
Abstract
Ankylosing spondylitis (AS), a common type of spondyloarthropathy, is a chronic inflammatory autoimmune disease that mainly affects spine joints, causing severe, chronic pain; additionally, in more advanced cases, it can cause spine fusion. Significant progress in its pathophysiology and treatment has been achieved in the last decade. Immune cells and innate cytokines have been suggested to be crucial in the pathogenesis of AS, especially human leukocyte antigen (HLA)‑B27 and the interleukin‑23/17 axis. However, the pathogenesis of AS remains unclear. The current study reviewed the etiology and pathogenesis of AS, including genome-wide association studies and cytokine pathways. This study also summarized the current pharmaceutical and surgical treatment with a discussion of future potential therapies.
Similar content being viewed by others

Introduction
Spondyloarthropathy (SpA) refers to a heterogeneous group of rheumatic diseases that present common clinical and genetic features, which are classified as peripheral or axial (axSpA) based on what parts of the body are predominantly affected. Ankylosing spondylitis (AS), a type of SpA, is an autoimmune disease that mainly involves spine joints, sacroiliac joints (SIJs) and their adjacent soft tissues, such as tendons and ligaments. In more advanced cases, this inflammation can lead to fibrosis and calcification, resulting in the loss of flexibility and the fusion of the spine, resembling “bamboo” with an immobile position. The main clinical manifestations include back pain and progressive spinal rigidity as well as inflammation of the hips, shoulders, peripheral joints and fingers/toes. In addition, there are extra-articular manifestations, such as acute anterior uveitis and inflammatory bowel disease (IBD). However, these extra-articular manifestations differ between East Asian and Caucasian populations. In a study involving 988 patients with ankylosing spondylitis in east Asia, only 0.4% developed inflammatory bowel disease.1 However, in some analyses performed in Western countries, ~5%–10% of patients with AS present with inflammatory bowel disease.2,3
The prevalence of AS has a clear correlation with the human leukocyte antigen (HLA)-B27 positive rate in specific populations. Studies have revealed that in HLA-B27-positive populations, the prevalence rate of AS is ~5%–6%.4 In a 2009 national survey in the United States, the prevalence of HLA-B27-positive populations varied in different ethnic communities, with 7.5%, 4.6%, and 1.1% in non-Hispanic whites, Mexican-Americans, and non-Hispanic blacks, respectively.5 In the literature, males reportedly account for the vast majority of cases of AS, while the incidence among men and women is similar in nonradiographic axial spondyloarthropathy (nr-axSpA), which refers to individuals meeting clinical criteria for axSpA without radiological evidence of sacroiliitis. A meta-analysis including eight studies including 2 236 patients with AS and 1 242 patients with nr-axSpA revealed that males accounted for 70.4% of AS patients and 46.5% of patients with nr-axSpA.6 Genetic susceptibility results have shown the following recurrent risk factors in different generations of relatives: monozygotic (MZ) twins, 63% (17/27); first-generation relatives, 8.2% (441/5 390); second-generation relatives, 1.0% (8/834); and third-generation relatives, 0.7% (7/997).7
Available criteria sets are frequently used in clinical practice to help clinicians make diagnoses. Currently, the most widely applied diagnostic classification of AS is the modified New York (mNY) criteria. In this classification system, a patient needs to meet at least one clinical criterion and the radiological diagnosis of AS. Another classification is Amor criteria and European Spondyloarthropathy Study Group (ESSG) criteria for diagnosing AS. In 2012, AV Tubergen discussed the different classification criteria sets for AS and SpA.8 In addition, the ASAS criteria, the dominant diagnostic criteria for axSpA, have gained popularity in Europe (2016 update of the ASAS-EULAR management recommendations for axial spondyloarthritis). As clinicians seemed to have difficulty differentiating AS and SpA, Joel D. Taurog established an algorithm for the diagnosis or exclusion of axSpA.9
The confusion in diagnosis and lack of disease-modifying therapeutics, including anti-TNF-α and anti-IL-17 treatment of AS, are largely due to the limited knowledge of the pathogenesis, which may involve immunity, heredity and other factors. In this paper, we reviewed the etiology of AS, current investigations of its pathogenesis and available treatments.
Etiology
As an autoimmune disease, AS develops through complex interactions between genetic background and environmental factors. Although significant progress has been achieved in the past decades, the etiology of AS remains unclear to some extent. To date, studies have revealed some factors that may be related to the occurrence of AS, including genetic background, immune reaction, microbial infection, and endocrinal abnormity.
Genetic background
Genetic factors have been acknowledged as crucial in the genesis of AS. The correlation between AS and genetics has been a perpetual topic since hereditary factors of AS were first confirmed within families in 1961.10 Twin studies have revealed significantly higher concordance between monozygotic twins (63%) than between dizygotic twins (23%). Genetic effects have been identified as pathogenic factors that contribute to over 90% of the population variance for AS manifestations.11,12 One of the most important genetic factors is major histocompatibility complex (MHC) class I allele HLA-B27, which was discovered in 1973.13 Despite the unclear pathomechanism, HLA-B27 has been associated with the prevalence of AS in different populations around the world.14 Studies have shown that 90%–95% of AS patients are HLA-B27 positive, while 1%–2% of HLA-B27-positive populations develop AS. This number increased to 15%–20% for those with an affected first-degree relative.15,16 The familial tendency of AS was remarkable with relative risks of 94, 25, and 4 for first-, second-, and third-degree relatives, respectively.17 In addition to the association with the genesis of AS, HLA-B27-positive patients showed a significantly lower average onset age and a higher prevalence of acute anterior uveitis than did HLA-B27-negative patients.18 HLA-B27 has a high degree of polymorphism. Over 100 subtypes have been identified thus far,19 with differing prevalence rates among different ethnicities, especially between those of East Asian and Caucasian descent. As reported, the most prevalent subtypes in AS are HLA-B2705 (Caucasian populations), HLA-B2704 (Chinese populations), and HLA-B2702 (Mediterranean populations).19,20,21,22 By contrast, two subtypes, HLA-B2706 and HLA-B2709, seem unrelated to AS.17,23,24 In addition, genetic influence is not alone in the development of AS. HLA-B27-transgenic rat studies on β2 microglobulin (β2m),25 a noncovalent part of the MHC-I complex, has proven that additional β2m reduces HLA-B27 misfolding and promotes arthritis and spondylitis, implying that B27 misfolding is associated with intestinal inflammation.26 This result suggested that abnormal β2m can coordinate with HLA-B27 in AS development, which may be explained by protein misfolding theories and will be discussed later in the pathogenesis section.
Even as the most emphasized genetic factor, the overall contribution of HLA-B27 to AS heritability is only ~20%,27 indicating that other genetic influences contribute to AS. With the progress of genome-wide association studies (GWASs) and other technologies, non-HLA-B27 and even non-HLA genes have been identified in AS in recent years; basically, the genetic differences between various ethnicities have been explored and compared in recent years. HLA-B60 is related to HLA-B27-negative AS and increases the disease susceptibility by 3–6-fold.28 An analysis in a Taiwanese population suggested that the interaction between HLA-B60 and HLA-B27 could be a better marker for the risk of AS susceptibility.29 HLA-B7,30 HLA-B16, HLA-B35,31,32 HLA-B38 and HLA-B3933 have also been associated with HLA-B27-negative AS in various ethnicities with unknown mechanisms. A study of 1 000 AS patients and 1 500 heathy individuals reported an initial association and independent replication of two new loci related to AS, ERAP1 (also known as ARTS1) and IL23R, in a North American sample and provided preliminary evidence for several non-MHC nonsynonymous single nucleotide polymorphisms (nsSNPs).34 Another study including GWAS of 2 053 AS patients and 5 140 controls presented SNPs in two gene deserts at 2p15 [rs10865331; combined P = 1.9 × 10(−19)] and 21q22 [rs2242944; P = 8.3 × 10(−20)], as well as in the genes ANTXR2 [rs4333130; P = 9.3 × 10(−8)] and IL1R2 [rs2310173; P = 4.8 × 10(−7)].35 Many GWASs have been performed in Caucasian and East Asian populations, focusing on genetic polymorphisms of AS susceptibility. One of these studies36 genotyped 69 non-MHC AS-associated SNPs and found that six loci (rs6759298, rs2297518, rs75301646, rs12615545, rs5837881, and rs27044) showed significant differences in both Caucasian and EA populations, with rs6759298 located in a ‘gene desert’ at chromosome 2p15 exhibiting the highest significance. However, genetic differences in several SNPs have been identified. For example, rs27980 and rs7711564 in ERAP1 previously identified in Caucasian descent were significantly associated with AS in Han Chinese, while no association was shown for ERAP1 SNPs such as rs27044.37,38,39 Similarly, IL23R (rs11209026), widely considered a major AS-associated SNP in Europe, showed no significance in a Chinese AS population.37 These studies on non-HLA-B27 genes and SNPs indicated a difference in the mechanism of disease pathogenesis between various ethnicities and may provide new insights into the pathogenesis and treatment of AS.
Immunological and microbial factors
AS is related to a series of autoimmune diseases, including IBD, anterior uveitis and psoriasis, which suggests that they may share a genetic basis and some common immunological processes. The differences observed in immune cells and cytokines in AS suggest the role of immunological effects in AS pathogenesis. In the peripheral blood of AS patients and healthy HLA-B27-positive controls, the levels of T cells secreting tumor necrosis factor (TNF)-α and interferon (IFN)-γ were reportedly lower. CD8+ T cells in AS patients tended to secrete more IL-10.40 Other findings have also demonstrated immunological influences in AS development, which is discussed in the following section.
Microbial infection acts as a triggering factor of the host innate immune system and AS development.41 HLA-B27 transgenic rats failed to develop features of SpA in a germ-free environment, which changed when commensal bacteria were introduced into the germ-free models,42,43 suggesting possible interactions between HLA-B27 and the microbiome. The gut microbiome, including Lachnospiraceae, Veillonellaceae, Prevotellaceae, Porphyromonadaceae, and Bacteroidaceae, showed significant differences in AS patients compared with that in healthy controls.44 Klebsiella pneumoniae acts as an opportunistic pathogen in the normal human gut, and studies have suggested that it may be an exacerbating agent in the autoimmune process of AS.45 Controversial results exist regarding the relationship between the fecal microbiome load, such as Klebsiella pneumoniae, and AS activity. Some scientists hypothesized that Klebsiella pneumoniae influences AS development indirectly through interplay with HLA-B27.46 In addition, gut microbiome infection is partly due to the relative deficiency of immune components, leading to immune responses of a higher intensity and longer duration.47
Other factors
Early in 1973, an etiological association between endocrine factors and AS was hypothesized because the presence of HLA-B27 and AS differed with sex.48 A study of 22 patients with AS detected testicular function and found a diminished testicular testosterone (T) reserve, elevated luteinizing hormone (LH) level, estradiol/testosterone ratio (E2:T) inversion and slightly increased estradiol (E2) level.49 The results in studies of ovarian function have also indicated sex hormone differences in menstruating and menopausal AS patients compared with those in matched healthy controls.50 It is reported that estradiol levels in patients with active AS are significantly lower than those in patients with inactive AS in the menstruation period. More observational results, such as male predominance, peak onset at young age and increased number of first manifestations after pregnancy, imply that sex hormones play a role in AS.51 Low levels of sex hormones, especially dehydroepiandrosterone sulfate (DHEAS), may also contribute to bone loss in patients with AS.52 A study examining the low-dose adrenocorticotropic hormone (ACTH) test (LDST) showed that after low-dose ACTH, the cortisol increment was significantly lower in AS patients than in controls (20.0 ± 4.4 vs 24 ± 2.2 microg/dl, P < 0.001).53 The subclinical glucocorticoid deficiency indicated an impaired hypothalamic-pituitary-adrenal (HPA) axis in AS patients, suggesting the involvement of the endocrine system in the etiology of AS.
Meta-analyses have suggested that vitamin D deficiency may be related to AS development. Vitamin D may play a protective role in AS based on a positive correlation between the serum vitamin D level and disease severity.54 There is a significant negative correlation between the vitamin D level and disease activity indicated by the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level.55 However, there are conflicts regarding the relationship between the vitamin D level and AS disease activity.55,56 The cause of low vitamin D levels in patients with AS is also largely unclear.
Pathogenesis
MHC genetics
The human MHC, also called the HLA complex, belongs to the cell-surface proteins acting in the process of acquired immunity. There are three subgroups in the MHC gene family: class I, II, and III. MHC class I encodes HLA-A, HLA-B, and HLA-C and is present on all nucleated human cells and platelets, presenting epitopes to T cell receptors (TCRs) on the surface of cytotoxic T lymphocytes (CTLs).57 The heterodimer MHC class I subgroup consists of a polymorphic heavy chain. The chain contains three domains, i.e., α1, α2, and α3. The α1 domain links noncovalently with the non-MHC molecule β2m, while α3 spans the plasma membrane and interacts with the CD8 coreceptor of T cells.58,59 The MHC class I complex can link to peptides of 8–10 amino acids in length via one cleft spaced by both α1 and α2, leading to the initiation and propagation of immune responses.59,60 A stable MHC molecule needs to be properly packaged and then folded in the cell organelle endoplasmic reticulum (ER) under guidance of chaperones (calreticulin and tapasin).57 Although the classic MHC class I contains one heavy chain, there are three different structures of MHC‐I, comprising cell-surface HLA‐B27 homodimers and intracellular and exosomal MHC‐I dimers.61 These components may function in distinct pathophysiological processes.
