococci (CHIPS) and staphylococcal complement inhibitor (SCIN), which modulate polymorphonuclear neutrophil (PMN) killing69 and alter macrophage phenotype from bactericidal M1 to anti-inflammatory M2 macrophages.70
SACs can begin to form as early as 4 days following infection.71 In early stages of SAC formation S. aureus coagulases, CoA, and von Willebrand factor-binding protein (vWbp) promote activation of prothrombin and cleavage of fibrinogen,71 while membrane-bound protein clumping factor A (ClfA) binds fibrinogen to promote the formation of a fibrin margin at the periphery of the abscess.72 The center of a SAC is comprised of live bacteria surrounded by viable and necrotic PMNs separated from healthy tissue by the fibrin margin lined with macrophages to prevent bacterial dissemination,64 which is easily detected by 7 days post-infection (Fig. 2a, b). As the SAC matures over weeks, immune cells are restricted to the periphery, where they cannot access bacteria that continue to replicate at the center of the abscess (Fig. 2c, d). M2 phenotype macrophages, which are necessary for resolution of inflammation by efferocytosis of cellular debris and neutrophil extracellular traps (NETs),73 are also restricted to the periphery of the abscess, allowing continued bacterial persistence within the abscess (Fig. 2e).
For these reasons, eradication of SACs cannot be accomplished by the immune system alone. Without intervention, SAC persistence within a host can lead to eventual rupture and subsequent bacterial dissemination to new, uncolonized tissue.63 Therefore, complete eradication of implant-associated infections relies, in part, on the careful surgical debridement of soft tissue and bone marrow to remove all abscesses harboring bacteria, along with local antimicrobial therapy to ensure elimination of all extracellular bacteria.
Glycocalyx on the implant
Implant-associated S. aureus osteomyelitis begins with bacterial cell adhesion to extracellular matrix components known as Microbial Surface Components Recognizing Adhesive Matrix Molecules (MSCRAMMs).74 As the acronym suggests, MSCRAMMs are proteins localized at the microbial cell surface that are capable of binding ligands found in the extracellular matrix, such as fibronectin, fibrinogen, and collagen.75 Adhesion of the bacterial cells to a substrate allows for subsequent proliferation and colonization of the region. The initial host innate immune response to bacterial colonization begins with the recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs),76 which stimulates the release of an array of pro-inflammatory cytokines, chemokines, antimicrobial peptides, and triggers neutrophil recruitment to the site of infection. At this time, neutrophils and other first responder innate immune cells are capable of targeting and sequestering planktonic bacterial cells by antibody-mediated opsonophagocytosis or killing by oxidative burst through the release of reactive oxygen species (ROS). At some point, PMN attack subsides when all planktonic bacteria are eliminated, or they become inaccessible in biofilms. One of the most notable virulence mechanisms of S. aureus is its ability to form biofilm matrices embedded with phenotypically altered bacterial cells.77 As an adaptive response to environmental stress, unicellular bacteria often form biofilms to survive in a protected, multi-cellular community that is resistant to host immune response and antibiotic therapy.
Biofilm formation occurs in four generalized stages including: (1) bacterial cell attachment, (2) proliferation, (3) biofilm maturation, and (4) detachment. First, bacteria will attach to a substrate, such as an implant material or segment of necrotic bone, via surface adhesin molecules (Fig. 2f). Then, the attached cells will proliferate to expand the bacterial population and begin producing a matrix of extracellular polymeric substances (EPS) to protect the multi-layered community (Fig. 2e). EPS is generally composed of self-produced polysaccharides, proteins, and nucleic acids (Fig. 2h), which encase the bacterial cells to mediate adhesion, provide mechanical stability and retain essential nutrients and enzymes.78 As the biofilm matures, it becomes a complex and heterogeneous structure containing protected pods of bacteria (Fig. 2i), as well as void spaces and intricate channels to facilitate nutrient and oxygen transfer through the bulk of the biofilm. Finally, biofilm detachment or dispersal is a key step in pathogenesis, enabling bacterial cell metastasis via fluid flow to new and uncolonized regions of the host (Fig. 2j).
S. aureus biofilms are regulated, in part, by the accessory gene regulator (agr) quorum sensing system. The agr regulon is a well-described two-component peptide quorum-sensing system that utilizes cell–cell communication to facilitate dynamic expression of an array of virulence genes, including those responsible for biofilm formation.79,80 During the initial phase of biofilm formation, the agr system responds to the local abundance of self-produced autoinducing peptides (AIPs)81,82 by expressing genes associated with colonization including protein A, coagulase, and fibronectin-binding protein83 resulting in robust biofilm formation.79 As the concentration of AIPs reaches a threshold, the agr system activates most temporally expressed virulence genes including, alpha-toxin, beta-hemolysin, Toxic shock syndrome toxin (TSST-1), and leukotoxin, thus activating biofilm dispersal.84,85
Biofilms confer long-term bacterial cell survival in hostile environments by a variety of mechanisms. First, biofilms protect bacterial cells from immune onslaught by providing a physical barrier that limits immune cell penetration, thereby preventing phagocytosis and ROS killing. Additionally, bacteria residing within biofilms are particularly pathogenic because of the extreme phenotypic diversity that exists among them, enabling antimicrobial resistance. S. aureus in a biofilm can display variable growth rates, altered oxygen, and nutrient dependence and acquired virulence mechanisms via horizontal gene transfer.86 For these reasons, bacteria within biofilms become very resistant to antimicrobial treatment and can survive drug dosing up to 1 000 times greater than their planktonic phenotype.87
In addition to conferring long-term bacterial survival, biofilms can cause immense damage to the surrounding host tissue. Bacterial infections have been shown to elicit bone resorption, both indirectly through host inflammatory factors and directly through bacterial factors. In the infected bone environment, PAMPs interact with innate toll-like receptors (TLRs) expressed on a variety of cell types and stimulate the release of inflammatory cytokines including: TNF, IL-1, and IL-6.60 This leads to activation and differentiation of osteoclasts, while stimulating increased RANKL production by osteoblasts shifting the RANKL/OPG ratio in favor of bone resorption.88 Recent studies have shown that host signaling through MyD88 and IL-1R is necessary for the control of infection but paradoxically contributes to pathological bone loss.89 Furthermore, ex vivo and in vitro studies have shown that various bacterial organisms have the ability independently degrade bone tissue in the absence of all host factors.90,91
Taken together, biofilm presents a significant concern to surgeons when treating implant-associated infections. It is clear that complete eradication of biofilm from implant hardware and associated necrotic tissue is of utmost importance for successful clearance of infection.
Unfortunately, physical removal of biofilm from implant hardware via irrigation or sonication has been met with limited success.92 Therefore, complete hardware exchange and extensive debridement remains the gold standard to reduce the risk of reinfection due to bacterial persistence in biofilms.
Colonization of the osteocyte-lacuno canalicular network (OLCN)
The third and most recently discovered reservoir of bacteria in chronic osteomyelitis is S. aureus colonization of the OLCN.93,94 This phenomenon was first observed in a systematic examination of infected bone by transmission electron microscopy (TEM) in an experimental murine model of chronic S. aureus osteomyelitis.93 TEM micrographs showed S. aureus invasion of canaliculi perpendicular to the medullary canal and subsequent colonization of lacunar spaces, devoid of osteocytes. These observations do not conform to the historical dogma that defined S. aureus as non-motile cocci ~1 μm in diameter, incapable of invading tissue at a submicron scale. Most recently, invasion of the OLCN has been confirmed in clinical bone samples from patients with diabetic foot ulcers and underlying osteomyelitis.94
S. aureus colonization of the OLCN was shown to be an active process rather than dormant persistence in an experiment that used BrdU labeling of actively dividing S. aureus at the leading edge of canalicular invasion.93 Based on the absence of any observable motility structures, such as flagella, cilia, or pseudopods, it is theorized that S. aureus is capable of invading the canaliculi of live bone with a novel motility mechanism. Our working model of OLCN invasion begins with the eradication of bone lining cells from cortical bone due to local inflammation and necrosis during the establishment of S. aureus infection (Fig. 2k). Next, we theorize that S. aureus can identify exposed submicron canalicular orifices via haptotaxis, triggering asymmetric binary fission in which deformed daughter cells are extruded. Widening and scalloping of colonized canaliculi is frequently observed, as the bacterial acid demineralizes the bone, and degradative enzymes consume the organic bone matrix for nutrients (Fig. 2l), a phenomenon also observed in ex vivo studies independent of host factors.90 Invasion proceeds with proliferation of S. aureus cells through canaliculi and lacunar spaces as osteocytes are killed by bacterial colonization (Fig. 2m). Unlike bacteria in the bone marrow that are surrounded by neutrophils, S. aureus inside the OLCN are surrounded by the dense mineral matrix of cortical bone and are completely inaccessible to immune cells (Fig. 2l). These observations suggest that S. aureus may be able to survive for decades within the OLCN with an inexhaustible supply of nutrients, while evading immune attack.
Recently, an in vitro platform was developed to mimic S. aureus invasion of submicron spaces, like that of the OLCN, using a nanoporous membrane.95 Surprisingly, this study found that S. aureus propagation through nanopores in vitro is not dependent on activation of the agr quorum sensing system. This result was validated in vivo where agr deletion did not impair the bacteria’s ability to invade the submicron-sized canalicular network of cortical bone despite global down-regulation of virulence-associated genes. Taken together, it is clear that continued research is warranted to determine the genetic mechanisms of S. aureus invasion of the OLCN.
The discovery of OLCN invasion is particularly concerning for the treatment of implant-associated osteomyelitis, because it is impossible to know if all segments of infected bone have been completely debrided. Debridement of bone is guided by the presence of necrotic “white” bone and healthy “red” bone. Directed by this method alone, S. aureus colonized bone could remain in the patient despite extensive debridement and eventually facilitate recurrence of infection following revision surgery. Additionally, deep invasion of cortical bone by bacteria occurs within 2 weeks in our mouse models of osteomyelitis, which suggests this hallmark of chronic infection may actually occur within the timescale of acute osteomyelitis.
Eradication of bacteria infecting the OLCN of bone remains an open question as it is currently not known if standard-of-care antibiotic therapy can be effective against S. aureus embedded within the bone matrix. Furthermore, it is possible that the bacteria within the OLCN exhibit an altered growth phenotype like that of SCVs making them tolerant to antimicrobial therapy. Recent studies have indicated that only select antimicrobials are effective against SCVs,96 thus additional experiments to confirm the efficacy of antibiotic therapy on S. aureus within the OLCN are needed.
Collectively, these novel findings suggest that invasion and propagation within the immune-privileged OLCN is a pathogenic mechanism that renders S. aureus infection of bone incurable. Further, the long-term colonization of cortical bone by slow growing bacteria seems to be the most likely explanation for infection recurrence after decades of quiescence.32,33,97 Considering this new information on distinct biofilms in chronic osteomyelitis, including SACs and colonization of the OLCN of live bone, a major unmet clinical need is a diagnostic that can identify the existence of bone infection with spatial resolution, and assess the completeness of debridement following revision surgery. Indeed the 2018 ICM Biofilm Workgroup deliberated on the question of, “Is the mapping of biofilm to a particular component or anatomical location an important consideration in management of implant-related infections?”8 Unfortunately, this group of experts concluded that, “At present, mapping of biofilms is only possible in the laboratory, not in the clinical setting. Therefore, it is of unknown clinical importance in relation to management of implant‐related infections.” Interested readers are encouraged to review these proceedings online.13
The role of S. aureus intracellular persistence in osteomyelitis
As mentioned above, the interspersed periods of bacterial quiescence in chronic osteomyelitis has been a perturbing problem that is readily documented over the years.32,33,97 A particularly interesting case of S. aureus osteomyelitis described a woman who was initially infected in her left femur as a child and treated surgically without antibiotic therapy.97 Approximately 75 years later the infection returned, and genetic analysis of the infecting pathogen revealed the S. aureus to be of the same strain from 75 years prior, maintaining its sensitivity to penicillin and oxacillin. This case demonstrates an interesting immune evasion strategy in which a single clonal strain of S. aureus can remain quiescent for long periods of time, then awaken decades later to cause serious osteomyelitis.
In addition to the mechanisms of osteomyelitis persistence described above, many groups have questioned the contribution of intracellular colonization of bone cells as a possible mechanism for quiescent osteomyelitis. The role of intracellular persistence in chronic osteomyelitis disease pathology remains a subject of intense debate because of a lack of compelling in vivo evidence.98,99
Intracellular persistence of S. aureus in a variety of host cell-types has been described in many disease settings.98,99,100 Current literature has shown that S. aureus is capable of surviving within professional and non-professional phagocytes including, macrophages, neutrophils, fibroblasts, keratinocytes, epithelial cells, and endothelial cells.101,102 Internalization can be triggered through initial attachment to host cell surface-associated proteins using MSCRAMMs. For example, fibronectin-binding proteins A and B (FnBP) have been shown to initialize internalization of S. aureus via fibronectin bridging to α5β1 integrins and subsequent cytoskeletal reorganization in endothelial cells.103 Once internalized, the bacteria can survive cell death by preventing fusion of the phagolysosome, escaping the endosome into the cytoplasm or resistance to cellular enzymes.104,105
In the context of osteomyelitis, osteoblasts have been the primary host cell investigated for intracellular persistence because of their long life-span and ultimate differentiation into osteocytes. Some groups hypothesize that osteoblasts106,107 can be infected intracellularly, resulting in persistence in chronic osteomyelitis, based on in vitro findings. These studies investigated many factors such as osteoblast cell signaling,108,109 induction of osteoblast apoptosis,110 the effect of antibiotics on intracellular S. aureus,107,111,112 and bacterial cell gene expression.113 Hamza et al. found that osteomyelitis could be induced in vivo by infecting the femurs of rats in an open femoral fracture model using osteoblasts that were previously intracellularly infected in vitro.114 The authors claim that this study provides direct in vivo evidence of osteomyelitis induction by intracellular infection alone. However, no difference between induction of osteomyelitis with intracellular and extracellular S. aureus were observed as establishment of infection required the exact same inoculum of 102 CFU S. aureus in both groups. Further, it cannot be determined if an infection was truly established by intracellularly infected osteoblasts, or if osteoblasts containing S. aureus simply lysed upon implantation in the host, releasing bacteria extracellularly for the establishment of infection.
