
Abstract
TGF-β 1–3 are unique multi-functional growth factors that are only expressed in mammals, and mainly secreted and stored as a latent complex in the extracellular matrix (ECM). The biological functions of TGF-β in adults can only be delivered after ligand activation, mostly in response to environmental perturbations. Although involved in multiple biological and pathological processes of the human body, the exact roles of TGF-β in maintaining stem cells and tissue homeostasis have not been well-documented until recent advances, which delineate their functions in a given context. Our recent findings, along with data reported by others, have clearly shown that temporal and spatial activation of TGF-β is involved in the recruitment of stem/progenitor cell participation in tissue regeneration/remodeling process, whereas sustained abnormalities in TGF-β ligand activation, regardless of genetic or environmental origin, will inevitably disrupt the normal physiology and lead to pathobiology of major diseases. Modulation of TGF-β signaling with different approaches has proven effective pre-clinically in the treatment of multiple pathologies such as sclerosis/fibrosis, tumor metastasis, osteoarthritis, and immune disorders. Thus, further elucidation of the mechanisms by which TGF-β is activated in different tissues/organs and how targeted cells respond in a context-dependent way can likely be translated with clinical benefits in the management of a broad range of diseases with the involvement of TGF-β.
Similar content being viewed by others

Introduction
The evolution of a multicellular organism into ever more complex life forms needs the establishment of communication and control among individual cells to maintain order in the organism. The basic physiological processes, including proliferation, differentiation, metabolism, and apoptosis, are intricately regulated by a dense signaling network that is elicited by cytokines, growth factors or polypeptide hormones. Among those polypeptide/hormone-induced signals, the transforming growth factor-β (TGF-β) family is particularly important.1
TGF-β 1–3 are unique multi-functional growth factors because they are present only in mammals, mainly secreted as a latent complex and immediately stored in the extracellular matrix (ECM).1, 2 The biological functions of TGF-β can only be delivered after ligand activation, which is intricately regulated in response to ECM perturbations.2,3,4 Hence, the TGF-β complex functions as a molecular sensor which responds to environmental perturbations by releasing an active TGF-β ligand, to promote or inhibit cell proliferation in a context-dependent manner. More importantly, activation of TGF-β in the right place at the right time is necessary to recruit stem/progenitor cells to participate in the tissue regeneration/remodeling process, whereas sustained abnormalities in TGF-β ligand expression, bioavailability, activation, receptor assemblage/stabilization, or post-transcriptional modifications will inevitably disrupt the normal physiology, and lead to pathobiology of major diseases either through the recruitment of excessive progenitors (as seen in osteoarthritis or Camurati–Engelmann disease), or trans-differentiation of resident cells to unfavorable lineage commitment (as seen in epithelial to mesenchymal transition during cancer metastasis or tissue/organ fibrosis).1,5,6,7,8
Understanding the mechanisms that underscore the temporal and spatial activation TGF-β, as well as how targeted cells contextually integrate the downstream signaling into coherent responses are essential to elucidate the central role of TGF-β in maintaining stem cell and tissue homeostasis. This may provide new insights into potential treatment of systemic or local disorders that are associated with abnormalities of TGF-β signaling.
Temporal and spatial activation of TGF-β is essential for tissue homeostasis
TGF-β proteins belong to the TGF-β superfamily, which consists of TGF-β1–3, the activins/inhibins/Müllerian-inhibiting substances (MIS), bone morphogenetic proteins (BMPs), Nodal, growth/differentiation factors (GDFs), and the distantly related glial cell line-derived neurotrophic factors (GDNF) family.9,10,11 TGF-β1–3 are present only in mammals. They are pleiotropic, regulate cell proliferation, migration, and differentiation during embryonic development, and have an essential role in maintaining tissue homeostasis in adults. In mammals, distinct genes encode TGF-β 1–3 isoforms, which are expressed in unique, occasionally overlapping patterns and can perform a variety of distinct functions in vivo.12,13,14 Initially cloned from human term placenta mRNA, TGF-β1 is the most abundant and ubiquitously expressed isoform.15 TGF-β1 has been identified in cartilage, endochondral, and intramembranous bone and skin during mouse development, thereby indicating its involvement in the development of these tissues/organs.16 TGF-β2, also known as glioblastoma-derived T-cell suppressor factor (G-TsF), was first discovered in human glioblastoma cells. During embryonic development, TGF-β2 is expressed by neurons and astroglial cells.17, whereas pathologically it is also involved in tumorigenesis by enhancing cell proliferation and reducing the host immune surveillance against tumor development.18 TGF-β3 was first identified from a cDNA library of a human rhabdomyosarcoma cell line. It has an essential role in the development of the palate and lungs, mainly through the regulation of epithelial–mesenchymal interactions during embryonic, fetal, and neonatal development.12,19 TGF-β3 is also possibly involved in the wound healing process, orchestrating an orderly migration of dermal and epidermal cells in injured skin.20
Although it was discovered more than 30 years ago, TGF-β, as a multi-functional cytokine, is still under major research in various fields ranging from embryonic development to adult organ physiology and pathobiology of major diseases, including cancer, organ fibrosis, cardiovascular diseases, and immunological abnormalities. Unlike most of the growth factors that are ready to function upon secretion, TGF-β is unique in that it is secreted as part of a latent complex that is stored in the extracellular matrix (ECM). Thereby, the magnitude and duration of TGF-β signaling is carefully controlled at many different levels, including the synthesis and activation of latent TGF-β isoforms, receptor activation and stability, and the activation and stability of intracellular Smad molecules and other downstream signaling molecules. Plenty of molecules have been identified as “TGF-β activators” whose mutation will lead to aberrant activation of TGF-β and ultimately pathological phenotypes. Although distal effects from circulating factors have been reported, TGF-β-mediated effects are usually restricted at the sites where the active ligand is released. Therefore, the temporal and spatial activation of this growth factor is critical for its context-dependent physiological effects in vivo. Considering the close relationship of TGF-β and ECM homeostasis, increasing evidence has indicated that TGF-β complex is more like a molecular sensor that responds instantly to ECM perturbations through the release of an active ligand that exerts physiological effects at a cellular level, thus ensuring normal tissue homeostasis.2 This section will first elaborate on the molecular basis of TGF-β latency and specific activation pathways that modulate its activation. This section will then further specify how the active TGF-β isoform functions alone or with the cross-talk of other environmental cues, balances the self-renewal of stem cells, assists with differentiation during normal physiological development, and how TGF-β acts as a pro-migratory factor to mobilize adult stem cells from their unique niche to repair damage and maintain normal tissue homeostasis.
Latent TGF-βs are deposited in ECM upon secretion
TGF-β family members are typically secreted and deposited in the ECM in its latent form, and their biological effects can only be delivered upon ligand activation. TGF-βs contain a characteristic cysteine-knot that is formed from multiple intra-chain disulfide bonds.9,10,11 Take TGF-β1 for example: the precursor peptide contains 390 amino acid (aa), including a signal peptide and a TGF-β1 pro-protein. This pro-protein (361 aa) is processed intracellularly by a furin-like convertase to generate an N-terminal latency-associated peptide (LAP, 249 aa), and a C-terminal mature TGF-β1.21,22,23,24,25,26 Both LAP and mature TGF-β1 form homodimers via disulfide bonds. After secretion, the LAP and TGF-β1 homodimers are further non-covalently associated as the small latent TGF-β1 complex (SLC). LAP-growth factor association is both necessary and sufficient to confer latency of TGF-β1–3, BMP-10, and GDF-8/myostatin. However, for BMP-4, -5, and -7, although LAP and the mature growth factor is also non-covalently associated, the complex is still active.27
In most cases, LAP of the SLC is further covalently associated with a latent TGF-β binding protein (LTBP) in the ECM, thus creating the large latent complex (LLC) that functions as an ECM reservoir of TGF-β. The LAP-LTBP association mainly functions to anchor the complex to ECM components such as fibrillin.2 LTBP is also involved in the proper folding and secretion of the SLC.28,29,30,31,32,33 To date, four LTBPs (LTBP1–4) have been identified, among which LTBP1, 3, and 4 are able to bind the SLC of all TGF-β isoforms.34 Therefore, although TGF-βs are abundant in the ECM.35,36,37, they are secreted and deposited in the latent form, and not able to induce downstream signaling to elicit biological effects.3,4
Latent TGF-βs are activated by various pathways in vivo
Although TGF-β ligand and receptors are ubiquitous in many types of cells, their biological effects are usually restricted at sites where the ligand is activated. Storage of inactive TGF-β in the matrix enables temporal and spatial regulation of TGF-β activation during tissue homeostasis Precise activation of latent TGF-β is a pre-requisite for it to function in the right locations within a specific time frame. In general, the activation of TGF-β requires the release of the LLC from the ECM and further proteolysis/deformation of LAP to release active TGF-β.38 Accumulating evidence has shown that TGF-β1 can be activated by plasmin, matrix metalloproteinases (MMPs), thrombospondin-1, lower pH, and reactive oxygen species.2 More importantly, TGF-β can also be activated by specific integrins that bind the Arg-Gly-Asp (RGD) sequence of LAPs. The integrin-RGD associate results in a contractile force-dependent conformational change of the latent complex, which releases TGF-β in its active form.39,40 In addition, a plethora of soluble extracellular agonists and antagonists coexist at the site where active TGF-β is released and further complicates the temporal and spatial access of the ligands to receptors.41
Until now, a variety of TGF-β activators have been reported. Most of these activators are also indicators of ECM perturbations. As TGF-β has profound effects on matrix homeostasis, it has been generally recognized not only as a cellular effector, but also as a potential sensor for environmental perturbations. Here we will describe well-recognized pathways that contribute to the in vivo activation of TGF-β. Inheritable genetic mutations that release excessive TGF-β from the ECM or induce over-production of the ligand will be specifically discussed in section “Genetic mutations in TGF-β signaling components cause bone-associated disorders”.
Proteolytic activation
Many proteases including plasmin and matrix metalloproteinases (e.g., MMP-2 and MMP-9) have been identified in vitro as TGF-β activators.42,43 Plasmin and MMP-2/9 are the primary enzymes involved in ECM degradation.44 Proteases can cleave the covalent bond between LAP and TGF-β peptide in the proLLC, thereby rendering the LLC activation competent. Proteases can also target the protease-sensitive hinge region of LTBP to liberate the LLC, which can then be further processed for activation.45 Proteases may directly cleave LAP to release TGF-β in its active form.46 The aforementioned enzymatic activation, couples matrix turnover with the generation of active TGF-β to maintain matrix homeostasis.47,48 More notably, plasminogen-null animals fail to replicate the pathology of TGF-β1-null animals, and the multisystem pathology of plasminogen-null animals can be alleviated by removal of fibrinogen.49 These observations suggest that plasmin is not solely responsible for the majority of the activation of TGF-β1 in vivo.
Activation by thrombospondin-1
Thrombospondin-1 (TSP-1) is a complex multi-functional glycoprotein which mediates cell-to-cell and cell-to-matrix interactions during multiple cellular events in a temporally regulated manner.50,51,52,53,54 TSP-1 has an important role in the wound healing process, regulating hemostasis, cell adhesion/migration/proliferation, ECM remodeling, and growth factor (e.g., TGF-β) activation.55 In addition to tissue repair, TSP-1 is also involved in tissue fibrosis, possibly by activating TGF-β. Either a blockage of TSP-1 activity or deletion of TSP-1 expression can attenuate pathological tissue fibrogenesis.56,57,58
The primary role of TSP-1 in modulating TGF-β activation is observed during injury, under stress, or in other pathologies involved with ECM perturbations. This phenomenon further supports the concept that the latent TGF-β complex embedded in the ECM functions as a sensor to environmental stimuli. TSP-1 will mobilize necessary molecular machineries to release TGF-β in its active form to meet the needs for tissue repair/remodeling, whereas an excessive response to ECM perturbation may super-activate TGF-β and exacerbate adversary effects such as fibrogenesis. Mechanistically, TSP-1 activates TGF-β by binding to specific sequences of the latent complex and inducing a conformational change to release active TGF-β.59,60 In the latent TGF-β complex, the RKPK sequence in the receptor-binding region of the mature TGF-β binds to the LSKL sequence at the amino terminus of the LAP, thus enabling ligand latency.40,61,62 TSP-1 activates TGF-β through the specific association of its type 1 repeats (TSRs) with LAP and the mature ligand. When the tryptophan-rich motifs (WSxW) present in each of the 3 TSRs of TSP-1 bind to the VLAL sequence in both LAP and the mature TGF-β ligand, it deforms the LAP-TGF-β complex by “inserting” a TSP-1 molecule. In addition, the KRFK sequence in the second TSR of TSP-1 can competitively bind to the LSKL sequence in the LAP and present to the receptor the mature TGF-β domain.63 In vivo evidence for the role of TSP-1 in TGF-β activation is shown by the fact that both TSP-1 and TGF-β1 null animals developed strikingly similar pathologies in multiple organs, particularly in the lungs and pancreas. During the perinatal period, administration of the KRFK peptide partially resolved the abnormal TSP-1 depletion phenotypes, specifically airway epithelial hyperplasia and pancreatic islet hyperplasia/acinar hypoplasia. In addition, wild-type mice treated with the LSKL blocking peptide in the perinatal period showed similar features to the TSP-1 knockout phenotype in both the airways and pancreas.64 Double knockout of β6 integrin and TSP-1 led to a phenotype different from either single knockout, characterized by cardiac degeneration, severe inflammation, and epithelial hyperplasia, which suggests a potential synergy between β6 integrin and TSP-1 in regulating latent TGF-β activation.65
The TSP-1-mediated TGF-β activation is observed in multi-organ fibrosis. Moreover, the expression of TSP-1 is induced by factors such as reactive oxygen species, high glucose, and angiotensin II which are closely associated with systemic diseases that have fibrotic end-organ involvement.66,67,68,69 Studies using TSP-1 antagonist peptides and diabetic TSP-1 knockout mice have demonstrated that TSP-1 is a major factor inducing fibrotic end-organ complications in diabetes.58,70,71 Treatment of diabetic mice with intraperitoneal injections of LSKL improved left ventricular function, and reduced Smad phosphorylation and cardiac fibrosis.70 Similarly, treatment with LSKL suppressed urinary TGF-β activity and improved markers of tubulointerstitial injury and podocyte function in diabetic mice.71 Moreover, evidence from several studies have demonstrated that TSP-1 can activate alveolar macrophage-dependent TGF-β in bleomycin-induced pulmonary fibrosis animal models, and either CD36 antagonist peptides or TSP-1 can reduce TGF-β activity and ameliorate pulmonary fibrosis.72,73
TSP-1-mediated TGF-β activation is also involved in the dermal wound healing process. The phenotype of excisional wound healing in the TSP-1 null mouse is consistent with a decrease in local TGF-β activation54, a delay in macrophage recruitment and capillary angiogenesis, and a persistence of inflammation, granulation tissue, and neovascularization.74 The TSP-1 null wound phenotype can be largely rescued by topical treatment with the KRFK activating peptide.74 KRFK treatment increased the TGF-β levels in these wounds and its effects were blocked by a pan-specific anti-TGF-β antibody. These data suggest that TSP-1 is essential for the local activation of TGF-β during injury and may affect the wound healing process. In addition, subcutaneous implantation of TSP-1-soaked sponges increased levels of active TGF-β and induced fibroblast migration.75 Overexpression of TSP-1 in scleroderma and in keloids induces increased TGF-β activity.76,77,78 All these data validate the involvement of TSP-1-induced TGF-β activation in dermal wound healing and sclerosis. However, how to modulate TSP-1 activity to avoid either defective or excessive wound repair processes in vivo remains to be determined.