HLA-B27
HLA-B27, basically belonging to the MHC-I surface protein encoded by the MHC B gene on chromosome 6, is the most essential gene that predisposes an individual to AS. HLA-B27 presents peptide antigens to T immunocytes of the human body defense process and is considered to be significantly linked to AS and associated inflammatory diseases. A study reviewed over 7500 endogenous peptides presented by the eight most frequent HLA–B27 allotypes (HLA–B2702 to HLA–B2709), suggesting that consensus-binding and selection motifs showed significant similarities and differences between various HLA–B27 allotypes.62 The connection between HLA-B27 and AS has not yet been fully elucidated, although it is widely accepted that the entire intracellular process of HLA-B27 formation needs to be considered. There are some prevailing theories regarding the mechanism, including the hypothesis of arthritogenic peptide, misfolding hypothesis, the hypothesis of molecular mimicry, as well as the hypothesis of the cell-surface HLA‐B27 homodimer.
Founded on the antigenic peptide presentation role of HLA class I molecules, the arthritogenic peptide hypothesis postulates that structurally exclusive peptide‐MHC complexes can directly initiate HLA‐B27-specific autoimmune responses by relying on the primary structure of antigen peptides.63 Some microbial peptides are similar to self-peptides in body tissues and can activate the response of certain HLA-B27-specific CD8+ T lymphocytes. The T lymphocytes react with these HLA-B27-peptide complexes, leading to autoreactivity and autoimmune disease.64,65 It has been suggested that cartilage, particularly the proteoglycan aggrecan,66 is the basic immunological target in SpA; however, currently, studies on such peptides have obtained inconsistent results. In addition, rats with HLA-B27-specific CD8- T lymphocytes still have AS, which means that more peptide mechanisms remain unelucidated.67 Additionally, a spectrometry study reviewed a large quantity of HLA-B27 subtypes and indicated that it is quantitative instead of qualitative changes in the peptide repertoire that may be more relevant to AS initiation and progression,62 further challenging the arthritogenic peptide hypothesis. The molecular mimicry hypothesis posits that the antigenic components of infectious bacterial pathogens partially resembling or cross-reacting with HLA molecules can stimulate CD8+ T lymphocytes, followed by responding to one HLA‐B27 relevant self‐peptide or the peptides directly produced by HLA‐B27.68 This hypothesis is largely based on previously identified amino acid structures of homologous origin between the HLA structure and specific sequences and previous results depicting cross-reactions among the HLA and some bacterial antigens.46 K. pneumoniae is a highlighted microorganism thought to participate in the pathogenesis of AS as a triggering and/or perpetuating factor.69 Some components in K. pneumoniae share structural likenesses with specific genetic or somatic sequences in humans and exhibit molecular mimicry in AS and other diseases. Similarly, molecular modeling suggested that a HLA-B27-derived dodecamer, a natural ligand of disease-associated B27 subtypes, was strikingly homologous to protein sequences from arthritogenic bacteria, particularly Chlamydia trachomatis, demonstrating the process of molecular mimicry of Chlamydial proteins.70 PulD-secreted pullulanase can cross-react with HLA-B27 and myosin, while pulA components can cross-react with type I, III, and IV collagens,46 proving the reasonability of the molecular mimicry hypothesis. These cross-reactions give rise to an amount of antibacterial antibodies that link to HLA molecules on immunocytes, chondrocytes and fibroblasts,71 further triggering a cascade of inflammatory reactions with the amount of cytokines, complement proteins, proteinases and the like produced.72 These sequential reactions lead to the genesis of arthritis and extra-articular or even systemic symptoms and signs of AS.
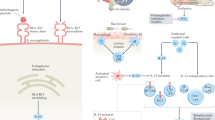
The mature HLA-B27 complex is a quaternary structure with three important components. The proper assembly and folding of HLA-B27 in the ER is essential for its function. After being synthesized as free heavy chains, HLA-B27 is then noncovalently linked and folded with β2m and antigenic peptide, followed by transport to the cell surface as a trimolecular complex.73 Nevertheless, HLA‐B27 exhibits a predisposition to misfolding and creating dimers and even multimers74; these characteristic changes may originate in its structure, which includes cysteine (C) at sites 67 (C67), 101 (C101), 164 (C164), and 325 (C325).75 Without correct folding, HLA-B27 would be produced and transmitted to the cell surface merely as homodimers consisting of heavy chains. The disease‐related structures of HLA‐B27, including HLA‐B2705, HLA‐B2704, and HLA‐B2702, have been found to exhibit a relatively lower rate of correct folding procedures compared with those of HLA‐B2706 and HLA‐B2709, which are generally not considered associated with AS.76 Due to cysteine residue C67 and other reasons, HLA-B27 tends to fold slower than other HLA alleles, and without proper folding, these defective HLA-B27 proteins continually gather in the ER.73 Improperly folded HLA-B27 proteins accumulate in the ER and activate autophagy and the interleukin (IL)-23/IL-17 pathway.73 Moreover, these misfolded molecules can interfere with ER function, leading to ER stress and even triggering the pro-inflammatory endoplasmic reticulum unfolded protein response (ERUPR), which further activates the IL-23/IL-17 pathway.73,77 However, conflicts also exist regarding whether the HLA-B27-activated ERUPR occurs in AS patients. The increased production of IL‐23 without significant ERUPR induction occurs in macrophages in AS.78 The disease‐related polymorphisms of the ERAP1 or HLA‐B27 locus would not change the ER stress intensities of AS,79 which also remained controversial in other studies conducted later.14 One possibility is that HLA-B27 misfolding results in autophagy and triggers the IL-23/IL-17 pathway instead of ERUPR.80 Further research is required for illumination of the connection of ERUPR and HLA-B27 during the development of AS.
HLA-B27 heavy chains tend to form homodimers without β2m via the disulfide bonds of the cysteine at C67.81 The dimeric HLA-B27 complexes, mostly found in the gut and synovium of patients, may contribute to the genesis of AS and some other SpAs. These HLA-B27 dimers could occur on antigen-presenting cells, thus stimulating IL-23 receptor + T lymphocytes to produce IL-17.82 The hypothesis of cell-surface HLA‐B27 homodimer formation suggests that HLA-B27 dimers might contribute to the development of AS. HLA‐B27 homodimers have been linked to receptors expressed on natural killer (NK) immunocytes, myelomonocytes and lymphocytes. The binding is realized via killer cell immunoglobulin‐like receptors (KIRs) and leucocyte immunoglobulin‐like receptors (LILRs), thus acting in the processes related to autoimmune disorders (Fig. 1).83,84 The 3 immunoglobulin domains and the long cytoplasmic tail 2 (KIR3DL2) receptor expressed by certain increased immune cells, including NK cells and Th17 cells, can recognize cell-surface HLA‐B27 homodimers via a greater affinity than that with the classic HLA‐B27 heterotrimers.85,86 The binding of KIR3DL2 with HLA-B27 homodimers was revealed to stimulate the survival and differentiation of KIR3DL2+CD4+ T lymphocytes in patients with SpA.87,88 Compared to KIR3DL2− lymphocytes, these T cells significantly increase cytokine output, including IL‐17, TNF‐α and INF-γ.85 These findings suggest that the aberrant HLA‐B27 homodimers function in AS pathogenesis.

Various functions of ER resident and cell surface HLA-B27 dimers. ER resident dimers might lead to ER stress and activate unfolded protein responses. Cell-surface dimers might be produced after the recycling of fully folded HLA-B27 cell surface molecules through the endocytic pathway and be re-expressed as dimers for presentation to receptors, such as KIR and LILR. Elevated propagation and existence of KIR3DL2+ CD4+ T lymphocytes and amplified IL-17 production in AS cases after stimulation with antigen-presenting cells expressing HLA-B27 homodimers were confirmed in earlier studies. These cells have largely been revealed to secrete TNF-α and IFN-γ. IL-17 has been shown to coordinate with TNF-α or IFN-γ in stimulating the release of inflammatory regulators and affecting bone construction to function in the development of AS. (Reprinted with permission of Elsevier, from chen et al.75)
Non-HLA-B27 MHC alleles
In addition to the widely accepted HLA-B27 genetics in AS, other non-HLA-B27 MHC alleles, including multiple MHC-I or II loci, have been associated with the pathogenesis of AS. These genes, including HLA-B40, HLA-B60, HLA-A, HLA-DRB1, HLA-DQA1, HLA-DPB1, or others, function in the development of AS via interaction with TCRs and KIRs expressed on NK cells and certain lymphocytes or participate in antigen presentation and other inflammatory processes.14 For example, HLA-G was proven to produce homodimers in the cell organelle endosomes through a completely folded β2m‐related form.89 Non-HLA-B27 genes may act independently or via linkage disequilibrium with HLA-B27. Additionally, HLA-A0201 tag SNP rs2394250 is reportedly related to AS independent of HLA-B27 for HLA-B27-positive or HLA-B27-negative AS patients.90 The mechanisms by which these non-HLA-B27 MHC genes affect AS remain largely unknown. Further study on the non-HLA-B27 MHC allele may offer insights into the pathogenesis of SpA and other related diseases.
Non-MHC genetics
In the past four decades, research on the pathogenesis of AS has been focused on MHC genes. However, the development of GWAS methods and the identification of a considerable number of genetic variants revealed the importance of non-MHC genes. Although the overall contribution of HLA-B27 is only ~20%, making HLA-B27 a major contributor, ~7% of the heritable risk originates from non-MHC variants.27,91,92 Therefore, non-MHC genetics also function in the pathogenesis of AS. Moreover, some pathways or mechanisms have been reported to be involved in the processes of AS in recent years.
ERAP1 and ERAP2
Three previously identified aminopeptidases were recognized as genetically related to AS vulnerability, including ERAP1 (coding for endoplasmic reticulum aminopeptidase 1 (ERAP1)), ERAP2 (coding for ERAP2), and NPEPPS (coding for puromycin-sensitive aminopeptidase (PSA)).90,93 Recent research has indicated that gene-to-gene interactions among HLA-B27 and ERAP1 and subsequent abnormal peptide presentation are likely relevant to the development of AS (Fig. 1).90,94 A case-control association study revealed that protective genetic variants were associated with reduced function of ERAP1 and ERAP2 and suppressed MHC-I expression on the cell surface.90 ERAP1 and ERAP2 variations may also reduce the speed of HLA-B27 folding by affecting the amount of relevant peptide accessible, thereby increasing ER stress and AS development.14
Both ERAP1 and ERAP2, genes at chromosome 5q15, participate in trimming peptides in the ER to nine amino acids for antigen presentation by HLA-I molecules, such as HLA-B27.95 In addition to the processing and presentation of antigens, ERAP1 can still trim several cytokine receptors on the cell surface, such as IL-1R2, TNFR1, and IL-6Rα, thus reducing their ability to conduct signals to cells, and the latter further affects inflammatory processes.96 These two genes are involved in the development of AS and other diseases. ERAP1 is reportedly associated with HLA‐B27− and HLA‐B40positive AS,95 while ERAP2 is related to HLA‐B27+ and HLA‐B27− AS.97 ERAP1 is also involved in the development of juvenile idiopathic arthritis, psoriasis, and Behçet’s disease, while ERAP2 is related to Crohn’s disease and psoriasis, as well as birdshot chorioretinopathy,79,91 with unclear mechanisms.
ERAP1, first reported in 200793 as a risk factor for AS, is now considered the second strongest gene associated with AS; together with HLA-B27, ERAP1 is accountable for 70% of genetic factors for familial AS.98 It has been found that ERAP1 polymorphisms influence the risk of AS for HLA-B27+ individuals.99 Considering its function, ERAP1 may act in association with molecules such as HLA-B27 and be concerned with aberrant peptide processing or mistaken antigen presentation, leading to a predisposition to AS (Fig. 2).99 To date, five SNPs of ERAP1 have been recognized, i.e., rs27044, rs30187, rs2287987, rs10050860, and rs174820,100 and research has shown robust epistasis among ERAP1 SNPs, such as rs30187, rs27044, and rs27037, and HLA-B27.101 Research has shown that loss-of-function polymorphisms in ERAP1 affect HLA-B27 free heavy chain expression and peptide dimerization or misfolding. This research also shows a larger amount of ERAP1 production in dendritic cells (DCs) in patients, implying that abnormal ERAP1 expression may count for AS development to some extent.102,103 ERAP2 can form a heterodimer and cleave peptides collaboratively with ERAP1. Moreover, the functional variants in ERAP2 have been identified as involved in AS development.90 For example, the ERAP2 SNP rs2248374 causes nonsense ERAP2 protein expression and gives rise to the reduced expression of MHC-I molecules on the cell surface of AS patients.104 These polymorphisms causing a decrease in ERAP1 activity as well as a deficiency in ERAP2 production have now been recognized as some kind of defensive variants against AS.90 Therefore, the suppression of ERAP1 and/or ERAP2 may have protective effects and a promising treatment for AS.

Demonstration of the possible role of HLA-B27 and ERAP1/2 in AS pathogenesis. HLA-B27 can present arthritogenic peptides to CD8+ T lymphocytes, which trigger AS initiation. Peptides enter the ER and are further trimmed by ERAP1 and ERAP2. Unusual peptides will be produced because of incorrect ERAP1 or ERAP2 trimming, leading to HLA-B27 free heavy chains (FHCs) and homodimers through endosomal recycling from the cell membrane and then to NK cell and Th17 cell activation by KIRs, particularly KIR3DL2. Abnormal peptide-HLA-B27 complexes gather in the ER, triggering UPR, ER stress, ER-associated protein degradation (ERAD) and autophagy. (Reprinted with permission of Elsevier, from Zohreh and colleagues110)
IL-23/IL-17 pathway
In AS, the first indication of IL-23/IL-17 relevance came from a GWAS study in 2007 that identified an IL-23 receptor (IL-23R) SNP related to AS pathogenesis (Fig. 3).34 In humans, the differentiation of Th17 cells may be triggered by IL-23, TGF-β, and IL-1β, among other inflammatory cytokines, and the differentiated immunocytes further generate IL-17A, IL-17F, IL-22, IL-26, and CCL20.105 Dysfunction of the IL-23/IL-17 pathway was identified in many diseases related to human immunological procedures, including psoriasis, IBD, rheumatoid arthritis and SpA106; additionally, IL-17 and IL-23 act as major cytokines for axSpA and psoriatic arthritis.107 Studies have shown higher serum levels of IL-23 and IL-17108 and the presence of IL-17+ cells in the facet joints in AS patients,109 suggesting that the innate immune system might be of greater relevance. Moreover, it has been shown that AS can be greatly ameliorated by blocking the IL-23/IL-17 pathway,105 further indicating a significant role in AS development. In AS, differentiated T lymphocytes can generate IL-17 and then trigger osteoclast activation, thus suppressing bone regeneration. Moreover, lymphocytes can produce IL-22 when exposed to IL-23 to stimulate osteoproliferation.110 This contradictory process may explain the coexistence of erosion and formation of bone for patients with AS.