Other bone cells such as cultured osteoclasts54 and osteocytes115 have been studied in vitro and demonstrate the ability to internalize S. aureus. While these studies may describe essential details on the interactions between bone cells and S. aureus, they do not necessarily provide direct evidence of intracellular infection in vivo.
The current research on intracellular persistence of S. aureus in osteomyelitis in vivo is still very limited. A small number of clinical case studies have shown intracellular persistence, where one study identified fibroblasts with internalized S. aureus in a PJI case,29 and another described a patient with recurrent osteomyelitis with intracellularly infected osteoblasts and osteocytes shown by TEM.33 The only experimental study that demonstrated intracellular infection of bone cells in vivo was performed 2 decades ago, in which embryonic chicks were infected with S. aureus and bone samples interrogated by TEM showed bone cells with internalized S. aureus.116
We have studied the pathogenesis of S. aureus chronic osteomyelitis extensively in experimental in vivo models, as well as in clinical cases.84,93,94,117 Throughout all in vivo studies, we failed to identify intracellularly infected live bone cells. Using TEM examination as described above, S. aureus invasion of the OLCN of cortical bone is consistently observed with exclusively dead or dying infected bone cells (Fig. 3a). As a result of these expansive studies, we conclude that intracellular persistence within osteoblasts, osteoclasts, and osteocytes is not a meaningful mechanism of chronic or recurrent osteomyelitis. In contrast, we routinely observed intracellular infection of leukocytes, so called “Trojan Horses,”100 in septic animals and patients. This occurs via S. aureus invasion without triggering a bactericidal respiratory burst, creating circulating cells that facilitate bacterial metastasis to immune privileged sites.118 Intracellular persistence within macrophages is particularly concerning because of this leukocyte’s longer life span and ability to travel through the host circulatory system.119

The role of S. aureus intracellular infection as a virulence mechanism in chronic osteomyelitis. Extensive TEM analyses of S. aureus-infected human bone samples (n > X) failed to identify significant evidence of viable bone cells (osteoblasts, osteoclasts, osteocytes) containing intracellular bacteria, while all S. aureus colonized OLCN contain necrotic osteocytes (OC) with extracellular bacteria (red arrows) within osteocyte lacunae (a, TEM ×15 000). In contrast, an acridine orange-stained smears of blood, harvested post-mortem from a patient that died from septic multiorgan failure, demonstrates both extracellular bacteria (orange) and colonized leukocytes (yellow cells) via fluorescent microscopy (b, ×20). c A higher power fluorescent image of the blood smear reveals a “Trojan horse” macrophage with cytoplasmic S. aureus, and acentric nucleus (fluorescent green). d TEM (×20 000) of this Trojan horse macrophage was performed via a “pop-off” technique, which confirmed intracellular S. aureus cocci within the cytoplasm, adjacent to the nucleus (yellow arrow)
In addition to persistent and recurrent infection, sepsis is a significant concern in implant-associated osteomyelitis. Approximately, 10% of PJI cases lead to sepsis and death,12 and S. aureus remains the leading and most deadly cause of bacteremia in the United States.120 In a postmortem examination study of patients who died from S. aureus septic multi-organ failure, fluorescent imaging of acridine orange-stained blood smears showed both extracellular bacteria as well as intracellularly colonized leukocytes (Fig. 3b). Higher magnification fluorescent and electron microscopy imaging revealed S. aureus survival within the cytoplasm of a Trojan Horse macrophage (Fig. 3c, d).
In a study directly comparing osteoblast and macrophage infection with S. aureus, researchers found that macrophages contained 100-fold more live bacteria than osteoblasts, and that osteoblasts were significantly less viable.121 This result suggests that macrophages may be a more likely host cell candidate for S. aureus intracellular persistence. Further, the same study found that S. aureus only survived within osteoblasts and macrophages for 7 and 5 days,121 respectively, suggesting that these cell types are likely not the source of persistent osteomyelitis years after initial infection.
It remains possible that intracellular infection could play a role in bone infection, however, that role is not immediately apparent because of the extremely limited amount of in vivo evidence. One could hypothesize that S. aureus may survive intracellularly for a brief period of time to avoid innate immune attack at early stages of infection, potentially providing direct access to exposed canaliculi on the bone surface. To that end, we are currently exploring the virulence mechanisms that may promote intracellular survival of S. aureus in the OLCN.
Collectively, it is apparent that additional in vivo studies must be performed to elucidate the specific role of S. aureus intracellular persistence in chronic osteomyelitis. A successful in vivo experiment investigating intracellular infection should be able to demonstrate that the host cells along with internalized bacterial cells, are both alive for a prolonged period of time. Further, a successful experiment should demonstrate that internalized bacterial cells have the same clonality as the initial infecting strain. Currently, most analyses of intracellular persistence are performed using TEM, and are therefore limited in their ability to define whether cells are alive or dead.122 Advances in in vivo imaging techniques123 combined with clinically relevant animal models124 may allow for more accurate longitudinal tracking of bacterial infections to possibly answer some outstanding questions related to intracellular persistence in osteomyelitis.
Classification of acute and chronic osteomyelitis
Many factors can increase the risk of osteomyelitis including: diabetes, obesity, malignancy, immune deficiencies, substance abuse, malnutrition, and trauma.41,125 Due to large variations in patient populations, bimodal age distribution (most patients are younger than 20 or older than 50) and variable clinical presentation, classification guidelines for selecting appropriate treatment are widely variable and not ubiquitously accepted.
Detection of osteomyelitis in the clinic relies on a combination of techniques including: clinical signs, radiology,126 and wound swabbing with organism culture or polymerase chain reaction (PCR) for species identification. Once detected, the diagnosis of osteomyelitis is commonly categorized into subjectively defined groups of sub-acute, acute, or chronic stages of disease severity.
In its simplest form, osteomyelitis classification will rely on disease duration to suggest a specific course of treatment. Acute osteomyelitis typically describes a recent bone infection that causes systemic inflammation.55 Chronic osteomyelitis describes bone infection of longer duration, with minimal systemic symptoms and the presence of key pathological features, such as a lymphoplasmacytic infiltrate, marrow fibrosis, and reactive new bone formation. Chronic stage osteomyelitis is sometimes defined as early as 4 weeks,50 since initial disease presentation to as late as 6 months127 and generally requires surgery.55 However, there is a lack of scientific rationale for the selection of a specific time-point causing a lack of consensus among medical professionals.
Other methods of classification, accessory to disease duration and clinical signs, include modalities of radiographic imaging.128,129 Radiography plays a vital role in the diagnosis of osteomyelitis and can be used to evaluate the extent of infection, the anatomy involved and even specific hallmarks of acute vs. chronic stage disease, such as the presence of a sinus tract, sequestrum (avascular necrotic bone), or involucrum (new bone formation).130 Unfortunately, typical radiographic techniques are limited in their accuracy when detecting low level disease, as well as in the presence of metallic implants, which decrease image quality with artifact.
When available, an intraoperative histopathological examination can be used to identify regions of acute infection in periprosthetic tissue specimens. Past studies have shown that acute inflammation defined by the presence of >1–10 polymorphonuclear leukocytes in several high-power fields of view (400×) can be a useful predictor for acute infection.131,132,133 However, the variability of osteomyelitis disease presentation extends beyond patient-to-patient differences to variability on a cellular level within a single lesion.
When a segment of infected bone is investigated histologically for hallmarks of acute and chronic inflammations (Fig. 4), it becomes clear that features of both acute and chronic osteomyelitis can be present in the same specimen. From a case of acute osteomyelitis in a tarsal bone under an infected ulcer in a diabetic patient, we can observe distinct characteristics of acute infection, as well as chronic inflammation within the same lesion (Fig. 4a). A region of acute infection can be identified by the presence of predominantly newly activated neutrophils, as well as fibrovascular granulation tissue around foci of dead bone (Fig. 4b). However, adjacent to this lesion, we can identify hallmarks of chronic inflammation where normal bone marrow is replaced by fibrosis, reactive bone formation, blood vessels and collections of lymphocytes and plasma cells (Fig. 4c, d). This case highlights the significant amount of heterogeneity that can exist within a single osteomyelitis lesion and is typical of many infected foot ulcers with underlying osteomyelitis that has been treated conservatively before surgery.

Histologic features of “acute” and “chronic” osteomyelitis exist in the same lesion. Hematoxylin and eosin-stained, paraffin-embedded, decalcified section of an infected metatarsal bone resected from a patient with a diabetic foot ulcer is shown, illustrating salient features of both acute and chronic osteomyelitis in the same bone. a Low power micrograph of the lesion in which most of the trabecular bone in this part of the metatarsus has been destroyed and replaced by an acute inflammatory reaction, consisting of neutrophils (*) and fibrovascular granulation tissue (black arrow) (scale bar = 1 mm). The inflammation extends to the bone beneath the articular cartilage (yellow arrow) and has destroyed much of the cortical bone (white arrow). Reactive new bone has formed in the lower part of the image, along with a chronic inflammatory and fibrovascular reaction. b A region of interest of acute inflammation (white box in a) is shown highlighting a fragment of dead cortical bone surround by neutrophils (black arrow), with an associated fibrinous exudate, which are hallmarks of acute osteomyelitis (scale bar = 25 mm). c A region of interest of chronic inflammation (black box in a) showing new bone formation (black arrow), and replacement of normal bone marrow with fibrovascular inflammatory tissue (boxed region) (scale bar = 50 mm). d This region of interest (boxed area in c) is presented at high power, showing blood vessels, osteoblasts rimming newly formed woven bone (bottom right), and collections of lymphocytes and plasma cells (arrows), which are characteristic of chronic osteomyelitis (scale bar = 25 mm)
Collectively, empiric definitions of acute and chronic stage osteomyelitis fail to accurately and comprehensively describe the extent of infection. Such classifications of acute vs. chronic stage disease significantly diminish guidance for treatment. For these reasons, we recommend that time-dependent classifications of osteomyelitis should be avoided because of extreme patient-to-patient variability, as well as pathophysiological variability at the cellular level.
Lack of definitive methods to effectively discriminate acute vs. chronic-stage bone infections without biopsy and histologic examination makes treatment protocols based on such classifications almost meaningless. It should be assumed at all stages of prosthesis infection that the patient is at risk for soft tissue abscess formation, biofilm formation on implant hardware, and cortical bone colonization by bacteria.
Alternative classification systems based on criteria, such as the condition of the host, extent of disease134 and previous disease admission135 have been proposed to guide and improve the success of disease treatment. While these classifications have achieved minimal traction and fail to achieve consensus among medical professionals, they demonstrate constructive progress in the definition of osteomyelitis. Therefore, although the time has come to update the clinical definitions of “acute versus chronic” bone infection, as this issue was identified as “the greatest research priority from 2018 International Consensus Meeting” (2018 ICM) on Musculoskeletal Infection,13 these key opinion leaders also found this issue to be the most controversial topic in this field. Remarkably, three attempts to draft a recommendation for the question, “What is the recommended time interval that would divide acute and chronic peri-PJI (4 weeks, 90 days, etc.)?” failed, and interested readers are encouraged to review these proceedings online.13 Thus, given the complexities that surround this issue, we cannot offer new definitions at this time. However, we concur with the 2018 ICM Workgroup that “these new definitions must incorporate our current understanding that the management of bone and implant‐related infections is inextricably intertwined with the biology of biofilms, infection of osteoblasts and osteocytes, and invasion of the osteocytic‐canalicular network by the bacteria.”13
Antibodies and S. aureus orthopedic infections
S. aureus has evolved numerous strategies to efficiently evade adaptive host defenses, which consist of cell-mediated responses dominated by T-cells and humoral antibody responses mediated by B-cells. Anti-S. aureus antibodies are prevalent in all humans due to life-long exposure to S. aureus by means of prior infections or asymptomatic carriage.136,137,138,139,140 However, the presence of these antibodies in the host (collectively termed humoral “immune proteome”) does not necessarily warrant protection against future infections. In fact, some individuals are more susceptible to S. aureus PJI than others likely due to the protective vs. susceptible nature of their immune proteome. Thus, a better understanding of the functional role of specific antibodies in S. aureus infections can aid in predicting outcomes, while providing targets for the development of novel therapies to combat S. aureus bone infection. In this section, we will discuss the utility of antibodies in diagnosing S. aureus orthopedic infections in addition to summarizing the discovery and development of antibody-based biologics to manage S. aureus infections.