Activation by integrins
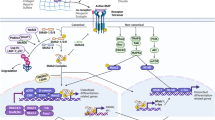
Integrins are dimeric cell-surface receptors composed of α- and β-subunits.79 They have been recently shown to have a central role in TGF-β activation.80 Current data show that at least two mechanisms are involved in the activation of latent TGF-β by integrins (Fig. 1). The first proposed mechanism is MMP-dependent. Specifically, integrins are suggested to spatially arrange MMPs, latent TGF-β and the TGF-β receptor in close proximity, which promotes further activation of latent TGF-β by proteolytically cleavage. The second mechanism is proteolytic action-independent, but more closely associated with cell traction forces that are directly transmitted to the LLC via integrin binding. The cellular contractile force can lead to conformational change of the latent complex, thus liberating TGF-β in its active form and/or presenting it to its receptor. It should be noted that both mechanisms are not mutually exclusive, and it is conceivable that cells can use either or both mechanisms at the same time depending on the specific organs or conditions.81

Epithelial cells activate TGF-β by enriching the latent complex through αvβ8-RGD association and recruiting membrane-bound matrix metalloproteinases (e.g., MMP-14) in proximity for further proteolytic cleavage ①. Active TGF-β can act on resident fibroblasts, inducing its trans-differentiation into myofibroblasts, which are the major contributor to excessive ECM (e.g., collagen) deposition and fibrosis. The myofibroblasts can further activate TGF-β in a contractile force-dependent manner through the αvβ6-RGD association ②. The active TGF-β can in turn act on epithelial cells, fibroblasts, and myofibroblasts in a paracrine/autocine manner, and thus form a feed-forward loop for a sustained TGF-β activation and fibrogenesis. Of note, sustained activation of TGF-β can also induce the epithelial–mesenchymal transition (EMT) of epithelial cells with the assistance of integrin α3β1, which forms a complex with TGF-β type I and II receptors (TβRI/II) and E-cadherin, facilitating β-catenin/Smad2 complex formation and nuclear translocation. LAP: latency-associated peptide, LTBP: latent TGF-β binding protein, SLC: small latent complex, LLC: large latent complex.
αvβ6 was the first integrin that was identified as a TGF-β activator.39 The mechanism of activation depends upon a direct interaction between αvβ6 and the RGD amino acid sequence of the prodomains (LAPs). The prodomains of TGF-β1 and TGF-β3 contain an RGD motif that is recognized by αv integrins. Mice with the integrin-binding RGD motif mutation show similar phenotypes to TGF-β1-null mice, such as multi-organ inflammation and defects in vasculogenesis, confirming the essential role of integrins in TGF-β activation.82
αvβ6 is normally expressed in epithelial cells at low levels.83 Inflammation or injury can increase the expression of αvβ6.84,85 Therefore, upregulation of αvβ6 and subsequent TGF-β activation in epithelial cells are believed to be a cellular response to suppress the perturbation such as inflammation. Consistent with the ability of β6 integrin to activate latent TGF-β and the pro-fibrotic effects of TGF-β, 86 in the mouse model of pulmonary fibrosis induced by belomycin, wild-type mice develop pulmonary inflammation with subsequent fibrosis, whereas integrin β6–/– mice show a minor fibrotic response in response to bleomycin.39 Moreover, TGF-β-targeted genes in the lungs of integrin β6–/–mice are not significantly induced by bleomycin compared to wild-type mice. These data indicate that the inflammatory stimulus upregulate the expression of αvβ6 and consequently induce excessive activation of TGF-β that results in fibrosis. As TGF-β markedly upregulates expression of αvβ6 by primary airway epithelial cells in vitro,87 it is likely that bleomycin triggers a feed-forward mechanism for coordinately upregulating integrin expression and TGF-β generation. We suggest that fibrosis is the result of a failure to interrupt this feed-forward loop that is perpetuated by persistent ECM perturbation after injury or inflammation.
Accumulating evidence has suggested the important role of force-resistance ECM in contractile force-dependent TGF-β activation. Activation by αvβ6 integrin requires LTBP-mediated incorporation of TGF-β into the ECM, and the association of the β6 cytoplasmic domain with the actin cytoskeleton.39,88,89 Furthermore, contractile force is necessary for TGF-β activation by myofibroblasts.81 Thus, tensile force exerted by integrins across the LTBP–prodomain–TGF-β complex is necessary to change the conformation of the prodomain and to free active TGF-β for receptor binding.81,88 A recent study by Shi et al. has solved the structure of latent TGF-β and provided mechanistic insights into latency and force-dependent activation by integrins. By using multi- and single-wavelength anomalous diffraction, they found that the two prodomains (LAPs) form a ring-like structure with arms forming a “straitjacket” that fasten each TGF-β monomer. The RGD motifs of LAPs locate to each shoulder, binding to the αv integrins. Upon applied tensile force, TGF-β1 is freed by the opening of the straitjacket, and is subsequently released from the prodomain and activated for receptor binding.40 At least four conditions need to be fulfilled to enable the cell traction-dependent integrin-mediated TGF-β1 activation: (1) the presence of the actin cytoskeleton to generate force and/or to provide mechanical resistance, (2) specific integrins that transmit this force on the LLC, (3) incorporation of latent TGF-β1 into the ECM as LLC, and (4) a second anchor point, i.e., an ECM that mechanically resists the cellular traction forces exerted to the LLC. Therefore, the activation of latent TGF-β1 is confined to cells expressing the appropriate integrin in a specific physiological/pathological context. Similar to contractile force-directed activation, a recent study from Coller’s group revealed that intravascular shear force was also able to activate latent TGF-β1 released from platelets.90 As TGF-β1 released from platelets during trauma or surgery might also contribute to the transient increase in plasma levels of plasminogen activator inhibitor-1 by activating endothelial cells,91,92,93,94,95,96,97 the shear force-induced TGF-β1 activation likely coordinates the process of platelet activation to arrest hemorrhage and the transient inhibition of fibrinolysis to allow an unopposed deposition of fibrin at the early stage of hemostasis. The shear force-activation model makes TGF-β1 a potential shear sensor as well as an effector. Hence, TGF-β1 may contribute to the vascular remodeling that occurs in response to changes in shear forces and maintain intravascular arterial shear within a limited range.98
In addition to force-directed activation, integrins can also activate latent TGF-β1 with the assistance of protease. αvβ8 is believed to be able to recruit membrane-bound MT1-MMP to the latent complex. This close proximity promotes activation of latent TGF-β1 by further proteolytic cleavage.99 Similarly, integrin αvβ3 has been proposed to act as a docking point for MMP-9 in metastatic breast cancer cells,100 and for MMP-2 in melanoma cells.101 In addition, integrins can cluster with the TGF-β-RII, thereby improving its availability to locally activate TGF-β1. Direct interaction with TGF-β-RII has been demonstrated for αvβ3 integrin upon stimulation with active TGF-β1 using a bioluminescence resonance energy transfer approach102 and immunoprecipitation. Interestingly, a similar interaction between different classes of latent TGF-β activator has also previously been suggested:103 the cell-surface-associated proteins (CD36 and TSP-1) concentrate latent TGF-β on the membrane where it is subsequently activated by plasmin. This cell surface-enrichment theory might also explain why mice that have null mutations in the genes encoding known protease activators thus far do not demonstrate any phenotype consistent with TGF-β deficiency. It is conceivable that protease activity is intricately modulated by its activators and inhibitors in vivo, as well as the surface concentration of proteases, and a proper spatial arrangement of latent TGF-β1, proteases and TGF-β receptor is a pre-requisite for the in vivo activation of TGF-β1 through proteolytic pathways.
Keeping in mind the two sets of activation mechanisms by integrins may help elucidate some “contradictory” data on integrin-mediated TGF-β activation in different tissues/organs or disease models. The involvement of β integrins in the activation of TGF-β is equivocal, mainly because the deletion of β integrins usually presents contradictory phenotypes in different disease models. Deletion of β6 integrin has been reported to protect mice from bile duct ligation-induced hepatic fibrosis,104 whereas global deletion of β3, β5, or β6 integrins or the conditional deletion of β8 integrins in hepatic stellate cells cannot protect mice from carbon tetrachloride-induced hepatic fibrosis.105 Because TGF-β activation in a bile duct ligation model is more likely to be contractile force-dependent, β6 anchorage to cytoskeleton is likely essential to ligand activation in the disease model, thereby conveying that β6 deletion is protective. Conversely, in carbon tetrachloride-induced hepatic fibrosis, excessive proteases are released due to extensive cytotoxic damage. In this case, integrin-mediated surface enrichment of proteases may be the major contributor to TGF-β activation, hence single deletion of β subunits is not sufficient to disrupt the superactivation of TGF-β. A similar theory can also explain the observation that integrin β6−/− mice have only minor lung fibrosis in response to bleomycin induction.105 Because the lungs are a highly contractile organ and its compliance is closely associated with the force-directed activation of TGF-β, β6 deletion directly disrupts intracellular anchorage, and thus may significantly retard the activation of TGF-β and lung fibrosis.
Activation by osteoclasts
Latent TGF-β present in conditioned medium can be activated by mild acid treatment (pH = 4.5),46 which probably denatures LAP and thus dissociates TGF-β. In vivo, osteoclasts generate a similar pH during bone resorption when an integrin-dependent sealing zone is generated between the bone and the cell.106 As the bone matrix deposited by osteoblasts contains abundant TGF-β in its latent form (~200 μg/kg),37,107 the acidic environment created by osteoclasts offers an ideal condition for TGF-β activation.108,109 Bone-conditioned medium harvested from bone cultures during bone resorption usually contains an increased level of active TGF-β, and the isolated osteoclasts are able to activate bone latent TGF-β in vitro.8,110 All this evidence indicates that latent TGF-β from surrounding bone tissue or stored in bone matrix becomes activated and released at this site during the bone resorption process. Alternatively, osteoclasts may also activate latent TGF-β by secretion of proteases in the absence of a low-pH environment. Protease action at a pH higher than the optimum for lysosomal enzyme activity may sufficiently retard enzyme activity to prevent degradation of TGF-β. It is therefore possible that osteoclast-mediated activation of latent TGF-β occurs outside the low-pH resorption lacuna, resulting in the presence of active TGF-β within the immediate environment of the bone resorption site.109 As the active TGF-β released during osteoclastic bone resorption is able to induce migration of osteogenic bone marrow mesenchymal stem cells (MSCs) to the bone resorption sites,8 osteoclast-mediated activation of TGF-β may therefore represent one of the mechanisms that couple bone resorption to new bone formation. Indeed, our recent study has shown that osteoclast-mediated release of active TGF-β1 is essential for the recruitment of MSCs to the bone resorption site during the parathyroid hormone (PTH)-induced bone remodeling process. By inhibiting osteoclast bone resorption with alendronate, osteoblast recruitment is uncoupled from PTH-induced bone resorption.111
Activation by reactive oxygen species (ROS)
Another potential mechanism for in vivo activation of TGF-β involves reactive oxygen species (ROS).112,113Barcellos-Hoff and her co-workers have shown that ionizing radiation increases the level of active TGF-β in exposed tissues, and that a ROS-generating metal ion-catalyzed ascorbate system is also able to activate recombinant latent TGF-β in vitro.112,113 ROS can stimulate the expression and secretion of TGF-β in a positive feedback loop in many types of cells, including hepatic stellate cells and hepatocytes.114,115 In addition, low level photodynamic therapy (10 J/cm2), which releases free radicals by light activation, has also been shown to increase active TGF-β when applied to cultured smooth muscle cells.116 It is currently believed that site-specific oxidation of LAP elicits a conformational change in the latent complex releasing free active TGF-β.112 ROS may also indirectly activate TGF-β through MMP activation.117 The activation of TGF-β in response to oxidative stress may reflect a need of the human body to produce TGF-β to maintain tissue homeostasis after perturbation such as inflammation. Indeed, LAP/TGF-β1 complex has been proposed to function as an oxidative stress sensor.118
Active TGF-βs bind specific receptors to elicit downstream signaling
Active TGF-β ligands signal by binding and bringing together two transmembrane serine-threonine kinases, known as receptor types I and II.119 In vertebrates, seven type I receptors [Activin-receptor like kinases (ALKs) 1–7] and five type II receptors have been identified so far.120 TGF-β superfamily ligands bind to and signal through specific type I and type II receptor complexes. Accessory receptors, including the type III receptor, TGF-β RIII (also known as betaglycan) and endoglin, have also been identified.10,121,122,123,124 Nevertheless, neither betaglycan nor endoglin is directly involved in intracellular TGF-β signaling due to the deficiency of a kinase domain. Instead, they affect the access of TGF-β ligand to its receptors, and consequently modulate the intracellular signaling.125,126 Betaglycan binds all three isoforms of TGF-β, with a particularly higher affinity for TGF-β2. However, endoglin binds TGF-β1 and TGF-β3 with identical affinity, and it has weak affinity for TGF-β2.127,128
Canonical signaling pathways (Smad-mediated signaling) of TGF-β
In most of the context, active TGF-β signals through a canonical (Smad-mediated) pathway. Upon ligand activation, a type II receptor phosphorylates its type I receptor partner, which then transmits the signal by phosphorylation of intracellular downstream substrates, i.e., Smads. Eight Smads (Smad1 to Smad8) have been identified in vertebrates.129 They have conserved Mad homology (MH)1 and MH2 domains connected by a linker region. The N-terminal MH1 domain has a β-hairpin loop which can bind to DNA, and the C-terminal MH2 domain mediates interaction with other molecules (e.g., receptors and other Smad isoforms).130 The linker region is subject to posttranslational modifications which affect interactions and the stability of Smad molecules. Upon ligand stimulation and subsequent activation by type II receptors, type I receptors transmit intracellular signaling through phosphorylation of downstream effector Smads.129,131,132 Specifically, Smad1/5/8 are activated by BMP receptors, whereas Smad2/3 are activated by TGF-β/activin/nodal receptors. These receptor-activated Smads (R-Smads) form heterotrimers with a common Smad (Smad4) shared by the TGF-β/activin/nodal and BMP signaling pathways, and translocate into the nucleus. The R-Smads, except for Smad2 which has two extra sequences inserted in the MH1 domain perturbing its DNA-binding affinity, can bind to preferred DNA sequences. The DNA sequence specificities of R-Smads add further diversity to the transcriptional responses of TGF-β signaling. Complexes of phosphorylated Smad2/3 and Smad4 bind to AGAC or its complement GTCT, known as a Smad-binding element (SBE).133,134 However, Smad4-pSmad1/5/8 complexes preferentially bind to GGCGCC or GGAGCC, known as the BMP-response element (BRE).135,136,137 It is noteworthy that although most of TGF-β signaling pathways go through phosphorylated R-Smads, not all transcriptional responses have Smad4 involvement. For example, in cultured epidermal keratinocytes, IκB kinase (IKK) of the classical nuclear factor κB (NF-κB) pathway recruits pSmad2/3 to a specific promoter region that drives cell differentiation.138 Data from recent studies also indicate that R-Smads can regulate miRNA processing in a Smad4-independent and RNA-sequence-specific manner by associating with the p68/Drosha/DGCR8 miRNA processing complex.139,140
Because the TGF-β superfamily signaling requires the interaction of type I and type II receptors, the interplay between the canonical BMP signaling pathway and the canonical TGF-β/activin signaling pathway has been noted.141 The type 1 BMP receptors (ALK2/3/6) + BMPR2 specifically transduce BMP signals; the type 1 Activin receptors (ALK4/7) specifically transduce signals from Activin/Activin-like ligands. In contrast, the type 2 receptors ACVR2A/B are shared between the BMP and Activin pathways and elicit activation of Smad1/5/8 or Smad2/3 in response to BMP or Activin-like ligands, respectively. TGF-β ligands elicit activation of Smad2/3 but do not share any receptors with BMPs or Activin/Activin-like ligands. In addition, TGF-βs and BMPs bind and assemble their receptors in a distinct manner. TGF-β binds TβR-II first and then crosslinks to TβR-I. This pattern was also adopted by activin,142 suggesting that TGF-βs/activins assemble their receptors in an ordered manner.143 Conversely, the BMPs and GDFs exhibit a much more heterogeneous pattern of crosslinking, with some binding to their receptors in a stepwise manner, whereas others exhibit weak affinity to a single receptor and instead crosslink to TβR-I and -II simultaneously.144,145,146,147,148,149 These findings indicate that the TGF-β superfamily members might differ in how they bind and assemble their receptors into signaling complexes.