IL-23/17 pathway in AS pathogenesis. The interplay of genetic and epigenetic influences, particularly Th17 and Th22 cells, with a few kinds of stress, such as mechanical stress, gut microbiota stress, and environmental triggers, gives rise to the production of pro-inflammatory molecules, including IL-17, IL-22, TNF-α, and IL-23. (Reprinted with permission of Elsevier, from Zohreh and colleagues110)
IL-23R genetic polymorphisms were considered related to AS vulnerability. The SNP rs11209026 leads to a nonsynonymous substitution of amino acid residue (R381Q) that considerably weakens IL-23R function, and the production of IL-17 can exert protective effects against AS inflammation and development.111 In addition to polymorphisms of the gene itself, variants in the vicinity of the IL12B locus, which encode the IL-12p40 subunit of IL-23, have been associated with AS.112 There are more IL-23/IL-17 pathway-associated loci, including IL-1R1, IL-2R, IL-6R, IL-12B, IL-27, STAT3, TYK2, CARD9, and RUNX3.92 A variant in the TYK2 locus is involved in AS pathogenesis by affecting TYK2 splicing with a strong odds ratio of 7.7.90 Based on these findings, it is reasonable to further concentrate on the connection of IL-23/IL-17 signaling and AS or possible therapeutic targeting molecules, including IL-1 and IL-23, and other downstream factors along the IL-23/IL-17 axis.
Lymphocyte activation and differentiation
Another important non-MHC genetic factor in the pathogenesis of AS is genes modulating the activation and differentiation of either CD4+ or CD8+ T lymphocytes. GWASs of SNPs identified several non-MHC genes that are concerned with the production as well as activation of lymphocytes and connected to AS, including RUNX3, EOMES, ZMIZ1, IL7, TBX21, and IL7R.90,112,113,114 Runt-related transcription factor 3 (RUNX3), which belongs to the transcription factor family crucial for regulating the expression of lineage-specific genes, can stimulate T cell differentiation to CD8+ T lymphocytes in thymopoiesis.115 RUNX3 polymorphisms have been linked to many human immune diseases or inflammatory processes, including systemic lupus erythematosus, psoriatic arthritis, and AS.116,117 Previous studies have revealed a connection between the RUNX3 polymorphism rs11249215 and AS in Caucasian,99 Han Chinese118 and Korean populations,119 and rs4648889 has been related to reduced RUNX3 expression in AS.120 RUNX3 stimulates eomesodermin expression encoded by the EOMES gene and is the transcription factor related to the differentiation of CD8.121 In addition to RUNX3, research has suggested a relationship between these AS-associated non-MHC genes and lymphocyte differentiation or activation. A study of IL7R α chain knockout mice indicated that IL7R participation in T lymphocyte production can trigger RUNX3 expression for immature T lymphocytes and their differentiation towards the CD8 lineage.122 Polymorphisms of molecules involved in the activation or suppression of lymphocytes, such as programmed cell death 1 (PDCD1), encoding PD-1, or T lymphocyte antigen 4 (CTLA-4), encoding CTLA-4, have been shown to influence the susceptibility to AS.106 These non-MHC gene variants function in the pathogenesis of AS via unknown mechanisms, and further investigations are needed regarding their pathways and possible clinical applications.
Immune cell and innate cytokines
AS is one type of seronegative spondyloarthritis, which is usually associated with chronic inflammation involving DCs, macrophages, NK cells and adaptive immune cells.123 These immune cells produce various innate cytokines that play a crucial role in the development of AS, as shown in Fig. 4. Human DCs located in lymphatic and nonlymphatic organs are divided into CD1c-positive (conventional DC1) or CD141-positive (conventional DC2) subsets.124,125 Another group of DCs, called plasmacytoid dendritic cells (pDCs), exhibit a plasma cell-like appearance and can produce CD56+, HLA-DR, derived dendritic cell antigen 2 (BDCA-2), Toll-like receptor 7 (TLR7), CD123, and TLR9 and can be distinguished from monocytes and conventional DCs by the lack of CD14 and CD11c expression.126,127 In addition to their function in the inborn and adaptive immune processes, these cells participate in B cell-mediated humoral immunity.127 Previous studies have shown elevated production of IL-1B and IL-6 in AS patients compared with that in normal subjects because a decrease in the number of circulating CD1c+ DCs increases the quantity of CD14-CD16+ mononuclear cells, which mediates the activation of CC chemokine receptor 6 (CCR6) expression.124,128 These processes trigger the Th17 immune response and IL-17 production, which are involved in autoimmune and inflammatory responses and are associated with clinical manifestations of AS.129,130 At the same time, CD1c-positive DCs can stimulate Th1 and Th2 responses. However, whether Th1 and Th17 cells act synergistically during the inflammatory process of AS is unclear. Some scholars believe that Th17 cells participate in the acute phase of inflammation,131,132 while Th1 cells function principally in prolonging or promoting late inflammatory reaction.133,134

Immunocytes are involved in the initiation, evolution, and regulation of AS. (Reprinted with permission of Elsevier, from Mohammad and colleagues123)
The frequency or quantity of macrophages for AS subjects is closely related to disease severity.130,135 Large numbers of CD68-positive macrophages or osteoclasts were observed in the sacroiliac joint lesions of AS patients.136 Previous research has confirmed the major role of CD163+ macrophages in inflamed body parts for SpA patients.137 Moreover, the number of macrophages was reduced after drug therapy in SpA patients.138 A previous study using HLA-B27/human β2m transgenic rats has shown that HLA-B27+ macrophages produced IL-23-based inflammatory factors, which contributed to the pathogenesis of enthesopathy.139 In the clinic, the expression of IL-23 was significantly increased in synovium tissues and serum in AS cases compared to that in healthy controls.20,140 Another animal experiment showed that lowering the level of macrophages has a protective effect against AS.141 The NK cell number is also higher in AS patients. It has been reported that the increased levels of IL-8 and SDF-1 in AS patients increase the expression of the NK cell-inhibitory receptor carcino-embryonic antigen-cell adhesion molecule (CEACAM1), thereby inhibiting NK cell activity. NK cells specifically recognize HLA-B27 molecules through the NK-inhibitory receptor KIR3DL1. Thus, these two types of cells are important in chemokine expression and development of AS.142
A few subgroups of CD4+ T lymphocytes participate in the development of AS. Wang et al.143 found significantly higher levels of IFN-γ-inducible protein 10 (IP-10/CXCL10), attracting Th1 cells and even causing an increased level of IFN-γ and TNF-α and aggressive inflammatory responses. Moreover, the Th2 cell response could be enhanced in AS patients due to the overexpression of the chemokine receptor CCR4 on CD4+ T cells.144 Raised serum TRAC and MDC levels in AS subjects are positively related to the migration of Th2 cells.143 An elevated level of Th1 density as well as an increased Th1/Th2 fraction was detected for cases with mild or severe AS.145 These results suggest that the adaptive reaction is turned on to attract more Th2 cells and restore the Th1/Th2 ratio.143 Th17 cells, whose differentiating process is regulated via the transcription factor signal transducer plus activator of transcription 3 (STAT3) and STAT5, mainly produce pro-inflammatory cytokines, including IL-17, IL-10, IL-21, and IFN-γ.146,147 Research has revealed elevated serum IL-17 and IL-23 concentrations for AS patients.148 It has been reported that in predisposed patients, the IL-23/IL-23 receptor complex results in increased IL-17 production by inducing the proliferation and terminal differentiation of Th17 cells.149 The IL-23/IL-17 axis causes inflammatory molecule secretion from fibroblasts, endothelial cells, DCs or macrophages, which induce joint destruction and inflammation that are observed in both rheumatoid arthritis and AS.140,146 The quantity of Th22 cells in peripheral circulation and the IL-22 concentrations in plasma were shown to increase without an obvious relation to AS severity.150 El-Zayadi et al.151 found that IL-22 could drive the propagation, migration and osteogenic differentiation of mesenchymal stem cells and as such, might be a novel cytokine boosting bone construction in SpA. Nevertheless, the level of IL-22 producing CD4+CD25highCD127low/- regulatory T cells (Tregs) was meaningfully lower for AS cases than for controls, but it is unclear whether the level of Tregs could be considered a predictive factor for disease severity or treatment outcome.152,153
Zhang et al.154 found that mononuclear cells in peripheral circulation in AS and HLA-B27-positive normal cohorts showed an elevated level of IL4+CD8+ T cells compared to those from HLA-B27-negative control cohorts, accompanied by an increased proportion of IL-4+ to IFN-γ+ cells. Previous studies have described that HLA-B27-restricted T cells could be responsive to self-antigens and arthritis-implicated antigens, resulting in autoimmune inflammation.155,156 Researchers have found that CD8+ T cells could respond to peptides LMP2 236-244 and VIP1R 400-408 for HLA-B2705 and HLA-B2709 individuals via this pathomechanism.157 CD8+ T cells could cause the direct lysis of target cells by CTLs via the secretion of perforin/granzyme or Fas/FasL signaling. These cells could also generate inflammatory products, including TNF-α, IFN-γ, and IL-17, to maintain chronic immune reactions for AS patients.158,159
Studies on B cells in AS have shown different results from those on T cells. An elevated number of plasma cells and plasma blasts has been found in the circulation and peripheral joints of AS patients.160,161 Based on current knowledge, B cells function as effectors possibly involved in the pathogenesis of AS through various mechanisms. First, B lymphocytes can differentiate into plasma cells that secrete antibodies and affect immune reactions, even triggering osteoclast genesis. Second, B lymphocytes produce cytokines such as IL-6 and receptor activator of nuclear factor kappa-B ligand (RANKL), which stimulate plasma cell formation and osteoclast genesis separately. Moreover, B lymphocytes can present antigens, further serving as costimulators during suitable activation of T lymphocytes. Last but not least, B lymphocytes help in forming germinal center-like ectopic lymphoid tissue for plasma cell generation.162
In conclusion, although multiple autoantibodies including anti-CD74 and anti-NOG/SOST have been found, the role for B cells is quite limited, and AS is still identified as a seronegative arthritis.
Treatments for AS
Pharmacological treatments
The aims of treating AS are to improve and maintain spinal flexibility and normal posture, relieve symptoms, decrease functional limitations, and reduce complications. The mainstays of pharmacological treatment involve nonsteroidal anti-inflammatory medications (NSAIDs) and TNF-α inhibitors (TNFis). Additional treatments include non-TNFi biologics (secukinumab), methotrexate, and sulfasalazine. Furthermore, the oral small molecule JAK inhibitors tofacitinib and filgotinib appear promising in clinical trials and may soon be approved for AS.163,164 Several guidelines for the management of AS have been issued by expert panels in France,165 Spain,166 Tukey,167 Canada,168,169 the United Kingdom,170 the United States171 and Europe,172 which are all based on systematic literature reviews. As shown in Fig. 5, there is substantial agreement among these strategies. For all AS patients, regardless of whether the disease is active or stable, physical therapy,173 exercise and abstaining from smoking174 are universally advised.

Drug treatment strategy for ankylosing spondylitis patients
NSAIDs, especially selective inhibitors of cyclooxygenase 2, are first-line treatments for patients with active AS. The determination of active disease is founded on laboratory (CRP/ESR), clinical and imaging (magnetic resonance imaging, MRI) findings.175 Compared with on-demand treatment, continuous NSAID treatment has shown no benefits in any clinical aspect,176 while hypertension and depression are more common among individuals undergoing continuous NSAID treatment. However, continuous use should be advised if symptom recurrence occurs after stopping or reducing the dose of NSAID drug.177,178 In adults with active AS, an appropriate trial consists of at least 2 kinds of NSAIDs, each administered over a minimum of 2 weeks at the maximum tolerated dosage, unless contraindicated.179 Nevertheless, the ‘lowest effective dose’ of NSAIDs has also been recommended in the National Institute for Health and Care Excellence (NICE) guidelines.170 No NSAID is recommended in terms of preferred efficacy. NSAID treatment should be chosen based on the patient’s history of NSAID application, comorbidities, and risk factors for adverse effects.171 Good responses to NSAIDs include a reduction in inflammatory back pain and functional improvement. An insufficient response to NSAID therapy is identified as active disease despite the administration of at least two different NSAIDs at the maximum anti-inflammatory dose and duration (at least two weeks for each). Intolerance or adverse effects are also involved. Analgesics, especially opioid-like drugs, may be added when NSAID treatment is unsuccessful or contraindicated.167,172
Despite treatment with NSAIDs, TNFis are advised for patients with high disease activity.171 An insufficient response to TNFis is defined as disease activity markers remaining over specific cut-offs (BASDAI ≥ 4 or AS Disease Activity Score (ASDAS) ≥ 2.1) over time. The ASDAS reflects the inflammatory condition better than the BASDAI, and its cut-offs are preferable. Biological agents should be used according to their indications and contraindications and patient comorbidities. No TNFi is recommended for preferred efficacy. Moreover, treatment with infliximab or adalimumab is preferred over treatment with etanercept for patients with IBD or frequently recurrent iritis.180,181 Predictors for a good response to TNFis are a short disease duration, patient age ≤40 years, the absence of enthesitis, positivity for HLA-B27, a good functional status, and a high CRP level.182 In a Swiss study, male sex was also identified as a good predictor for TNF response.183 Contraindications for TNFis include the presence of an active infection, tuberculosis, advanced heart failure, lupus, multiple sclerosis and cancer. In patients with active AS and who have contraindications for TNFis, sulfasalazine or pamidronate is recommended over non-TNFi biologics, such as abatacept and tocilizumab.171 When AS patients fail to respond to the first TNFi, treatment with a second biologic should be advised. The different biologics can be an IL-17 inhibitor (IL-17i) or a different TNFi.184 The treatment should be changed if no significant improvement occurs after application for 3 months. If a 6-month trial results in no clinical remission or decrease in disease severity, the treatment must be changed. After failure treatment of the first TNFi, a second TNFi with a lower efficacy can also be effective.184 However, before switching the treatment, it is essential to reconsider the indications of the first TNFi. Given the circumstances of a primary lack of efficacy, primary failure might be due to an incorrect diagnosis rather than drug resistance. Furthermore, the symptoms and signs may result from either a different or concomitant condition. Biologics may fail in AS patients with concomitant vertebral fracture or degenerative disc disease. In patients with persistent remission, the tapering of TNFi or IL-17i treatment can be considered.185 The period of remission should be at least 6 months. Ideally, tapering can be continued to zero (withdrawal). However, tapering only very slowly and allowing sufficient time for remission are suggested before the next step in the tapering process.