Antibodies as diagnostic and prognostic markers of orthopedic infections
Deep-rooted infections such as implant-associated osteomyelitis present a diagnostic predicament because patients typically present with non-specific symptoms, including pain, swelling of the joint, and fever. Definitive diagnosis of the infection requires obtaining intraoperative specimens or via image-guided biopsy procedures, which are expensive, invasive, and traumatic. Most clinicians rely on inflammatory cell counts and on markers, such as the C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) for diagnosing S. aureus orthopedic infections. However, these techniques are not pathogen-specific, nor are they anatomically specific. Importantly, they cannot accurately assess if a patient is responding to treatment.
Blood-based approaches describing anti-S. aureus humoral immune response in physiologic and pathologic conditions provide superior alternatives for diagnosing S. aureus infections. Indeed, several groups including ours have attempted to describe blood-serum-based S. aureus-specific antibodies during colonization, bacteremia, and orthopedic infections.138,139,141,142,143,144 Utilizing a multi-antigen Luminex immunoassay with immunodominant antigens belonging to distinct functional classes, such as iron acquisition proteins, cell division proteins, secreted toxins, and cell attachment proteins, we demonstrated that: (1) S. aureus orthopedic infections can be diagnosed using antibody-based immunoassays, (2) anti-S. aureus antibody responses against certain antigens predominate during PJI, and (3) single antigen and/or a combination of antigens are useful predictors of ongoing S. aureus orthopedic infection.144 For instance, our studies illustrated that antibody responses against iron-sensing determinant proteins (IsdA, IsdB, IsdH) could be predictors of infection outcomes in patients with S. aureus PJI, and that higher serum anti-IsdA and anti-IsdB IgG levels in patients were associated with increased mortality.144 In contrast, IgG levels in serum against a cell division protein called autolysin (Atl) was protective against S. aureus infections in patients undergoing orthopedic surgeries.117,143
The clinical utility of serum-based immunoassays for the diagnosis of microbial infections cannot be discounted. However, high levels of pre-existing anti-S. aureus antibodies are present in most patients due to asymptomatic carriage.136,139 Additionally, prior exposure due to infection confounds reliable diagnosis of S. aureus infections using this method. One way to obviate this problem is by examining pathogen-specific antibodies in recently stimulated circulating plasmablasts in the patient’s blood.
As adaptive responses develop during an infection, pathogen-specific B-cells stimulated in germinal centers of lymph nodes proliferate, secrete their antibodies and enter the circulation. These plasmablasts, often called antibody-secreting cells (ASCs), emerge early in an infection and are circulating in the blood as long as the infecting pathogen remains active.145,146,147 This attribute makes ASCs and their specific antibodies perfect biomarkers for ongoing infections, successful (or failed) therapy, and recurrence. Though ASCs have been explored in multiple settings,145,146,147 they have yet to be exploited in general medical practice for infection diagnosis and prognosis. Here, we describe a simple approach developed by our group to utilize ASCs for diagnosing S. aureus orthopedic infections.
Peripheral blood mononuclear cells (PBMC), which include newly activated ASCs, are harvested from whole blood and washed extensively to remove pre-existing serum immunoglobulins. Subsequently, the ASCs are cultured in vitro to allow their expression and secretion of antibodies in response to any ongoing infection (Fig. 5a). The resulting media enriched for newly synthesized antibodies (MENSA) represent a measurable and accessible way to detect acute immune responses to ongoing infections. MENSA levels are direct indicators of ongoing infections as they develop and as the infections wane under the attack of the host’s immune responses.

A diagnostic and prognostic immunoassay for the measurement of anti-S. aureus antibody levels in patients with osteomyelitis. a Schematic illustration of production of serum and isolation of medium enriched for newly synthesized anti-S. aureus antibodies (MENSA) from peripheral mononuclear cells of patients with osteomyelitis. b Anti-S. aureus antibody levels in serum and MENSA were determined using a custom bead-based multiantigen Luminex immunoassay developed by our group. Here, we examined anti-S. aureus IgG responses in serum and MENSA of diabetic foot infections (DFI) patients undergoing foot salvage antimicrobial therapy (FST). The change in antibody titers over the course of FST of a representative patient whose DFI was negative for S. aureus, a patient with S. aureus infection that responded to FST, and a patient with S. aureus DFI who failed FST is presented here. Remarkably, MENSA levels faithfully reflected the S. aureus infection over time while serum levels remained unchanged (see Oh et al.148 for more details)
In a recent study involving the management of foot salvage therapy (FST) for diabetic foot infections (DFI), we demonstrated that our immunoassay utilizing MENSA can accurately monitor treatment response and detect persistent or recurrent infections that are invisible in a serum response.148 Anti-S. aureus IgG levels in MENSA decreased with successful FST, and rose with re-infection, while IgG levels in serum remained unchanged throughout the FST treatment period (Fig. 5b). These results demonstrate that MENSA evaluation could be used as a prognostic tool to guide clinical decisions in orthopedic infections. Additionally, tracking changes within the DFI microbiome may be a promising tool for monitoring treatment response.149
Antibodies as biologics for combatting orthopedic infections
Battling the tenacious S. aureus with antibiotics is a challenge because the pathogen has extraordinary abilities to develop resistance against antibiotics.150 Immunotherapies may be effective adjuvants with antibiotics for combating hard-to-treat osteomyelitis. Over the past two decades, numerous potential S. aureus vaccine targets received preclinical validation,151,152 yet none turned out to be effective in providing protection against S. aureus infections in humans.153 For instance, a phase 2 clinical trial of an IsdB active vaccine (Merck’s V710), in which 8000 patients undergoing heart valve replacement were randomized to placebo or the vaccine, had to be stopped due to a five-fold increase in fatal and adverse outcomes in the vaccinated individuals due to S. aureus bacteremia.154 This clinical trial and several others were either terminated abruptly by the FDA or showed no efficacy in humans. Major factors contributing to the failure of functional active vaccines against S. aureus include: (1) use of antigen targets that produce antibodies, which opsonize bacteria but fail to induce cell lysis or phagocytosis, (2) limited biological activity against the targets, (3) overreliance on murine models for preclinical validation, and (4) lack of strategies to combat site-specific infection such as osteomyelitis.104,151,152,153,155,156,157
Passive immunization involving monoclonal antibodies (mAbs) is becoming a more attractive immunotherapy to treat S. aureus osteomyelitis. A benefit of passive immunization with mAbs is their high antigen-neutralizing specificity. Additionally, mAbs can be administered locally to the site of infection, and production of mAb at scale is not cost prohibitive. Several anti-S. aureus mAb passive immunization agents have been evaluated in human clinical trials. These include mAbs that target fibrinogen-binding cell surface protein (ClfA),158 cell wall components (lipoteichoic acid (LTA), poly-N-acetylated glucosamine (PNAG))159,160 and secreted toxin, such as α-hemolysin (MEDI4893 and AR-301).161,162,163,164 Unfortunately, these first-generation mAb biologics, despite their promise of safety and tolerability in humans, demonstrated limited efficacy in clinical trials. Their inadequate success could again be attributed to limited biological activity against the target, their inability to effectively mediate phagocytic killing, and the array of different mechanisms utilized by S. aureus to circumvent components of host immune system.165,166
The second generation of antibody-based biologics, currently in preclinical development, target multiple antigens with essential functions for S. aureus immunoevasion, colonization, intracellular growth and persistence within the host cells. Targeting leukocidin toxins that selectively kill immune cells is key to attenuating S. aureus virulence. Therefore, several multivalent mAbs that can neutralize these toxins are currently under development. For instance, Rouha and colleagues described one such multivalent mAb that targets α-hemolysin and four leukocidins (HlgAB, HlgCB, LukED, PVL). This broadly toxin-neutralizing mAb was shown to provide protection against S. aureus in murine sepsis and pneumonia models.167 Other neutralizing antibodies that target essential S. aureus immunoevasion mechanisms are also under preclinical testing. These include mAbs that target the B-cell antigen staphylococcal protein A,168,169 the immunodominant surface protein IsaA,170 and the immune-stimulatory staphylococcal superantigen SEB.171,172 Interestingly, multivalent mAbs that simultaneously target and neutralize several of the aforementioned toxins and immunoevasion proteins are under preclinical development by various pharmaceutical companies and research institutions.
It has recently been acknowledged that blocking the ability of S. aureus to colonize, persist, and grow intracellularly within different host cells, such as macrophages, may be a viable strategy to reduce S. aureus burden in human disease. We discovered that certain PJI patients who have high serum IgG levels against a cell division protein called Atl ended up recovering fully from MRSA PJI, whereas those that had lower anti-Atl IgG levels often had morbid clinical outcomes.143 To further investigate this intriguing phenomenon, we developed mAbs against the glucosaminidase (Gmd) subunit of Atl. Not surprisingly, we observed that anti-Gmd mAb can inhibit S. aureus cellular functions, such as binary fission, induce megacluster formation, and opsonophagocytosis by macrophages, and protect mice from S. aureus implant-associated osteomyelitis.117,173
Based on the recent transformative clinical success of biologic checkpoint inhibitor cancer immunotherapy, it is likely that antibody-based biologics offer similar promise for treating S. aureus osteomyelitis. Encouragingly, there are several anti-S. aureus mAb biologics currently under preclinical and clinical development. Combined with our advances in understanding the pathophysiology of S. aureus, we optimistically predict that, in time, mAb-based biologics will be routinely utilized to prevent and treat S. aureus osteomyelitis.
Novel antibiotic therapies to combat osteomyelitis
The recurring and resilient nature of osteomyelitis, as well as its affiliated high patient morbidity, extended hospitalization, and costly healthcare expenses, have prompted research efforts to develop novel therapeutics to improve upon the standards used today to treat osteomyelitis. These efforts have primarily focused on developing and investigating implant coatings that inhibit bacterial adhesion, prevent biofilm formation, and provide bactericidal activity, as well as therapeutics for the local delivery of antimicrobials in dead space management. These strategies to combat osteomyelitis are essential to consistently achieve favorable patient outcomes.
Implant coatings
As discussed previously, S. aureus has the ability to attach to and colonize both the tissues of the musculoskeletal system and orthopedic implants.84,85,174,175 S. aureus first adheres to a substrate by binding to host proteins, including fibrinogen via Clumping factor A and B (ClfA, ClfB), fibronectin via fibronectin-binding proteins A and B (FnBPA, FnBPB), and collagen via collagen adhesin (Can).75 Then subsequent colonization, and expression of various virulence factors (i.e. exotoxins, enterotoxins, adhesive factors, etc.)176,177 prompt a detrimental inflammatory reaction.130,178 In order to halt the ultimate development of PJI, implants are treated with physical and chemical modifications to prevent the first and cardinal step of infection, bacterial adherence. Yet, it is important to note that implant coatings primarily reduce a patient’s susceptibility to infection and are not effective in treating an established infection.
