
Abstract
Carboxyl terminus of Hsp70-interacting protein (CHIP or STUB1) is an E3 ligase and regulates the stability of several proteins which are involved in different cellular functions. Our previous studies demonstrated that Chip deficient mice display bone loss phenotype due to increased osteoclast formation through enhancing TRAF6 activity in osteoclasts. In this study we provide novel evidence about the function of CHIP. We found that osteoblast differentiation and bone formation were also decreased in Chip KO mice. In bone marrow stromal (BMS) cells derived from Chip−/− mice, expression of a panel of osteoblast marker genes was significantly decreased. ALP activity and mineralized bone matrix formation were also reduced in Chip-deficient BMS cells. We also found that in addition to the regulation of TRAF6, CHIP also inhibits TNFα-induced NF-κB signaling through promoting TRAF2 and TRAF5 degradation. Specific deletion of Chip in BMS cells downregulated expression of osteoblast marker genes which could be reversed by the addition of NF-κB inhibitor. These results demonstrate that the osteopenic phenotype observed in Chip−/− mice was due to the combination of increased osteoclast formation and decreased osteoblast differentiation. Taken together, our findings indicate a significant role of CHIP in bone remodeling.
Similar content being viewed by others


Introduction
Bone remodeling is a dynamic process, involving a balance between the activity of bone-forming osteoblasts, derived from mesenchymal progenitor cells, and the activity of bone-resorbing osteoclasts, derived from hematopoietic stem cells.1 A disruption of this balance leads to many bone and joint diseases, especially metabolic bone diseases. Although the coupling of bone resorption and bone formation has long been recognized, the mechanism mediating this fundamental process in skeletal homeostasis is not fully understood. The understanding of the regulatory mechanisms of osteoclast formation and osteoblast function is critical for the rational drug design for the treatment of metabolic bone disorders.
Osteoclasts are derived from hematopoietic stem cells which undergo proliferation and differentiation to become multinucleated bone-resorbing osteoclasts.2 The process of osteoclastogenesis is regulated by several cytokines such as RANKL and TNF-α,3 which activate NF-κB signaling.2 TNF-α receptor-associated factors (TRAFs) are intracellular adapter proteins, which play critical roles in NF-κB signaling.4,5 In TRAF family members, TRAF2, TRAF5, and TRAF6 have been shown to activate NF-κB signaling.6 TRAF2 and TRAF5 interact with TNF-α receptors and mediate TNF-α-induced osteoclast formation. In Traf2 deficient cells, TNF-α-induced osteoclast formation was severely impaired.7 Similar results were also found in Traf5-deficient bone marrow stromal (BMS) cells.8 Therefore, both TRAF2 and TRAF5 are essential for TNF-α-induced osteoclast formation.7,8 Moreover, TRAF6 interacts with RANK to activate NF-κB signaling which in turn regulates osteoclast formation.2,9,10,11 Multiple TRAF family members, including TRAFs 2, 3, 5, and 6, have been demonstrated to play an important role in regulation of NF-κB signaling.4,5,6,7,8,9,10,11 In previous studies, we examined the effect of CHIP on TRAF6 protein stability.12 In the present studies we further examined the effects of CHIP on protein stability of TRAFs 2, 3, and 5. We found that in addition to TRAF6, CHIP also induced the degradation of TRAF2 and TRAF5, but not TRAF3.
Osteoblast is a cell type derived from mesenchymal progenitor cells and is responsible for bone formation. Osteoclast and osteoblast are two major cell types involved in bone remodeling in trabecular bone. Several previous studies have demonstrated that NF-κB signaling plays critical roles in regulation of osteoclast and osteoblast activities in bone. Activation of NF-κB signaling promotes osteoclast formation13,14,15,16 and inhibits osteoblast function.17,18,19,20,21,22
CHIP is an E3 ligase, which has been reported to promote ubiquitination and degradation of several substrates involving in different signaling pathways.23,24,25 In vivo findings demonstrated that Chip deficiency causes increased sensitivity to stress associated hyperthermia,26 leading to a markedly reduced life span in mice.27 In addition, Chip−/− mice developed severe defects in heart and neural tissues.28,29 Moreover, Chip−/− mice exhibit defects in motor sensory, cognitive, and reproductive function.30 Although the mechanisms of NF-κB regulation during bone remodeling have been extensively investigated, the roles of CHIP in regulation of NF-κB signaling and osteoblast function has not been demonstrated. Our previous study demonstrated that Chip−/− mice develop osteopenic phenotype due to increased osteoclast formation.12 In the present studies, we investigated the roles of CHIP in regulation of protein stability of multiple TRAF family members, NF-κB signaling, and osteoblast function. Our findings provide new insights into the role of CHIP in regulation of osteoclast and osteoblast functions.