Local injections of glucocorticoids seem to be an option for treating enthesopathy and arthritis. Glucocorticoid injections into involved peripheral joints, sacroiliac joints, or entheses could provide immediate symptom relief. Previous studies have shown that partly because of the increased risks of osteoporosis, hyperlipidemia and insulin resistance, long-term treatment with systemic glucocorticoids is relatively contraindicated. A recent study reported that AS patients achieved relief from signs and symptoms after short-term treatment with high doses of glucocorticoids (50 mg/day).186 In patients with peripheral arthritis as a comorbidity, conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), such as methotrexate, leflunomide, and sulfasalazine, should be considered but not in those with isolated axSpA or enthesitis.172 Methotrexate treatment has not been proven effective in AS patients without peripheral arthritis, regardless of administration of a TNFi.
Other biologics include rituximab (a monoclonal antibody against CD20+ B cells), ustekinumab (a monoclonal antibody against IL-12/23) and secukinumab (a monoclonal antibody against IL-17). If TNFi fails to treat AS, rituximab may be an alternative approach.187 In AS patients with concomitant moderate to severe psoriasis, ustekinumab treatment reportedly achieved safe and significant improvements.188 Ustekinumab can also reduce arthritis, enthesitis, dactylitis, and skin lesions and improve function.189 In a prospective clinical trial, ustekinumab was associated with a reduction in signs and symptoms in active AS and was well tolerated.190 However, a recent study did not demonstrate the efficacy of ustekinumab (anti-IL12p40) and risankizumab (anti-IL23p19) in the treatment of axial SpA.191,192 At the moment, the mechanism of blocking IL-23, which does not play a role in SpA treatment, remains uncovered.193 IL-17A and IL-22 could be suppressed in anti-IL23R in prophylactic experiments194 and were comparable to those after therapeutic anti-IL23R treatment. Thus, the initiation instead of the persistence of experimental SpA may depend on IL-23 signaling. Secukinumab targets IL-17 and is effective in patients with TNFi failure.195 In a phase 3 trial, secukinumab showed salient efficacy in AS patients with an insufficient response or contraindications to TNFis.196 In another phase 3 trial, secukinumab was verified to provide sustained efficacy in signs, symptoms and physical function in subjects with AS over 3 years.197 A study cohort of Taiwanese patients indicated that secukinumab was also well tolerated in Asian patients, with a safety profile consistent with that reported in the overall study population.198
In patients with stable AS, using NSAID treatment on-demand is recommended. Continuing treatment with TNFi alone is suggested rather than treatment with TNFi and NSAID or DMARD.199 The continued use of NSAIDs or DMARDs has uncertain therapeutic effects with increased risks of gastrointestinal, cardiovascular, renal and hematological toxicity.200
Surgical treatments
Untreated AS can cause spinal deformity, with more than 30% of AS patients suffering from thoracolumbar kyphosis.201 Corrective osteotomy and stabilization are very common in surgical procedures and are recommended under certain conditions,171 such as adult patients suffering severe kyphosis or advanced hip arthritis. This procedure has a perioperative mortality rate of 4% and permanent neurologic sequelae rate of 5%.202 This surgery is confirmed to contribute to preventing the natural processes of progressive deformity, reducing pain caused by muscle fatigue, improving disability, restoring the global balance and horizontal axis of view, and improving respiratory and digestion function.203,204,205,206,207,208,209,210
Osteotomy procedures for the treatment of kyphotic deformity can be fundamentally differentiated into closing- vs. opening-wedge osteotomy (CWO/OWO) procedures.210 In 1945, OWO was first introduced by Smith-Petersen et al.211 and has subsequently been modified by several surgeons.212,213 For correction of the kyphotic deformity of the lumbar spine, this technique creates a gap in laminae and spinous processes, forcing manual extension of the lumbar spine. The complication rate is relatively high due to the sharp lordotic angle and the elongation of the anterior column occurring in this procedure, which can cause serious vascular and neurological injuries.214,215 To solve this problem, Wilson et al.216 first presented poly-segmental wedge osteotomy (PWO) in 1949, which was improved by Zielke in the 1980s.217,218,219,220 Correction was achieved via multiple CWOs in the posterior lumbar spine to generate a rather harmonious opening of the anterior disc spaces with tempered posterior shortening spanning. Zielke and his coworkers, different from the former procedure, advocated PWO with internal fixation using Harrington rods, laminar hooks, and later transpedicular screws. Nevertheless, on the instrumentation, implant failure has been described in >40% of patients due to limited mobility of the intervertebral disc and strong effect.221 Thus, the postoperative satisfaction rates were far from the expected rates.221,222,223
In 1963, monosegmental CWO was first introduced by Scudese and Calabro224 and thoroughly developed by Ziwjan in 1982225 and Thomasen in 1985.226 During this procedure, the posterior elements of one vertebra, in combination with the posterior wedge of the vertebral body, are resected to achieve correction by passive extension of the lumbar spine. Similar to PWO, internal fixation is also needed to enhance immediate stability. In this technique, correction is achieved. However, the postoperative satisfaction and complication rates of CWO seem to be better than those of OWO or PWO.226,227
Based on these three surgical procedures, surgical techniques for treating kyphosis have been constantly improved. Closing–opening-wedge osteotomy (COWO), particularly beneficial in cervical spine surgery, was first introduced by Kawahara et al.228 to overcome some limitations and simultaneously combine the benefits of CWO and OWO. Posterior column resection is performed in a manner similar to that of CWO, with the tip of the wedge at the vertebral midsagittal point. A plane is osteotomized anteriorly from this point parallel to the endplates or the anterior cortex is fractured and the anterior column is opened during osteotomy closure.210 This procedure has been shown to reduce localized kyphosis from an average of 67–18° at 2.2- to 7.5-year follow-ups.228 Ji et al.206 and Bourghli et al.229 have shown similar results; in addition, spinal cord shortening and aorta lengthening were well tolerated in all patients.
Regarding the incidence of spine fractures, it is estimated to be 4 times greater in patients with AS than in the general population,230 largely because of the combination of rigidity and osteoporosis that develop in these patients.231 Spine fracture can be severe even after minor trauma232,233; 75% of these fractures occur in the cervical spine, particularly at the C5-T1 cervicothoracic junction.234,235 Because these fractures often follow minor trauma (47%)236 and occur in patients who typically already have long-standing back pain, the diagnosis is often delayed. To make matters worse, these fractures can be missed on X-rays and often require CT for diagnosis.237 Neurological deterioration during the initial hospitalization after spinal fractures in the context of AS is common, and the 1-year mortality rate is high.238 There is a consensus on the optimal therapeutic treatment of spine fractures in patients with AS. Most reports in the literature describing the surgical treatment of spine fracture in AS patients have been limited to case reports and smaller series.239,240 Complete spinal cord paralysis persisting for more than a few hours does not seem to benefit from surgery.241 A retrospective study conducted by Backhaus et al.236 included 119 patients with 129 spine fractures due to AS. Sixty-one patients (51%) developed either incomplete or complete paraplegia. Furthermore, revision surgery due to implant loosening or insufficient stabilization was required in 15% of patients. For some patients, the risks would outweigh the benefits. Therefore, evidence for routine surgical interventions is limited. For patients with threatening or definite neurological deficiencies, surgery may be considered by experienced surgeons.
Cauda equina syndrome (CES) is a rare complication of long-standing AS. Neurological symptoms occur insidiously and have a poor prognosis without effective treatment. AS-related CES may be due to chronic arachnoiditis and dural fibrosis leading to diminished cerebrospinal fluid (CSF) resorption with dural sac dilation and diverticula formation.68,242 Leaving these patients untreated or treated with steroids alone is inappropriate. Surgical treatment of dural ectasia, either by lumboperitoneal shunting or laminectomy, may improve neurological dysfunction or halt the progression of neurological defects.243
Hip abnormality is commonly recognized by rheumatologists in AS patients and affects approximately one-fourth to one-third of AS patients.244 Proposed by the ACR, based on evidence, arthroplasty is the best treatment option for patients with advanced hip arthritis and severe hip pain who often would otherwise experience progressive limitations in mobility and reliance on opiates.171 The annual trends of total hip arthroplasty (THA) in AS patients have significantly declined, which may be attributed to improvements in medical management that delay the time from disease onset to THA.245
There have been many studies on THA, although THA could be a difficult procedure in patients with AS. Based on current reports, THA can generate satisfactory results, greatly improving hip joint function and relieving pain without significant complications.246,247,248,249,250 Data from 54 AS patients who underwent 81 THAs between 2008 and 2014 were retrospectively analyzed by Xu et al.246 Significant improvements in the Harris hip score (HHS), visual analog scale (VAS) score, and range of motion (ROM) were found postoperatively, with favorable prosthetic localization observed by radiographic evaluations; all of these findings indicated acceptable short- and mid-term benefits. A total of 181 hips of 103 patients were included in another large study with a follow-up period of over 27 years conducted by Joshi et al.251 A satisfactory survival rate (71%) and no functional impairment or reankylosis were reported, demonstrating the long-term improvement of hip function for AS patients.
Nevertheless, evidence for the efficacy of THA in AS patients is based only on observational studies and case series. Controversies persist concerning the surgical timing, implant selection, intraoperative management strategies, and heterotopic ossification (HO) prophylaxis. Concomitant severe hip and spinal deformity is particularly challenging to treat, and there is no consensus on which deformity to repair first.246,252,253,254,255,256 Prior to THA, some people advocate that a spinal osteotomy should be performed to reduce the risk of hip dislocation.246,253,254,255 Others agree with the assertion that a THA performed first would contribute to placement of the patient in a stable prone position to facilitate corrective spine osteotomy.256 Another debate has been focused on the superiority of cementless or cemented components in AS patients. Cemented prosthetic components are recommended for AS patients with severe osteoporosis who should be treated with revised total hip replacement (THR) to ensure a good fit of the prosthesis to the canal.251,253,257 Additionally, the failure rate of THR using cemented components (5%) is lower than that of THR with cementless components (28%).253 In contrast, cementless prostheses are preferred in AS patients, especially young patients, without significant morphological changes in the proximal femurs because this strategy can reduce the difficulty of future revisions while allowing bone ingrowth to enhance the durability of the implant.258,259
HO is the main postoperative complication of joint arthroplasty, with high occurrence rates ranging between 4 and 74.7%.253 There are no clear recommendations on perioperative precautionary measures for HO after THR in AS patients. Previous studies have suggested the use of NSAIDs, such as indomethacin, and radiation therapy, both preoperatively and postoperatively, to prevent HO,253,258,260,261 while others believed that the risk of prevention outweighed the benefits.246,251,262 To minimize the adverse effects, Feng et al.248 shortened the medication time by application of indomethacin for 2 weeks as prophylaxis. No patients who received perioperative prophylaxis developed reankylosis by HO in this study.248 Although radiation therapy has been proven effective for preventing HO, another retrospective study including 129 hips of 91 Asian AS patients conducted by Weng et al.263 showed no significant difference between the postoperative single-fraction radiotherapy group and the control group (P = 0.210), suggesting that postoperative radiation may not be necessary in Asian patients.
Conclusions and perspectives
Ankylosing spondylitis is a painful and debilitating disease, with considerable socioeconomic burdens. In 2002, the yearly mean total (direct and productivity) costs of AS were US$6,720 per patient in the USA and Euro 9 462 per patient in Europe.264 There is no effective disease-modifying treatment largely due to the unclear pathogenesis. This review focuses on the etiology, pathogenesis and treatment progress of AS. The etiology of ankylosing spondylitis may be related to genetic background, immune function, pathogenic infection, endocrine abnormalities and other factors. HLA-B27 is the gene crucial to the development of ankylosing spondylitis, while the non-HLA-B27 gene is also important in its pathogenesis. Genetic differences between various ethnicities, particularly East Asian and Caucasian descent, have been noted through GWASs and may shed light on therapeutic strategies. Certain autoimmune diseases and microbial infections also contribute to ankylosing spondylitis, suggesting that they may have a common immune and genetic basis. In addition, the possible causes include hormone abnormalities and changes in vitamin D levels.