Silver is often studied as an antimicrobial coating due to its broad spectrum activity against Gram-positive and Gram-negative bacteria, fungi, and viruses.179 As a result of its nonspecific biocidal activity, silver has been used in various industrial, healthcare, and domestic applications most commonly in the topical chemoprophylaxis of burns either as a topical cream or antimicrobial wound dressing.180 The mechanism of action of silver is a result of biologically active ions released from its surface producing a toxic effect.181,182 Silver toxicity arises from three mechanisms. First, silver ions bound to the cell wall create morphological irregularities causing cell lysis and release of intracellular contents.183 Second, silver ions bound to sulfhydryl groups of proteins or DNA impair essential metabolic pathways and inhibit DNA replication.184 And lastly, silver has been observed to produce excessive ROS causing oxidative damage.185 These mechanisms of action also prevent biofilm formation by inhibiting the production of exopolysaccharides, which is a required prerequisite for biofilm formation.186
Due to its successful application in various fields, silver has been used to directly coat orthopedic implants, added to bone cement187,188 and incorporated into hydroxyapatite (HA) coatings of replacements.189,190 However, the true efficacy and safety of silver is still debated in orthopedic applications. Incorporation of silver nanoparticles into bone cement has shown increased antibacterial activity against various strains, including Staphylococcus epidermidis, methicillin-resistant S. epidermis (MRSE), and MRSA, compared to gentamicin-impregnated bone cement.187 While bone cement eluting silver nanoparticles does not appear to be cytotoxic to fibroblast cells, silver salt-eluting bone cement is confirmed to be highly cytotoxic.191,192 A study investigating the direct application of silver to Kirschner-wires (K-wires) inserted into S. aureus-infected femoral canals of rabbits found no difference in bacterial colonization between the coated and non-coated K-wires.189 However, the incorporation of silver into HA coatings of metal implants has shown more favorable antibacterial outcomes, with the added benefit of HA composition for improved osseointegration.193,194,195,196 Akiyama et al. investigated the antimicrobial activity of titanium rods coated with HA or silver (Ag)-HA in a murine model of MRSA osteomyelitis and found that Ag-HA-coated rods showed increased antimicrobial activity and increased bone formation compared to HA-coated rods.193
Clinical studies have also shown that coating implants with silver substantially reduces implant-associated osteomyelitis. Wafa et al. reported in a case-controlled study that silver-coated implants had a 11.8% infection rate compared to 22.4% for the uncoated-control group.197 Similarly, in another case-controlled study, Hardes et al. reported a 5.9%–17.6% infection reduction in patients with bone sarcoma treated with silver-coated implants.198 While these results appear promising, a serious concern for the application of silver in osteomyelitis treatment is systemic192,199 and cellular200,201 toxicity, documented in clinical cases of neuropathy192 and myoclonic status epilepticus.199 Because of silver toxicity observed in vitro and in vivo, additional studies are needed to investigate ideal formulations and effective concentrations for the safe clinical use of silver.202
In addition to silver, antibiotics have also been thoroughly investigated for incorporation into implant coatings. Antibiotics are the clinical standard for both systemic and local treatment of various infections caused by a broad-spectrum of pathogens and unlike silver, are not considered highly cytotoxic. Therefore, prophylactic coating of implant hardware with antibiotics is a rational approach for infection prevention. Coating hardware with antibiotics enables the local delivery of high concentrations of drug that would otherwise be toxic if delivered systemically. Numerous coating techniques and antibiotics have been investigated to treat S. aureus-related bone infections. The most common strategy for antibiotic coating is the attachment of biocompatible synthetic polymers loaded with antibiotics to the surface of implants. Commonly used polymers are poly(d,l-lactide) (PDLLA),203,204,205,206,207 polylactic acid (PLA),208 polylactic-co-glycolic acid (PLGA),209,210 poly(β-amino esters)/poly(acrylic acid),211 and poly(ethylene glycol)-poly(lactic-co-caprolactone) (PEG-PLC).212 Alternative drug-coating techniques include coprecipitation and covalent bonding of drug to implant.213,214,215 Gentamicin is the most studied antibiotic in coatings because of its application in the clinic for treating methicillin-sensitive S. aureus (MSSA) bone-related infections.203,204,206,207,208,210,211,213,214,216,217,218 Vancomycin, commonly used to treat MRSA infections, has also been incorporated into implant coatings.212,215,219 Additional antibiotics such as fosfomycin,204 doxycycline,209 minocycline,20 rifampin,20 colistin, daptomycin, and cefoxitin have also been incorporated into implant coatings.205 Studies using antibiotic-loaded implant coatings have primarily investigated the efficacy of prophylactic treatment in periprosthetic models of infection, consisting of rodents (mouse and rat) rabbits, and sheep. Regardless of the drug, antibiotic-coated implants generally showed a 0.3–4.5 log-reduction of colonized bacteria (MSSA or MRSA) in bone and 0.35–4.5 log-reduction on implants in comparison to a non-coated control.204,207,211,213,217,218,219 Diefenbeck et al. demonstrated that plasma chemically oxidized titanium rods coated with gentamicin and gentamicin+tannic acid showed 100% and 90% protection in a rat tibia infection model.214 Additionally, Metsemakers et al. demonstrated highly favorable results in a rabbit infection model, where doxycycline-coated intramedullary nails had 100% protection against MSSA (JAR060131; doxycycline-susceptible clinical isolate) and 43% protection against MRSA (LUH15101; doxycycline-resistant clinical isolate).209 To date, only one Level 2 randomized-controlled study investigating antibiotic-loaded implant coatings has been performed. In this study, the antibiotic loaded polymer described as Defensive Antibacterial Coating; DAC® was evaluated for its ability to minimize infection in trauma cases.220 This 253 patient multi-center European study observed a 4.6% rate of surgical site infections in the uncoated implant control group and zero infections in patients treated with DAC®-coated implants. Additionally, no adverse side-effects were observed. The positive outcomes of this clinical study prompt the need for further investigation of the efficacy of antibiotic coated implants.
An essential governing factor in infection management is the drug-release kinetics, which must be assessed in vitro. Ideally a biphasic drug-release profile is warranted, consisting of a bolus drug release to immediately deliver high concentrations of antibiotics to eradicate any bacteria, followed by a sustained drug release above the minimum inhibitory concentration that will kill any remaining bacteria (i.e. bacteria emerging from biofilms). Alternative strategies have also investigated the conjugation of bisphosphonates to conventional antibiotics, such as fluoroquinolones221,222,223 and glycopeptides,224 for systemic or local delivery to bone infection sites offering drug depot release kinetics near the bone surface.225 However, an alarming issue associated with prophylactic antibiotic implant coatings is the risk of antimicrobial-resistant strains. Antibiotic resistance is estimated to causes 10 million deaths/year by the year 2050.226,227 This suggests that significant attention should be directed to the development of novel antimicrobial classes to circumvent the constantly evolving bacterial resistance mechanisms.
In addition to silver and antibiotics, groups have explored the use of antimicrobial peptides (AMPs), bacteriophages (phages), and biofilm dispersal agents228 in implant coatings to prevent bacterial attachment and colonization. Various interesting coating strategies have been explored to bind these antimicrobials to implant hardware. Although these topics are beyond the scope of this review, the interested reader is referred to other comprehensive reviews.229,230
Dead space management
As discussed previously, implant-associated osteomyelitis commonly occurs in TJR or trauma cases where the insertion of implants is attributed to the contiguous spread of bacteria and infection. The clinical gold standard for treating implant-associated osteomyelitis is a two-stage surgical revision utilizing PMMA bone cement spacers for the local delivery of antibiotics. Antibiotic-eluting PMMA bone cement spacers are a necessity due to the limited blood flow to the site of infection and the ability to deliver high concentrations of select antibiotics with low systemic exposure.231 These spacers remain within the patient for 1–10 weeks and are typically removed in a second procedure, where bone grafting is performed with the installation of new hardware stabilization or external fixation to enable fracture healing.232,233,234,235,236 The numerous factors involved in treating infection after fracture fixation, such as the unpredictable nature of bone healing and infection, make each patient a unique case. Despite this unpredictability, reported treatment success rates vary between 70% and 90% as defined by osseous union and infection management.237,238,239 To fully understand the variance in success rates of these procedures, a closer examination of the antibiotic-laden spacers is needed.
The primary advantage of using antibiotic-laden PMMA is the delivery of high concentrations of drug directly to the infection site, which cannot be otherwise achieved by systemic administration due to off-target systemic toxicity. Gentamicin and vancomycin are the most commonly used antibiotics in PMMA spacers for implant-associated osteomyelitis.240 These antibiotics are typically chosen because of their broad-spectrum activity, compatibility with polymerization of PMMA, minimal host tissue toxicity, and widespread bioavailability.241,242 Despite these characteristics, a universal caveat of antibiotic-laden PMMA spacers is their poor drug release kinetics. In vitro studies demonstrate that only 5%–8% of the total antibiotic incorporated is released and in vivo studies assessing gentamicin release from PMMA also confirm that only 5%–18% of total drug is eluted.243,244,245,246,247,248,249 Additionally, drug release profiles of antibiotics from PMMA demonstrate an initial burst release within the first 24 h followed by a rapid decrease in release rates.250,251 This is detrimental because ideally a cement spacer remains in situ for 6 weeks and the subtherapeutic antibiotic concentrations can lead to microorganism resistance to the incorporated drug.252,253 The non-eluting surface of spacers can also be a substrate for bacterial colonization and biofilm formation.254 Furthermore, the limited number of antibiotics compatible for incorporation into PMMA are not fully effective against the various microbiological defenses and subpopulations of microorganisms commonly associated with implant-associated osteomyelitis.
These shortcomings of PMMA have set the stage for research efforts to develop an improved vehicle for local drug administration and dead space management. The main design criteria for an improved vehicle are: (1) enhanced drug elution kinetics for a biphasic drug-release profile (i.e. burst release followed by sustained release), and (2) a vehicle that is both biodegradable and osteoconductive. Drug-containing scaffolds that are biodegradable and osteoconductive enable the reduction of the required two-stage revision for PMMA to a single-stage revision, where the drug-containing scaffold not only successfully manages the infection, but also enables bone healing of the defect. Currently, various materials have been successfully loaded with antibiotics for the local delivery of antibiotics clinically, including allograft bone, bioactive glass, calcium sulfates, calcium phosphates, collagen implants, and demineralized bone matrix.255,256,257,258,259,260,261 Other biomaterials have been developed and investigated in preclinical models ranging from natural polymers, synthetic polymers, ceramics, and composite materials, which have been previously reviewed.262
A limiting factor in developing novel biomaterials for the management of dead space in osteomyelitis is the establishment of small animal models with clinical validity. Models utilizing internal fixation plates have only partially been able to partially exchange stabilizing screws with dead space management or complete hardware exchange with no dead space management.263,264 Although larger animal models exist, mouse models are preferred due to cost-effectiveness and ability to assess genetic traits for susceptible hosts using genetically modified lines. To date, only one mouse model exists where complete hardware exchange has been performed in a septic critical-sized defect.265 In this mouse model of implant-associated osteomyelitis, a mid-diaphyseal infection was established by insertion of a titanium screw inoculated with a bioluminescent strain of MSSA (Xen36).265 After 7 days of establishing the infection, examination yielded peri-implant and subcutaneous abscesses, as well as biofilm formation on the screw. At that time, a revision surgery was performed removing the infected screw and debriding any pathological soft tissue. New surgical hardware (polyether ether ketone (PEEK) fixation plate and four screws) were then installed and a 3 mm osteotomy was performed to remove the infected mid-diaphysis of the femur. The resulting defect enabled implantation of an antibiotic-impregnated spacer to assess infection management.
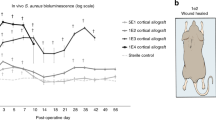
In a follow-up study, 3D-printed calcium phosphate scaffolds (CaPS) with incorporated sitafloxacin and rifampin were implemented into this single-stage revision mouse model of femoral implant-associated osteomyelitis.266 A dual coating of PLGA enhanced the mechanical strength and elution kinetics of the scaffolds. Drug-release kinetics exhibited a biphasic release in which an initial burst release was observed within the first 48 h followed by sustained zero-order release, which maintained the local concentration at ~ 900× the minimum inhibitory concentration of each drug. Examination of the concomitant delivery and independent local delivery of sitafloxacin and rifampin from 3D printed CaPS within the mouse infection model revealed improved infection management between the 3D printed CaPS with incorporated sitafloxacin and rifampin and the clinical control, gentamicin-laden PMMA bone cement. Reduced bacterial colonization rates were observed at both 3 and 10 weeks post-revision surgery for the 3Dprinted CaPS. Furthermore, a significant increase in bone formation was observed for 3D printed CaPS with incorporated rifampin in comparison to mice treated with gentamicin-laden bone cement, which underwent an additional revision surgery to insert a 3D printed CaPS with no antibiotics for bone healing.
Collectively, emerging technologies used to create biodegradable and controlled-release antibiotic-laden spacers have demonstrated the ability to improve upon the infection management of the clinical gold standard of antibiotic impregnated PMMA. The additional osteoconductive elements of these spacers enable a reduction of the required two-stage revision to single-stage. Future work must focus on refining the bone-healing potential of these scaffolds by addition of osteoinductive elements to enhance bone regeneration in preparation for definitive large animal studies and clinical trials.
Conclusion
Osteomyelitis has remained the bane of orthopedic surgery for over a half century, and while there have been many advances in our understanding of the pathophysiological consequences of bone infection during this time, there have been no changes to standard of care treatments. Of note is that while it is well-established that eradication of bacteria from all reservoirs of biofilm is imperative for curative treatment in the clinic, the nature of these bacterial reservoirs in chronic osteomyelitis have been poorly understood. Specifically, the recent discovery of S. aureus invasion of the OLCN warrants future experiments to elucidate the mechanism of invasion, and to determine how this privileged environment renders standard of care antibiotic therapies ineffective. Additionally, due to a lack of in vivo studies, the role of intracellular persistence in chronic osteomyelitis remains unclear and warrants further validation studies.
The recent breakthroughs in cancer immunotherapy, most notably checkpoint inhibitors, begs the question if similar advances could be made for osteomyelitis. Now that an important first step has been made by defining the host immune proteome against S. aureus, efforts are warranted to translate this new information into diagnostics and vaccines that can better detect ongoing infections, assess response to therapy, and develop adjuvants to antibiotic therapy. In particular, we find that MENSA has great potential to identify the pathogenic organism, distinguish acute versus chronic infection, and assess the patient’s response to therapy.
Finally, the time has come to formally investigate the use of non-FDA-approved antibiotic bone cement, as a means to cure osteomyelitis via bolus high-dose delivery of antimicrobials. In addition to the lack of level 1 clinical evidence to support this practice, it is now known that this foreign body material is rapidly colonized by biofilm bacteria after the initial release of the antibiotics. Moreover, rationally designed, custom 3D-printed antibiotic impregnated spacer technologies have emerged that can achieve both initial high-dose bolus release, and sustained antibiotic release at levels above the MIC. As these technologies may lead to standardized single-stage revisions for bone infection, their clinical investigation is warranted.
References
Klenerman, L. A history of osteomyelitis from the Journal of Bone and Joint Surgery: 1948 to 2006. J. Bone Jt. Surg. Br. Vol. 89, 667–670 (2007).
Peltola, H., Pääkkönen, M., Kallio, P., Kallio, M. J. & Group, O.-S. A. S. Short-versus long-term antimicrobial treatment for acute hematogenous osteomyelitis of childhood: prospective, randomized trial on 131 culture-positive cases. Pediatr. Infect. Dis. J. 29, 1123–1128 (2010).