Results
Bone mass is decreased in Chip −/− mice
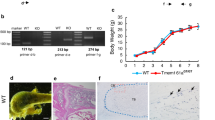
To determine the contribution of CHIP in skeletal development, we performed whole skeletal Alizarin red/Alcian blue staining using postnatal 3-day-old wild-type (WT) and ChipKO mice. The sizes of the skeleton of Chip−/− mice were much smaller than those of WT littermates and Chipheterozygous KO mice (Chip+/-) (Fig. 1a). We also analyzed the appearance of 1-month-old Chip−/− mice. The sizes of Chip−/− mice were also smaller than WT and Chip+/- KO mice (Fig. 1b). The X-ray radiographic analysis showed that the length of the spine was shorter and bone density of vertebral bone was reduced in 1-month-old Chip−/− mice compared to WT and Chip+/- KO mice (Fig. 1c). To further confirm changes in the spine of Chip−/− mice, Safranin O/Fast green staining was performed on vertebral sections. The results showed that vertebral bone volume in spine was significantly reduced in 1-month-old Chip−/− mice compared to WT littermates (Fig. 1d). These results indicate that Chip deficiency in mice results in a bone loss phenotype.

Bone mass is decreased in ChipKO mice. a The skeleton of 3-day-old newborn mice was stained with Alizarin red and Alcian blue. The size of Chip−/− mice was significantly smaller than those of WT and Chip+/- mice. b The picture showed the appearance of 1-month-old Chip KO mice (Chip−/− and Chip+/-) and WT littermate. The size of Chip−/− mice was smaller than that of WT and Chip+/- mice. c X-ray radiographic analysis showed that bone density of the spine of 1-month-old Chip−/− mice was lower compared to that of WT and Chip+/- mice (indicated by red arrowhead). d Representative images of Safranin O/fast green stained sections of vertebrae of 1-month-old Chip−/− mouse and its WT littermate showing reduced bone mass in vertebral bone of Chip−/− mice (indicated by yellow arrowhead).
Alterations of osteoclast formation and osteoblast function in Chip −/− mice
The osteopenic phenotype observed in Chip−/− mice could be due to either increased bone resorption or reduced bone formation. To determine the effect of Chip deficiency on bone formation, bone histology and the calcein double labeling assays were performed. The results showed that the bone volume and bone formation rates were significantly reduced in Chip−/− mice (Fig. 2a-d). In addition, we also found that osteoblast numbers were significantly reduced in Chip−/− mice (Fig. 2e). To determine the role of CHIP in bone resorption, we performed TRAP staining using histological sections of tibiae from 1-month-old WT and Chip−/− mice. The data revealed that TRAP positive osteoclast numbers were significantly increased in Chip−/− mice (Fig. 2f, g). These results suggest that the bone loss phenotype observed in Chip−/− mice could be due to the combination of increased bone resorption and decreased bone formation. To further determine changes in osteoblast differentiation, we isolated BMS cells from 1-month-old WT and Chip−/− mice, and examined Ki67 expression by real-time PCR and performed alkaline phosphatase (ALP) and Alizarin red staining assays. The results showed that Ki67 expression was significantly reduced and ALP activity and mineralized bone matrix formation were dramatically decreased in Chip-deficient BMS cells compared to BMS cells isolated from WT mice (Fig. 2h, i). These results further demonstrated that osteoblast function was reduced in Chip−/− mice.

Reduction in bone formation and osteoblast function in Chip−/− mice. a, b Representative images of Alcian blue/H&E Orange G stained tibiae sections from 1-month-old WT and Chip−/− mice showed reduced bone volume in long bone (indicated by yellow arrowhead). b The quantification of bone volume was performed. c, d Calcein double labeling of trabecular bones in WT and Chip−/− mice that were injected with calcein 4 and 1 day before the mice were sacrificed. Pictures of trabecular bone in tibiae were taken under the fluorescence microscope. Chip−/− mice showed a decrease in bone formation rates (d). e Results of histomorphometric analysis also showed that osteoblast numbers were also significantly reduced in Chip−/− mice. f, g TRAP staining was performed using tibial sections of 1-month-old WT and Chip−/− mice. TRAP-positive cells were indicated by blue arrowheads. The numbers of TRAP-positive osteoclasts were higher in Chip−/− mice (g). h, i Bone marrow stromal (BMS) cells were isolated from 1-month-old WT and Chip−/− mice, and were cultured with ascorbic acid and β-glycerophosphate for 3 days for Ki67 expression (h) and ALP staining (i). The BMS cells were also cultured with osteoblast differentiation medium in the presence of BMP-2 (50 ng·mL-1) for 2 weeks and Alizarin red staining was performed (i). Ki67 expression, ALP activity, and mineralized bone matrix formation were significant decreased in Chip−/− mice. *P < 0.05, **P < 0.01, unpaired Student’s t-test, n = 6.
CHIP regulates protein stability of multiple TRAF family members
Our previous study showed that CHIP promotes TRAF6 degradation and inhibits RANKL-induced NF-κB signaling.12 To further determine the mechanism by which CHIP regulates TRAF6 signaling, we performed a series of in vitro experiments to determine if CHIP affects TRAF6 interaction with IKK using 293 T cells. We found that CHIP directly interacts with TRAF6 (Fig. 3a). IL-1β enhanced CHIP/TRAF6 interaction (Fig. 3b). Expression of CHIP significantly inhibited TRAF6/IKK-β interaction in 293 T cells (Fig. 3c). These results suggest that CHIP may inhibit TRAF6/IKK-β interaction by inducing TRAF6 degradation.