The pathogenesis of AS is very complex. Current studies suggest that it may be the result of a variety of complicated mechanisms. In the pathogenesis of AS, various hypotheses, such as the false folding hypothesis, attempt to elucidate the role of HLA-B27. Other non-HLA-B27 genes may influence the occurrence of ankylosing spondylitis through immune function and gene interaction. Overexpression of ERAP1 and ERAP2 is also considered an important pathogenesis of AS. Many factors and related genes in this pathway are constantly being discovered. In addition, related research has focused on the disorder of the IL-23/IL-17 axis and the abnormality of lymphocyte activation and differentiation. Most immune cells and cytokines reportedly participate in the pathogenesis of AS. In particular, the IL-23/IL-17 pathway plays a crucial role in the development of the disease. At present, the pathogenesis of AS is considered to mainly involve immune T cells, while B cells are also slightly involved. There are some studies on the pathogenesis of AS mediated by B cells, and related studies may be strengthened in the future. Alternatively, researchers may continue to explore the correlation among cytokines and immune cells and diseases to predict the occurrence, development and severity of disease. Although the pathogenesis of ankylosing spondylitis is not yet clear, the existing research results can have a certain guiding significance for clinical practice.
The treatment of AS is mainly composed of drug and surgical treatment. In clinic, NSAIDs and TNF-alpha inhibitors are the main drugs for AS. Moreover, interleukin receptor blockers and some drugs that inhibit new bone formation have received increased attention and become the focus of future research, including IL-6 receptor inhibitor sarilumab and Wnt signal pathway inhibitors. Once AS is not effectively controlled, more severe deformities may appear, and surgical treatment is needed. For severe spinal deformities, spinal surgery is required. For sacroiliac joint lesions, total hip arthroplasty is needed. Treatment of AS has always been the center of research all over the world. Currently, the research hotspot of surgical treatment of AS focuses on the choice of surgical path, perioperative management, and the prevention and treatment of complications, especially HO. With scientific and technological development, early diagnosis and effective treatment of AS will be possible. The socioeconomic burden of AS will be significantly reduced, and patients' suffering will be alleviated.
References
Wang, C.-R. et al. Rare occurrence of inflammatory bowel disease in a cohort of Han Chinese ankylosing spondylitis patients- a single institute study. Sci. Rep. 7, 13165–13165 (2017).
Lindström, U., Olofsson, T., Wedrén, S., Qirjazo, I. & Askling, J. Impact of extra-articular spondyloarthritis manifestations and comorbidities on drug retention of a first TNF-inhibitor in ankylosing spondylitis: a population-based nationwide study. RMD open 4, e000762–e000762 (2018).
Moltó, A. & Nikiphorou, E. Comorbidities in spondyloarthritis. Front. Med. 5, 62–62 (2018).
Reveille, J. D. & Weisman, M. H. The epidemiology of back pain, axial spondyloarthritis and HLA-B27 in the United States. Am. J. Med. Sci. 345, 431–436 (2013).
Reveille, J. D., Hirsch, R., Dillon, C. F., Carroll, M. D. & Weisman, M. H. The prevalence of HLA-B27 in the US: data from the US National Health and Nutrition Examination Survey, 2009. Arthritis Rheum. 64, 1407–1411 (2012).
de Winter, J. J., van Mens, L. J., van der Heijde, D., Landewe, R. & Baeten, D. L. Prevalence of peripheral and extra-articular disease in ankylosing spondylitis versus non-radiographic axial spondyloarthritis: a meta-analysis. Arthritis Res. Ther. 18, 196 (2016).
Brown, M. A., Laval, S. H., Brophy, S. & Calin, A. Recurrence risk modelling of the genetic susceptibility to ankylosing spondylitis. Ann. Rheum. Dis. 59, 883–886 (2000).
van Tubergen, A. & Weber, U. Diagnosis and classification in spondyloarthritis: identifying a chameleon. Nat. Rev. Rheumatol. 8, 253–261 (2012).
Taurog, J. D., Chhabra, A. & Colbert, R. A. Ankylosing spondylitis and axial spondyloarthritis. N. Engl. J. Med. 375, 1303 (2016).
de Blecourt, J., Polman, A. & de Blecourt-Meindersma, T. Hereditary factors in rheumatoid arthritis and ankylosing spondylitis. Ann. Rheum. Dis. 20, 215–220 (1961).
Brown, M. A. et al. Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum. 40, 1823–1828 (1997).
Reveille, J. D. The genetic basis of ankylosing spondylitis. Curr. Opin. Rheumatol. 18, 332–341 (2006).
Brewerton, D. A. et al. Ankylosing spondylitis and HL-A 27. Lancet 1, 904–907 (1973).
Reveille, J. D. An update on the contribution of the MHC to AS susceptibility. Clin. Rheuma. 33, 749–757 (2014).
Reveille, J. D. The genetic basis of spondyloarthritis. Ann. Rheum. Dis. 70, i44–i50 (2011).
Brown, M. A. Genetics of ankylosing spondylitis. Curr. Opin. Rheumatol. 22, 126–132 (2010).
Taurog, J. D. The mystery of HLA-B27: if it isn't one thing, it's another. Arthritis Rheum. 56, 2478–2481 (2007).
Feldtkeller, E., Khan, M. A., van der Heijde, D., van der Linden, S. & Braun, J. Age at disease onset and diagnosis delay in HLA-B27 negative vs. positive patients with ankylosing spondylitis. Rheumatol. Int. 23, 61–66 (2003).
Khan, M. A. Polymorphism of HLA-B27: 105 subtypes currently known. Curr. Rheumatol. Rep. 15, 362 (2013).
Uchanska-Ziegler, B., Ziegler, A. & Schmieder, P. Structural and dynamic features of HLA-B27 subtypes. Curr. Opin. Rheumatol. 25, 411–418 (2013).
Sorrentino, R., Bockmann, R. A. & Fiorillo, M. T. HLA-B27 and antigen presentation: at the crossroads between immune defense and autoimmunity. Mol. Immunol. 57, 22–27 (2014).
Wucherpfennig, K. W. Presentation of a self-peptide in two distinct conformations by a disease-associated HLA-B27 subtype. J. Exp. Med. 199, 151–154 (2004).
D'Amato, M. et al. Relevance of residue 116 of HLA-B27 in determining susceptibility to ankylosing spondylitis. Eur. J. Immunol. 25, 3199–3201 (1995).
Koh, W. H. & Boey, M. L. Ankylosing spondylitis in Singapore: a study of 150 patients and a local update. Ann. Acad. Med. Singap. 27, 3–6 (1998).
Tran, T. M. et al. Additional human beta2-microglobulin curbs HLA-B27 misfolding and promotes arthritis and spondylitis without colitis in male HLA-B27-transgenic rats. Arthritis Rheum. 54, 1317–1327 (2006).
Weinreich, S. S., HoebeHewryk, B., vanderHorst, A. R., Boog, C. J. P. & Ivanyi, P. The role of MHC class I heterodimer expression in mouse ankylosing enthesopathy. Immunogenetics 46, 35–40 (1997).
Breban, M., Said-Nahal, R., Hugot, J. P. & Miceli-Richard, C. Familial and genetic aspects of spondyloarthropathy. Rheum. Dis. Clin. North Am. 29, 575–594 (2003).
Wei, J. C., Tsai, W. C., Lin, H. S., Tsai, C. Y. & Chou, C. T. HLA-B60 and B61 are strongly associated with ankylosing spondylitis in HLA-B27-negative Taiwan Chinese patients. Rheumatology 43, 839–842 (2004).
Wei, J. C. et al. Interaction between HLA-B60 and HLA-B27 as a better predictor of ankylosing spondylitis in a Taiwanese population. PLoS ONE 10, e0137189 (2015).
Khan, M. A., Kushner, I. & Braun, W. E. A subgroup of ankylosing spondylitis associated with HLA-B7 in American blacks. Arthritis Rheum. 21, 528–530 (1978).
Khan, M. A., Kushner, I. & Braun, W. E. B27-negative HLA-BW16 in ankylosing spondylitis. Lancet 1, 1370–1371 (1978).
Wagener, P., Zeidler, H., Eckert, G. & Deicher, H. Increased frequency of HLA-Bw62 and Bw35 CREG antigens in HLA-B27 negative ankylosing spondylitis. Z. Rheumatol. 43, 253–257 (1984).
Yamaguchi, A. et al. Association of HLA-B39 with HLA-B27-negative ankylosing spondylitis and pauciarticular juvenile rheumatoid arthritis in Japanese patients. Evidence for a role of the peptide-anchoring B pocket. Arthritis Rheum. 38, 1672–1677 (1995).
Consortium, W. T. C. C. et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 39, 1329–1337 (2007).
Australo-Anglo-American Spondyloarthritis Consortium (TASC) et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat. Genet. 42, 123–127 (2010).
Liu, J. et al. Genetic association of non-MHC region with ankylosing spondylitis in a Chinese population. Ann. Rheum. Dis., https://doi.org/10.1136/annrheumdis-2018-214625 (2018).
Davidson, S. I. et al. Association of ERAP1, but Not IL23R, With Ankylosing Spondylitis in a Han Chinese Population. Arthritis Rheum.-Us 60, 3263–3268 (2009).
Chen, C. & Zhang, X. ERAP1 variants are associated with ankylosing spondylitis in East Asian population: a new Chinese case-control study and meta-analysis of published series. Int. J. Immunogenet. 42, 168–173 (2015).
Lee, Y. H. & Song, G. G. Associations between ERAP1 polymorphisms and susceptibility to ankylosing spondylitis: a meta-analysis. Clin. Rheumatol. 35, 2009–2015 (2016).
Rudwaleit, M. et al. Low T cell production of TNFalpha and IFNgamma in ankylosing spondylitis: its relation to HLA-B27 and influence of the TNF-308 gene polymorphism. Ann. Rheum. Dis. 60, 36–42 (2001).
Sieper, J., Braun, J. & Kingsley, G. H. Report on the fourth international workshop on reactive arthritis. Arthritis Rheum. 43, 720–734 (2000).
Taurog, J. D. et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J. Exp. Med. 180, 2359–2364 (1994).
Rath, H. C. et al. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J. Clin. Invest 98, 945–953 (1996).
Costello, M. E. et al. Brief report: intestinal dysbiosis in ankylosing spondylitis. Arthritis Rheuma. 67, 686–691 (2015).
Gomez-Simmonds, A. & Uhlemann, A. C. Clinical implications of genomic adaptation and evolution of carbapenem-resistant Klebsiella pneumoniae. J. Infect. Dis. 215, S18–S27 (2017).
Zhang, L. et al. The association of HLA-B27 and Klebsiella pneumoniae in ankylosing spondylitis: a systematic review. Micro. Pathog. 117, 49–54 (2018).
Sieper, J. & Braun, J. Pathogenesis of spondylarthropathies. Arthritis Rheum. 38, 1547–1554 (1995).
Schlosstein, L., Terasaki, P. I., Bluestone, R. & Pearson, C. M. High association of an HL-A antigen, W27, with ankylosing spondylitis. N. Engl. J. Med. 288, 704–706 (1973).
Tapiaserrano, R. et al. Testicular function in active ankylosing-spondylitis-therapeutic response to human chorionic-gonadotropin. J. Rheuma. 18, 841–848 (1991).
Jimenezbalderas, F. J., Tapiaserrano, R., Maderocervera, J. I., Murrieta, S. & Mintz, G. Ovarian-function studies in active ankylosing-spondylitis in women-clinical-response to estrogen therapy. J. Rheuma. 17, 497–502 (1990).
Gooren, L. J., Giltay, E. J., van Schaardenburg, D. & Dijkmans, B. A. Gonadal and adrenal sex steroids in ankylosing spondylitis. Rheum. Dis. Clin. North Am. 26, 969–987 (2000).
Aydin, T., Karacan, I., Demir, S. E. & Sahin, Z. Bone loss in males with ankylosing spondylitis: its relation to sex hormone levels. Clin. Endocrinol. 63, 467–469 (2005).
Kebapcilar, L. et al. Impaired hypothalamo-pituitary-adrenal axis in patients with ankylosing spondylitis. J. Endocrinol. Invest 33, 42–47, https://doi.org/10.1007/BF03346548 (2010).
Cai, G. et al. Vitamin D in ankylosing spondylitis: review and meta-analysis. Clin. Chim. Acta 438, 316–322 (2015).
Pokhai, G. G., Bandagi, S. & Abrudescu, A. Vitamin D levels in ankylosing spondylitis: does deficiency correspond to disease activity? Rev. Bras. Reum. 54, 330–334 (2014).
Almanea, S., Miller, W. H., Siebert, S. & Derakhshan, M. H. Serum vitamin D in ankylosing spondylitis and axial spondylitis: a systematic review and meta-analysis. Rheumatology 57, key075.401 (2018).
Madden, D. R. The three-dimensional structure of peptide-MHC complexes. Annu. Rev. Immunol. 13, 587–622 (1995).
Toh, H. et al. Changes at the floor of the peptide-binding groove induce a strong preference for Proline at position 3 of the bound peptide: Molecular dynamics simulations of HLA-A*0217. Biopolymers 54, 318–327 (2000).
Nguyen, T. T. et al. Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1. Nat. Struct. Mol. Biol. 18, 604–613 (2011).
Yewdell, J. W. DRiPs solidify: progress in understanding endogenous MHC class I antigen processing. Trends Immunol. 32, 548–558 (2011).
Alvarez-Navarro, C. & de Castro, J. A. L. ERAP1 structure, function and pathogenetic role in ankylosing spondylitis and other MHC-associated diseases. Mol. Immunol. 57, 12–21 (2014).
Schittenhelm, R. B., Tc, L. K. S., Wilmann, P. G., Dudek, N. L. & Purcell, A. W. Revisiting the arthritogenic peptide theory: Quantitative not qualitative changes in the peptide repertoire of HLA-B27 allotypes. Arthritis Rheuma. 67, 702–713 (2015).