Steiner, C., Andrews, R., Barrett, M., Weiss, A. US Agency for Healthcare Research and Quality; 2012. HCUP projections: mobility/orthopedic procedures 2003 to 2012. Report# 2012-03 (2014).
Kremers, H. M. et al. Prevalence of total hip and knee replacement in the United States. J. Bone Jt. Surg. Am. Vol. 97, 1386 (2015).
Elek, S. D. Experimental staphylococcal infections in the skin of man. Ann. N. Y. Acad. Sci. 65, 85–90 (1956).
Zimmerli, W., Waldvogel, F. A., Vaudaux, P. & Nydegger, U. E. Pathogenesis of foreign body infection: description and characteristics of an animal model. J. Infect. Dis. 146, 487–497 (1982).
Steckelberg, J. M. & Osmon, D. R. Prosthetic joint infections in Infections Associated with Indwelling Medical Devices. 3rd edn, 173–209 (American Society of Microbiology, 2000).
Saeed, K., et al. The 2018 International Consensus Meeting on Musculoskeletal Infection: Summary from the Biofilm Workgroup and consensus on biofilm related musculoskeletal Infections. J. Orthop. Res. 37, 1007–1017 (2019).
Gahukamble, A. D. et al. Propionibacterium acnes and Staphylococcus lugdunensis cause pyogenic osteomyelitis in an intramedullary nail model in rabbits. J. Clin. Microbiol. 52, 1595–1606 (2014).
Stulberg, J. J. et al. Adherence to Surgical Care Improvement Project measures and the association with postoperative infections. JAMA 303, 2479–2485 (2010).
Kurtz, S. M. et al. Infection burden for hip and knee arthroplasty in the United States. J. Arthroplast. 23, 984–991 (2008).
Cram, P. et al. Total knee arthroplasty volume, utilization, and outcomes among Medicare beneficiaries, 1991–2010. JAMA 308, 1227–1236 (2012).
Schwarz, E. M., et al. The 2018 International Consensus Meeting on Musculoskeletal Infection: research priorities from the General Assembly Questions. J. Orthop. Res. 37, 997–1006 (2019).
Rosas, S. et al. Season of the year influences infection rates following total hip arthroplasty. World J. Orthop. 8, 895 (2017).
Azzam, K., McHale, K., Austin, M., Purtill, J. J. & Parvizi, J. Outcome of a second two-stage reimplantation for periprosthetic knee infection. Clin. Orthop. Relat. Res. 467, 1706–1714 (2009).
Kurtz, S. M., Lau, E., Watson, H., Schmier, J. K. & Parvizi, J. Economic burden of periprosthetic joint infection in the United States. J. Arthroplast. 27, 61–5. e1 (2012).
Arciola, C. R., An, Y., Campoccia, D., Donati, M. & Montanaro, L. Etiology of implant orthopedic infections: a survey on 1027 clinical isolates. Int. J. Artif. Organs 28, 1091–1100 (2005).
Walter, G., Kemmerer, M., Kappler, C. & Hoffmann, R. Treatment algorithms for chronic osteomyelitis. Dtsch. Arzteblatt Int. 109, 257–264 (2012).
Pulido, L., Ghanem, E., Joshi, A., Purtill, J. J. & Parvizi, J. Periprosthetic joint infection: the incidence, timing, and predisposing factors. Clin. Orthop. Relat. Res. 466, 1710–1715 (2008).
Darouiche, R. O. Treatment of infections associated with surgical implants. N. Engl. J. Med. 350, 1422–1429 (2004).
Kaplan, S. L. Recent lessons for the management of bone and joint infections. J. Infect. 68, S51–S56 (2014).
Lowy, F. D. Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532 (1998).
Kluytmans, J., Van Belkum, A. & Verbrugh, H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin. Microbiol. Rev. 10, 505–520 (1997).
Otto, M. Staphylococcus aureus toxins. Curr. Opin. Microbiol. 17, 32–37 (2014).
Otto, M. Targeted immunotherapy for staphylococcal infections. BioDrugs 22, 27–36 (2008).
Hall-Stoodley, L., Costerton, J. W. & Stoodley, P. Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2, 95–108 (2004).
Ricciardi, B. F. et al. Staphylococcus aureus evasion of host immunity in the setting of prosthetic joint infection: biofilm and beyond. Curr. Rev. Musculoskelet. Med. 11, 389–400 (2018).
Tuchscherr, L. et al. Staphylococcus aureus small-colony variants are adapted phenotypes for intracellular persistence. J. Infect. Dis. 202, 1031–1040 (2010).
Sendi, P. et al. Staphylococcus aureus small colony variants in prosthetic joint infection. Clin. Infect. Dis. 43, 961–967 (2006).
Pendleton, J. N., Gorman, S. P. & Gilmore, B. F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 11, 297–308 (2013).
Hemmady, M. V., Al-Maiyah, M., Shoaib, A. & Morgan-Jones, R. L. Recurrence of chronic osteomyelitis in a regenerated fibula after 65 years. Orthopedics. 30, 403–404 (2007).
Gallie, W. First recurrence of osteomyelitis eighty years after infection. J. Bone Jt. Surg. Br. Vol. 33, 110–111 (1951).
Bosse, M. J., Gruber, H. E. & Ramp, W. K. Internalization of bacteria by osteoblasts in a patient with recurrent, long-term osteomyelitis: a case report. JBJS 87, 1343–1347 (2005).
Brodie, B. C. Pathological researches respecting the diseases of joints. Med Chir. Trans. 4, 210–280 (1813).
Costerton, J. W., Geesey, G. G. & Cheng, K. J. How bacteria stick. Sci. Am. 238, 86–95 (1978).
Buchholz, H. W., Elson, R. A. & Heinert, K. Antibiotic-loaded acrylic cement: current concepts. Clin. Orthop. Relat. Res. 190, 96–108 (1984).
Parvizi, J., Gehrke, T., Mont, M. A. & Callaghan, J. J. Introduction: Proceedings of International Consensus on Orthopedic Infections. J. Arthroplasty 34, S1–S2 (2019).
Bryan, A. J. et al. Irrigation and debridement with component retention for acute infection after hip arthroplasty: improved results with contemporary management. JBJS 99, 2011–2018 (2017).
Kuiper, J. W., Willink, R. T., Moojen, D. J. F., van den Bekerom, M. P. & Colen, S. Treatment of acute periprosthetic infections with prosthesis retention: review of current concepts. World J. Orthop. 5, 667 (2014).
Kuiper, J. W. et al. Prosthetic joint-associated infections treated with DAIR (debridement, antibiotics, irrigation, and retention): analysis of risk factors and local antibiotic carriers in 91 patients. Acta Orthop. 84, 380–386 (2013).
Minassian, A. M. et al. Chronic osteomyelitis of the pubic bones following radiotherapy for urological malignancy. J. Clin. Urol. 10, 213–219 (2017).
Crockarell, J. R., Hanssen, A. D., Osmon, D. R. & Morrey, B. F. Treatment of infection with debridement and retention of the components following hip arthroplasty. J. Bone Jt. Surg. Am. 80, 1306–1313 (1998).
Zimmerli, W., Widmer, A. F., Blatter, M., Frei, R. & Ochsner, P. E. Role of rifampin for treatment of orthopedic implant-related staphylococcal infections: a randomized controlled trial. Foreign-Body Infection (FBI) Study Group. JAMA 279, 1537–1541 (1998).
Osmon, D. R. et al. Diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin. Infect. Dis. 56, e1–e25 (2013).
Sukeik, M., Patel, S. & Haddad, F. S. Aggressive early debridement for treatment of acutely infected cemented total hip arthroplasty. Clin. Orthop. Relat. Res. 470, 3164–3170 (2012).
Holmberg, A., Thórhallsdóttir, V. G., Robertsson, O., W-Dahl, A. & Stefánsdóttir, A. 75% success rate after open debridement, exchange of tibial insert, and antibiotics in knee prosthetic joint infections: Report on 145 cases from the Swedish Knee Arthroplasty Register. Acta Orthop. 86, 457–462 (2015).
Tsang, S. J., Ting, J., Simpson, A. & Gaston, P. Outcomes following debridement, antibiotics and implant retention in the management of periprosthetic infections of the hip: a review of cohort studies. Bone Jt. J. 99, 1458–1466 (2017).
Kim, J. G., Bae, J. H., Lee, S. Y., Cho, W. T. & Lim, H. C. The parameters affecting the success of irrigation and debridement with component retention in the treatment of acutely infected total knee arthroplasty. Clin. Orthop. Surg. 7, 69–76 (2015).
Bedair, H. et al. The Mark Coventry Award: diagnosis of early postoperative TKA infection using synovial fluid analysis. Clin. Orthop. Relat. Res. 469, 34–40 (2011).
Tsukayama, T. D., Estrada, R. & Gustilo, R. B. Infection after total hip arthroplasty. A study of the treatment of one hundred and six infections. J. Bone. Joint. Surg. Am. 78, 512–523 (1996).
Toms, A., Davidson, D., Masri, B. & Duncan, C. The management of peri-prosthetic infection in total joint arthroplasty. J. bone Jt. Surg. Br. Vol. 88, 149–155 (2006).
Zimmerli, W., Trampuz, A. & Ochsner, P. E. Prosthetic-joint infections. New Engl. J. Med. 351, 1645–1654 (2004).
Baker, P. et al. Patient reported outcome measures after revision of the infected TKR: comparison of single versus two-stage revision. Knee Surg. Sports Traumatol. Arthrosc. 21, 2713–2720 (2013).
Vaishya, R., Agarwal, A. K., Rawat, S. K., Singh, H. & Vijay, V. Is single-stage revision safe following infected total knee arthroplasty? A critical review. Cureus 9, e1629 (2017).
Kronig, I., Vaudaux, P., Suvà, D., Lew, D. & Uçkay, I. Acute and chronic osteomyelitis. In Clinical Infectious Disease. 2nd edn, 448–453 (2015).
Hanssen, A. D. Prophylactic use of antibiotic bone cement: an emerging standard—in opposition. J. Arthroplast. 19, 73–77 (2004).
Guelcher, S. A. et al. Dual-purpose bone grafts improve healing and reduce infection. J. Orthop. Trauma 25, 477–482 (2011).
Barth, R. E., Vogely, H. C., Hoepelman, A. I. & Peters, E. J. ‘To bead or not to bead?’ Treatment of osteomyelitis and prosthetic joint-associated infections with gentamicin bead chains. Int. J. Antimicrob. Agents 38, 371–375 (2011).
Cochran, A. R., Ong, K. L., Lau, E., Mont, M. A. & Malkani, A. L. Risk of reinfection after treatment of infected total knee arthroplasty. J. Arthroplast. 31, 156–161 (2016).
Nishitani, K., Bello-Irizarry, S. N., de Mesy Bentley, K., Daiss, J. L. & Schwarz, E. M. The role of the immune system and bone cells in acute and chronic osteomyelitis. Osteoimmunology 2nd edn, 283–295 (2016).
Moran, G. J., Amii, R. N., Abrahamian, F. M. & Talan, D. A. Methicillin-resistant Staphylococcus aureus in community-acquired skin infections. Emerg. Infect. Dis. 11, 928 (2005).
Kobayashi, S. D., Malachowa, N. & DeLeo, F. R. Pathogenesis of Staphylococcus aureus abscesses. Am. J. Pathol. 185, 1518–1527 (2015).
Cheng, A. G., DeDent, A. C., Schneewind, O. & Missiakas, D. A play in four acts: Staphylococcus aureus abscess formation. Trends Microbiol. 19, 225–232 (2011).
Cheng, A. G. et al. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 23, 3393–3404 (2009).
Yipp, B. G. et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 18, 1386–1393 (2012).
Cheung, G. Y. & Otto, M. The potential use of toxin antibodies as a strategy for controlling acute Staphylococcus aureus infections. Expert Opin. Ther. Targets 16, 601–612 (2012).
Kim, H. K., Thammavongsa, V., Schneewind, O. & Missiakas, D. Recurrent infections and immune evasion strategies of Staphylococcus aureus. Curr. Opin. Microbiol. 15, 92–99 (2012).
Zecconi, A. & Scali, F. Staphylococcus aureus virulence factors in evasion from innate immune defenses in human and animal diseases. Immunol. Lett. 150, 12–22 (2013).
Rooijakkers, S. H. et al. Early expression of SCIN and CHIPS drives instant immune evasion by Staphylococcus aureus. Cell. Microbiol. 8, 1282–1293 (2006).
Thurlow, L. R. et al. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 186, 6585–6596 (2011).
Cheng, A. G. et al. Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLoS Pathog. 6, e1001036 (2010).
Farnsworth, C. W. et al. Adaptive upregulation of Clumping Factor A (ClfA) by S. aureus in the obese, type 2 diabetic host mediates increased virulence. Infect. Immun. IAI, 01005–01016 (2017).
Nakazawa, D. et al. The responses of macrophages in interaction with neutrophils that undergo NETosis. J. Autoimmun. 67, 19–28 (2016).
Patti, J. M., Allen, B. L., McGavin, M. J. & Höök, M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 48, 585–617 (1994).
Foster, T. J., Geoghegan, J. A., Ganesh, V. K. & Höök, M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 12, 49 (2014).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Costerton, J. W., Stewart, P. S. & Greenberg, E. Bacterial biofilms: a common cause of persistent infections. Science 284, 1318–1322 (1999).
Flemming, H.-C. & Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 8, 623–633 (2010).