CHIP attenuates IL-1β-induced TRAF6/IKK-β complex formation. a Flag-TRAF6 and Flag-p65 were transfected into 293 T cells with or without Myc-CHIP. The immunoprecipitates were prepared with the anti-Myc antibody followed by the Western blot analysis. The data showed that CHIP interacts with TRAF6, but not p65. b IL-1β enhances CHIP/TRAF6 interaction. 293 T cells were transfected with the Flag-TRAF6 and Myc-CHIP as indicated and treated with IL-1β. The cells lysates were immunoprecipitated with the anti-Myc antibody followed by the Western blot analysis using the anti-Flag antibody. c HA-IKK-β and Flag-TRAF6 were co-transfected with Myc-CHIP into 293 T cells. IP assay was performed using the anti-Flag antibody followed by the Western blot analysis using the HA antibody. The TRAF6/IKK-β interaction was attenuated by CHIP. To prevent TRAF6 degradation, proteasome inhibitor MG-132 was added to the cell culture in the TRAF6/IKK-β co-IP assay.
Given that TRAF2, TRAF3, and TRAF5 belong to the same TRAF family and play critical roles in TNF-α-induced NF-κB signaling, we next examined if CHIP affects stability of TRAF2, TRAF3, and TRAF5. The results of Western blot analysis showed that the protein levels of TRAF2 were decreased when CHIP expression was increased in 293 T cells. In contrast, the mutant CHIP (H260Q), which lacks the E3 ligase ability, failed to affect steady state protein levels of TRAF2 (Fig. 4a). We further examined the half-life of TRAF2 protein by assessing changes in TRAF2 protein levels after treating cells with protein synthesis inhibitor cycloheximide (CHX). The results showed that the half-life of TRAF2 protein was greatly reduced by WT CHIP but not by the mutant CHIP (H260Q) (Fig. 4b). These results indicate that CHIP is a negative regulator of TRAF2 protein. We also examined the proteasome-dependency of CHIP-induced TRAF2 degradation and found that CHIP-mediated TRAF2 degradation could be reversed by the addition of proteasome inhibitor, MG-132 (Fig. 4c), suggesting that CHIP-induced TRAF2 degradation is proteasome-dependent. In addition, protein levels of TRAF5 were also decreased when CHIP expression is increased (Fig. 4d). The IP result demonstrated that TRAF5 was co-precipitated by CHIP in 293 T cells (Fig. 4e). To determine if CHIP-induced TRAF5 degradation is ubiquitin/proteasome-dependent, we treated cells with proteasome inhibitor MG-132. The results showed that CHIP-induced TRAF5 degradation could be reversed by the treatment of MG-132 (Fig. 4f). In contrast, CHIP has no significant effect on TRAF3 degradation (data not shown). Taken together, CHIP regulates protein stability of multiple TRAF family members.

CHIP promotes TRAF2 and TRAF5 degradation. a CHIP promotes T RAF2 degradation. Myc-TRAF2 was co-transfected with increasing concentrations of Myc-CHIP or mutant Myc-CHIP (H260Q) in 293 T cells. 24 h after transfection, the cell lysates were collected and samples were subjected to Western blot analysis. TRAF2 protein levels were decreased when cells were transfected with increasing concentrations of CHIP expression plasmid. b 293 T cells were transfected with TRAF2 and WT or mutant CHIP expression plasmids and treated with cycloheximide (CHX) for different periods of time as indicated. Transfection of WT CHIP, but not mutant CHIP, accelerated TRAF2 degradation. c TRAF2 was transfected with or without CHIP in 293 T cells. Steady state protein levels of TRAF2 were reduced by co-transfection with CHIP. The CHIP-mediated TRAF2 degradation was inhibited by the treatment with proteasome inhibitor MG-132. d Myc-TRAF5 and increasing concentrations Myc-CHIP expression plasmids were co-transfected into 293 T cells. CHIP promoted TRAF5 degradation. e HA-CHIP and Myc-TRAF5 were co-transfected into 293 T cells. Cell lysates were incubated with the anti-HA antibody and immunoprecipitates were then subjected to Western blot analysis. The results showed that TRAF5 interacts with CHIP. f TRAF5 was transfected with or without CHIP in 293 T cells. Steady state protein levels of TRAF5 were reduced by co-transfection with CHIP. The CHIP-mediated TRAF5 degradation was inhibited by the treatment with proteasome inhibitor MG-132.