Chatzikyriakidou, A., Voulgari, P. V. & Drosos, A. A. What is the role of HLA-B27 in spondyloarthropathies? Autoimmun. Rev. 10, 464–468 (2011).
Faham, M. et al. Discovery of T cell receptor beta motifs specific to HLA-B27-positive ankylosing spondylitis by deep repertoire sequence analysis. Arthritis Rheuma. 69, 774–784 (2017).
de Castro, J. A. L. The HLA-B27 peptidome: building on the cornerstone. Arthritis Rheum. 62, 316–319 (2010).
Wolfgang, K. et al. Identification of novel human aggrecan T cell epitopes in HLA-B27 transgenic mice associated with spondyloarthropathy. J. Immunol. 173, 4859–4866 (2004).
Lin, A., Guo, X., Inman, R. D. & Sivak, J. M. Ocular inflammation in HLA-B27 transgenic mice reveals a potential role for MHC class I in corneal immune privilege. Mol. Vis. 21, 131–137 (2015).
Antoniou, A. N., Lenart, I. & Guiliano, D. B. Pathogenicity of misfolded and dimeric HLA-B27 molecules. Int. J. Rheumatol. 2011, 486856 (2011).
Rashid, T. & Ebringer, A. Ankylosing spondylitis is linked to Klebsiella–the evidence. Clin. Rheuma. 26, 858–864 (2007).
Manuel, R. et al. Molecular mimicry of an HLA-B27-derived ligand of arthritis-linked subtypes with chlamydial proteins. J. Biol. Chem. 277, 37573–37581 (2002).
Ryu, K. H. et al. Tonsil-derived mesenchymal stromal cells: evaluation of biologic, immunologic and genetic factors for successful banking. Cytotherapy 14, 1193–1202 (2012).
Ciccia, F., Rizzo, A. & Triolo, G. Subclinical gut inflammation in ankylosing spondylitis. Curr. Opin. Rheumatol. 28, 89–96 (2016).
Colbert, R. A., DeLay, M. L., Layh-Schmitt, G. & Sowders, D. P. HLA-B27 misfolding and spondyloarthropathies. Adv. Exp. Med Biol. 649, 217–234 (2009).
Colbert, R. A., Tran, T. M. & Layh-Schmitt, G. HLA-B27 misfolding and ankylosing spondylitis. Mol. Immunol. 57, 44–51 (2014).
Chen, B. et al. Role of HLA-B27 in the pathogenesis of ankylosing spondylitis (review). Mol. Med. Rep. 15, 1943–1951 (2017).
Antoniou, A. N., Ford, S., Taurog, J. D., Butcher, G. W. & Powis, S. J. Formation of HLA-B27 homodimers and their relationship to assembly kinetics. J. Biol. Chem. 279, 8895–8902 (2004).
Turner, M. J. et al. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J. Immunol. 175, 2438–2448 (2005).
Zeng, L., Lindstrom, M. J. & Smith, J. A. Ankylosing spondylitis macrophage production of higher levels of interleukin-23 in response to lipopolysaccharide without induction of a significant unfolded protein response. Arthritis Rheum. 63, 3807–3817 (2011).
Kenna, T. J. et al. Disease-associated polymorphisms in ERAP1 do not alter endoplasmic reticulum stress in patients with ankylosing spondylitis. Genes Immun. 16, 35–42 (2015).
Colbert, R. A., Delay, M. L., Layh-Schmitt, G. & Sowders, D. P. HLA-B27 misfolding and spondyloarthropathies. Prion. 3, 15–26 (2009).
Chen, B., Li, D. & Xu, W. Association of ankylosing spondylitis with HLA-B27 and ERAP1: pathogenic role of antigenic peptide. Med Hypotheses 80, 36–38 (2013).
Ranganathan, V., Gracey, E., Brown, M. A., Inman, R. D. & Haroon, N. Pathogenesis of ankylosing spondylitis—recent advances and future directions. Nat. Rev. Rheumatol. 13, 359–367 (2017).
Allen, R. L., Raine, T., Haude, A., Trowsdale, J. & Wilson, M. J. Cutting edge: leukocyte receptor complex-encoded immunomodulatory receptors show differing specificity for alternative HLA-B27 structures. J. Immunol. 167, 5543–5547 (2001).
Tam, L. S., Gu, J. & Yu, D. Pathogenesis of ankylosing spondylitis. Nat. Rev. Rheumatol. 6, 399–405 (2010).
Bowness, P. et al. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J. Immunol. 186, 2672–2680 (2011).
Chan, A. T., Kollnberger, S. D., Wedderburn, L. R. & Bowness, P. Expansion and enhanced survival of natural killer cells expressing the killer immunoglobulin-like receptor KIR3DL2 in spondylarthritis. Arthritis Rheum. 52, 3586–3595 (2005).
Giles, J. et al. HLA-B27 homodimers and free H chains are stronger ligands for leukocyte Ig-like receptor B2 than classical HLA class I. J. Immunol. 188, 6184–6193 (2012).
Wong-Baeza, I. et al. KIR3DL2 binds to HLA-B27 dimers and free H chains more strongly than other HLA class I and promotes the expansion of T cells in ankylosing spondylitis. J. Immunol. 190, 3216–3224 (2013).
Lynch, S. et al. Novel MHC class I structures on exosomes. J. Immunol. 183, 1884–1891 (2009).
International Genetics of Ankylosing Spondylitis Consortium. et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet. 45, 730–738 (2013).
Brown, M. A., Kenna, T. & Wordsworth, B. P. Genetics of ankylosing spondylitis–insights into pathogenesis. Nat. Rev. Rheumatol. 12, 81–91 (2016).
Ellinghaus, D. et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. 48, 510–518 (2016).
Wellcome Trust Case Control Consortium. et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 39, 1329–1337 (2007).
Graham, D. S. C. et al. Association of IRF5 in UK SLE families identifies a variant involved in polyadenylation. Hum. Mol. Genet 16, 579–591 (2007).
Kanaseki, T., Blanchard, N., Hammer, G. E., Gonzalez, F. & Shastri, N. ERAAP synergizes with MHC class I molecules to make the final cut in the antigenic peptide precursors in the endoplasmic reticulum. Immunity 25, 795–806 (2006).
Cui, X. et al. Identification of ARTS-1 as a novel TNFR1-binding protein that promotes TNFR1 ectodomain shedding. J. Clin. Invest 110, 515–526 (2002).
Robinson, P. C. et al. ERAP2 is associated with ankylosing spondylitis in HLA-B27-positive and HLA-B27-negative patients. Ann. Rheum. Dis. 74, 1627–1629 (2015).
Tsui, F. W. et al. Association of an ERAP1 ERAP2 haplotype with familial ankylosing spondylitis. Ann. Rheum. Dis. 69, 733–736 (2010).
Evans, D. M. et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat. Genet. 43, 761–767 (2011).
Vitulano, C., Tedeschi, V., Paladini, F., Sorrentino, R. & Fiorillo, M. T. The interplay between HLA-B27 and ERAP1/ERAP2 aminopeptidases: from anti-viral protection to spondyloarthritis. Clin. Exp. Immunol. 190, 281–290 (2017).
Wang, C. M. et al. ERAP1 genetic variations associated with HLA-B27 interaction and disease severity of syndesmophytes formation in Taiwanese ankylosing spondylitis. Arthritis Res. Ther. 14, R125 (2012).
Kochan, G. et al. Crystal structures of the endoplasmic reticulum aminopeptidase-1 (ERAP1) reveal the molecular basis for N-terminal peptide trimming. Proc. Natl Acad. Sci. USA 108, 7745–7750 (2011).
Campbell, E. C., Fettke, F., Bhat, S., Morley, K. D. & Powis, S. J. Expression of MHC class I dimers and ERAP1 in an ankylosing spondylitis patient cohort. Immunology 133, 379–385 (2011).
Andres, A. M. et al. Balancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentation. Plos Genet 6, e1001157 (2010).
van den Berg, W. B. & McInnes, I. B. Th17 cells and IL-17 a–focus on immunopathogenesis and immunotherapeutics. Semin Arthritis Rheu 43, 158–170 (2013).
Mahmoudi, M., Aslani, S., Nicknam, M. H., Karami, J. & Jamshidi, A. R. New insights toward the pathogenesis of ankylosing spondylitis; genetic variations and epigenetic modifications. Mod. Rheuma. 27, 198–209 (2017).
Paine, A. & Ritchlin, C. T. Targeting the interleukin-23/17 axis in axial spondyloarthritis. Curr. Opin. Rheumatol. 28, 359–367 (2016).
Mei, Y. et al. Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clin. Rheuma. 30, 269–273 (2011).
Appel, H. et al. Analysis of IL-17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res. Ther. 13, R95 (2011).
Babaie, F. et al. The role of gut microbiota and IL-23/IL-17 pathway in ankylosing spondylitis immunopathogenesis: new insights and updates. Immunol. Lett. 196, 52–62 (2018).
Di Meglio, P. et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS ONE 6, e17160 (2011).
Danoy, P. et al. Association of variants at 1q32 and STAT3 with ankylosing spondylitis suggests genetic overlap with Crohn's disease. Plos Genet 6, e1001195 (2010).
Reveille, J. D. et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat. Genet. 42, 123–U147 (2010).
Lau, M. C. et al. Genetic association of ankylosing spondylitis with TBX21 influences T-bet and pro-inflammatory cytokine expression in humans and SKG mice as a model of spondyloarthritis. Ann. Rheum. Dis. 76, 261–269 (2017).
Woolf, E. et al. Runx3 and Runx1 are required for CD8 T cell development during thymopoiesis. Proc. Natl Acad. Sci. USA 100, 7731–7736 (2003).
Apel, M. et al. Variants in RUNX3 contribute to susceptibility to psoriatic arthritis, exhibiting further common ground with ankylosing spondylitis. Arthritis Rheum. 65, 1224–1231 (2013).
Wang, W. et al. RUNX3 gene polymorphisms are associated with clinical features of systemic lupus erythematosus in Chinese Han population. J. Dermatol Sci. 80, 69–71 (2015).
Lian, Z. et al. Analysis of PPARGC1B, RUNX3 and TBKBP1 polymorphisms in Chinese Han patients with ankylosing spondylitis: a case-control study. PLoS ONE 8, e61527 (2013).
Cho, S. M., Jung, S. H. & Chung, Y. J. A variant in RUNX3 is associated with the risk of ankylosing spondylitis in Koreans. Genom. Inform. 15, 65–68 (2017).
Vecellio, M. et al. The genetic association of RUNX3 with ankylosing spondylitis can be explained by allele-specific effects on IRF4 recruitment that alter gene expression. Ann. Rheum. Dis. 75, 1534–1540 (2016).
Yagi, R. et al. The transcription factor GATA3 actively represses RUNX3 protein-regulated production of interferon-gamma. Immunity 32, 507–517 (2010).
Franke, A. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 42, 1118–1125 (2010).
Rezaiemanesh, A. et al. Immune cells involved in the pathogenesis of ankylosing spondylitis. Biomed. Pharmacother. 100, 198–204 (2018).
O'Keeffe, M., Mok, W. H. & Radford, K. J. Human dendritic cell subsets and function in health and disease. Cell. Mol. Life Sci. 72, 4309–4325 (2015).
Guilliams, M. et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat. Rev. Immunol. 14, 571–578 (2014).
Zang, M., Chen, K., Qu, C. & Brinckjensen, N. S. Monocyte-derived dendritic cells: targets as potent antigen-presenting cells for the design of vaccines against infectious diseases. Int. J. Infect. Dis. 19, 1–5 (2014).
McKenna, K., Beignon, A. S. & Bhardwaj, N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J. Virol. 79, 17–27 (2005).
Wright, P. B. et al. Ankylosing spondylitis patients display altered dendritic cell and T cell populations that implicate pathogenic roles for the IL-23 cytokine axis and intestinal inflammation. Rheumatology 55, 120–132 (2016).
Zambrano-Zaragoza, J. F., Agraz-Cibrian, J. M., González-Reyes, C., Durán-Avelar, M. J. & Vibanco-Pérez, N. Ankylosing spondylitis: from cells to genes. Int. J. Inflam. 2013, 501653 (2013).
Talpin, A. et al. Monocyte-derived dendritic cells from HLA-B27+ axial spondyloarthritis (SpA) patients display altered functional capacity and deregulated gene expression. Arthritis Res. Ther. 16, 417 (2014).
Paul, B. et al. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J. Immunol. 186, 2672–2680 (2011).
Lei, Z. et al. Increased frequencies of Th22 cells as well as Th17 cells in the peripheral blood of patients with ankylosing spondylitis and rheumatoid arthritis. Plos ONE 7, e31000 (2012).
Leonardo, L. C. et al. In vivo peripheral blood proinflammatory T cells in patients with ankylosing spondylitis. J. Rheumatol. 39, 830–835 (2012).
Coffre, M. et al. T.5. Th1 versus Th17 Cells in Ankylosing Spondylitis. Clin. Immunol. 131, S47–S47 (2009).
Kruithof, E. et al. Synovial histopathology of psoriatic arthritis, both oligo-and polyarticular, resembles spondyloarthropathy more than it does rheumatoid arthritis. Arthritis Res. Ther. 7, R569 (2005).
Braun, J. et al. Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthritis Rheum. 38, 499–505 (2010).
Mulherin, D., Fitzgerald, O. & Bresnihan, B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. 39, 115–124 (2010).
Barry, B. et al. Synovial tissue sublining CD68 expression is a biomarker of therapeutic response in rheumatoid arthritis clinical trials: consistency across centers. J. Rheuma. 36, 1800–1802 (2009).
Rezaiemanesh, A. et al. Ankylosing spondylitis M-CSF-derived macrophages are undergoing unfolded protein response (UPR) and express higher levels of interleukin-23. Mod. Rheuma. 27, 862–867 (2017).