Notably, although TGF-β does not share or compete for receptors with BMPs, both strongly induce phosphorylation of Smad1/5/8 in many different cell types, including fibroblasts, endothelial cells, epithelial cells, and epithelium-derived cancer cells.150,151,152,153,154 Despite this common phosphorylation event, TGF-β cannot induce BMP-like transcriptional responses. Grönrooset al.155 found that although TGF-β was able to stimulate the phosphorylation of Smad1/5/8 in parallel with the classical induction of Smad2/3 phosphorylation, pSmad1/5 and pSmad3 formed complexes readily binding to BMP-responsive elements and mediated TGF-β-induced transcriptional repression on BMP responses. Therefore, Smad3 has an important role in restricting the TGF-β signaling to the canonical transcriptional output and effectively prevents TGF-β from eliciting BMP-like “off-target” responses
The DNA-binding affinity of Smad complexes is not strong. Hence, they need to interact and cooperate with other DNA sequence-specific transcription factors to target the specific downstream genes.156 The requirement of DNA-binding co-factors that either activate or repress transcription results in a context-dependent and cell type-specific response.129,157 The forkhead-box family member FoxH1 (previously known as Fast1) was the first identified transcription factor that facilitates Smad-mediated transcription. The Foxh1–Smad2/3–Smad4 complex binds to a composite site known as the “activin response element” (ARE) on target differentiation genes in embryonic cells.158 Accredited to the advancement from ChIP-seq, various families of DNA-binding transcription factors that interact with Smads have been identified. These transcription factors cooperate with Smad complexes, targeting a specific subset of TGF-β responsive genes for coordinated regulation of cellular activities.159 Among the many factors utilizing Smad complexes as transcriptional co-factors, FOXH1,160 EOMES,161 OCT4,162 and NANOG163,164 are particularly involved in stem cells, whereas MYOD1 and PU.1 are more relevant to muscle cells and Pro-B cells, respectively. These findings support the notion that the availability of cell type-specific co-factors determine the cellular response to TGF-β signaling by providing context and directing the transcriptional activity of Smad proteins. Smad-mediated assembly of basal transcription machinery is also dependent on chromatin conformation, and thus Smads interact with and recruit various chromatin-modifying enzymes to DNA.165,166 Smad2/3 can interact with the histone acetyltransferases CBP/p300 and recruit the basal transcription machinery, thus initiate transcription from the associated promoter.167,168 Alternatively, depending on the context, the Smad complex can also recruit histone deacetylases (HDAC1/3/4/5/6) to remove acetyl residues on histone tails, and thus condenses chromatin and represses transcription.169 A well-recognized model through which the Smad complex orchestrates with chromatin-modifying enzymes to gain access to DNA by associating with co-transcription factors to maintain stem cell homeostasis will be discussed in section "TGF-β signaling in embryonic stem cells".
In the unstimulated state, Smad proteins interact with components of the Ran GTPase export/import system170 and the nuclear pore complex,171 resulting in the formation of a highly dynamic equilibrium in which unphosphorylated Smad proteins constantly shuttle between the nucleus and the cytoplasm.172 Upon phosphorylation of the R-Smads and the formation of the heteromeric complex with co-Smad in the cytoplasm, the increased import rate and decreased export rate of the trimer lead to its accumulation in the nucleus. This increased nuclear retention is mediated by transcriptional co-factors such as TAZ and YAP, which are the downstream effectors of the Hippo pathway. This cross talk links the TGF-β pathway to the Hippo pathway and sensing of cell density and cell polarity.173,174 Protein phosphatases (e.g., PPMA1 or SCPs) can dephosphorylate the R-Smads, leading to the disruption of the trimer, and eventually turn off Smad signaling.175,176
Surface receptors are regulated by endocytosis and degraded by SMURF2 and other HECT E3 ligases.177 Inhibitory Smads (I-Smad) such as Smad6 and 7 are transcriptional targets of TGF-β superfamily signaling and bind to activated receptors competing with R-Smad binding and recruiting the SMURF ubiquitin ligases, thus establishing a classical negative feedback loop.178,179 Activated R-Smad proteins could also be degraded via the proteasome by ubiquitination via HECT E3 ligases such as SMURF1,2, NEDD4L, and WWP2.180,181,182 R-Smad proteins contain multiple PY motifs in the linker region.181 Serine/threonine and proline residues of these PY motifs can be phosphorylated by ERK, GSK3,183 and CDK8 and 9,184 thereby interacting with WW domains of HECT E3. R-Smads are subsequently degraded by the proteasome and the transcriptional activity is terminated. This provides a platform in which the duration of TGF-β family signaling integrates with other pathways such as IGF, FGF, and WNT.
TGF-β signaling can also be fine-tuned by association with other factors. Our recent study has shown that PTH, which regulates calcium homeostasis and bone metabolism by binding to and activating a G protein-coupled receptor, is able to induce the recruitment and co-localization of TβRII with β-arrestin, an adaptor protein involved in PTH receptor endocytosis, thus mediating the internalization of TβRII–PTH1R as a complex in osteoblasts.185 The interaction of PTH and TGF-β signaling at the membrane receptor level may have significant physiological importance in maintaining tissue homeostasis, especially in coupling bone resorption to bone formation. We have demonstrated that the anabolic action of PTH on bone is dependent on active TGF-β1 released by PTH-mediated osteoclastic bone resorption.111 However, over-production of active TGF-β ligand in the local microenvironment may blunt the migration of MSCs to the bone resorption sites for coupled bone formation.8 Through the endocytosis of the TβRII–PTH1R complex, PTH provides surveillance of the over-activation of TGF-β signaling so as to ensure proper MSC migration mediated by local gradient of TGF-β.
Smad-independent signaling pathways
The Smad-independent signaling pathways of TGF-β are generally considered as important effector pathways for tyrosine kinase receptors.131,186,187 TGF-β activates these non-Smad pathways through interactions of signaling mediators with the type I/II receptors, either directly or through adaptor proteins. The Smad-mediated downstream gene expression may also activate non-Smad pathways. TGF-β can directly activate the Ras–Raf–MEK–ERK/MAPK pathway through the interaction of ShcA and the TGF-β receptor complex. In response to TGF-β, TGF-β type I receptor mediates tyrosine phosphorylation of ShcA, which then recruits Grb2 and Sos, to form a complex, initiating Ras activation and consequently ERK/MAPK signaling cascade.188 TGF-β can also activate TAK1 through TRAF6, an ubiquitin ligase, which interacts with the TGF-β receptor complex, leading to induction of p38 and JNK MAPK signaling.189,190 TGF-β also modulates the activities of the small GTPase proteins Rho, Rac, and Cdc42, which regulate cytoskeletal organization and gene expression,191,192,193 however the exact mechanism still remains to be explored. TGF-β-activated RhoA can activate its downstream targets ROCK and LIM kinase.194 TGF-β activates Akt through PI3K, 195,196 and consequently, initiates signaling pathways, e.g., through mTOR, that have roles in cell survival, growth, migration, and invasion.197,198 The roles of TGF-β-induced, Smad-independent signaling in stem cells are still unclear and remain to be elucidated.
Cross talk with other pathways
TGF-β can cross talk with several other signaling pathways at the level of ligands, receptors, agonists and antagonists, and thus elicits a context-dependent biological effect to meet the specific needs during development or tissue repair.199
Wnt signaling
Wnt is implicated in stimulation of cell proliferation during embryonal development and tumorigenesis. Key molecules in the Wnt signaling pathway are the transcription factors β-catenin, T-cell factor (TCF), and lymphoid enhancer factor (LEF). Smads form complexes with both LEF1200 and β-catenin,201,202 which enhance the induction of epithelial–mesenchymal transition (EMT). In addition, Smad7 forms a complex with β-catenin, which is important for TGF-β-induced apoptosis.203 The cross talk between TGF-β superfamily and Wnt signaling pathways has an essential role in dictating stem cell homeostasis in concert with combinatorial activities of other signaling pathways. A typical example of how the cross talk between Nodal/Activin/Smad2/3, ERK/MAPK, and Wnt/GSK3β/β-catenin pathways affects the balance of self-renewal and differentiation status of ESCs has recently been described by Singh et al.204 Specifically, activation of PI3K/Akt signaling establishes conditions where Activin A/Smad2/3 performs a pro-self-renewal function by activating target genes, such as Nanog. Although in the absence of PI3K signaling, Wnt effectors are activated by ERK targeting GSK3β, and function in conjunction with Smad2/3 to promote differentiation. This signaling paradigm with convergence on Smad2/3 is believed to have far-reaching implications for cell fate decisions during early embryonic development.
Parathyroid hormone
Parathyroid hormone regulates calcium homeostasis and bone metabolism by binding to and activating a G protein-coupled receptor. TβRII forms a complex with and phosphorylates the PTH receptor, which modulates the internalization of the receptor complex. Specifically, PTH induces the recruitment of TβRII as an endocytic activator, which phosphorylates the cytoplasmic domain of PTH1R and facilitates PTH-induced endocytosis of the PTH1R-TβRII complex, and consequently results in downregulation of TGF-β signaling.185
Notch signaling
The Notch pathway specifies cell fate determination during development. TGF-β induces several Notch receptor ligands, including Jagged1,205,206 and Notch signaling induces TGF-β.207 The cooperation between TGF-β and Notch signaling enhances EMT. However, there are reports that in certain cell types, e.g., esophageal epithelial cells, Notch signaling counteracts EMT by induction of miR200 that targets ZEB and TGF-β.208
Tyrosine kinase receptors
A major pathway induced by tyrosine kinase receptors is the Ras pathway. Cooperation between Ras and TGF-β signaling is particularly important during EMT.209 In hepatocarcinoma cells, TGF-β induces both platelet-derived growth factor (PDGF) and PDGF receptors, which enhances PI3K and β-catenin signaling and promotes the survival and invasion of cancer cells.210 Enhanced PI3K signaling also activates Akt, which phosphorylates and activates Twist, promoting EMT.211
Hippo
The Hippo pathway senses cell density and controls cell growth via the transcriptional regulators TAZ and YAP. TAZ/YAP binds Smad complexes and sequesters them in the cytoplasm in high-density cell cultures, thereby attenuating TGF-β signaling.173 Moreover, the Crumbs polarity complex interacts with TAZ/YAP and promotes their phosphorylation and cytoplasmic retention. Disruption of the Crumbs complex enhances TGF-β signaling and promotes EMT.174
Active TGF-βs induce migration of mesenchymal stem cells
A normal tissue repair or remodeling process not only requires the transient amplification and differentiation of adult stem/progenitor cells, but also the proper migration of these stem/progenitor cells to the sites needed.212,213,214,215,216 Latent TGF-βs are generally considered as molecular sensors2 that respond to perturbations of the ECM by releasing active TGF-βs as pro-migratory factors, thus mobilizing and recruiting adult stem cells to participate in tissue repair/remodeling. Active TGF-βs are released from the perturbed ECM like many other pro-migratory factors in response to injury or inflammation. Although these other factors regulate mobilization of hematopoietic stem cells (HSCs) and epithelial progenitor cells (EPCs),217,218,219 TGF-βs mediate the migration of MSCs from peripheral blood or surrounding tissue to be integrated into the injured/remodeling tissues.