CHIP inhibits TRAF-mediated NF-κB signaling
Since CHIP regulates TRAF2 and TRAF5 protein levels and TRAF2 and TRAF5 play an important role in TNF-α-induced NF-κB signaling, we then examined whether CHIP affects TNF-α-induced I-κBα phosphorylation and p65 nuclear translocation. The results showed that overexpression of CHIP markedly inhibited the TNF-α-induced I-κBα phosphorylation (Fig. 5a). Notably, the total I-κBα protein levels were increased after CHIP transfection. This is probably because the phosphorylation-induced I-κBα degradation is decreased in CHIP-expressing cells. Next, we performed Western blot analysis using the fractions of the cytoplasm and nuclear proteins and examined changes in p65 localization. The results demonstrated that overexpression of CHIP prevented p65 nuclear translocation when the cells were treated with TNF-α for 40 min (Fig. 5b). These results suggest that CHIP inhibits TNF-α-induced I-κBα phosphorylation. To obtain additional evidence about the role of CHIP in p65 nuclear translocation, we performed immunostaining assay and found that transfection of CHIP inhibited p65 nuclear translocation (Fig. 5c).

CHIP inhibits TRAF2 and TRAF5-mediated NF-κB signaling. a Hela cells stably transfected with HA-CHIP or empty vector were stimulated by TNF-α at indicated time points. The effects of CHIP on endogenous levels of I-κBα and phosphorylated I-κBα were examined. CHIP inhibited TNFα-induced I-κBα phosphorylation. b After treatment with TNF-α in Hela cells stably transfected with HA-CHIP or empty vector, p65 protein levels in the cytoplasm (Cyto) and nucleus were detected. CHIP inhibited p65 nuclear translocation. c 293 T cells were transfected with or without Myc-CHIP. After treatment with IL-1β for 30 min, the cells were stained with the anti-Myc antibody (green) or anti-p65 antibody (red). The nuclear was stained by DAPI (blue). CHIP inhibited p65 nuclear translocation. The white dotted circles in b3 and d3 showed the nuclear region of cells with or without Myc-CHIP transfection. Scale bar: 10 μm. d Luciferase assay was performed in bone marrow stromal (BMS) cells transfected with indicated plasmids and treated with TNF-α for 8 h. CHIP inhibited TNF-α-induced NF-κB signaling. *P < 0.05, **P < 0.01, one-way ANOVA followed by Tukey Post-Hoc test, n = 4, compared between control and TNF-α groups; ##P < 0.01, one-way ANOVA followed by Tukey Post-Hoc test, n = 4, compared between empty vector and CHIP transfection groups. e–g NF-κB reporter construct was transfected into BMS cells with TRAF2, TRAF5 or TRAF6. CHIP inhibited TRAF2, TRAF5 or TRAF6-induced NF-κB signaling. **P < 0.01, one-way ANOVA followed by Tukey Post-Hoc test, n = 4, compared between empty vector and TRAF2, TRAF5 or TRAF6 transfected groups; #P < 0.05, ##P < 0.01, one-way ANOVA followed by Tukey Post-Hoc test, n = 4, compared between TRAF2, TRAF5 or TRAF6 transfection alone or with CHIP co-transfection group.
To further determine the role of CHIP in NF-κB signaling, we performed NF-κB luciferase assay. The NF-κB reporter construct was transfected with HA-CHIP or empty vectors into BMS cells isolated from 1-month-old WT mice treated with or without TNF-α. The results showed that NF-κB reporter activity was significantly increased by the treatment of TNF-α. However, NF-κB activity was significantly inhibited by the transfection of CHIP (Fig. 5d). To determine the role of CHIP in TRAF2, TRAF5, and TRAF6-regulated NF-κB activity, we transfected TRAF2, TRAF5 or TRAF6 expression plasmids with or without CHIP into BMS cells. The result showed that the TRAF2, TRAF5, and TRAF6-induced NF-κB reporter activity was significantly inhibited by CHIP co-expression (Fig. 5e-g). Taken together, these results suggest that CHIP negatively regulates TRAF2, TRAF5, and TRAF6-induced NF-κB activity.
CHIP regulates osteoblast gene expression
In an effort to investigate the role of CHIP in osteoblast differentiation, we first examined changes in levels of several proteins critical for osteoblast differentiation, such as β-catenin, Runx2, and Smad3. Previous in vitro studies have shown that CHIP regulates protein stability of these proteins in different cell types.23,25,31 Surprisingly, the protein levels of these molecules were not significantly changed in Chip-deficient BMS cells (data not shown). We next examined expression of a panel of osteoblast marker genes by the real-time PCR assay. The mRNA levels of Osterix(Osx) and alkaline phosphatase (Alp)were significantly decreased in BMS cells derived from 1-month-old Chip−/− mice (Fig. 6a, b). As a consequence, the expression of early osteoblast differentiation markers, such as type I collagen(Col1) and osteopontin(OPN), were also significantly decreased in Chip-deficient BMS cells (Fig. 6c, d). The expression levels of late stage osteoblast marker genes, such as bone sialoprotein(BSP) and osteocalcin(OC), were also significantly reduced in Chip-deficient BMS cells (Fig. 6e, f). These data suggest that osteoblast differentiation was impaired in Chip−/− mice.