Jethwa, H. & Bowness, P. The interleukin (IL)-23/IL-17 axis in ankylosing spondylitis: new advances and potentials for treatment. Clin. Exp. Immunol. 183, 30–36 (2016).
Kashiwagi, N., Nakano, M., Saniabadi, A. R., Adachi, M. & Yoshikawa, T. Anti-inflammatory effect of granulocyte and monocyte adsorption apheresis in a rabbit model of immune arthritis. Inflammation 26, 199–205 (2002).
Azuz-Lieberman, N. et al. The involvement of NK cells in ankylosing spondylitis. Int. Immunol. 17, 837–845 (2005).
Wang, J. et al. Circulating levels of Th1 and Th2 chemokines in patients with ankylosing spondylitis. Cytokine 81, 10–14 (2016).
Yang, P. T. et al. Increased CCR4 expression on circulating CD4+ T cells in ankylosing spondylitis, rheumatoid arthritis and systemic lupus erythematosus. Clin. Exp. Immunol. 138, 342–347 (2010).
Wang, C., Liao, Q., Hu, Y. & Zhong, D. T lymphocyte subset imbalances in patients contribute to ankylosing spondylitis. Exp. Ther. Med. 9, 250–256 (2015).
Konya, C., Paz, Z., Apostolidis, S. A. & Tsokos, G. C. Update on the role of interleukin 17 in rheumatologic autoimmune diseases. Cytokine 75, 207–215 (2015).
Torchinsky, M. B. & Blander, J. M. T helper 17 cells: discovery, function, and physiological trigger. Cell. Mol. Life Sci. 67, 1407–1421 (2010).
Xueyi, L. et al. Levels of circulating Th17 cells and regulatory T cells in ankylosing spondylitis patients with an inadequate response to anti-TNF-alpha therapy. J. Clin. Immunol. 33, 151–161 (2013).
Sherlock, J. P., Buckley, C. D. & Cua, D. J. The critical role of interleukin-23 in spondyloarthropathy. Mol. Immunol. 57, 38–43 (2014).
Zhang, L. et al. Increased frequencies of Th22 cells as well as Th17 cells in the peripheral blood of patients with ankylosing spondylitis and rheumatoid arthritis. Plos ONE 7, e31000 (2012).
El-Zayadi, A. A. et al. Interleukin-22 drives the proliferation, migration and osteogenic differentiation of mesenchymal stem cells: a novel cytokine that could contribute to new bone formation in spondyloarthropathies. Rheumatology 56, 488–493 (2017).
Zhao, S. S., Hu, J. W., Wang, J., Lou, X. J. & Zhou, L. L. Inverse correlation between CD4+ CD25high CD127low/- regulatory T-cells and serum immunoglobulin A in patients with new-onset ankylosing spondylitis. J. Int. Med. Res. 39, 1968–1974 (2011).
Liao, H. T., Lin, Y. F., Tsai, C. Y. & Chou, C. T. Regulatory T cells in ankylosing spondylitis and the response after adalimumab treatment. Jt. Bone Spine 82, 423–427 (2015).
Zhang, L., Jarvis, L. B., Baek, H. J. & Gaston, J. S. Regulatory IL4+CD8+ T cells in patients with ankylosing spondylitis and healthy controls. Ann. Rheum. Dis. 68, 1345–1351 (2009).
Gao, X. M., Wordsworth, P. & McMichael, A. Collagen-specific cytotoxic T lymphocyte responses in patients with ankylosing spondylitis and reactive arthritis. Eur. J. Immunol. 24, 1665–1670 (2010).
Ugrinovic, S., Mertz, A., Wu, P., Braun, J. & Sieper, J. A single nonamer from the Yersinia 60-kDa heat shock protein is the target of HLA-B27-restricted CTL response in Yersinia-induced reactive arthritis. J. Immunol. 159, 5715–5723 (1997).
Fiorillo, M. T., Maragno, M., Butler, R., Dupuis, M. L. & Sorrentino, R. CD8+ T-cell autoreactivity to an HLA-B27–restricted self-epitope correlates with ankylosing spondylitis. J. Clin. Invest 106, 47–53 (2000).
Yang, L. et al. JAK2 inhibitor combined with DC-activated AFP-specific T-cells enhances antitumor function in a Fas/FasL signal-independent pathway. Onco Targets Ther. 9, 4425–4433 (2016).
Akane, K., Kojima, S., Mak, T., Shiku, H. & Suzuki, H. CD8+CD122+CD49dlow regulatory T cells maintain T-cell homeostasis by killing activated T cells via Fas/FasL-mediated cytotoxicity. Proc. Natl Acad. Sci. USA 113, 2460–2465 (2016).
Eghtedari, A. A., Davis, P. & Bacon, P. A. Immunological reactivity in ankylosing spondylitis. Circulating immunoblasts, autoantibodies, and immunoglobulins. Ann. Rheum. Dis. 35, 155–157 (1976).
Lin, Q. et al. Value of the peripheral blood B-cells subsets in patients with ankylosing spondylitis. Chin. Med. J. 122, 1784–1789 (2009).
Quaden, D. H., de Winter, L. M. & Somers, V. Detection of novel diagnostic antibodies in ankylosing spondylitis: an overview. Autoimmun. Rev. 15, 820–832 (2016).
van der Heijde, D. et al. Tofacitinib in patients with ankylosing spondylitis: a phase II, 16-week, randomised, placebo-controlled, dose-ranging study. Ann. Rheum. Dis. 76, 1340–1347 (2017).
van der Heijde, D. et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): results from a randomised, placebo-controlled, phase 2 trial. Lancet (Lond., Engl.) 392, 2378–2387 (2018).
Wendling, D. et al. 2018 update of French Society for Rheumatology (SFR) recommendations about the everyday management of patients with spondyloarthritis. Jt. Bone Spine 85, 275–284 (2018).
Torre-Alonso, J. C. et al. Identification and management of comorbidity in psoriatic arthritis: evidence- and expert-based recommendations from a multidisciplinary panel from Spain. Rheumatol. Int. 37, 1239–1248 (2017).
Bodur, H. et al. Turkish league against rheumatism consensus report: recommendations for management of axial spondyloarthritis. Arch. Rheumatol. 33, 1–16 (2018).
Rohekar, S. et al. 2014 Update of the Canadian rheumatology association/spondyloarthritis research consortium of Canada treatment recommendations for the management of spondyloarthritis. Part II: specific management recommendations. J. Rheuma. 42, 665–681 (2015).
Rohekar, S. et al. 2014 update of the Canadian rheumatology association/spondyloarthritis research consortium of Canada treatment recommendations for the management of spondyloarthritis. Part I: principles of the management of spondyloarthritis in Canada. J. Rheuma. 42, 654–664 (2015).
National Institute for Health and Care Excellence. Spondyloarthritis in Over 16s: Diagnosis and Management. (National Institute for Health and Care Excellence, UK, 2017).
Ward, M. M. et al. American college of rheumatology/spondylitis association of america/spondyloarthritis research and treatment network 2015 recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Rheuma. 68, 282–298 (2016).
van der Heijde, D. et al. 2016 update of the ASAS-EULAR management recommendations for axial spondyloarthritis. Ann. Rheum. Dis. 76, 978–991 (2017).
Dagfinrud, H., Kvien, T. K. & Hagen, K. B. The cochrane review of physiotherapy interventions for ankylosing spondylitis. J. Rheuma. 32, 1899–1906 (2005).
Dulger, S. et al. How does smoking cessation affect disease activity, function loss, and quality of life in smokers with ankylosing spondylitis? J. Clin. Rheumatol. https://doi.org/10.1097/RHU.0000000000000851 (2018).
Smolen, J. S. et al. Treating axial spondyloarthritis and peripheral spondyloarthritis, especially psoriatic arthritis, to target: 2017 update of recommendations by an international task force. Ann. Rheum. Dis. 77, 3–17 (2018).
Sieper, J. et al. Effect of continuous versus on-demand treatment of ankylosing spondylitis with diclofenac over 2 years on radiographic progression of the spine: results from a randomised multicentre trial (ENRADAS). Ann. Rheum. Dis. 75, 1438–1443 (2016).
Kroon, F., Landewe, R., Dougados, M. & van der Heijde, D. Continuous NSAID use reverts the effects of inflammation on radiographic progression in patients with ankylosing spondylitis. Ann. Rheum. Dis. 71, 1623–1629 (2012).
Wanders, A. et al. Nonsteroidal antiinflammatory drugs reduce radiographic progression in patients with ankylosing spondylitis: a randomized clinical trial. Arthritis Rheum. 52, 1756–1765 (2005).
Sieper, J. et al. Efficacy and safety of infliximab plus naproxen versus naproxen alone in patients with early, active axial spondyloarthritis: results from the double-blind, placebo-controlled INFAST study, Part 1. Ann. Rheum. Dis. 73, 101–107 (2014).
Gao, X. et al. Clinical and economic burden of extra-articular manifestations in ankylosing spondylitis patients treated with anti-tumor necrosis factor agents. J. Med. Econ. 15, 1054–1063 (2012).
Cobo-Ibanez, T., Ordonez, M. D. C., Munoz-Fernandez, S., Madero-Prado, R. & Martin-Mola, E. Do TNF-blockers reduce or induce uveitis? Rheumatology 47, 731–732 (2008).
Deodhar, A. & Yu, D. Switching tumor necrosis factor inhibitors in the treatment of axial spondyloarthritis. Semin. Arthritis Rheu. 47, 343–350 (2017).
Hebeisen, M. et al. Response to tumor necrosis factor inhibition in male and female patients with ankylosing spondylitis: data from a swiss cohort. J. Rheumatol. 45, 506–512 (2018).
Lie, E. et al. Effectiveness of switching between TNF inhibitors in ankylosing spondylitis: data from the NOR-DMARD register. Ann. Rheum. Dis. 70, 157–163 (2011).
Cantini, F., Niccoli, L., Cassara, E., Kaloudi, O. & Nannini, C. Duration of remission after halving of the etanercept dose in patients with ankylosing spondylitis: a randomized, prospective, long-term, follow-up study. Biologics 7, 1–6 (2013).
Haibel, H. et al. Efficacy of oral prednisolone in active ankylosing spondylitis: results of a double-blind, randomised, placebo-controlled short-term trial. Ann. Rheum. Dis. 73, 243–246 (2014).
Song, I. H. et al. Different response to rituximab in tumor necrosis factor blocker-naive patients with active ankylosing spondylitis and in patients in whom tumor necrosis factor blockers have failed: a twenty-four-week clinical trial. Arthritis Rheum. 62, 1290–1297 (2010).
McInnes, I. B. et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet 382, 780–789 (2013).
Kavanaugh, A. et al. Effect of ustekinumab on physical function and health-related quality of life in patients with psoriatic arthritis: a randomized, placebo-controlled, phase II trial. Curr. Med. Res. Opin. 26, 2385–2392 (2010).
Denis, P., Hermann, K. G. A., Johanna, C., Joachim, L. & Joachim, S. Ustekinumab for the treatment of patients with active ankylosing spondylitis: results of a 28-week, prospective, open-label, proof-of-concept study (TOPAS). Ann. Rheum. Dis. 73, 817–823 (2014).
Deodhar, A. et al. Three multicenter, randomized, double-blind, placebo-controlled studies evaluating the efficacy and safety of ustekinumab in axial spondyloarthritis. Arthritis Rheuma. 71, 258–270 (2019).
Baeten, D. et al. Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann. Rheum. Dis. 77, 1295–1302 (2018).
Siebert, S., Millar, N. L. & Mcinnes, I. B. Why did IL-23p19 inhibition fail in AS: a tale of tissues, trials or translation?. Ann. Rheum. Dis. 78, 1015–1018, https://doi.org/10.1136/annrheumdis-2018-213654 (2019).
Tok, M. N. V. et al. The initiation, but not the persistence, of experimental spondyloarthritis is dependent on interleukin-23 signaling. Front. Immunol. 9, 1550 (2018).
Baeten, D. et al. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N. Engl. J. Med. 373, 2534–2548 (2015).
Braun, J. et al. Effect of secukinumab on clinical and radiographic outcomes in ankylosing spondylitis: 2-year results from the randomised phase III MEASURE 1 study. Ann. Rheum. Dis. 76, 1070–1077 (2017).
Baraliakos, X. et al. Long-term effects of interleukin-17A inhibition with secukinumab in active ankylosing spondylitis: 3-year efficacy and safety results from an extension of the Phase 3 MEASURE 1 trial. Clin. Exp. Rheum. 36, 50–55 (2017).
Wei, C. C. et al. Efficacy and safety of secukinumab in Asian patients with active ankylosing spondylitis: 52-week pooled results from two phase 3 studies. Int. J. Rheum. Dis. 20, 589–596 (2017).
Baraliakos, X. et al. Safety and efficacy of readministration of infliximab after longterm continuous therapy and withdrawal in patients with ankylosing spondylitis. J. Rheuma. 34, 510–515 (2007).
Ravindran, V., Scott, D. L. & Choy, E. H. A systematic review and meta-analysis of efficacy and toxicity of disease modifying anti-rheumatic drugs and biological agents for psoriatic arthritis. Ann. Rheum. Dis. 67, 855–859 (2008).
Kubiak, E. N., Moskovich, R., Errico, T. J. & Di Cesare, P. E. Orthopaedic management of ankylosing spondylitis. J. Am. Acad. Orthop. Surg. 13, 267–278 (2005).
van Royen, B. J. & de Gast, A. Lumbar osteotomy for correction of thoracolumbar kyphotic deformity in ankylosing spondylitis. A structured review of three methods of treatment. Ann. Rheum. Dis. 58, 399–406 (1999).