Boles, B. R. & Horswill, A. R. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog. 4, e1000052 (2008).
Periasamy, S. et al. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl Acad. Sci. USA 109, 1281–1286 (2012).
George, E. A. & Muir, T. W. Molecular mechanisms of agr quorum sensing in virulent staphylococci. Chembiochem 8, 847–855 (2007).
Recsei, P. et al. Regulation of exoprotein gene expression in Staphylococcus aureus by agr. Mol. Gen. Genet. 202, 58–61 (1986).
Salam, A. M. & Quave, C. L. Targeting virulence in Staphylococcus aureus by chemical inhibition of the accessory gene regulator system in vivo. MSphere 3, e00500–e00517 (2018).
Nishitani, K. et al. Quantifying the natural history of biofilm formation in vivo during the establishment of chronic implant‐associated Staphylococcus aureus osteomyelitis in mice to identify critical pathogen and host factors. J. Orthop. Res. 33, 1311–1319 (2015).
Gillaspy, A. F. et al. Role of the accessory gene regulator (agr) in pathogenesis of Staphylococcal osteomyelitis. Infect. Immun. 63, 3373–3380 (1995).
Savage, V. J., Chopra, I. & O'Neill, A. J. Staphylococcus aureus biofilms promote horizontal transfer of antibiotic resistance. Antimicrob. Agents Chemother. 57, 1968–1970 (2013).
Mah, T.-F. C. & O'Toole, G. A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 9, 34–39 (2001).
Redlich, K. & Smolen, J. S. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 11, 234 (2012).
Putnam, N. E. et al. MyD88 and IL-1R signaling drive antibacterial immunity and osteoclast-driven bone loss during Staphylococcus aureus osteomyelitis. PLoS Pathog. 15, e1007744 (2019).
Junka, A. et al. Bad to the Bone: on in vitro and ex vivo microbial biofilm ability to directly destroy colonized bone surfaces without participation of host immunity or osteoclastogenesis. PLoS ONE 12, e0169565 (2017).
Junka, A. F. et al. Microbial biofilms are able to destroy hydroxyapatite in the absence of host immunity in vitro. J. Oral. Maxillofac. Surg. 73, 451–464 (2015).
Urish, K. L., DeMuth, P. W., Craft, D. W., Haider, H. & Davis, C. M. III Pulse lavage is inadequate at removal of biofilm from the surface of total knee arthroplasty materials. J. Arthroplast. 29, 1128–1132 (2014).
de Mesy Bentley, K. L., et al. Evidence of Staphylococcus aureus deformation, proliferation, and migration in canaliculi of live cortical bone in murine models of osteomyelitis. J. Bone Miner. Res. 32, 985–990 (2017).
de Mesy Bentley, K. L., MacDonald, A., Schwarz, E. M. & Oh, I. Chronic osteomyelitis with Staphylococcus aureus deformation in submicron canaliculi of osteocytes: a case report. Jbjs Case Connect. 8, e8 (2018).
Masters, E. A. et al. An in vitro platform for elucidating the molecular genetics of S. aureus invasion of the osteocyte lacuno-canalicular network during chronic osteomyelitis. Nanomed. Nanotechnol. Biol. Med. e102039 (2019).
Trombetta, R. P., Dunman, P. M., Schwarz, E. M., Kates, S. L. & Awad, H. A. A high-throughput screening approach to repurpose FDA-approved drugs for bactericidal applications against Staphylococcus aureus small-colony variants. mSphere 3, e00422-18 (2018).
Libraty, D. H., Patkar, C. & Torres, B. Staphylococcus aureus reactivation osteomyelitis after 75 years. New Engl. J. Med. 366, 481–482 (2012).
Lowy, F. D. Is Staphylococcus aureus an intracellular pathogen? Trends Microbiol. 8, 341–343 (2000).
Garzoni, C. & Kelley, W. L. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 17, 59–65 (2009).
Garzoni, C. & Kelley, W. L. Return of the Trojan horse: intracellular phenotype switching and immune evasion by Staphylococcus aureus. EMBO Mol. Med. 3, 115–117 (2011).
Hébert, A., Sayasith, K., Sénéchal, S., Dubreuil, P. & Lagacé, J. Demonstration of intracellular Staphylococcus aureus in bovine mastitis alveolar cells and macrophages isolated from naturally infected cow milk. FEMS Microbiol. Lett. 193, 57–62 (2000).
Clement, S. et al. Evidence of an intracellular reservoir in the nasal mucosa of patients with recurrent Staphylococcus aureus rhinosinusitis. J. Infect. Dis. 192, 1023–1028 (2005).
Edwards, A. M., Potts, J. R., Josefsson, E. & Massey, R. C. Staphylococcus aureus host cell invasion and virulence in sepsis is facilitated by the multiple repeats within FnBPA. PLoS Pathog. 6, e1000964 (2010).
Uribe-Querol, E. & Rosales, C. Control of phagocytosis by microbial pathogens. Front. Immunol. 8, 1368 (2017).
Fraunholz, M. & Sinha, B. Intracellular Staphylococcus aureus: live-in and let die. Front. Cell. Infect. Microbiol. 2, 43 (2012).
Ellington, J. K. et al. Intracellular Staphylococcus aureus. A mechanism for the indolence of osteomyelitis. J. Bone Jt. Surg. Br. 85, 918–921 (2003).
Ellington, J. K. et al. Intracellular Staphylococcus aureus and antibiotic resistance: implications for treatment of Staphylococcal osteomyelitis. J. Orthop. Res. 24, 87–93 (2006).
Ellington, J. K., Elhofy, A., Bost, K. L. & Hudson, M. C. Involvement of mitogen-activated protein kinase pathways in Staphylococcus aureus invasion of normal osteoblasts. Infect. Immun. 69, 5235–5242 (2001).
Reott, M. A.Jr., Ritchie-Miller, S. L., Anguita, J. & Hudson, M. C. TRAIL expression is induced in both osteoblasts containing intracellular Staphylococcus aureus and uninfected osteoblasts in infected cultures. FEMS Microbiol. Lett. 278, 185–192 (2008).
Ning, Rd., Zhang, Xl., Li, Qt. & Guo, Xk. The effect of Staphylococcus aureus on apoptosis of cultured human osteoblasts. Orthop. Surg. 3, 199–204 (2011).
Mohamed, W. et al. Intracellular proliferation of S. aureus in osteoblasts and effects of rifampicin and gentamicin on S. aureus intracellular proliferation and survival. Eur. Cell Mater. 28, 258–268 (2014).
Valour, F. et al. Antimicrobial activity against intra-osteoblastic Staphylococcus aureus. Antimicrob. Agents Chemother. 59, 2029–2036 (2015).
Valour, F. et al. Delta-toxin production deficiency in Staphylococcus aureus: a diagnostic marker of bone and joint infection chronicity linked with osteoblast invasion and biofilm formation. Clin. Microbiol. Infect. 21, 568. e1–e11 (2015).
Hamza, T. et al. Intra-cellular Staphylococcus aureus alone causes infection in vivo. Eur. Cells Mater. 25, 341 (2013).
Yang, D. et al. Novel insights into Staphylococcus aureus deep bone infections: the involvement of osteocytes. mBio 9, e00415–e00418 (2018).
Reilly, S., Hudson, M., Kellam, J. & Ramp, W. In vivo internalization of Staphylococcus aureus by embryonic chick osteoblasts. Bone 26, 63–70 (2000).
Varrone, J. J. et al. Passive immunization with anti-glucosaminidase monoclonal antibodies protects mice from implant-associated osteomyelitis by mediating opsonophagocytosis of Staphylococcus aureus megaclusters. J. Orthop. Res. 32, 1389–1396 (2014).
Thwaites, G. E. & Gant, V. Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat. Rev. Microbiol. 9, 215 (2011).
Muraille, E., Leo, O. & Moser, M. TH1/TH2 paradigm extended: macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front. Immunol. 5, 603 (2014).
Shorr, A. F. et al. Healthcare-associated bloodstream infection: a distinct entity? Insights from a large US database. Crit. Care Med. 34, 2588–2595 (2006).
Hamza, T. & Li, B. Differential responses of osteoblasts and macrophages upon Staphylococcus aureus infection. BMC Microbiol. 14, 207 (2014).
Webb, L. et al. Osteomyelitis and intraosteoblastic Staphylococcus aureus. J. Surg. Orthop. Adv. 16, 73–78 (2007).
Cassat, J. E. et al. Integrated molecular imaging reveals tissue heterogeneity driving host-pathogen interactions. Sci. Transl. Med. 10, eaan6361 (2018).
Reizner, W. et al. A systematic review of animal models for Staphylococcus aureus osteomyelitis. Eur. Cells Mater. 27, 196 (2014).
Lazzarini, L., Mader, J. T. & Calhoun, J. H. Osteomyelitis in long bones. JBJS 86, 2305–2318 (2004).
Pineda, C., Espinosa, R. & Pena, A. Radiographic imaging in osteomyelitis: the role of plain radiography, computed tomography, ultrasonography, magnetic resonance imaging, and scintigraphy. Semin. Plastic Surg. 23, 080–089 (2009).
Hotchen, A. J., McNally, M. A. & Sendi, P. The classification of long bone osteomyelitis: a systemic review of the literature. J. Bone Jt. Infect. 2, 167 (2017).
Baltensperger, M. et al. Is primary chronic osteomyelitis a uniform disease? Proposal of a classification based on a retrospective analysis of patients treated in the past 30 years. J. Cranio-Maxillofac. Surg. 32, 43–50 (2004).
Dym, H. & Zeidan, J. Microbiology of acute and chronic osteomyelitis and antibiotic treatment. Dent. Clin. 61, 271–282 (2017).
Kavanagh, N. et al. Staphylococcal osteomyelitis: disease progression, treatment challenges, and future directions. Clin. Microbiol. Rev. 31, e00084-17 (2018).
Lonner, J. H., Desai, P., Dicesare, P. E., Steiner, G. & Zuckerman, J. D. The reliability of analysis of intraoperative frozen sections for identifying active infection during revision hip or knee arthroplasty. JBJS 78, 1553–1558 (1996).
Feldman, D. S., Lonner, J. H., Desai, P. M. & Zuckerman, J. D. The role of intraoperative frozen sections in revision total joint arthroplasty. JBJS 77, 1807–1813 (1995).
Athanasou, N., Pandey, R., De Steiger, R., Crook, D. & Smith, P. M. Diagnosis of infection by frozen section during revision arthroplasty. J. Bone Jt. Surg. Br. Vol. 77, 28–33 (1995).
Cierny Iii, G., Mader, J. T. & Penninck, J. J. The classic: a clinical staging system for adult osteomyelitis. Clin. Orthop. Relat. Res. (1976–2007). 414, 7–24 (2003).
Waldvogel, F. A., Medoff, G. & Swartz, M. N. Osteomyelitis: a review of clinical features, therapeutic considerations and unusual aspects. New Engl. J. Med. 282, 260–266 (1970).
Holtfreter, S. et al. Human immune proteome in experimental colonization with Staphylococcus aureus. Clin. Vaccin. Immunol. 16, 1607–1614 (2009).
Holtfreter, S., Kolata, J. & Broker, B. M. Towards the immune proteome of Staphylococcus aureus—the anti-S. aureus antibody response. Int. J. Med. Microbiol. 300, 176–192 (2010).
Verkaik, N. J. et al. Heterogeneity of the humoral immune response following Staphylococcus aureus bacteremia. Eur. J. Clin. Microbiol. Infect. Dis. 29, 509–518 (2010).
Verkaik, N. J. et al. Induction of antibodies by Staphylococcus aureus nasal colonization in young children. Clin. Microbiol Infect. 16, 1312–1317 (2010).
van Belkum, A. et al. Co-evolutionary aspects of human colonisation and infection by Staphylococcus aureus. Infect. Genet Evol. 9, 32–47 (2009).
van den Berg, S. et al. A multiplex assay for the quantification of antibody responses in Staphylococcus aureus infections in mice. J. Immunol. Methods 365, 142–148 (2011).
Dryla, A. et al. Comparison of antibody repertoires against Staphylococcus aureus in healthy individuals and in acutely infected patients. Clin. Diagn. Lab Immunol. 12, 387–398 (2005).
Gedbjerg, N. et al. Anti-glucosaminidase IgG in sera as a biomarker of host immunity against Staphylococcus aureus in orthopaedic surgery patients. J. Bone Jt. Surg. Am. 95, e171 (2013).
Nishitani, K. et al. A diagnostic serum antibody test for patients with Staphylococcus aureus osteomyelitis. Clin. Orthop. Relat. Res. 473, 2735–2749 (2015).
Carter, M. J., Mitchell, R. M., Meyer Sauteur, P. M., Kelly, D. F. & Truck, J. The antibody-secreting cell response to infection: kinetics and clinical applications. Front Immunol. 8, 630 (2017).
Lee, F. E., Falsey, A. R., Halliley, J. L., Sanz, I. & Walsh, E. E. Circulating antibody-secreting cells during acute respiratory syncytial virus infection in adults. J. Infect. Dis. 202, 1659–1666 (2010).
Lee, F. E. et al. Circulating human antibody-secreting cells during vaccinations and respiratory viral infections are characterized by high specificity and lack of bystander effect. J. Immunol. 186, 5514–5521 (2011).