CHIP regulates osteoblast marker gene expression. a–h mRNA levels of osteoblast and osteoclast marker genes were examined in bone marrow stromal (BMS) cells derived from 1-month-old WT and Chip−/− mice. Expression of osteoblast marker genes were significantly decreased in Chip-deficient BMS cells. In contrast, Rankl expression was significantly increased in Chip-deficient BMS cells. *P < 0.05, **P < 0.01, unpaired Student’s t-test, n = 4.
In this study we also examined the mRNA expression of osteoprotegerin (OPG) and Rankl in BMS cells. We found that OPG expression was slightly reduced in Chip-deficient BMS cells (Fig. 6g). In contrast, Rankl expression was significantly increased in Chip-deficient BMS cells (Fig. 6h). These results suggest that reduction in osteoblast differentiation contributes to the bone loss phenotype observed in Chip−/− mice.
To further determine the specific role of CHIP in osteoblast function, we generated Chipflox/flox mice. We isolated BMS cells from 1-month-old Chipflox/flox mice and infected these cells with Ad-CMV-Cre virus. The results showed that in vitro deletion of Chip in BMS cells significantly decreased CHIP protein levels (Fig. 7a) and inhibited expression of osteoblast marker genes, including Osx, Col1a1, OPN, BSP,and OC, in Chip-deficient BMS cells (Fig. 7b-f).

Osteoblast differentiation is inhibited in Chip-deficient BMS cells. Bone marrow stromal (BMS) cells were isolated from 1-month-old Chipflox/flox mice and infected with Ad-CMV-Cre or Ad-CMV-GFP (control virus) and cultured in the presence of osteoblast differentiation medium for 3 days. Expression of CHIP protein was examined by Western blotting (a) and expression of osteoblast marker genes was examined by real-time PCR (b–f). The results showed that expression of osteoblast marker genes, including osterix(Osx),type I collagen(Col1a1), osteopontin(OPN), bone sialoprotein(BSP) and osteocalcin(OC) was significantly decreased in Chip-deficient BMS cells. *P < 0.05, unpaired Student’s t-test, n = 4.
CHIP-regulated osteoblast function is NF-κB-dependent
To determine if Chip-deficiency mediated inhibition of osteoblast differentiation is NF-κB-dependent, we isolated BMS cells from 1-month-old Chipflox/flox mice and infected these cells with Ad-CMV-Cre or Ad-CMV-GFP (control virus) and treated cells with or without NF-κB inhibitor, BP-1-102. We examined changes in expression of several osteoblast marker genes in these cells and found that treatment with NF-κB inhibitor significantly reversed the inhibitory effects on the expression of osteoblast marker genes observed in Chip-deficient BMS cells (Fig. 8). BP-1-102 inhibits nuclear NF-κB retention both in vitro and in vivo, thereby attenuating NF-κB activation.32 These results suggest that CHIP regulates osteoblast differentiation through a NF-κB-dependent mechanism.

Inhibition of NF-κB signaling reverses the inhibitory effects of osteoblast differentiation observed in Chip-deficient BMS cells. Bone marrow stromal (BMS) cells were isolated from 1-month-old Chipflox/flox mice and infected with Ad-CMV-Cre or Ad-CMV-GFP (control virus) and cultured with osteoblast differentiation medium for 3 days in the presence or absence of NF-κB inhibitor, BP-1-102. Expression of osteoblast marker genes, including Osx, Col1a1, BSP,and OC was inhibited in Chip-deficient BMS cells. Addition of NF-κB inhibitor (BP-1-102) reversed inhibitory effects of expression of osteoblast marker genes observed in Chip-deficient BMS cells. *P < 0.05, **P < 0.01, one-way ANOVA followed by Tukey Post-Hoc test, n = 4, compared between WT and Chip-deficient BMS cells. #P < 0.05, one-way ANOVA followed by Tukey Post-Hoc test, n = 4, compared between Chip-deficient BMS cells treated with or without BP-1-102.
Discussion
Bone remodeling is a highly coordinated process involving bone resorption and bone formation. The dynamic process of bone remodeling was impaired in Chip−/− mice, suggesting that CHIP plays a critical role in bone remodeling. Our previous findings showed that the bone loss phenotype observed in Chip−/− mice is due to abnormally increased osteoclast formation and bone resorption. In the present studies we provided novel evidence that in addition to the enhanced osteoclast formation, Chip deficiency in mice also leads to reduced bone formation.
NF-κB and its signaling molecules play critical roles in regulation of osteoclast formation13,14,15,16 and osteoblast function,17,18,19,20,21,22 and TRAF family members are critical mediators in NF-κB signaling.7,8,9,10 Our previous study showed that TRAF6 protein levels are increased in Chip-deficient osteoclasts and we have characterized the effect of CHIP on regulation of TRAF6 protein stability.12 In the present studies, we further investigated the role of CHIP in TRAF6/IKKβ interaction and TRAF2 and TRAF5 protein stability and functions. We found that 1) CHIP interferes with TRAF6/IKKβ interaction; 2) CHIP induces TRAF2 and TRAF5 protein degradation; 3) CHIP inhibits TRAF2 and TRAF5-induced NF-κB signaling. These results suggest that CHIP functions as a critical regulator in NF-κB signaling through regulation of multiple TRAF family members.