The normal adult bone undergoes continual remodeling by precisely coordinating the activities of osteoblasts and osteoclasts. Osteoblasts derived from bone marrow MSCs deposit calcified bone matrix; while osteoclasts, which are multinucleated cells derived from macrophages/monocytes in the HSC lineage, resorb bone.220,221 Factors released from bone matrix during osteoclastic bone resorption orchestrate migration of MSCs to the resorptive surfaces of the bone. Particularly, osteoclastic bone resorption releases and activates TGF-β1 previously stored in the bone matrix, which recruits bone MSCs to the active bone remodeling sites through the canonical pSmad2/3 signaling pathway. TGF-β recruits MSCs in a gradient-dependent manner, i.e., osteoclastic bone resorption induce activation of TGF-β1, which diffuses from the bone resorption site and acts as a chemoattractant for BMSCs. Osteoblastic progenitors sense the TGF-β1 gradient and subsequently migrate to the bone resorption site, where they are induced to differentiate into osteoblasts in response to other environmental factors such as bone matrix-derived insulin-like growth factor 1 (IGF-1).222 Interestingly, either over-activation or inhibition of TGF-β signaling that distorts the local TGF-β gradient may impede the migration of MSCs to the normal bone remodeling surfaces. With this hypothesis, we have delineated the pathogenesis of Camurati–Engelmann disease (CED), in which mutations in the LAP cause conformational dissociation,223 resulting in increased release of activated TGF-β1 and distortion of the resorption-induced TGF-β1 gradients. Owing to the inadequate recruitment of BMSC to sites of resorption, poor-quality bone is formed with unfilled resorbed areas and haphazard sclerotic areas.8 This theory has also been expanded to explain the pathogenesis of osteoarthritis, in which enhanced osteoclastic bone resorption caused by joint instability results in the release of excessive TGF-β1. The pathologically high level of TGF-β1 ligand in the tibial subchondral bone distorts the physiological TGF-β1 gradients, leading to osteoblastic progenitor aggregation in the bone marrow with compromised bone formation capability (“osteoid islet”). The aberrant bone formation in the subchondral bone in turn causes uneven distribution of stress on the articular cartilage, and in a feed-forward manner leads to cartilage degeneration.6 In addition, we have also demonstrated in vivo that the anabolic action of PTH on bone is dependent on the active TGF-β1 released during osteoclastic bone resorption. By inhibition of osteoclastic bone resorption with alendronate, the depleted active TGF-β1 released from bone matrix is insufficient to recruit MSCs to the proper resorptive sites, thus impairing the anabolic action of PTH on bone.111
Active TGF-β also controls the mobilization and recruitment of MSCs to participate in tissue repair. A recent study of ours has shown that TGF-βs were activated in the vascular matrix in both rat and mouse models of mechanical injury of arteries. The active TGF-β released from the injured vessels induced the migration of MSCs and the cascade expression of monocyte chemotactic protein-1 (MCP-1), which amplified the signal for migration. Specifically, sustained activation of TGF-β was observed in peripheral blood, and Sca1+CD29+CD11b−CD45− MSCs, of which 91% were also Nestin+, were mobilized to peripheral blood and migrated to the remodeling arteries. The MSCs were noted to differentiate into endothelial cells for re-endothelialization and myofibroblastic cells to form thick neointima. Intravenous injection of recombinant active TGF-β1 in uninjured mice was also sufficient to rapidly mobilize MSCs into circulation. Blockade of TGF-β signaling with TGF-β type I receptor kinase inhibitor significantly attenuated the mobilization and recruitment of MSCs to the injured arteries.224 These findings strongly indicate that TGF-β is an injury-activated messenger essential for the mobilization and recruitment of MSCs to participate in tissue repair/remodeling.
Consistently, another recent study of ours on the pathogenesis of asthma demonstrates the involvement of TGF-β1 in bone marrow MSCs migration. By using a cockroach allergen-induced asthma mouse model, we found increased MSCs and TGF-β1 activation and its downstream signaling in lungs of CRE (cockroach extract)-treated mice. Further in vitro trans-well assay confirmed that TGF-β1 released from allergen-activated epithelium functions as the primary chemoattractant that induces MSCs migration. Consistently, by either intravenous injection of GFP+ MSCs (sorted from bone marrow of Nestin-GFP mice) to the CRE-treated mice, or directly immunizing Nestin-GFP mice with CRE, we observed significantly increased accumulation of GFP+ MSCs in the asthma airways. Importantly, the airway accumulation of MSCs was significantly attenuated by systemic administration of TGF-β1 neutralization antibody. Taken together, we believe that TGF-β1 is a primary pro-migratory factor released into the circulation from the injured vessels of the CRE-challenged lung tissue. It mediates the mobilization of MSCs to the circulation and further recruits these cells to the perturbed airways in asthma, likely to participate in tissue repair.225
Interestingly, one elegantly performed study by Mao’s group has also demonstrated the cell homing capacity of TGF-β3 in the functional regeneration of the articular surface of the rabbit synovial joint.226 By replacing the excised proximal humeral condyles of skeletally mature rabbits with cell-free bio-scaffolds spatially infused with TGF-β3-adsorbed hydrogel, weight-bearing and locomotion of rabbits were resumed 3–4 weeks after surgery. Histological and mechanical analysis of the joint revealed that the TGF-β3-infused bio-scaffolds had recruited more cells than did spontaneous cell migration without TGF-β3. As a result, TGF-β3-infused bio-scaffolds were fully covered with avascular hyaline cartilage and integrated with regenerated subchondral bone that had well defined blood vessels 4 months after surgery. On the contrary, TGF-β3-free bio-scaffolds had only scattered cartilage formation with compromised compressive and shear properties. It should be noted the that the lineage of the recruited endogenous cells was not delineated. However, this study further underscores the importance of TGF-β-mediated cell homing in tissue regeneration.
In addition to mobilization and recruitment of MSCs towards wounds, TGF-β also mediates homing of bone marrow-derived human MSCs to glioma stem cells (GSCs).227 By using glioma models, Shinojima et al. found that TGF-β attracts BM-hMSCs via TGF-β receptors (TGFβR). Intravascularly administered BM-hMSCs home to GSC xenografts that express TGF-β. BM-hMSCs carrying the oncolytic adenovirus Delta-24-RGD prolonged the survival of TGF-β-secreting GSC xenografts, and this effect was abrogated by inhibition of TGFβR on BM-hMSCs. These data show that TGF-β/TGFβR axis can mediate the tropism of BM-hMSCs for GSCs, and TGF-β may serve as a predictor for patients in whom BM-hMSC delivery could be effective.227
Context-dependent TGF-β signaling balances stem cells self-renewal and differentiation
Stem cells are long-lived cells functioning to make and replenish the differentiated cells that are lost through normal stress and injury. Stem cells are also capable of replenishing themselves, a process known as self-renewal. Extensive efforts have been made to identify factors that determine the self-renewal and differentiation of stem cells. Stem cells receive signals from their surrounding environment (niche) and galvanize intracellular transduction pathways, which deliver information to its genome via activated transcription factors. These transcription factors cooperate with co-activators and chromatin remodelers, intricately balancing the proliferation and cell fate of stem cells. In addition to pro-migratory effect, the pleiotropic effects of TGF-β have an essential role in balancing the self-renewal and differentiation of stem cells. Because TGF-β is abundantly stored in ECM in the latent form, its temporal and spatial activation and intracellular communication with other signaling pathways (e.g., Wnts signaling) should always be considered, so as to better delineate its context-dependent role in the determination of cell fate.
TGF-β signaling in embryonic stem cells
Embryonic stem cells (ESCs) are pluripotent stem cells derived from the inner cell mass of the blastocyst.228 ESCs can differentiate into all cell types in the body (pluripotent), whereas adult stem cells can generate only a limited number of cell types (multipotent). In addition, ESCs are capable of proliferating indefinitely.229 Hence, ESCs are useful tools for both research and regenerative medicine. Many of the responses of stem cells to TGF-β family ligands are regulated by Smad-mediated transcription activation or repression of key genes. Smads cooperate with master regulators of cell differentiation or pluripotency.160,162,163,164,230,231,232,233,234,235 In stem cells, some genes are in an active state within the euchromatin, and their Smad binding sites are accessible to incoming Smad4–RSmad complexes. In this case, TGF-β- or BMP-activated Smads increase or decrease RNA polymerase II (Pol II) action and the transcriptional magnitude of these genes. Nodal signaling modulates cell homeostasis (e.g., SerpinE1) or Smad pathway feedback related genes (e.g., Smad7) in this mode of action. Nodal signal-driven Smad complexes, with the assistance of other DNA-binding cofactors, readily bind to the Smad binding sites, thus upregulating or downregulating the basal activity of these genes. However, most genes that control master regulators of stem cell differentiation are in a quiescent but “poised” state, which can be switched to rapid transcription in response to differentiation signals given the chromatin repressive marks are erased. The nature of the inaccessible poised state implies that activation of ESC differentiation genes by the TGF-β/Smad pathway may be different from that of those readily accessible homeostasis genes. In general, chromatin structure modification that allows the access of pSmad2/3–Smad4–FOXH1 complex to the AREs is pre-requisite for TGF-β/Smad regulation. A typical model explaining how Smad complexes gain access to the AREs of poised master regulator genes has been proposed recently.236 Goosecoid (Gsc) and Mixl1 are two master genes for mesendodermal differentiation of ESCs. The promoters of these two genes are “poised”, with Pol II being paused at the transcription start site and kept from active transcription by a chromatin compacting complex of H3K9me3 and HP1. In response to Nodal/Activin signaling, the downstream pSmad2/3 forms a complex with tripartite motif 33 (TRIM33, also known as TIF1g/ectodermin), and elicits an active chromatin conformation with an added acetylation mark at histone lysine 18 by histone acetyltransferase p300. The pSmad2/3–TRIM33 complexes then translocate to the nucleus, recognize histone marks, displace HP1, and consequently allow the access of Smad4–pSmad2/3 to the AREs within the Gsc and Mixl1 promoters.236 The complex further recruits additional transcriptional regulators, such as FOXH1, further generating the requisite active chromatin conformation and initiating Gsc and Mixl1 transcription. In parallel, Activin/Nodal regulate genes involved in essential cellular functions and homeostasis (such as Smad7 and SerpinE1, which are not in a “poised” state) in a TRIM33-independently way, in which pSmad2/3–Smad4–FOXH1 directly binds the AREs of these genes.129 The result of these events is that Nodal switches Gsc and Mixl1 from the poised state to the activated state, and by orchestrating with other induced functional genes, triggers mesendodermal differentiation. In addition, TRIM33 has been observed to mediate Smad4 ubiquitination,237,238 thus probably providing a negative feedback activity for the inactivation of Smad4 and signal turnover. Specifically, the Pol II kinases CDK8 and CDK9 phosphorylate Smads complexes at an interdomain linker region to activate transcription. In the process, ubiquitin ligases recognize the phosphorylated linker, leading to proteasome-mediated turnover of Smad proteins and signaling attenuation.184,239 In addition, Smad4 can be directly inactivated by poly-(ADP)-ribosylation, which provides another mechanism for decommissioning Smad4 in transcriptional complexes.240
The regulation model of Smads-dependent TGF-β singling may help to better understand the contextual role of TGF-β in balancing stem cell pluripotency and differentiation. The “core transcriptional factors” NANOG, SOX2, and OCT4 form an interactive network that induces pluripotency in ESCs.241,242 This triad mediates chromatin-modifying complexes to establish repressive marks coexisted with activating marks that poise chromatin for abrupt transcription of differentiation genes.242 BMP signaling directs Smad1 to co-occupy the genome with leukemia inhibitory factor (LIF)-activated signal transducer and activator of transcription 3 (STAT3), OCT4, SOX2, and NANOG at sites with activating mark H3K4me3, and thus stimulates self-renewal of ESC.229 The consequently activated genes, including Oct4, Sox2, Nanog, and Id3,243 in turn form a feed-forward cycle. In response to Nodal, the OCT4 complex also activates Nanog and the Nodal negative feedback regulators, Smad7 and Lefty1, Lefty2,162 by directing Smad3 to neighboring sites,162 and thus maintains the self-renewal and pluripotency of ESCs. In the absence of the pluripotency enforcing factor LIF, ESCs respond to autocrine signals and differentiate into mesendodermal cells of the primitive streak and ectodermal cells. Nodal signaling drives mesendodermal differentiation by inducing the expression of poised Gsc and Mixl1 through the pSmad2/3–TRIM33-mediated mechanisms as detailed above.236 The resulting induction of Gsc and Mixl1 commits primitive embryo cells to mesendodermal fates.236 In summary, BMP activates Smad1, which co-occupies the genome with LIF-activated STAT3 and the core pluripotency triad OCT4–SOX2–NANOG, thus stimulating ESCs self-renewal. When contextual self-renewal signals attenuate, the poised chromatin marks provide an entry point for Smad3 complexes to activate differentiation genes.
Undoubtedly, signaling cross talk between Smad2/3 pathway and other signaling pathways also have an important role in dictating the stem cell homeostasis. The complex cross talk between Nodal/Activin/pSmad2/3, ERK/MAPK, and canonical Wnt/GSK3β/β-catenin pathways204 has provide a paradigm for cell fate decisions during early embryonic development. Therefore, even though stem cells have receptors which enable them to respond to TGF-βs and other growth factors, their responses are determined by the integration of both intrinsic (e.g., distinct master regulators present during development) and extrinsic (e.g., ligand activation, competition or cross talk of various niche factors) factors. As TGF-β and BMP antagonize each other and cross talk with other pathways, even subtle differences can elicit profound or contradictory effects. It is now well-recognized that the master regulators expressed by a specific stem cell lineage decide the genes to be regulated by pSmad2/3–Smad4. The concept may be especially pertinent in explaining why stem cells at distinct developmental stages may respond differently to a collection of extracellular cues. Because the abundance of cell-type-specific master genes increases upon differentiation, the master regulators at a specific lineage stage will competitively interact with Smad2/3,242 and ultimately lead to a context-dependent phenotype. In addition, some master regulators may themselves be targets of a signaling pathway, the order in which a cell receives a series of signals also matters. Hence, a spatial and temporal profiling of signaling at the single-cell level is necessary and will lead to promising findings.244
TGF-β signaling in tissue-specific stem cells
Adult stem cells are undifferentiated cells found throughout the body after development. Some adult stem cells are perpetually active, such as intestinal stem cells, whereas others are quiescent, such as stem cells of the hematopoietic system, hair follicles, and mammary gland. Upon specific environmental stimulations, quiescent stem cells can re-enter the cell cycle in response to specific environmental cues, and give rise to lineage-specific progenitors which then differentiate to make functional tissue. TGF-β superfamily members participate in most of these steps, and universally in most tissues. TGF-β superfamily members balance active proliferation and reversible cell cycle exit, thus maintaining reservoirs of stem cells that respond quickly to external changes. Comprehensive coverage of TGF-β superfamily signaling in all adult stem cells is beyond the scope of this review. Here, we discuss briefly only a few of the well-studied adult stem cells whose proliferation and quiescence have been shown with clear TGF-β involvement. Of note, since we propose that the bone remodeling process is a representative model to demonstrate how TGF-β signaling orchestrates with other environment-derived cues to determine the fate of MSCs at the correct sites, the role of TGF-β as a coupling factor for site-directed differentiation of MSCs/progenitors during the bone remodeling process will be particularly discussed in section "TGF-β is the major coupler of bone resorption to formation".