Oh, I. et al. Tracking anti-Staphylococcus aureus antibodies produced in vivo and ex vivo during foot salvage therapy for diabetic foot infections reveals prognostic insights and evidence of diversified humoral immunity. Infect. Immun. 86, e00629-18 (2018).
MacDonald, A., Brodell, J. Jr., Daiss, J., Schwarz, E. & Oh, I. Evidence of differential microbiomes in healing versus non-healing diabetic foot ulcers prior to and following foot salvage therapy. J. Orthop. Res. 37, 1596–1603 (2019).
Foster, T. J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 41, 430–449 (2017).
Fowler, V. G. Jr. & Proctor, R. A. Where does a Staphylococcus aureus vaccine stand? Clin. Microbiol. Infect. 20(Suppl. 5), 66–75 (2014).
Proctor, R. A. Challenges for a universal Staphylococcus aureus vaccine. Clin. Infect. Dis. 54, 1179–1186 (2012).
Bagnoli, F., Bertholet, S. & Grandi, G. Inferring reasons for the failure of Staphylococcus aureus vaccines in clinical trials. Front. Cell. Infect. Microbiol. 2, 16 (2012).
Fowler, V. G. et al. Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: a randomized trial. JAMA 309, 1368–1378 (2013).
Giersing, B. K., Dastgheyb, S. S., Modjarrad, K. & Moorthy, V. Status of vaccine research and development of vaccines for Staphylococcus aureus. Vaccine 34, 2962–2966 (2016).
Salgado-Pabón, W. & Schlievert, P. M. Models matter: the search for an effective Staphylococcus aureus vaccine. Nat. Rev. Microbiol. 12, 585 (2014).
Warren, H. S. et al. Mice are not men. Proc. Natl. Acad. Sci. USA 112, E345–E345 (2015).
Hall, A. E. et al. Characterization of a protective monoclonal antibody recognizing Staphylococcus aureus MSCRAMM protein clumping factor A. Infect. Immun. 71, 6864–6870 (2003).
Weisman, L. E. et al. Phase 1/2 double-blind, placebo-controlled, dose escalation, safety, and pharmacokinetic study of pagibaximab (BSYX-A110), an antistaphylococcal monoclonal antibody for the prevention of staphylococcal bloodstream infections, in very-low-birth-weight neonates. Antimicrob. Agents Chemother. 53, 2879–2886 (2009).
Weisman, L. E. et al. A randomized study of a monoclonal antibody (pagibaximab) to prevent staphylococcal sepsis. Pediatrics 128, 271–279 (2011).
Oganesyan, V. et al. Mechanisms of neutralization of a human anti-alpha-toxin antibody. J. Biol. Chem. 289, 29874–29880 (2014).
Hua, L. et al. MEDI4893* promotes survival and extends the antibiotic treatment window in a Staphylococcus aureus immunocompromised pneumonia model. Antimicrob. Agents Chemother. 59, 4526–4532 (2015).
Yu, X. Q. et al. Safety, tolerability, and pharmacokinetics of MEDI4893, an investigational, extended-half-life, anti-Staphylococcus aureus alpha-toxin human monoclonal antibody, in healthy adults. Antimicrob. Agents Chemother. 61, e01020-16 (2017).
Francois, B. et al. Safety and tolerability of a single administration of AR-301, a human monoclonal antibody, in ICU patients with severe pneumonia caused by Staphylococcus aureus: first-in-human trial. Intensive Care Med. 44, 1787–1796 (2018).
Foster, T. J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 3, 948–958 (2005).
Thammavongsa, V., Kim, H. K., Missiakas, D. & Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 13, 529–543 (2015).
Rouha, H. et al. Five birds, one stone: neutralization of alpha-hemolysin and 4 bi-component leukocidins of Staphylococcus aureus with a single human monoclonal antibody. MAbs 7, 243–254 (2015).
Thammavongsa, V., Rauch, S., Kim, H. K., Missiakas, D. M. & Schneewind, O. Protein A-neutralizing monoclonal antibody protects neonatal mice against Staphylococcus aureus. Vaccine 33, 523–526 (2015).
Varshney, A. K. et al. A natural human monoclonal antibody targeting Staphylococcus Protein A protects against Staphylococcus aureus bacteremia. PLoS ONE 13, e0190537 (2018).
van den Berg, S. et al. A human monoclonal antibody targeting the conserved staphylococcal antigen IsaA protects mice against Staphylococcus aureus bacteremia. Int. J. Med. Microbiol. 305, 55–64 (2015).
Karauzum, H. et al. Synthetic human monoclonal antibodies toward staphylococcal enterotoxin B (SEB) protective against toxic shock syndrome. J. Biol. Chem. 287, 25203–25215 (2012).
Dutta, K. et al. Mechanisms mediating enhanced neutralization efficacy of staphylococcal enterotoxin B by combinations of monoclonal antibodies. J. Biol. Chem. 290, 6715–6730 (2015).
Varrone, J. J., Li, D., Daiss, J. L. & Schwarz, E. M. Anti-glucosaminidase monoclonal antibodies as a passive immunization for methicillin-resistant Staphylococcus aureus (MRSA) orthopaedic infections. Bonekey Osteovision 8, 187–194 (2011).
Christensen, G. D. & Simpson, W. A. Gram-positive bacteria: Pathogenesis of staphylococcal musculoskeletal infections. (eds J. L, Esterhai, A. G, Gristina & R, Poss) In Musculoskeletal Infection. 57–78 (AAOS: Park Ridge, 1992).
An, Y. H. et al. Rapid quantification of staphylococci adhered to titanium surfaces using image analyzed epifluorescence microscopy. J. Microbiol. Methods 24, 29–40 (1995).
Zetola, N., Francis, J. S., Nuermberger, E. L. & Bishai, W. R. Community-acquired meticillin-resistant Staphylococcus aureus: an emerging threat. Lancet Infect. Dis. 5, 275–286 (2005).
Alexander, E. H. & Hudson, M. C. Factors influencing the internalization of Staphylococcus aureus and impacts on the course of infections in humans. Appl. Microbiol. Biotechnol. 56, 361–366 (2001).
Lew, D. P. & Waldvogel, F. A. Osteomyelitis. Lancet 364, 369–379 (2004).
Nair, L. S. & Laurencin, C. T. Nanofibers and nanoparticles for orthopaedic surgery applications. J. Bone Jt. Surg. Am. 90(Suppl. 1), 128–131 (2008).
Phillips, P. L. et al. Antimicrobial dressing efficacy against mature Pseudomonas aeruginosa biofilm on porcine skin explants. Int. Wound J. 12, 469–483 (2015).
Brennan, S. A. et al. Silver nanoparticles and their orthopaedic applications. Bone Jt. J. 97-b, 582–589 (2015).
Chaloupka, K., Malam, Y. & Seifalian, A. M. Nanosilver as a new generation of nanoproduct in biomedical applications. Trends Biotechnol. 28, 580–588 (2010).
Yamanaka, M., Hara, K. & Kudo, J. Bactericidal actions of a silver ion solution on Escherichia coli, studied by energy-filtering transmission electron microscopy and proteomic analysis. Appl. Environ. Microbiol. 71, 7589–7593 (2005).
Thurman, R. B., Gerba, C. P. & Bitton, G. The molecular mechanisms of copper and silver ion disinfection of bacteria and viruses. Crit. Rev. Environ. Control 18, 295–315 (1989).
Park, H. J. et al. Silver-ion-mediated reactive oxygen species generation affecting bactericidal activity. Water Res. 43, 1027–1032 (2009).
Gurunathan, S., Han, J. W., Kwon, D.-N. & Kim, J.-H. Enhanced antibacterial and anti-biofilm activities of silver nanoparticles against Gram-negative and Gram-positive bacteria. Nanoscale Res. Lett. 9, 373 (2014).
Alt, V. et al. An in vitro assessment of the antibacterial properties and cytotoxicity of nanoparticulate silver bone cement. Biomaterials 25, 4383–4391 (2004).
Prokopovich, P., Leech, R., Carmalt, C. J., Parkin, I. P. & Perni, S. A novel bone cement impregnated with silver-tiopronin nanoparticles: its antimicrobial, cytotoxic, and mechanical properties. Int. J. Nanomed. 8, 2227–2237 (2013).
Sheehan, E., McKenna, J., Mulhall, K. J., Marks, P. & McCormack, D. Adhesion of Staphylococcus to orthopaedic metals, an in vivo study. J. Orthop. Res. 22, 39–43 (2004).
Liu, X., Mou, Y., Wu, S. & Man, H. C. Synthesis of silver-incorporated hydroxyapatite nanocomposites for antimicrobial implant coatings. Appl. Surf. Sci. 273, 748–757 (2013).
Sudmann, E. et al. Systemic and local silver accumulation after total hip replacement using silver-impregnated bone cement. Med Prog. Technol. 20, 179–184 (1994).
Vik, H., Andersen, K. J., Julshamn, K. & Todnem, K. Neuropathy caused by silver absorption from arthroplasty cement. Lancet 1, 872 (1985).
Akiyama, T. et al. Silver oxide-containing hydroxyapatite coating has in vivo antibacterial activity in the rat tibia. J. Orthop. Res. 31, 1195–1200 (2013).
Chen, W. et al. In vitro anti-bacterial and biological properties of magnetron co-sputtered silver-containing hydroxyapatite coating. Biomaterials 27, 5512–5517 (2006).
Chen, W. et al. Antibacterial and osteogenic properties of silver-containing hydroxyapatite coatings produced using a sol gel process. J. Biomed. Mater. Res. Part A. 82A, 899–906 (2007).
Fielding, G. & Bose, S. SiO2 and ZnO dopants in three-dimensionally printed tricalcium phosphate bone tissue engineering scaffolds enhance osteogenesis and angiogenesis in vivo. Acta Biomater. 9, 9137–9148 (2013).
Wafa, H. et al. Retrospective evaluation of the incidence of early periprosthetic infection with silver-treated endoprostheses in high-risk patients: case-control study. Bone Jt. J. 97-b, 252–257 (2015).
Hardes, J. et al. Reduction of periprosthetic infection with silver-coated megaprostheses in patients with bone sarcoma. J. Surg. Oncol. 101, 389–395 (2010).
Mirsattari, S. M., Hammond, R. R., Sharpe, M. D., Leung, F. Y. & Young, G. B. Myoclonic status epilepticus following repeated oral ingestion of colloidal silver. Neurology 62, 1408–1410 (2004).
Braydich-Stolle, L., Hussain, S., Schlager, J. J. & Hofmann, M. C. In vitro cytotoxicity of nanoparticles in mammalian germline stem cells. Toxicol. Sci. 88, 412–419 (2005).
Kim, S. et al. Oxidative stress-dependent toxicity of silver nanoparticles in human hepatoma cells. Toxicol. In Vitro 23, 1076–1084 (2009).
Liu, W. et al. Impact of silver nanoparticles on human cells: effect of particle size. Nanotoxicology 4, 319–330 (2010).
Schmidmaier, G., Kerstan, M., Schwabe, P., Sudkamp, N. & Raschke, M. Clinical experiences in the use of a gentamicin-coated titanium nail in tibia fractures. Injury 48, 2235–2241 (2017).
Gulcu, A. et al. Fosfomycin addition to poly(d,l-lactide) coating does not affect prophylaxis efficacy in rat implant-related infection model, but that of gentamicin does. PLoS ONE 11, e0165544 (2016).
Back, D. A. et al. Testing of antibiotic releasing implant coatings to fight bacteria in combat-associated osteomyelitis—an in-vitro study. Int. Orthop. 40, 1039–1047 (2016).
Vester, H., Wildemann, B., Schmidmaier, G., Stockle, U. & Lucke, M. Gentamycin delivered from a PDLLA coating of metallic implants: In vivo and in vitro characterisation for local prophylaxis of implant-related osteomyelitis. Injury 41, 1053–1059 (2010).
Lucke, M. et al. Gentamicin coating of metallic implants reduces implant-related osteomyelitis in rats. Bone 32, 521–531 (2003).
Raschke, M., Vordemvenne, T. & Fuchs, T. Limb salvage or amputation? The use of a gentamicin coated nail in a severe, grade IIIc tibia fracture. Eur. J. Trauma Emerg. Surg. 36, 605–608 (2010).
Metsemakers, W. J. et al. A doxycycline-loaded polymer-lipid encapsulation matrix coating for the prevention of implant-related osteomyelitis due to doxycycline-resistant methicillin-resistant Staphylococcus aureus. J. Control. Release 209, 47–56 (2015).
Price, J. S., Tencer, A. F., Arm, D. M. & Bohach, G. A. Controlled release of antibiotics from coated orthopedic implants. J. Biomed. Mater. Res. 30, 281–286 (1996).
Min, J. et al. Designer dual therapy nanolayered implant coatings eradicate biofilms and accelerate bone tissue repair. ACS Nano 10, 4441–4450 (2016).
Li, D. et al. The immobilization of antibiotic-loaded polymeric coatings on osteoarticular Ti implants for the prevention of bone infections. Biomater. Sci. 5, 2337–2346 (2017).
Liu, D., He, C., Liu, Z. & Xu, W. Gentamicin coating of nanotubular anodized titanium implant reduces implant-related osteomyelitis and enhances bone biocompatibility in rabbits. Int. J. Nanomed. 12, 5461–5471 (2017).