Osteoblast/osteoclast cross talk regulates bone formation and bone resorption with remarkable precision. For example, the key regulator for osteoclast bone resorption is RANKL, which expressed by activated osteoblasts.33 Its action on the RANK receptor in osteoclasts is regulated by OPG, a decoy receptor, which is also expressed in osteoblasts.34 In Chip-deficient BMS cells, Rankl expression was significantly increased (Fig. 6j). These results demonstrate that in addition to the direct regulation of bone resorption via promoting TRAF degradation, CHIP also indirectly enhances osteoclast formation via controlling osteoblast/osteoclast cross talk.
Although previous studies demonstrated that CHIP regulates protein stability of β-catenin, Runx2, and Smad3 in vitro,23,25,31 in the present studies, we did not detect significant changes in the steady state protein levels of these molecules, except slight reduction of β-catenin protein levels in Chip-deficient BMS cells (data not shown). It is known that activation of NF-κB signaling inhibits osteoblast function;17,18,19,20,21,22 however, the detail mechanism remains undefined. Recent studies showed that transcriptional enhancers of cell type specific genes could serve as signal integration hubs binding to signal-dependent transcription factors, such as NF-κB, and lineage-determining transcription factors, such as Runx2, to control the gene expression program and determine cell differentiation fate.35,36 This may explain why NF-κB regulates osteoblast function without altering Runx2 protein levels.
It is well known that NF-κB signaling plays an important role in inflammation and in rheumatoid arthritis (RA).37,38 In addition to the inflammation, activation of NF-κB signaling also leads to bone loss by upregulation of osteoclast formation and inhibition of osteoblast function. In the present studies, we demonstrated that CHIP inhibits NF-κB signaling by inducing the degradation of multiple TRAF family members, including TRAFs 2, 5, and 6. In Chip−/− mice osteoclast formation was increased12 and osteoblast differentiation was inhibited, demonstrated by the present studies. These findings suggest that CHIP may play an important role in the development of RA disease. Expression or function of CHIP may be inhibited during RA development, leading to activation of NF-κB signaling and bone loss phenotype in joint tissues due to increased osteoclast formation and reduced osteoblast function. To further determine the specific role of CHIP in joint tissues, we have recently generated Chipflox/flox mice. Using tissue-specific knockout approach, we will further dissect the specific effects of CHIP in specific cell populations in bone and cartilage tissues, including mesenchymal stem cells (MSCs), osteoblast and osteoclast lineage cells, and in synovial cells.
In summary, in this study we demonstrate that bone loss phenotype observed in Chip−/− mice is due to the combination of increased bone resorption and reduced bone formation. Since abnormal increase in osteoclast formation and reduced osteoblast function may lead to bone and joint diseases, our findings suggest that CHIP may serve as a novel target for the drug development to treat bone loss associated diseases.
Materials and methods
Mice
Chip knockout (KO) mice were obtained from NIH. The first three coding exons of the Chip gene were targeted by homologous recombination. Both wild-type (WT) and Chip−/− mice were maintained in a C57BL/6 and 129SvEv background. Chipflox/flox mice were generated in Nanjing Biomedical Research Institute of Nanjing University, Nanjing, China. In these mice the Chip gene was floxed at the flanking sites of exon 1 and exon 3.
Quantitative real-time PCR
Total RNA was extracted from BMS cells which were isolated from 1-month-old Chip−/− mice and WT littermates or BMS cells derived from Chipflox/flox mice infected with Ad-CMV-Cre or Ad-CMV-GFP (control virus). cDNA was synthesized from 1 μg of RNA using the iScript cDNA synthesis kit (Bio-Rad). Real-time PCR analysis was performed using primers for detecting osteoblast and osteoclast marker genes, including osterix (Osx), type I collagen(Col1),alkaline phosphatase(Alp), OPN,osteocalcin(OC), osteoprotegerin(Opg), receptor activator of NF-κB ligand (Rankl), and Ki67.
Luciferase assays
BMS cells were transiently transfected with pGL3/NF-κB-luc reporter and expression plasmids as indicated. The luciferase assay was performed to examine the effect of CHIP in NF-κB signaling.
Histology
Tibiae and vertebrae were harvested and fixed in 10% NB-formalin for 3 days and decalcified for 14 days in 14% EDTA and then paraffin-embedded. Three micrometers sections were cut and Alcian blue/H&E Orange G and Safranin O/Fast green staining and TRAP staining were performed.
Plasmids and reagents
pCMV/Myc-TRAF6, TRAF2, and TRAF5 was provided by Dr. Lingqiang Zhang (State Key Laboratory of Proteomics, Beijing, China). pEFNeo/HA-CHIP, pRKIM/Myc-CHIP, and pRKIM/Myc-CHIP (H260Q) were generated in our lab as described previously.23 I-κBα, phosphor-I-κBα (Ser32) and anti-α-tubulin (3873p) were purchased from Cell Signaling Technology (Beverly, MA). Anti-TRAF6 (H-274) and anti-p65 (C-20) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-CHIP antibody was developed in our lab.16 MG132 was purchased from Millipore (Billerica, MA). Cyclohexanone (CHX) was purchased from Amresco (AMRESCO Inc., OH).