Mesenchymal stem cells
Mesenchymal stem cells are multipotent cells existing in various adult tissues including muscle, adipose tissue, connective tissue, bone marrow and teeth, blood, placenta and umbilical cord.245,246 The differentiation potential of MSCs depends on the niche where they locate. Proliferation of human MSCs can be stimulated by Wnt or TGF-β signaling.247,248,249 TGF-β1 induces Smad3-dependent nuclear accumulation of β-catenin, thereby stimulating MSC proliferation. On the other hand, BMP2 antagonizes Wnt3a signaling and inhibits proliferation of MSCs through interaction of Smad1/5 with Dishevelled-1.250 In addition to proliferation, TGF-β signaling also directs the differentiation fate of MSCs.251 BMPs can induce differentiation of MSCs into chondroblasts or osteoblasts in vitro. TGF-β and activin also promote chondroblast differentiation at early stages, whereas TGF-β inhibits osteoblast maturation at late stages in differentiation.251 Hence, inhibition of TGF-β/activin signaling strongly enhances osteoblast maturation.252 These inhibitory effects of TGF-β/activin signaling on MSC differentiation are possibly mediated by induction of expression of inhibitory Smads, such as Smad6, which in turn represses BMP signaling.253 In addition, BMP7 has been shown to induce the generation of brown fat from MSCs in the absence of the normally required hormonal induction.254 TGF-β is also involved in cardiomyocyte lineage differentiation of MSCs. In human MSCs, TGF-β treatment induces the expression of cardiomyocyte markers including α-smooth muscle actin, myocardin, and calponin 1, along with the Notch ligand, Jagged 1. Increased expression of these genes is Smad3- and Rho kinase-dependent. Prevention of Jagged 1 expression blocks the expression of cardiomyocyte genes, suggesting that Jagged 1 has an important role in TGF-β-induced expression of cardiomyocyte marker genes.255 These studies implicate that the microenvironment is critical for the induction of MSC differentiation into different lineages. Considering the complexity of the bone marrow niche and the involvement of multiple cell-secreted or bone matrix-derived factors in the determination of cell fate, it is more likely that TGF-β functions as a pleiotropic growth factor that reactivates quiescent stem cells into a transient amplification in response to environmental cues (e.g., ECM perturbation). In the meantime, TGF-β acts as a pro-migratory factor recruiting MSCs to the sites where the cell fate is ultimately determined in consultation with other niche-specific factors to meet the tissue need.
Hematopoietic stem cells
HSCs are stem cells found in bone marrow, being able to differentiate into all blood cell types. TGF-β signaling has an important role in regulating the quiescence of HSCs.256 In cell culture, TGF-β signaling-deficient HSCs have a higher proliferative capacity, whereas the quiescence and maintenance of HSCs depend on TGF-β signaling.257,258 Furthermore, the response of HSCs to TGF-β stimulation is biphasic. High concentrations of TGF-β inhibit HSC proliferation, whereas low concentrations of TGF-β stimulate its proliferation.259 As TGF-β is produced in a latent form by various cells, the mechanism of activation is critical for the regulation of HSC quiescence. Non-myelinating Schwann cells have been shown to mediate activation of TGF-β, suggesting that glial cells maintain HSC quiescence by limiting activation of latent TGF-β as components of a bone marrow niche.260 Notably, each subtype of HSCs responds distinctively to the TGF-β signaling. For example, TGF-β signaling leads to different effects on myeloid-biased (My-) and lymphoid-biased (Ly-) HSC subtypes.261
Neural stem cells
Neural stem cells (NSCs) are stem cells giving rise to neural progenitor cells and eventually to neurons, astrocytes and oligodendrocytes.262 Targeted inactivation of TGF-β type II receptor gene in the mid/hind brain in the developing stage enhanced the self-renewal of mouse NSCs, resulting in an enlarged midbrain. In the meantime, inactivation of TGF-β type II receptor was accompanied with ectopic expression of FGF and Wnt ligands suggesting that TGF-β signaling may control the size of the midbrain by antagonizing FGF and Wnt signaling, and consequently inhibits NSC self-renewal.263
In the adult central neural system, NSCs reside in the sub-granular zone of the dentate gyrus and in the sub-ventricular zone adjacent to the lateral ventricles.264 Neurogenesis in this region is regulated by different factors at the level of cell proliferation, fate determination, and survival. TGF-β signaling has an important role in the maintenance and proliferation of NSCs.263,264 In the case of brain lesions or neurodegeneration, TGF-β1 is upregulated and activated in astroglial, neuronal and microglia cells,17,265,266,267 and it coordinates cellular responses associated with either beneficial or detrimental effects on the neurogenesis depending on the cellular context.268,269,270,271 For example, TGF-β is able to induce the synthesis of type 1 Plasminogen activator inhibitor by astrocytes through Smad3-dependent pathway, and thus protects hippocampal, cerebellar, and cortical neurons against N-methyl-d-aspartate toxicity.272
Hair follicle stem cells
Epithelial hair follicle stem cells (HFSCs) are stem cells residing in a specific niche of the hair follicle, referred to as the bulge.273 In adult mice, hair follicles undergo dynamic, synchronized phases of growth (anagen), degeneration (catagen), and rest (telogen). Throughout the telogen phase, HFSCs are quiescent. Their quiescence is maintained in part by BMPs provided from the inner layer of non-stem niche cells274 and from surrounding dermal fibroblasts and adipocytes.275 In the normal niche, BMP signaling must be transiently lowered in favor of transient amplification and lineage commitment of HFSCs, but then it must be restored to the normal level to maintain the quiescence of HFSCs. In addition to BMP, TGF-β also has a role in the telogen phase through induction of apoptosis.276,277 Of note, targeted inactivation of Tgfb1, 2, and 3, respectively, results in differential effects on embryonic hair follicle development. Tgfb2−/− mice exhibit a profound delay in hair follicle morphogenesis, characterized by a 50% reduced number of hair follicles.278 Mechanistically, TGF-β2, which is primarily produced by dermal papillae, possibly stimulates HFSC proliferation by counteracting BMP-mediated quiescence in the niche.244 Moreover, TβRII-deficient HFSCs display elevated pSmad1 and BMP signaling and delayed hair cycle entry, further suggesting the antagonism of TGF-β and BMP signaling in the determination the HFSC proliferation and cell fate. The antagonism between TGF-β and BMP signaling is probably through Tmeff1, which can block BMP2-mediated mesoderm induction in Xenopus embryos.279 Induction of TGF-β2 signaling and inhibition of BMP signaling in activated hair germ progenitors are normally accompanied with Tmeff1 upregulation,244 and TβRII mutation diminishes Tmeff1. Knockdown of Tmeff1 in wild-type HFSCs leads to abrogation of TGF-β2-mediated suppression of BMP signaling, and delays hair follicle regeneration. Moreover, Tmeff1 diminishes the response of wild-type HFSCs to BMP signaling in vitro, in a fashion similar to that of TGF-β2.244
Skeletal muscle stem cells
Muscle stem cells (MuSCs) are stem cells isolated from skeletal muscle with myogenic potential in response to environmental cues.280,281,282 In the adult, quiescent MuSCs reside between muscle fibers and surrounding basement membranes. BMP4 signals from emerging tendons impact the behavior of a subpopulation of dividing MuSCs at the tips of fetal skeletal muscle.283 Upregulation of BMP signaling promotes proliferation of MuSCs, whereas blockade of BMP signaling results in fewer MuSCs.283 Upon muscle injury, quiescent MuSCs proliferate and differentiate into myoblasts, and fuse to form de novo multinucleated myofibers.284 As the muscle ages, its regenerative capacity declines, possibly due to diminished activation of the Notch pathway.285,286 In addition, aged muscle produces excessive TGF-β, which induces erroneously high levels of pSmad3 in resident MuSCs and disrupts their regenerative capacity, whereas attenuation of canonical TGF-β signaling in aged, injured muscle restores MuSC activity.287 These findings indicate a shift from active Notch to active TGF-β/pSmad3 signaling in the MuSC niche with age, and the antagonism between TGF-β and Notch has an essential role in controlling MuSC proliferation. Of note, active Notch reduces TGF-β/pSmad3-dependent upregulation of cyclin-dependent kinase (CDK) inhibitors p15, p16, p21, and p27.287 These findings suggest that an age-specific interaction between TGF-β and Notch signaling controls CDK inhibitor levels in MuSCs and in turn governs tissue regenerative capacity upon muscle injury.
Taken together, the knowledge regarding how TGF-β signals acts with other contextual environmental cues to determine stem cell fates is not only fundamental to stem cell biology, but could useful for regenerative medicine. As the goal of regenerative medicine is to replace malfunctioning cells/tissues/organs with the competent ones, targeted manipulation of these signals to direct stem cell differentiation into specific cell type will undoubtedly contribute to future applications of stem cells in regenerative medicine.
Inhibition of TGF-β enhances reprogramming of somatic cells
Adult somatic cells can be forced to reprogram into induced pluripotent stem cells (iPSCs) by ectopically expressing certain transcription factors. The classical iPSC techniques was pioneered by forced expression of the four “Yamanaka factors”, Oct4, Sox2, Klf4, and c-Myc in the mouse embryonic fibroblasts and thus reset the differentiation clock of these cells back to the pluripotent state equivalent to a blastocyst.288 Many other reprogramming techniques or conditions have been developed afterward, mainly aimed to decrease the risk of genomic insertions of exogenous reprogramming factors or to increase the efficiency of reprogramming process. Because the four “Yamanaka” factors orchestrate to inhibit the TGF-β signaling pathway that induces epithelial–mesenchymal transition (EMT) through Snails, factors that antagonize TGF-β signaling are believed to enhance reprogramming. Indeed, small molecules that can selectively inhibit TGF-β type I receptor kinases enhances iPSC induction and can replace the requirement of Sox2 for iPSC induction. Inhibition of TGF-β signaling in partially reprogrammed iPSCs even induces Nanog expression and ultimately promotes full reprogramming.289,290,291 Like small molecules, Smad7, one of the I-Smad proteins can also replace Sox2 to enhance the reprogramming process.292 Conversely, either treating reprogramming iPSCs with TGF-β, or introducing an activated TGF-β type I receptor decreases reprogramming efficiency.290,293 Furthermore, expression of miRNAs that inhibits TGF-β- signaling and EMT enhances iPSC reprogramming.293,294,295 Therefore, TGF-β signaling suppresses somatic cell reprogramming possibly by induction of EMT, although it is important for ESC self-renewal. On the contrary, BMP signaling can induce the mesenchymal–epithelial transition (MET) process, a reversal process to EMT, and thus counteracts TGF-β stimulation in some contexts and promotes reprogramming into iPSCs.292,296
TGF-β is the major coupler of bone resorption to formation
The musculoskeletal system is dynamic in that it undergoes continual adaptations during vertebrate life to attain and preserve skeletal size, shape, microstructure and to regulate mineral homeostasis. In addition, its matrix is a major reservoir of growth factors whose bioavailability and temporal and spatial activation modulate the balance between normal physiology and pathology of tissue/organs involved. Skeletal homeostasis is maintained by intricate regulation of the bone remodeling process. A typical bone remodeling cycle consists of three distinct phases: (a) initiation phase, during which osteoclasts are formed and resorb damaged bone; (b) reversal phase, the transition of osteoclast to osteoblast activity; and (c) formation phase, when osteoblasts rebuild an equivalent amount of bone to that resorbed.1 Termination of osteoclast bone resorption and recruitment/differentiation of MSCs are generally recognized as essential steps in the reversal phase.[1] Our recent findings, in combination with those from others, have clearly demonstrated that the bone remodeling process is an optimal model to unveil the mechanisms of how pro-migratory factors present or released in the stem cell niches are orchestrated to mobilize/recruit stem cells/progenitors for a rapid but also transient amplification and differentiation, thus maintaining or restoring homeostasis of involved tissues/organs.
Bone remodeling is the driving force for the evolution of the terrestrial skeleton
Multiple adaptations were needed for animals to evolve from aquatic to terrestrial life, and a coupled bone remodeling is critical for this process. Osteoblastic bone formation exists in all vertebrates. However, osteoclasts and bone resorption only occur permanently in amphibians and terrestrial animals; whereas the expression of primitive bone resorbing cells and/or osteoclasts are species-dependent in fish, occurring at either specific developmental stages or in times of physiologic stress, such as transitioning from sea to either brackish or fresh water.297,298,299,300 A major skeletal difference between aquatic and terrestrial animals is that terrestrial vertebrates have to carry their body weight. A lighter skeleton affords a quick, efficient ability to enhance survival. Bone remodeling, with the evolutionary development of osteoclasts, therefore solved the issues of skeletal weight and a mechanism to regulate mineral metabolism through bone resorption.297 However, to continue to maintain the integrity of the skeleton, temporal and spatial coordination of osteoblast bone formation with osteoclast bone resorption also became essential in terrestrial vertebrates. Quite a few cells have been identified to participate in the highly coordinated bone remodeling process.