Diefenbeck, M. et al. Gentamicin coating of plasma chemical oxidized titanium alloy prevents implant-related osteomyelitis in rats. Biomaterials 101, 156–164 (2016).
Stewart, S. et al. Vancomycin-modified implant surface inhibits biofilm formation and supports bone-healing in an infected osteotomy model in sheep: a proof-of-concept study. J. Bone Jt. Surg. Am. Vol. 94, 1406–1415 (2012).
Folsch, C. et al. Systemic antibiotic therapy does not significantly improve outcome in a rat model of implant-associated osteomyelitis induced by Methicillin susceptible Staphylococcus aureus. Arch. Orthop. Trauma Surg. 136, 585–592 (2016).
Folsch, C. et al. Coating with a novel gentamicinpalmitate formulation prevents implant-associated osteomyelitis induced by methicillin-susceptible Staphylococcus aureus in a rat model. Int Orthop. 39, 981–988 (2015).
Moskowitz, J. S. et al. The effectiveness of the controlled release of gentamicin from polyelectrolyte multilayers in the treatment of Staphylococcus aureus infection in a rabbit bone model. Biomaterials 31, 6019–6030 (2010).
Jennings, J. A. et al. Antibiotic-loaded phosphatidylcholine inhibits staphylococcal bone infection. World J. Orthop. 7, 467–474 (2016).
Alaee, F. et al. General assembly, prevention, operating room—surgical technique: Proceedings of International Consensus on Orthopedic Infections. J. Arthroplast. 34, S139–S146 (2019).
Herczegh, P. et al. Osteoadsorptive bisphosphonate derivatives of fluoroquinolone antibacterials. J. Med. Chem. 45, 2338–2341 (2002).
Houghton, T. J. et al. Linking bisphosphonates to the free amino groups in fluoroquinolones: preparation of osteotropic prodrugs for the prevention of osteomyelitis. J. Med. Chem. 51, 6955–6969 (2008).
Sedghizadeh, P. P. et al. Design, synthesis, and antimicrobial evaluation of a novel bone-targeting bisphosphonate-ciprofloxacin conjugate for the treatment of osteomyelitis biofilms. J. Med. Chem. 60, 2326–2343 (2017).
Tanaka, K. S. et al. Synthesis and in vitro evaluation of bisphosphonated glycopeptide prodrugs for the treatment of osteomyelitis. Bioorg. Med. Chem. Lett. 20, 1355–1359 (2010).
Zhang, S., Gangal, G. & Uludağ, H. ‘Magic bullets’ for bone diseases: progress in rational design of bone-seeking medicinal agents. Chem. Soc. Rev. 36, 507–531 (2007).
Li, Y. et al. Directed vaccination against pneumococcal disease. Proc. Natl. Acad. Sci. USA 113, 6898–6903 (2016).
BlackR. E., Cousens, S. & Johnson, H. L. et al. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet 375, 1969–1987 (2010).
Masters, E., Harris, M. & Jennings, J. Cis-2-decenoic acid interacts with bacterial cell membranes to potentiate additive and synergistic responses against biofilm. J. Bacteriol. Mycol. 3, 1031–1038 (2016).
Chouirfa, H., Bouloussa, H., Migonney, V. & Falentin-Daudre, C. Review of titanium surface modification techniques and coatings for antibacterial applications. Acta Biomater. 83, 37–54 (2019).
Moriarty, T. F. et al. Orthopaedic device-related infection: current and future interventions for improved prevention and treatment. EFORT Open Rev. 1, 89–99 (2016).
Gogia, J. S., Meehan, J. P., Di Cesare, P. E. & Jamali, A. A. Local antibiotic therapy in osteomyelitis. Semin. Plast. Surg. 23, 100–107 (2009).
Walenkamp, G. H., Kleijn, L. L. & de Leeuw, M. Osteomyelitis treated with gentamicin-PMMA beads: 100 patients followed for 1–12 years. Acta Orthop. Scand. 69, 518–522 (1998).
Hake, M. E. et al. Local antibiotic therapy strategies in orthopaedic trauma: practical tips and tricks and review of the literature. Injury 46, 1447–1456 (2015).
Tsang, S. T. et al. Exchange nailing for nonunion of diaphyseal fractures of the tibia: our results and an analysis of the risk factors for failure. Bone Jt. J. 98-b, 534–541 (2016).
Seebach, E. et al. Mesenchymal stromal cell implantation for stimulation of long bone healing aggravates Staphylococcus aureus induced osteomyelitis. Acta Biomater. 21, 165–177 (2015).
Mohapatra, N. & Jain, S. Antibiotic laden bone cement in chronic osteomyelitis. J. Orthop. Traumatol. Rehabil. 9, 74–77 (2017).
Berkes, M. et al. Maintenance of hardware after early postoperative infection following fracture internal fixation. JBJS 92, 823–828 (2010).
Tschudin-Sutter, S. et al. Validation of a treatment algorithm for orthopaedic implant-related infections with device-retention-results from a prospective observational cohort study. Clin. Microbiol. Infect. 22, 457.e1–e9 (2016).
Metsemakers, W. J. et al. Infection after fracture fixation: current surgical and microbiological concepts. Injury 49, 511–522 (2018).
Bistolfi, A. et al. Antibiotic-loaded cement in orthopedic surgery: a review. ISRN Orthop. 2011, 290851 (2011).
Rathbone, C. R., Cross, J. D., Brown, K. V., Murray, C. K. & Wenke, J. C. Effect of various concentrations of antibiotics on osteogenic cell viability and activity. J. Orthop. Res. 29, 1070–1074 (2011).
Shiels, S. M., Tennent, D. J., Akers, K. S. & Wenke, J. C. Determining potential of PMMA as a depot for rifampin to treat recalcitrant orthopaedic infections. Injury 48, 2095–2100 (2017).
Picknell, B., Mizen, L. & Sutherland, R. Antibacterial activity of antibiotics in acrylic bone cement. J. Bone Jt. Surg. Br. 59, 302–307 (1977).
Hoff, S. F., Fitzgerald, R. H. Jr. & Kelly, P. J. The depot administration of penicillin G and gentamicin in acrylic bone cement. J. Bone Jt. Surg. Am. 63, 798–804 (1981).
Chohfi, M. et al. Pharmacokinetics, uses, and limitations of vancomycin-loaded bone cement. Int. Orthop. 22, 171–177 (1998).
Penner, M. J., Duncan, C. P. & Masri, B. A. The in vitro elution characteristics of antibiotic-loaded CMW and Palacos-R bone cements. J. Arthroplast. 14, 209–214 (1999).
van de Belt, H. et al. Gentamicin release from polymethylmethacrylate bone cements and Staphylococcus aureus biofilm formation. Acta Orthop. Scand. 71, 625–629 (2000).
Torholm, C., Lidgren, L., Lindberg, L. & Kahlmeter, G. Total hip joint arthroplasty with gentamicin-impregnated cement. A clinical study of gentamicin excretion kinetics. Clin. Orthop. Relat. Res. 181, 99–106 (1983).
Bunetel, L., Segui, A., Cormier, M., Percheron, E. & Langlais, F. Release of gentamicin from acrylic bone cement. Clin. Pharmacokinet. 17, 291–297 (1989).
Moojen, D. J. et al. In vitro release of antibiotics from commercial PMMA beads and articulating hip spacers. J. Arthroplast. 23, 1152–1156 (2008).
Anagnostakos, K., Wilmes, P., Schmitt, E. & Kelm, J. Elution of gentamicin and vancomycin from polymethylmethacrylate beads and hip spacers in vivo. Acta Orthop. 80, 193–197 (2009).
Jiranek, W. A., Hanssen, A. D. & Greenwald, A. S. Antibiotic-loaded bone cement for infection prophylaxis in total joint replacement. J. Bone Jt. Surg. Am. 88, 2487–2500 (2006).
Lewis, G. Properties of antibiotic-loaded acrylic bone cements for use in cemented arthroplasties: a state-of-the-art review. J. Biomed. Mater. Res. Part B 89, 558–574 (2009).
Ma, D. et al. Viable bacteria persist on antibiotic spacers following two-stage revision for periprosthetic joint infection. J. Orthop. Res. 36, 452–458 (2017).
Ferguson, J. Y. et al. The use of a biodegradable antibiotic-loaded calcium sulphate carrier containing tobramycin for the treatment of chronic osteomyelitis: a series of 195 cases. Bone Jt. J. 96-b, 829–836 (2014).
Borkhuu, B. et al. Antibiotic-loaded allograft decreases the rate of acute deep wound infection after spinal fusion in cerebral palsy. Spine 33, 2300–2304 (2008).
Romano, C. L. et al. A comparative study of the use of bioactive glass S53P4 and antibiotic-loaded calcium-based bone substitutes in the treatment of chronic osteomyelitis: a retrospective comparative study. Bone Jt. J. 96-b, 845–850 (2014).
Itokazu, M., Aoki, T., Nonomura, H., Nishimoto, Y. & Itoh, Y. Antibiotic-loaded porous hydroxyapatite blocks for the treatment of osteomyelitis and postoperative infection. A preliminary report. Bulletin 57, 125–129 (1998).
Letsch, R., Rosenthal, E. & Joka, T. [Local antibiotic administration in osteomyelitis treatment—a comparative study with two different carrier substances]. Aktuel-. Traumatol. 23, 324–329 (1993).
Rupprecht, S. et al. Antibiotic-containing collagen for the treatment of bone defects. J. Biomed. Mater. Res. Part B 83, 314–319 (2007).
Bibbo, C. & Patel, D. V. The effect of demineralized bone matrix-calcium sulfate with vancomycin on calcaneal fracture healing and infection rates: a prospective study. Foot Ankle Int. 27, 487–493 (2006).
Inzana, J. A., Schwarz, E. M., Kates, S. L. & Awad, H. A. Biomaterials approaches to treating implant-associated osteomyelitis. Biomaterials 81, 58–71 (2016).
Inzana, J. A., Schwarz, E. M., Kates, S. L. & Awad, H. A. A novel murine model of established Staphylococcal bone infection in the presence of a fracture fixation plate to study therapies utilizing antibiotic-laden spacers after revision surgery. Bone 72, 128–136 (2015).
Yokogawa, N. et al. Immunotherapy synergizes with debridement and antibiotic therapy in a murine 1-stage exchange model of MRSA implant-associated osteomyelitis. J. Orthop. Res. 36, 1590–1598 (2018).
Trombetta, R. P., de Mesy Bentley, K. L., Schwarz, E. M., Kates, S. L., & Awad, H. A. A murine femoral ostectomy model with hardware exchange to assess antibiotic-impregnated spacers for implant-associated osteomyelitis. Eur. Cell Mater. 37, 431–443 (2019).
Trombetta, R. P. et al. Calcium phosphate spacers for the local delivery of sitafloxacin and rifampin to treat orthopedic infections: efficacy and proof of concept in a mouse model of single-stage revision of device-associated osteomyelitis. Pharmaceutics 11, 94 (2019).
Li, D. et al. Quantitative mouse model of implant‐associated osteomyelitis and the kinetics of microbial growth, osteolysis, and humoral immunity. J. Orthop. Res. 26, 96–105 (2008).
Acknowledgements
E.M.S., S.L.K. and H.A.A. are supported by grants from AOTrauma, Clinical Priority Program (Davos, Switzerland), and NIH NIAMS (P50 AR072000 and P30 AR069655). G.M. is supported by grants from NIH NIAMS P30 AR069655 pilot and AO Trauma Research Fellowship (Davos, Switzerland). B.F.B. is supported by grants from the NIH (R01 AR43510 and R01 AG049994). Y.M. is supported by grants from Amedica Inc. H.I. is supported by grants from Bristol-Myers, Astellas, and Asahi-Kasei. S.N.B. was supported by grants from the NIH (P30 AR061307 and T32 AR53459). I.O. is supported by the Goldstein Award from the Department of Orthopaedics, University of Rochester, Rochester, NY and the NIH (R21 AR074571 and R21 AR073321). C.X. is supported by grants from the NIH (R21 AR073321, R21 AR500710 and P30 AR069655-01 Pilot). J.L.D. is supported by grants from the NIH (R21 AI119646). The authors would like to the URMC Electron Microscope Shared Resource Laboratory. The content of this review is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or other funding sources.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Masters, E.A., Trombetta, R.P., de Mesy Bentley, K.L. et al. Evolving concepts in bone infection: redefining “biofilm”, “acute vs. chronic osteomyelitis”, “the immune proteome” and “local antibiotic therapy”. Bone Res 7, 20 (2019). https://doi.org/10.1038/s41413-019-0061-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-019-0061-z
This article is cited by
-
Antibiofilm activity of marine microbial natural products: potential peptide- and polyketide-derived molecules from marine microbes toward targeting biofilm-forming pathogens
Journal of Natural Medicines (2024)
-
Efficacy of lysostaphin-coated titanium plates on implant-associated MRSA osteitis in minipigs
European Journal of Trauma and Emergency Surgery (2024)
-
The use of combination therapy for the improvement of colistin activity against bacterial biofilm
Brazilian Journal of Microbiology (2024)
-
In vitro activity of exebacase against methicillin-resistant Staphylococcus aureus biofilms on orthopedic Kirschner wires
BMC Research Notes (2023)
-
Antimicrobials in polymethylmethacrylate: from prevention to prosthetic joint infection treatment: basic principles and risk of resistance
Arthroplasty (2023)