Cell culture
293 T cells and Hela cells were cultured in DMEM supplemented with 10% fetal calf serum (FCS) at 37 °C under 5% CO2. In CHIP/TRAF5 co-IP assay, proteasome inhibitor MG-132 was added (10 μM). BMS cells were isolated from 1-month-old Chipflox/flox mice and cultured with αMEM and 10% FCS. BMS cells were then infected with Ad-CMV-Cre or Ad-CMV-GFP (control virus) in the presence of osteoblast differentiation medium for 3 days.39 Expression of osteoblast marker genes was examined by real-time PCR assay. For mineralized bone matrix formation assay, BMS cells were cultured with osteoblast differentiation medium with BMP-2 (50 ng·mL-1) for 2 weeks. Alizerin red staining was then performed. NF-κB inhibitor, BP-1-102, was obtained from Calbiochem (EMD Millipore, Billerica, MA).
Immunoprecipitation and Western blot analysis
The immunoprecipitation (IP) and Western blot assays were performed as previously described.12,23
Alizarin red staining
BMS cells were isolated from femur and tibia of 1-month-old WT and Chip−/− mice (or Chipflox/flox mice infected with Ad-CMV-Cre or Ad-CMV-GFP), and were cultured with αMEM supplemented with 10% FCS and treated with ascorbic acid (50 μg·mL-1) and β-glycerophosphate (10 mmol·L-1) and BMP-2 (50 ng·mL-1) for 2 weeks. At the end of the cell culture, BMS cells were fixed in 10% formalin and Alizarin red staining was performed.40,41
Calcein labeling
Calcein (20 mg·kg-1, i.p.) labeling was performed 1 and 4 days before 1-month-old WT and Chip−/− mice were sacrificed. At the end of the experiments, tibiae were removed, fixed in 75% ethanol, and embedded in plastic. Transverse sections were cut at 5-μm thickness and unstained sections were viewed under fluorescent microscope.40,42
References
Hadjidakis, D. J. & Androulakis, I. I. Bone remodeling. Ann. N. Y. Acad. Sci. 1092, 385–396 (2006).
Teitelbaum, S. L. & Ross, F. P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet 4, 638–649 (2003).
Teitelbaum, S. L. Osteoclasts; culprits in inflammatory osteolysis. Arthritis Res. Ther. 8, 201 (2006).
Wu, M., Wang, Y., Deng, L., Chen, W. & Li, Y. TRAF family member-associated NF-κB activator (TANK) induced by RANKL negatively regulates osteoclasts survival and function. Int. J. Biol. Sci. 8, 1398–1407 (2012).
Maruyama, K., Kawagoe, T., Kondo, T., Akira, S. & Takeuchi, O. TRAF family member-associated NF-κB activator (TANK) Is a negative regulator of osteoclastogenesis and bone formation. J. Biol. Chem. 287, 29114–29124 (2012).
Darnay, B. G., Haridas, V., Ni, J., Moore, P. A. & Aggarwal, B. B. Characterization of the intracellular domain of receptor activator of NF-kappaB (RANK). Interaction with tumor necrosis factor receptor-associated factors and activation of NF-kappab and c-Jun N-terminal kinase. J. Biol. Chem. 273, 20551–20555 (1998).
Kanazawa, K. & Kudo, A. TRAF2 is essential for TNF-alpha-induced osteoclastogenesis. J. Bone Miner. Res. 20, 840–847 (2005).
Kanazawa, K., Azuma, Y., Nakano, H. & Kudo, A. TRAF5 functions in both RANKL- and TNFα-induced osteoclastogenesis. J. Bone Miner. Res. 18, 443–450 (2003).
Ye, H. et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature 418, 443–447 (2002).
Kadono, Y. et al. Strength of TRAF6 signalling determines osteoclastogenesis. EMBO Rep. 6, 171–176 (2005).
Walsh, M. C. & Choi, Y. Biology of the TRANCE axis. Cytokine Growth Factor Rev. 14, 251–263 (2003).
Li, S. et al. CHIP/STUB1 regulates osteoclast formation through degradation of TRAF6. Arthritis Rheumatol. 66, 1854–1863 (2014).
Franzoso, G. et al. Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 11, 3482–3496 (1997).
Boyce, B. F., Yao, Z. & Xing, L. Functions of NF-κB in bone. Ann. N. Y. Acad. Sci. 1192, 367–375 (2010).
Otero, J. E., Chen, T., Zhang, K. & Abu-Amer, Y. Constitutively active canonical NF-κB pathway induces severe bone loss in mice. PLoS One 7, e38694 (2012).
Abu-Amer, Y. NF-κB signaling and bone resorption. Osteoporos. Int. 24, 2377–2386 (2013).