The osteocyte is the most abundant cell population in mineralized bone tissue.301 It has been considered as the prominent candidate to sense dynamic changes in mechanical loading, and to initiate the remodeling process. Osteocytes are cocooned in fluid-filled cavities (lacunae) within the mineralized bone,301 and possess long dendrite-like processes that extend throughout canaliculi (tunnels) within the mineralized matrix. Osteocytes interact with each other and osteoblasts via these dendrite-like processes,302 functioning as a mechanosensor to coordinate activities of other bone cells.303 Recent studies have revealed that osteocytes expressed higher concentrations of receptor activator of nuclear factor kappa B ligand (RANKL) than mature osteoblasts and bone marrow stromal cells, further underscoring the essential role of osteocytes to initiate bone remodeling via osteoclastogenesis.304,305 In addition, osteocytes regulate bone remodeling by expressing sclerostin (SOST),306,307,308,309 which negatively regulates bone formation by inhibiting BMP and WNT signaling in osteoblast lineage cells.310,311,312
Other cells present in the bone marrow also dynamically cross talk with osteoblast and osteoclast via either direct cell contact or soluble signals, thus modulating bone homeostasis during normal physiology.313 Mature B cells produce 50% of total bone marrow-derived osteoprotegerin (OPG), a decoy receptor for RANKL. Indeed, mice lacking B cells present an osteoporotic bone phenotype.314 T cells have been reported to promote OPG production by B cells, possibly through CD40 ligand (CD40L) to CD40 co-stimulation.314 Consequently, T-cell-deficient nude mice, CD40 KO mice, and CD40L KO mice display diminished bone marrow OPG production and osteoporotic phenotype.314 Megakaryocytes derived from HSCs can also express RANKL and OPG and secrete an unknown soluble anti-osteoclastic substance,315 and thus participate in the regulation of bone remodeling. Osteal macrophages (Osteomacs) are a special subtype of macrophage residing on or within endosteal and periosteal surfaces. Osteomacs have an important role in musculoskeletal development, homeostasis and repair.316 Osteomacs form a canopy which generates a unique microenvironment to facilitate “coupled” osteoclast resorption and osteoblast formation in a temporary anatomical structure known as “basic multicellular units” (BMUs).317 Osteomacs are required for full functional differentiation of osteoblasts in vitro. Depletion of macrophages results in complete loss of osteoblast bone-forming surface evidencing that osteomacs are also required to maintain mature osteoblasts.318
Mesenchymal stem cells are recruited by bone matrix TGF-β to couple bone resorption and formation
Osteoblasts are derived from bone marrow MSCs (also referred to as bone marrow stromal cells or skeletal stem cells).221 Ex vivo mouse MSCs can be identified based on the absence of hematopoietic and endothelial markers and the presence of PDGFRa.319,320,321 PDGFRa+ Sca-1−CD45−Ter119−cells also express high levels of HSC niche factors Cxcl12 and Stem cell factor (Scf),319,321 hence they not only contribute to osteoblast and adipocyte lineage,321 but also constitute the cellular component of the HSC niche. Cells that express the Nestin-GFP transgene (Nes-GFP) contain all of the mesenchymal progenitor activity (fibroblastic colony forming units; CFU-F) in mouse bone marrow.322,323 These Nes-GFP+ cells segregate with distinct vessels in vivo. The Nes-GFP bright cells, which exclusively locate along arterioles, are much rarer than the reticular Nes-GFP dim cells largely associated with sinusoids. The Nes-GFP bright cells are quiescent, contain the most CFU-F, and closely associate with HSC quiescence and maintenance in the bone marrow.322 Notably, a recent study reported that the number of both Nes-GFP bright and dim cells significantly decreased in adult endochondral bone, and only a small number of Nes-GFP dim cells were detectable in the adult bone marrow.324 These data indicate that Nestin+ MSCs may be more relevant to the endochondral ossification during development rather than the skeletal remodeling process in adult, or the expression of Nestin is transient in the MSCs. Another recent study by the Morrison group has demonstrated that Leptin Receptor (LepR) is a marker that highly enriches bone marrow MSCs accounting for 94% of adult bone marrow CFU-Fs.325 LepR+ cells are Scf-GFP+, Cxcl12−DsRedhigh, and Nes-GFP dim. They emerge postnatally and give rise to most bone and adipocytes formed in adult bone marrow. LepR+ cells are normally quiescent, but they transiently proliferate to participate in bone regeneration after irradiation or fracture.325 Therefore, LepR+ cells may represent the major cellular source of bone and adipocytes in adult bone marrow, and possibly account for the largest proportion of MSCs that participate in the bone remodeling process and maintain the HSCs in adult bone marrow.326
To overcome the challenge during evolution of vertebrate animals transitioning from ocean to land, cytokines and growth factors were likely selected to deposit in the bone matrix during bone formation. A large reservoir of factors in the bone matrix becomes available during osteoclastic bone resorption to induce recruitment and differentiation of MSCs and limit further osteoclastic resorption, thereby coupling bone resorption and formation. A critical step of coupled bone remodeling is to recruit MSCs to the bone resorption sites. Vertebrate TGF-βs 1–3, which appear to have no counterparts in nematodes (Caenorhabditis elegans) or insects (Drosophila),327 may have evolved to serve as the major coupling factor that coordinates the dynamic bone remodeling process of vertebrate mammals after they transitioned from ocean to land (Fig. 2). A more complex mechanical loading to the terrestrial vertebrates may promote the release of active TGF-β from the bone matrix328 for a coupled bone remodeling. In addition, osteoclastic bone resorption evolved by terrestrial vertebrates have an essential role in activating TGF-β during bone remodeling. Osteoclasts can form an integrin-dependent sealing zone between the bone and the cell,106 generating an acidic environment ideal for the activation of latent TGF-β richly deposited in the bone matrix.37,107,108,109 Osteoclasts can also activate latent TGF-β by secretion of proteases in the absence of a low-pH environment,109 leading to the proteolytic cleavage of LTBP1329 and enrichment of active TGF-β within the immediate environment of the bone resorption site. More importantly, our recent work has clearly demonstrated that TGF-β1, as one of the most abundant bone matrix cytokines,330 is activated during osteoclastic bone resorption and induces migration of MSCs to the bone remodeling sites via Smad-dependent signaling.8 TGF-β can also promote the migration of MSCs via JNK pathway and the induced expression of MCP-1, although these findings have only been reported in an artery injury model.224 In parallel, other growth factors released from bone matrix during resorption, such as IGF-1, promote osteoblast differentiation of MSCs.222 Studies of the effects of TGF-β1 on osteoclast activity show that high concentrations and prolonged exposure to active TGF-β1 inhibit migration of osteoclast precursors (macrophages/monocytes).331 Hence, the gradient of TGF-β1 generated at the resorptive site inhibits further recruitment of osteoclast precursors and avoids excessive bone resorption. This is particularly important for coupled bone remodeling since continuously elevated osteoclastic activity reduces the quality of bone and results in pathologic conditions.

TGF-β1 is released from the bone matrix and activated during osteoclast-mediated bone resorption, creating a gradient. TGF-β1 induces migration of MSCs to the bone remodeling sites to couple bone resorption and formation. The bone-resorptive microenvironment also provides signals (e.g., IGF-1) that direct the lineage-specific differentiation of MSCs. In addition, PTH orchestrates signaling of local factors and thus regulates cellular activities, including those of MSCs, T cells, and other PTH-responsive cells in the bone marrow to coordinate bone remodeling
Interestingly, osteoblast bone formation still exists in vertebrates that have defective osteoclasts function to release sufficient bone matrix-derived growth factors, indicating that osteoclasts per se may also be able to produce coupling factor(s).332 These potential coupling factors released by osteoclast include Sphingosine 1-phosphate, platelet-derived growth factor-bb (PDGF-bb), and the EphB4.ephrin-B2 bidirectional signaling complex. Sphingosine 1-phosphate is secreted by osteoclasts, and functions to recruit osteoblast and promote survival of mature osteoblast.333 Osteoclasts can also secret PDGF-bb, which not only induces migration and osteogenic differentiation of MSCs,334 but also controls osteoblast chemotaxis via PDGF-bb/PDGFR-beta signaling.335 More importantly, our recent findings have shown that PDGF-bb secreted by preosteoclasts is able to induces the formation of CD31(hi)Emcn(hi) vessel, a specific vessel subtype that functions to couple angiogenesis and osteogenesis during bone modeling and remodeling.336 EphB4 receptors are expressed on osteoblasts, whereas the ligand ephrin-B2 is expressed by osteoclasts. Signaling through EphB4 receptors induces osteoblastic differentiation, whereas signaling through ephrin-B2 inhibits osteoclastogenesis.337 The bidirectional signaling complex of EphB4.ephrin-B2 functions to activate osteoblastic bone formation and inhibit osteoclastic bone resorption simultaneously, and this is particularly critical at the transition point of bone remodeling. Notably, the direct cell contact between osteoblasts and osteoclasts within the BMU is not always possible, and osteoblastic bone formation could sustain long after osteoclasts vacate the resorption site. This fact suggests that both contact-dependent and contact-independent signals are involved in a coupled bone remodeling process.
Bone remodeling dynamically changes the bone marrow microenvironment
The self-renewal, transient amplification, or differentiation of bone marrow MSCs are dynamically regulated by the bone marrow microenvironment.338,339,340 External signals produce transcription responses that allow cells to respond to cues from their environment at a certain magnitude and duration. In the reversal phase of bone remodeling, the bone-resorptive microenvironment provides signals that inhibit bone resorption and promote bone formation by recruiting MSCs and inducing osteoblastic differentiation. Multiple cytokines, growth factors, and minerals are released from the bone matrix or secreted by local cells during the bone remodeling. For example, IGF-1 released from the bone matrix can stimulate osteoblast differentiation of MSCs by activation of mammalian target of rapamycin (mTOR) through the PI3K/Akt pathway.222 Semaphorin 4D (SEMA4D), which is expressed on the cell surface of osteoclasts, binds to its receptor, Plexin-B1, on osteoblasts to inhibit the RhoA/Rho-associated protein kinase (ROCK) pathway.341 The ROCK pathway normally phosphorylates IRS-1, a key factor in the PI3K/Akt/mTOR pathway.341 Therefore, osteoclast expressing SEMA4D prohibits MSC differentiation via cell-to-cell contact, creating a boundary between bone resorption and formation. Thereby, the dynamic changes in the bone marrow microenvironment result in the coordination of the reversal phase during coupled bone remodeling.
In addition, the elasticity of the bone matrix has an important role, with a stiffer matrix directing differentiation of MSCs into osteoblasts.330 At fresh bone resorption sites, the bone mineral matrix without lining cells is exposed, providing a stiff elastic microenvironment in favor of osteoblastic differentiation. Blood vessels are in close proximity to bone remodeling, and a complex interrelationship between angiogenesis and osteogenesis has been reported.342 Basic fibroblast growth factor (FGF2) is a potent mitogenic factor that can promote angiogenesis by inducing endothelial cell proliferation, migration, and expression of necessary angiogenic factors including proteases, growth factors, and integrins.343,344,345,346 TGF-β has been shown to enhance production FGF2 in osteoblasts.347,348,349 In aortic endothelial cells, TGF-β stimulation of an ALK5/TβRII-Smad2 complex enhances expression of vascular endothelial growth factor (VEGF),350 which is a strong pro-angiogenic factor. Whether a similar mechanism of action exists in cells in the bone remodeling site remains to be investigated. HSCs are present in the same niche as MSCs.351 Although MSCs and osteoblast precursors can influence the fate of HSCs by producing several key maintenance factors including CXC chemokine ligand 12 (CXCL12), angiopoietin 1(ANGPT1), KIT ligand (KITL), and vascular cell adhesion molecule 1 (VCAM1),323,351,352,353 the opposite remains unknown.
PTH serves as endocrine regulator of skeletal TFG-β to control bone homeostasis in terrestrial vertebrates
The parathyroid gland evolved in amphibians,354 and represents the transition of aquatic to terrestrial life. PTH is the major endocrine hormone produced by the parathyroid gland, which regulates calcium homeostasis. Interestingly, permanent emergence of osteoclasts and bone resorption is also observed as vertebrates transitioned to land, 297,298,299,300 and favors survival by lightening skeletal weight and a ready source of calcium. In addition to the critical role of PTH mobilizing calcium in times of need, PTH has also been shown to orchestrate signaling of local bone factors including TGF-β, Wnts, BMP, and IGF-1, integrating systemic control of bone remodeling.185,355,356,357,358,359,360,361,362 (Fig. 2)
PTH orchestrates the signaling of many local factors that determine the fate of MSCs. Endocytosis of growth factors and G protein-coupled receptors is known to integrate different signaling pathways.363 PTH can induce the recruitment of TβRII as an endocytic activator.185 TβRII directly phosphorylates the cytoplasmic domain of the PTH type 1 receptor (PTH1R) and facilitates PTH-induced endocytosis of a PTH1R-TβRII complex, resulting in downregulation of TGF-β effects, likely limiting further MSC recruitment. Concomitantly, PTH stimulates the commitment of MSCs to the osteoblast lineage by enhancing bone morphogenetic protein (BMP) and Wnt signaling. Low-density lipoprotein-related protein 6 (LRP6) serves as a co-receptor in the canonical Wnt pathway364 and interacts with BMP signaling.365 PTH has been shown to recruit LRP6 as a co-receptor, which recruits axin from the cytoplasm to stabilize β-catenin.360 PTH binding to PTH1R can also induce endocytosis of a PTH1R/LRP6 complex, resulting in enhanced BMP-pSmad1 downstream signaling, and ultimately promotes osteoblastic differentiation of MSCs.362 Deleting LRP6 in mature osteoblasts in mice results in a loss of anabolic response to PTH.366 PTH can expand Nestin-positive MSC populations,323,352,367 although the precise mechanism of action remains unclear. PTH-enhancement of MSC transient amplification, differentiation, and function is a part of the integration of the signaling networks of local factors for the spatial-temporal regulation of bone remodeling.
PTH can modify other cells in the bone marrow microenvironment. Intermittent PTH treatment increases the number of osteoblast by converting lining cells to active osteoblasts.368 PTH stimulates bone marrow CD8+ T cells to produce large amounts of Wnt10b, which activates Wnt signaling in MSCs and osteoblast precursors, thus increasing osteoblast proliferation and differentiation.369 PTH has also been shown to improve the bone marrow microenvironment by spatially relocating small blood vessels in proximity to sites of new bone formation, possibly via upregulation of VEGF-A and neuropilin-1 and -2.370 The consequent proximity of blood vessels allows more efficient delivery of nutrients to support new bone formation.