Allouch, H., Shousha, M. & Böhm, H. Operationen bei ankylosierender spondylitis (morbus bechterew). Z. Rheumatol. 76, 848–859 (2018).
Qian, B. P., Mao, S. H., Jiang, J., Wang, B. & Qiu, Y. Mechanisms, predisposing factors, and prognosis of intraoperative vertebral subluxation during pedicle subtraction osteotomy in surgical correction of thoracolumbar kyphosis secondary to ankylosing spondylitis. Spine 42, E983–E990 (2016).
He, A. et al. One-stage surgical treatment of cervical spine fracture-dislocation in patients with ankylosing spondylitis via the combined anterior-posterior approach. Medicine 96, e7432 (2017).
Ji, M. et al. Change in abdominal morphology after surgical correction of thoracolumbar kyphosis secondary to ankylosing spondylitis: a computed tomographic study. Spine 40, 1244–1249 (2015).
Lin, B., Zhang, B., Li, Z. & Li, Q. Corrective surgery for deformity of the upper cervical spine due to ankylosing spondylitis. Indian J. Orthop. 48, 211–215 (2014).
Kiaer, T. & Gehrchen, M. Transpedicular closed wedge osteotomy in ankylosing spondylitis: results of surgical treatment and prospective outcome analysis. Eur. Spine J. 19, 57–64 (2010).
Etame, A. B., Than, K. D., Wang, A. C., La, M. F. & Park, P. Surgical management of symptomatic cervical or cervicothoracic kyphosis due to ankylosing spondylitis. Spine 33, 559–564 (2008).
Koller, H., Mayer, M., Hempfing, A. & Koller, J. Osteotomies in ankylosing spondylitis: where, how many, and how much? Eur. Spine J. 27, 70–100 (2018).
Smith-Petersen, M., Larson, C. & Aufranc, O. Osteotomy of the spine for correction of flexion deformity in rheumatoid arthritis. Clin. Orthop. Relat. Res. 27, 6–9 (1969).
La Chapelle, E. H. Osteotomy of the lumbar spine for correction of kyphosis in a case of ankylosing spondylarthritis. J. Bone Jt. Surg. Am. 28, 851–858 (1946).
Goel, M. K. Vertebral osteotomy for correction of fixed flexion deformity of the spine. J. Bone Jt. Surg. Am. 50, 287–294 (1968).
Klems, H. & Friedebold, G. Rupture of the abdominal aorta following a corrective spinal operation for ankylopoeitic spondylitis. Z. Orthop. Ihre Grenzgeb. 108, 554–563 (1971).
Weatherley, C., Jaffray, D. & Terry, A. Vascular complications associated with osteotomy in ankylosing spondylitis: a report of two cases. Spine 13, 43–46 (1988).
Wilson, M. J. & Turkell, J. H. Multiple spinal wedge osteotomy its use in a case of Marie-Strumpell spondylitis. Am. J. Surg. 77, 777–782 (1949).
Chen, P. Q. Correction of kyphotic deformity in ankylosing spondylitis using multiple spinal osteotomy and Zielke's VDS instruments. Taiwan Yi Xue Hui Za Zhi. 87, 692–699 (1988).
Hehne, H. J., Becker, H. J. & Zielke, K. Spondylodiscitis in kyphotic deformity of ankylosing spondylitis and its healing affected by dorsal correction osteotomies. Report of 33 patients. Z. Orthop. Ihre Grenzgeb. 128, 494–502 (1990).
Simmons, E. H. The surgical correction of flexion deformity of the cervical spine in ankylosing spondylitis. Clin Orthop Relat Res. 86, 132–143, https://doi.org/10.1097/00003086-197207000-00019 (1972).
Püschel, J. & Zielke, K. Corrective surgery for kyphosis in bekhterev's disease—indication, technique, results (author's transl). Z. Orthop. Ihre Grenzgeb. 120, 338–342 (1982).
van Royen, B. J., de Kleuver, M. & Slot, G. H. Polysegmental lumbar posterior wedge osteotomies for correction of kyphosis in ankylosing spondylitis. Eur. Spine J. 7, 104–110 (1998).
Hehne, H. J., Zielke, K. & Bohm, H. Polysegmental lumbar osteotomies and transpedicled fixation for correction of long-curved kyphotic deformities in ankylosing spondylitis. Rep. 177 cases. Clin. Orthop. Relat. Res. 258, 49–55 (1990).
Halm, H., Metz-Stavenhagen, P., Schmitt, A. & Zielke, K. Surgical treatment of kyphotic spinal deformities in ankylosing spondylitis using the Harrington compression system: long-term results based on the MOPO scales in the framework of a retrospective questionnaire. Z. Orthop. Ihre Grenzgeb. 133, 141–147 (1995).
Scudese, V. A. & Calabro, J. J. Vertebral wedge osteotomy. Correction of rheumatoid (ankylosing) spondylitis. JAMA 186, 627–631 (1963).
Ziwjan, J. L. The treatment of flexion deformities of the spine in Bechterew disease. Beitr. Orthop. Traumatol. 29, 195–199 (1982).
Thomasen, E. Vertebral osteotomy for correction of kyphosis in ankylosing spondylitis. Clin. Orthop. Relat. Res. 194, 142–152 (1985).
Thiranont, N. & Netrawichien, P. Transpedicular decancellation closed wedge vertebral osteotomy for treatment of fixed flexion deformity of spine in ankylosing spondylitis. Spine 18, 2517–2522 (1993).
Kawahara, N. et al. Closing-opening wedge osteotomy to correct angular kyphotic deformity by a single posterior approach. Spine 26, 391–402 (2007).
Bourghli, A. et al. Modified closing-opening wedge osteotomy for the treatment of sagittal malalignment in thoracolumbar fractures malunion. Spine J. 15, 2574–2582 (2015).
Finkelstein, J. A., Chapman, J. R. & Mirza, S. Occult vertebral fractures in ankylosing spondylitis. Spinal Cord. 37, 444–447 (1999).
Whang, P. G. et al. The management of spinal injuries in patients with ankylosing spondylitis or diffuse idiopathic skeletal hyperostosis: a comparison of treatment methods and clinical outcomes. J. Spinal Disord. Tech. 22, 77–85 (2009).
Hunter, T., Forster, B. & Dvorak, M. Ankylosed spines are prone to fracture. Can. Fam. Physician 41, 1213–1216 (1995).
Daveyranasinghe, N. & Deodhar, A. Osteoporosis and vertebral fractures in ankylosing spondylitis. Curr. Opin. Rheumatol. 25, 509–516 (2013).
Hitchon, P. W., From, A. M., Brenton, M. D., Glaser, J. A. & Torner, J. C. Fractures of the thoracolumbar spine complicating ankylosing spondylitis. J. Neurosurg. 97, 218–222 (2002).
Mitra, D., Elvins, D. M., Speden, D. J. & Collins, A. J. The prevalence of vertebral fractures in mild ankylosing spondylitis and their relationship to bone mineral density. Rheumatology 39, 85–89 (2000).
Backhaus, M. et al. Spine fractures in patients with ankylosing spondylitis: an analysis of 129 fractures after surgical treatment. Orthopade 40, 917–920 (2011). 922–914.
Bernd, L., Blasius, K. & Lukoschek, M. Spinal fractures in ankylosing spondylitis. Z. Orthop. Ihre Grenzgeb. 130, 59–63 (1992).
Schiefer, T. K. et al. In-hospital neurologic deterioration following fractures of the ankylosed spine: a single-institution experience. World Neurosurg. 83, 775–783 (2015).
Ma, J., Wang, C., Zhou, X., Zhou, S. & Jia, L. Surgical therapy of cervical spine fracture in patients with ankylosing spondylitis. Medicine 94, e1663 (2015).
An, S. B. et al. Surgical outcomes after traumatic vertebral fractures in patients with ankylosing spondylitis. J. Korean Neurosurg. Soc. 56, 108–113 (2014).
Apple, D. F. & Anson, C. Spinal cord injury occurring in patients with ankylosing spondylitis: a multicenter study. Orthopedics 18, 1005–1011 (1995).
Ea, H. K., Liote, F., Lot, G. & Bardin, T. Cauda equina syndrome in ankylosing spondylitis: successful treatment with lumboperitoneal shunting. Spine 35, E1423–E1429 (2010).
Ahn, N. U. et al. Cauda equina syndrome in ankylosing spondylitis (the CES-AS syndrome): meta-analysis of outcomes after medical and surgical treatments. J. Spinal Disord. 14, 427–433 (2001).
Vander, C. B. et al. Hip involvement in ankylosing spondylitis: epidemiology and risk factors associated with hip replacement surgery. Rheumatology 49, 73–81 (2010).
Bloom, L. et al. Have the yearly trends of total hip arthroplasty in ankylosing spondylitis patients decreased? Surg. Technol. Int. 31, 327–332 (2017).
Xu, J., Zeng, M., Xie, J., Wen, T. & Hu, Y. Cementless total hip arthroplasty in patients with ankylosing spondylitis: a retrospective observational study. Medicine 96, e5813 (2017).
He, C. et al. The effect of total hip replacement on employment in patients with ankylosing spondylitis. Clin. Rheuma. 35, 2975–2981 (2016).
Feng, D. X. et al. Bilaterally primary cementless total hip arthroplasty for severe hip ankylosis with ankylosing spondylitis. Orthop. Surg. 8, 352–359 (2016).
Ye, C. et al. Cementless bilateral synchronous total hip arthroplasty in ankylosing spondylitis with hip ankylosis. Int. Orthop. 38, 2473–2476 (2014).
Xu, B. G. et al. Medium-term follow-up outcomes of total hip arthroplasty for patients with ankylosing spondylitis. Zhongguo Gu Shang 26, 1052–1056 (2013).
Joshi, A. B., Markovic, L., Hardinge, K. & Murphy, J. C. Total hip arthroplasty in ankylosing spondylitis: an analysis of 181 hips. J. Arthroplast. 17, 427–433 (2002).
Bisla, R. S., Ranawat, C. S. & Inglis, A. E. Total hip replacement in patients with ankylosing spondylitis with involvement of the hip. J. Bone Jt. Surg. Am. 58, 233–238 (1976).
Tang, W. M. & Chiu, K. Y. Primary total hip arthroplasty in patients with ankylosing spondylitis. J. Arthroplast. 15, 52–58 (2000).
Bangjian, H., Peijian, T. & Ju, L. Bilateral synchronous total hip arthroplasty for ankylosed hips. Int. Orthop. 36, 697–701 (2012).
Zheng, G. Q., Zhang, Y. G., Chen, J. Y. & Wang, Y. Decision making regarding spinal osteotomy and total hip replacement for ankylosing spondylitis: experience with 28 patients. Bone Jt. J. 96-B, 360–365 (2014).
Kim, K. T. et al. Surgical treatment of "chin-on-pubis" deformity in a patient with ankylosing spondylitis: a case report of consecutive cervical, thoracic, and lumbar corrective osteotomies. Spine 37, E1017–E1021 (2012).
Yang, P., Wang, C. & Wang, K. Effect of morphological changes in proximal femur on prosthesis selection of total hip arthroplasty in patients with ankylosing spondylitis. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 20, 448–450 (2006).
Bhan, S., Eachempati, K. K. & Malhotra, R. Primary cementless total hip arthroplasty for bony ankylosis in patients with ankylosing spondylitis. J. Arthroplast. 23, 859–866 (2008).
Lee, S. H., Lee, G. W., Seol, Y. J., Park, K. S. & Yoon, T. R. Comparison of outcomes of total hip arthroplasty between patients with ankylosing spondylitis and avascular necrosis of the femoral head. Clin. Orthop. Surg. 9, 263–269 (2017).
Wang, W., Huang, G., Huang, T. & Wu, R. Bilaterally primary cementless total hip arthroplasty in patients with ankylosing spondylitis. BMC Musculoskelet. Disord. 15, 344 (2014).
Schmalzried, T. P., Amstutz, H. C. & Dorey, F. J. Nerve palsy associated with total hip replacement. Risk factors and prognosis. J. Bone Jt. Surg. Am. 73, 1074–1080 (1991).
Brinker, M. R., Rosenberg, A. G., Kull, L. & Cox, D. D. Primary noncemented total hip arthroplasty in patients with ankylosing spondylitis. Clinical and radiographic results at an average follow-up period of 6 years. J. Arthroplast. 11, 802–812 (1996).
Weng, H. K. et al. Total hip arthroplasty for patients who have ankylosing spondylitis: is postoperative irradiation required for prophylaxis of heterotopic ossification? J. Arthroplast. 30, 1752–1756 (2015).
Boonen, A. Socioeconomic consequences of ankylosing spondylitis. Clin. Exp. Rheumatol. 20, S23–S26 (2002).
Acknowledgements
This work was supported by the Beijing Natural Science Foundation youth project (7184325) and the China Postdoctoral Foundation NO.62 general program.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, W., He, X., Cheng, K. et al. Ankylosing spondylitis: etiology, pathogenesis, and treatments. Bone Res 7, 22 (2019). https://doi.org/10.1038/s41413-019-0057-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-019-0057-8
This article is cited by
-
Cost Effectiveness of Tofacitinib for the Treatment of Active Ankylosing Spondylitis in Greece
Clinical Drug Investigation (2024)
-
“Long-term MRI findings in Ankylosing spondylitis patients treated with TNF inhibitors for a decade”
Rheumatology International (2024)
-
Role of endoplasmic reticulum aminopeptidase-1 gene polymorphism (rs13167972) in occurrence susceptibility of ankylosing spondylitis in a sample of Iraqi male patients
Molecular Biology Reports (2024)
-
The oral microbiome in autoimmune diseases: friend or foe?
Journal of Translational Medicine (2023)
-
RNA sequencing and bioinformatics analysis of differentially expressed genes in the peripheral serum of ankylosing spondylitis patients
Journal of Orthopaedic Surgery and Research (2023)