Krum, S. A., Chang, J., Miranda-Carboni, G. & Wang, C.-Y. Novel functions for NFκB: inhibition of bone formation. Nat. Rev. Rheumatol. 6, 607–611 (2010).
Chang, J. et al. NF-κB inhibits osteogenic differentiation of mesenchymal stem cells by promoting β-catenin degradation. Proc. Natl Acad. Sci. USA 110, 9469–9474 (2013).
Yao, Z. et al. NF-κB RelB negatively regulates osteoclast differentiation and bone formation. J. Bone Miner. Res. 29, 866–877 (2014).
Yu, B. et al. Non-canonical Wnt4 prevents skeletal aging and inflammation by inhibiting NF-κB. Nat. Med. 20, 1009–1017 (2014).
Swarnkar, G., Zhang, K., Mbalaviele, G., Long, F. & Abu-Amer, Y. Constitutive activation of IKK2/NF-κB impairs osteogenesis and skeletal development. PLoS One 9, e91421 (2014).
Pacios, S. et al. Osteoblast lineage cells play an essential role in periodontal bone loss through activation of nuclear factor-Kappa B. Sci. Rep. 5, 16694 (2015).
Li, X. et al. CHIP promotes Runx2 degradation and negatively regulates osteoblast differentiation. J. Cell Biol. 181, 959–972 (2008).
Li, L. et al. CHIP mediates degradation of Smad proteins and potentially regulates Smad-induced transcription. Mol. Cell Biol. 24, 856–864 (2004).
Xin, H. et al. CHIP controls the sensitivity of transforming growth factor-beta signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J. Biol. Chem. 280, 20842–20850 (2005).
Dai, Q. et al. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 22, 5446–5458 (2003).
Min, J. N. et al. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol. Cell Biol. 28, 4018–4025 (2008).
Zhang, C., Xu, Z., He, X. R., Michael, L. H. & Patterson, C. CHIP, a cochaperone/ubiquitin ligase that regulates protein quality control, is required for maximal cardioprotection after myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 288, 2836–2842 (2005).
Dickey, C. A. et al. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J. Neurosci. 26, 6985–6996 (2006).
Shi, C. H. et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum. Mol. Genet. 23, 1013–1024 (2013).
Kajiro, M. et al. The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat. Cell Biol. 11, 312–319 (2009).
Zhang, X. et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc. Natl Acad. Sci. USA 109, 9623–9628 (2012).
Li, J. et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl Acad. Sci. USA 97, 1566–1571 (2000).
Simonet, W. S. et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89, 309–319 (1997).
Trompouki, E. et al. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell 147, 577–589 (2011).
Mullen, A. C. et al. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 147, 565–576 (2011).
Alves, C. H., Farrell, E., Vis, M., Colin, E. M. & Lubberts, E. Animal models of bone loss in inflammatory arthritis: from cytokines in the bench to novel treatments for bone loss in the bedside—a comprehensive review. Clin. Rev. Allergy Immunol. 51, 27–47 (2016).
van Loo, G. & Beyaert, R. Negative regulation of NF-κB and its involvement in rheumatoid arthritis. Arthritis Res. Ther. 13, 221 (2011).
Oh, C.-D. et al. The rock inhibitor immortalizes rat nucleus pulposus and annulus fibrosus cells: establishment of intervertebral disc cell lines with novel approaches. Spine (Phila Pa 1976) 41, 255–261 (2016).
Yan, Y. et al. Axin2 controls bone remodeling through the beta-catenin-BMP signaling pathway in adult mice. J. Cell Sci. 122(Pt 19), 3566–3578 (2009).
Huang, J., Zhao, L., Xing, L. & Chen, D. MicroRNA-204 regulates Runx2 protein expression and mesenchymal stem cell differentiation. Stem Cells 28, 357–364 (2010).
Zhao, M. et al. Smurf1 inhibits osteoblast differentiation and bone formation in vitro and in vivo. J. Biol. Chem. 279, 12854–12859 (2004).
Acknowledgements
We would like to express our gratitude to Ms. Lily Yu for her help on processing and staining histological samples. This work was supported by National Institutes of Health Grants, R01 AR054465, R01 AR070222, and R01 AR070222 to DC. This work was also partially supported by the grants of Natural Science Foundation of China (NSFC) to TW (grants No. 81301531 and 81572104). This work was partially supported by the grant from Shenzhen Science and Technology Innovation Committee, China (grant No. JCYJ20160331114205502) to DC and the grant from Shenzhen Development and Reform Committee, China for Shenzhen Engineering Laboratory of Orthopedic Regenerative Technologies to DC.
Author information
Authors and Affiliations
Contributions
T.W. and D.C. contributed to the experimental design, data interpretation, and manuscript preparation and revision. T.W., S.L., and D.Y. carried out experiments. G-Q.Z., P.X.M., Z.C., and G.X. contributed to the data interpretation.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, T., Li, S., Yi, D. et al. CHIP regulates bone mass by targeting multiple TRAF family members in bone marrow stromal cells. Bone Res 6, 10 (2018). https://doi.org/10.1038/s41413-018-0010-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-018-0010-2