Taken together, TGF-β is a critical factor in the temporal and spatial coupling of bone remodeling. PTH, the hormone with evolutionary significance, may act as the major endocrine regulator of bone remodeling by orchestrating the signaling of many pathways and directing the fate of MSC.
Aberrant TGF-β signaling leads to multiple pathologies
Temporal and spatial regulation of TGF-β activation is essential in maintenance of tissue homeostasis and regeneration of the damaged tissue by recruiting stem/progenitor cells to the right place at the right time, whereas sustained abnormal activation of TGF-β may lead to pathological conditions due to excessive recruitment of stem/progenitor cells and their subsequent differentiation. Here, we discuss multiple pathologies associated with abnormal TGF-β signaling. Typical skeletal disorders are taken as examples to demonstrate how abnormalities of TGF-β signaling lead to loss of site-directed recruitment of MSCs that causes uncoupled bone remodeling phenotypes. Other diseases with well-documented TGF-β involvement, such as tissue/organ fibrosis and bone metastases of cancer are also discussed in this section.
TGF-β-induced osteoid islets in the subchondral bone initiate osteoarthritis
Osteoarthritis (OA) is a degenerative joint disease characterized by progressive articular cartilage degradation, subchondral bone sclerosis, reduced mobility, and debilitating joint pain. Although it is still under debate whether OA starts in cartilage or bone,371 subchondral bone sclerosis has now been recognized as a major contributor to the cartilage degeneration by generating unevenly distributed stress on the articular cartilage, leading to its gradual deterioration.372,373 Recent evidence has demonstrated that the onset of OA is associated with increased bone remodeling in the early stage,374,375 and subsequent slow turnover/densification of the subchondral plate and complete loss of cartilage.376,377,378 Notably, subchondral sclerosis without precedent stage of increased bone remodeling does not lead to progressive OA in experimental models.376,379 Therefore, both early stage increased bone loss, and the late-stage subchondral densification are important for the pathogenesis of cartilage degeneration in OA. Importantly, our recent findings have shown that this spatial and temporal separation of subchondral bone phenotype is likely initiated by the abnormal activation of TGF-β.6
Bone is constantly remodeled or modeled in response to changes in mechanical loading, particularly, when joint stability is decreased such as occurs during aging or with ligament injury or obesity. As osteocytes are the principal mechanosensors, aberrant mechanical loading may result in alterations in the release of signaling molecules such as OPG, RANKL, and sclerostin to increase osteoclast activity and turnover rate in OA subchondral bone. In post-surgery of anterior cruciate ligament transection (ACLT) OA mice, osteoclastic bone resorption in the subchondral bone was significantly increased as early as 7 days. The excessive release of active TGF-β1 to the bone marrow caused by osteoclastic bone resorption recruits Nestin+ MSCs to form marrow osteoid islets. Notably, osteoclastic bone resorption was temporally and spatially separated from TGF-β1-induced recruitment of MSCs in the marrow, resulting in aberrant bone formation. The uncoupled bone remodeling process in subchondral bone alters its micro-architecture and eventually compromises the functional integrity of “subchondral bone-articular cartilage complex”.380 The notion was further supported by the development of OA-like changes in the CED mouse model with aberrant activation of TGF-β1.6
Biomechanical factors have an essential role in the degenerative process of articular cartilage, which is maintained in a mechanically active environment. The subchondral bone volume and subchondral bone plate (SBP) thickness fluctuate substantially in ACLT rodent models. In human osteoarthritis joints, SBP is markedly thicker relative to those of healthy subjects.376 Computerized stimulation models of human knee OA suggest that expansion of 1–2% subchondral bone significantly changes the distribution of articular cartilage stress. In addition, the formation of osteoid islets and aberrant bone formation induced by TGF-β1 changes micro-architecture of subchondral bone.6 Although intermittent articular loading seems to be necessary for normal cartilage metabolism, abnormal loading patterns mediated by TGF-β1 may irregularly induce progressive cartilage degeneration.381 Cell death, water content and fibronectin content in the cartilage explants are increased in a load duration and magnitude-dependent manner.382 Chondrocytes in the superficial zone are more vulnerable to repetitive mechanical loading than those in the deeper layer of articular cartilage.383,384 Vigorous cyclic loading also leads to cartilage matrix damage such as breakage of collagen fiber and proteoglycan depletion possibly due to increased MMP-3.385 Therefore, the fluctuation of the mechanical property of subchondral bone inevitably affects its capacity to dissipate the mechanical stimuli from the joint surface and consequently leads to cartilage degeneration in OA. The loss of cartilage integrity will in turn increase the overload of the joint, leading to subchondral bone sclerosis as the joint attempts to adapt to the increased loads. Ultimately, this positive feedback loop causes progressive deterioration of articular cartilage in clinically evident OA.376
In addition to induction of aberrant bone formation in the subchondral bone of OA, excessive activation of TGF-β can affect multiple joint tissues. TGF-β signaling is crucial in normal cartilage development and the maintenance of articular chondrocyte homeostasis in synovial joints.386,387 However, recent findings by van der Kraan et al.388 have shown that ageing process may switch the TGF-β signaling in chondrocytes from the canonical anabolic ALK5-Smad2/3 pathway to the catabolic ALK1-Smad1/5/8 pathway, suggesting that excessive activation of TGF-β in aged individuals might actually exacerbate cartilage deterioration. Excessive TGF-β also induces synovial fibrosis and osteophyte formation,389,390 which are common features of osteoarthritis and also closely associated with its progression.
Genetic mutations in TGF-β signaling components cause bone-associated disorders
TGF-β and its family members are multi-functional growth factors that have critical roles in development and maintenance of the skeleton. Various pathological skeletal phenotypes are consequent to mutations in genes encoding ligands, receptors, and signaling molecules of the TGF-β family. The TGF-β related diseases form an important subgroup of skeletal dysplasia,391 which covers both monogenic and polygenic diseases involving the skeletal system. The monogenic skeletal disease is caused by a single gene mutation and belongs to relatively simple and traceable Mendelian disease. In contrast, the polygenic skeletal disease is multifactorial (e.g., osteoporosis). Its phenotype is determined by combined and concerted effects of a group of genes (susceptibility genes) and the environment. Hence, its inheritance is complex and less predictable. These diseases give us clues to delineate the roles of TGF-β in the skeleton in vivo as well as physiological mechanisms controlling the skeletal system. Here we discuss representative monogenic and polygenic bone disorders associated with TGF-β mutations with a focus on their significance for understanding mechanisms regulating the skeletal system.
Monogenic bone disorders
In general, most if not all monogenic TGF-β-related skeletal disorders share a common pathogenesis. A specific gene mutation leads to excessive and sustained production or activation of TGF-β ligand (Fig. 3), which distorts the normal bone remodeling process. The temporally and spatially regulated physiological TGF-β gradient may be disrupted and lead to excessive recruitment of MSCs or altered downstream targeted effects. Skeletal phenotypes with uncoupled bone remodeling appearing as sclerosis result in addition to other specific end-organ effects.

① Mutations in genes involved in the synthesis/assembly of extracellular matrix (ECM), e.g., Fibrillin-1 (FBN1), cause compromised matrix sequestration of the large latent complex of TGF-β and excessive TGF signaling, ultimately resulting in genetic disorders such as Marfan syndrome (MFS) and stiff skin syndrome (SSKS). ② Mutations in the region encoding latency-associated peptide (LAP) increase the release of active TGF-β, and cause Camurati–Engelmann disease (CED). ③ Mutations in genes encoding TGF-β type I and II receptors (TβRI/II) lead to compensatory synthesis of TGF-β ligand, and cause Loeys–Dietz syndrome (LDS). ④ Mutations in smads repressor, such as SKI, super-activate TGF-β signaling and causes Shprintzen–Goldberg syndrome (SGS) phenotypes. VSMC vascular smooth muscle cells
Camurati–Engelmann disease (CED) is characterized by a fusiform thickening of the diaphysis of the long bones and skull. CED is caused by mutations in the TGF-β1 gene, resulting in premature activation of TGF-β1.392,393,394,395 Approximately 11 different TGF-β1 mutations have been identified from CED-afflicted families.396,397 All mutations are in the region encoding LAP, which either destabilize disulfide bridging of LAP or affect secretion of the protein, leading to enhanced TGF-β1 signaling. Patients with CED show a typical uncoupled bone remodeling phenotype, characterized with decreased trabecular connectivity despite normal osteoblast and osteoclast numbers.397,398 The conditioned medium collected from cells expressing the CED mutant TGF-β1 shows significantly increased ratio of active/total TGF-β1, and it hyperactively induces the migration of MSCs.8 However, the targeted recruitment of MSCs to the bone remodeling site is likely disrupted due to loss of the TGF-β gradient.
In addition to CED, several other genetic disorders with skeletal manifestations including Marfan syndrome (MFS), Loeys–Dietz syndrome (LDS), Shprintzen–Goldberg syndrome (SGS), and neurofibromatosis type 1 (NF1) also involve aberrant TGF-β signaling. MFS is caused by mutations in the fibrillin-1 encoding gene (FBN1) and often results in aortic dilatation, myopia, bone overgrowth, and joint laxity.399,400,401,402 Fibrillin-1 is deposited in the ECM and interacts directly with latent TGF-β-binding proteins (LTBPs), keeping TGF-β sequestered and unable to exert its biological activity. In MFS, secondary to mutated structural fibrillin-1, excessive activation of TGF-β in the lungs, heart valves, and aorta causes the pathological features.400,403 LDS is caused by inactivating mutations in genes encoding TβRI and TβRII.404 Physical manifestations of LDS include arterial aneurysms, hypertelorism, bifid uvula/cleft palate and bone overgrowth resulting in arachnodactyly, joint laxity, and scoliosis. Although the exact mechanism is still unclear, TGF-β signaling is elevated in affected tissues of LDS despite the inactivating mutation.404 A recent study by Dietz and coworkers405 suggests that vessel smooth muscle cells (VSMCs) that carry the LDS mutation may compensate for their signaling deficiency by upregulating the expression of TGF-β ligands. Indeed, tissue from LDS patients or mice shows increased expression of TGF-β1 regardless of etiologies.406,407 The upregulated TGF-β1 production by specific cell types (e.g., VSMCs) could lead to paracrine overdrive of TGF-β signaling in neighboring cells despite the presence of mutant receptors. In addition, infiltrating CD45+ cells in the aortic root adventitia as the disease progresses, might also contribute to excessive TGF-β1 production.405 SGS is caused by mutations in the Sloan-Kettering Institute (SKI) proto-oncoprotein, 408,409 and results in similar physical features as MFS plus craniosynostosis. SKI negatively regulates Smad-dependent TGF-β signaling by impeding Smad2 and Smad3 activation, preventing nuclear translocation of the SMAD4 complex and inhibiting TGF-β target gene output by competing with p300/CBP for SMAD binding and recruiting transcriptional repressor proteins, such as mSin3A and HDACs.410,411,412 The neurocutaneous syndrome, neurofibromatosis type 1 (NF1), has skeletal features including kyphoscoliosis, osteoporosis, and tibial pseudoarthrosis. The Nf1flox/−;Col2.3Cre+ mouse model that closely recapitulates the skeletal abnormalities found in the human NF1 disease has hyperactive TGF-β1 signaling.413 The exact mechanisms underlying mutant neurofibromin-associated hyperactivation of TGF-β signaling remain unknown, particularly in relation to the osseous defects.
Polygenic bone disorders
Osteoporosis is the pathological decrease of bone tissue leading to an increased risk of fracture, mainly in spine (vertebral body), distal radius, and femoral neck. It has been estimated that osteoporosis results in more than 1.3 million osteoporotic fractures per year in the US, and more than 40% of postmenopausal women are reported to have sustained a fracture.414,415 Osteoporosis is classified as primary and secondary osteoporosis based on its etiology. Primary osteoporosis consists of postmenopausal and senile osteoporosis. Postmenopausal osteoporosis is the most common bone and joint disease in women after menopause. Secondary osteoporosis results from various diseases relating to bone metabolism, or prolonged use of medications such as glucocorticoids. The underlying mechanism in all cases of osteoporosis is an imbalance between bone resorption and bone formation. Three main mechanisms interplay and underlie the development of bone fragility, including an inadequate peak bone mass, excessive bone resorption, and inadequate bone formation during remodeling.416
Polymorphisms in several genes associated with bone mass or osteoporotic fracture have been identified by population-based and case-controlled studies. Candidate genes include TGF-β, vitamin D receptor (VDR), estrogen receptor (ER), and type I collagen.417 Specific TGF-β gene polymorphisms that are associated with BMD and/or osteoporotic fractures have been identified.418,419 A C/T polymorphism which causes a proline-leucine substitution at amino acid 10 in the TGF-β1 encoding region has been identified. The C allele is associated with increased BMD and a reduced osteoporotic fractures risk in postmenopausal Japanese women. In addition, the C allele is associated with circulating levels of TGF-β1, suggesting that the C allele may influence protein secretion or stability.420 However, the underlying molecular mechanisms are still unclear. Of note, subsequent large-scale association studies in Japanese421 and other ethnic populations422,423 have not been able to replicate the association.
Sustained TGF-β activation results in organ fibrosis
Progressive fibrosis in tissues/organs, such as the liver, lung, kidney, heart, bone, and skin, is a major health burden and cause of patient suffering. It has been well-recognized that fibrogenesis is not a unique pathological process but the consequence of excessive tissue repair. A central event in tissue repair is the release of cytokines in response to injury. Accumulating data has evidenced that TGF-β can initiate and terminate tissue repair, and its sustained production/activation result in the development of tissue/organ fibrosis.
Excessive or sustained production/activation of TGF-β is a key molecular mediator of tissue fibrosis (Fig. 1). The pro-fibrogenic effect of TGF-β is mainly attributed to its capacity to attract fibroblasts and to stimulate their proliferation. Excessive activation of TGF-β also causes pulmonary and hepatic fibrosis by inducing EMT in alveolar epithelial cells and trans-differentiation of quiescent hepatic stellate cells into myofibroblasts, respectively.424,425 In addition, EMT may also contribute to TGF-β-induced cardiac fibrosis.426 Moreover, our recent data support the concept that TGF-β may promote the migration of either local resident MSCs or bone marrow MSCs to the injured tissues by local elevated levels of active TGF-β. The recruited MSCs can differentiate into myofibroblasts under sustained TGF-β stimulation, further contributing to the fibrosis/sclerosis of involved tissue/organs.224,225 Here, we elaborate on the specific roles had by TGF-β in the fibrosis of major organs.
Kidney
Kidney is particularly vulnerable to the consequences of fibrosis, largely due to its intricate anatomical architecture and filtrating function. Chronic kidney diseases (CKD) are characterized by the accumulation of ECM in the glomeruli (glomerulosclerosis) and/or the tubular interstitium (tubulointerstitial fibrosis). TGF-β has been identified as an important pathogenic factor in the course of CKD.
Accumulating evidence suggests a causal relationship between elevated level of TGF-β1 and the accumulation of ECM during glomerulosclerosis. In vitro incubation of normal glomeruli or mesangial cells with TGF-β1 led to accumulation of ECM characterized by increased production of extracellular-matrix proteins, suppressed protease activity, and increased integrin expression.427,428,429 Injection either a TGF-β1 neutralizing antiserum or a proteoglycan that binds TGF-β1 in nephritic rats prevented the increased production of matrix proteins by the glomeruli and blocked the accumulation of matrix.430,431 More importantly, overexpression of the TGF-β1 gene in rat kidneys led to rapid development of glomerulosclerosis.432 TGF-β can cause podocytopenia by inducing podocyte apoptosis and detachment from the glomerular basement membrane, thus initiating the development of glomerulosclerosis.433,434 TGF-β also induces mesangial expansion caused by mesangial cell hypertrophy, proliferation (and eventually apoptosis), and ECM accumulation.435,436,437 TGF-β also induces endothelial-to-mesenchymal transition (EndoMT) of glomerular endothelial cells through the Smad3 pathway,438,439,440 giving rise to glomerular myofibroblasts, a major source of ECM.
Tubulointerstitial fibrosis is characterized by excessive accumulation of ECM in the tubular interstitium, and is considered a central event in the progression of CKD, regardless of etiology. In addition, TGF-β mediates several key tubular pathological events, including fibroblast proliferation, EMT in tubular epithelial cells, fibroblast ECM production, and epithelial cell death, cumulatively leading to interstitial fibrosis.441,442
Myofibroblasts have been recognized as the dominant collagen-producing cells in organ fibrosis.443,444 A recent study elegantly performed by LeBleu et al.445 has delineated the origin of myofibroblasts in renal fibrosis. It has been shown that 50% of the myofibroblasts in renal fibrosis arise from local resident fibroblasts, 35% from bone marrow mesenchymal cells, 10% from the endothelial-to-mesenchymal transition, and 5% from the epithelial-to-mesenchymal transition program. Importantly, targeted knockout of Tgfbr2 in αSMA+ cells significantly reduced the myofibroblasts derived through differentiation from bone marrow cells, further indicating the role of TGF-β in the recruitment of bone marrow MSCs and the induction of their differentiation into myofibroblasts.
Liver
Liver fibrosis is the final consequence of many chronic liver injuries/diseases446 such as cirrhosis and hepatocellular carcinoma, which are leading causes of morbidity and mortality worldwide. Regardless of the inciting etiology, all the chronic liver diseases converge on a common pathophysiology, progressing from moderate to severe inflammation, next to fibrosis and finally to cirrhosis.
Multiple studies suggest that TGF-β is involved in many kinds of chronic liver disease (CLD). In liver-biopsy CLD specimens, the amount of TGF-β1 mRNA is positively correlated with that of type I collagen mRNA.447 TGF-β1 mRNA concentrations in the liver positively correlate with serum concentrations of peptide fragments of type III collagen and the histological activity of the liver disease. More importantly, TGF-β1 protein is specifically detected by immunohistochemical staining in areas of fibrosis.448 A similar pathophysiologic effect of increased levels of TGF-β occur in induced animal models for hepatic fibrosis.449 In a conditional transgenic mouse model with tetracycline-regulated expression of TGF-β1 in liver, fibrosis progressed to an intermediate state during the upregulation of TGF-β1 expression.450 Furthermore, the fibrogenic process can be attenuated simply by blockade of TGF-β signaling.451
TGF-β is believed to trigger a series of important cellular events related to fibrogenesis and repair in liver diseases.452,453 Most liver cells are sensitive to TGF-β, which initiates both the canonical Smad-mediated and the non-canonical Smad-independent downstream signals. In the early stages of liver fibrogenesis, excessive activation of TGF-β may impair liver regeneration454 and amplify hepatocyte apoptosis.455 Hepatic stellate cells are then activated and initiate fibrosis.456 During the development of fibrosis, TGF-β directly activates the quiescent hepatic stellate cells through the canonical Smad-dependent pathway, inducing its trans-differentiation into myofibroblasts, which deposit excessive ECM.457 TGF-β also has an essential role during the inflammatory process linked to liver fibrosis by mediating the terminal differentiation of regulatory T cells, negative regulators of inflammation. TGF-β induces cell death and EMT of hepatocytes, further contributing to the ECM deposition and fibrosis. Activation of liver sinusoidal endothelial cells and neoangiogenesis are also partially induced by TGF-β. TGF-β can induce a feed-forward loop through the production of ROS458 and eventually contributes to liver fibrosis.
Lung
The involvement of TGF-β in pulmonary fibrosis has been evidenced in both animal models and humans with pulmonary diseases. Rats induced with bleomycin for the development of fibrosis demonstrate significantly elevated levels of total lung TGF-β1 compared to the non-induced controls. Importantly, increases of TGF-β1 precede the synthesis of collagens, fibronectin, and proteoglycans.459 In people with idiopathic pulmonary fibrosis, TGF-β1 is increased in alveolar walls at the sites where ECM is accumulated.460 In addition, TGF-β mRNA level in bronchoalveolar cells obtained from patients with autoimmune diseases and lung fibrosis is 10 time higher than bronchoalveolar cells of normal subjects or patients with asthma.461 Alveolar macrophages are believed to produce the majority of TGF-β1 during the process of fibrogenesis.462 The increased TGF-β level in lung tissue can induce the trans-differentiation of resident cells to myofibroblasts, which secret the most ECM during fibrosis. Our recent data reveals that excessive TGF-β1 produced by allergen-challenged lung tissue can be released into circulation, and it mobilizes and recruits bone marrow MSCs to the perturbed airways. Those recruited MSCs may eventually differentiate into myofibroblasts under sustained TGF-β1 stimulation, further contributing to the lung fibrosis.225
Skin
Skin fibrosis is usually observed as a local manifestation of systemic connective tissue disorders such as systemic scleroderma,463 or a complication secondary to skin injury such as hypertrophic scars from burns.464 Regardless of etiology, increased amounts of TGF-β1 have a critical role in skin fibrosis. Of note, a self-limited acute injury will only elicit a transient activation of TGF-β for a desired action; whereas a repeated injury may override the normal termination signals of TGF-β activation, creating a sustained increase in TGF-β signaling, and blindly lead to the progressive deposition of ECM and skin fibrosis by either stimulating resident fibroblasts proliferation, or inducing trans-differentiation into myofibroblasts through EMT process.465
Sustained activation of TGF-β1 signaling caused by genetic mutations can result in familial skin fibrosis such as stiff skin syndrome (SSKS), which is characterized by hard, thick skin, limited joint mobility, and flexion contractures. Lipodystrophy and muscle weakness have also been reported occasionally.466 SSKS is caused by mutations specifically localized to the fourth transforming growth factor-β-binding protein-like domain (TB4) of fibrillin-1, which encodes the RGD motif, through which fibrillin-1 binds cell-surface integrins αvβ3, α5β1, and αvβ6.466,467 Recent findings from Dietz’s group have demonstrated that mouse lines harboring analogous amino acid substitutions in fibrillin-1 recapitulate aggressive skin fibrosis that is prevented by integrin-modulating therapies and reversed by antagonism of TGF-β.468 The proposed mechanism is that SSKS mutations promote increased deposition of abnormal microfibrillar aggregates that fail to make contact with neighboring cells but retain the ability to bind to the TGF-β LLC, resulting in an increased concentration of latent TGF-β in tissues of SSKS.466,467 The high dermal concentration of TGF-β in SSKS may then allow a sustained enhanced signaling state, possibly by mechanical traction-based activation of the excessive amounts of latent TGF-β in the stiffened dermis, resulting in a similar feed-forward mechanism as observed in fibrosis.469
TGF-β functioning as a key factor in fibrogenesis offers a promising target for the development of new therapeutic agents for the fibrotic conditions associated with excessive production/activation of TGF-β. Injection of TGF-β neutralizing antibody has been proven to be effective in the treatment of fibrosis in kidney,430 skin,470 lung,471 brain,472 joint,473 and arterial wall.474 Injection of TGF-β neutralizing antibody significantly reduced the synthesis of matrix proteins and the deposition of plasminogen activator inhibitor-type I in the glomeruli, and blocked the accumulation of mesangial matrix in nephritic rats. Treatment with TGF-β1 neutralizing antibody also substantially reduced the collagen content of dermal wounds and minimized the scar formation. Anti-TGF-β1 treatment also worked well at the site of brain injury by reducing fibrous scar tissue and inflammation. Anti-TGF-β1 could alleviate synovial inflammation and delay bone and synovial destruction in arthritic joints. In addition, injection of anti-TGF-β1 neutralizing antibody to the rats with carotid-artery injury suppressed the accumulation of matrix that underlies the development of intimal hyperplasia and restenosis. Similarly, our recent data obtained from mouse model of wire-induced injury of femoral artery have shown that intravenous administration of TβRI inhibitor (SB-505124) significantly diminished the formation of neointima in injured arteries compared to vehicle-treated control.224 These consistent therapeutic successes in animal models underscore the translational potential of TGF-β1 modulation in the treatment of organ fibrosis.
Bone matrix-derived TGF-β promotes skeletal metastases
Cancer diagnosed at an early stage can usually be effectively treated with some combination of surgery, radiation, hormonal, and chemotherapies. However, treatment options for metastatic or recurrent cancer with acceptable outcomes are still limited. Bones are a common place for metastasis. Skeletal metastasis of cancer is a complex process, including cancer cells escaping the primary site, circulating to distant sites, evading the host immune response, and proliferating into the metastatic site.475 Tumor cells could generate adhesive molecules that mediate binding to marrow stromal cells and bone matrix. The interactions between tumor cells and bone promote the production of angiogenic and bone resorbing factors by tumor, and further enhance tumor growth in bone.476 Of note, the bone microenvironment houses abundant growth factors including TGF-β, IGF-1, and -2, FGFs, PDGFs and BMPs.477 During tumor-induced osteoclastic bone resorption, these factors are released and enriched in the bone microenvironment. These bone matrix-derived factors, particularly TGF-β, can act back upon the tumor to facilitate further tumor expansion and enhance cytokine production, and also upon osteoblasts to suppress bone formation, further promoting tumor growth and metastasis in bone.478
In cancer, TGF-β can be either a tumor suppressor or a promoter depending on the temporal stage of the disease.479 In the early stage of tumors initiation, TGF-β limits the growth of tumor cells through its antiproliferative and proapoptotic actions.480 Conversely, during tumor progression, TGF-β acts as a tumor-promoter by inducing proliferation, angiogenesis, and immunosuppression, and thus promotes invasion and metastasis of cancer.481 The loss of function of TGF-β signaling also contributes to certain tumor types. Mutations in TβRII are frequently detected in colon cancer, gastric tumors, and gliomas. These mutations may result in DNA repair defects and cancer predisposition, likely due to cellular escape from TGF-β-mediated growth surveillance.482,483,484,485 Loss-of-function mutations in TβRI have been observed in ovarian cancers, metastatic breast cancers, pancreatic carcinomas, and T-cell lymphomas.486,487,488,489 In addition, mutations in the TGF-β signaling components, such as Smad proteins mutations have been detected in several carcinomas. Smad2 mutations have been identified in human colorectal cancer and lung cancers.490,491 Smad4 mutations have been detected in pancreatic carcinomas and familial juvenile polyposis,492,493 and Smad2 and Smad4 double mutations have been detected in hepatocellular carcinoma.494 Moreover, Smad3 is frequently downregulated in cancer and inactivating mutations have also been reported.495,496,497 TGF-β can antagonize BMP-like responses by the formation of pSmad1/5–pSmad3 complex specifically binding to BRE and suppressing the transcription of downstream genes.155 Inactivation of Smad3 in cancer alleviates the antagonism of TGF-β on BMP responses, and enables TGF-β to induce BRE downstream ID genes,155 which are potent tumor promoters normally suppressed by canonical TGF-β signaling.481 The induction of ID genes thus deregulates tumor cell proliferation and confers invasiveness, angiogenesis, and metastasis.498
In advanced cancer, cells lose TGF-β suppressive effects resulting in compensatory over-production of TGF-β. Elevated level of TGF-β in serum is often observed in the later stages of cancer patient, and is associated with increased invasiveness and a poor clinical outcome of cancer.499 TGF-β signaling can act on multiple cells in the local microenvironment of bone, and consequently enhance tumor growth and invasiveness.479,500 Specifically, TGF-β could induce the expression, secretion and activation of MMPs that mediate the migration of endothelial cells, thus promoting tumor angiogenesis.500,501 TGF-β could also indirectly induce the expression of the pro-angiogenic factors such as VEGF and connective tissue growth factor (CTGF) in fibroblastic and epithelial cells,502,503 and thus further contributes to the angiogenesis and invasiveness of tumor.504,505 Inhibition of TGF-β signaling with TGF-β-neutralizing antibodies can suppress angiogenesis in human breast and prostate cancer,506 further validating the critical role of TGF-β as a pro-angiogenic factor during tumor. Furthermore, excessive production of tumor-derived TGF-β could suppress T-lymphocytes and natural killer cells, leading to cellular escape of cancer cells from cytotoxic T lymphocyte clearance.507 Target blockade of TGF-β signaling in T-cells results in eradication of tumors in mice challenged with live tumor cells,508 indicating the suppressive action of TGF-β signaling on the T-cell mediated antitumor immunity.
TGF-β is also involved in the EMT of cancer,7,509 which is essential to increase tumor cell mobility and invasiveness closely related to metastasis. TGF-β interacts with other oncogenic pathways to maintain the mesenchymal phenotype of tumor cells by downregulating E-cadherin and upregulating mesenchymal genes.510 Smads, Ras, Rho, ERK MAPK, p38 MAPK, and Wnt signaling pathways have been implicated in the TGF-β-induced EMT.7 TGF-β activates transcriptional factors such as Snail and Slug to regulate EMT.511,512 Particularly, SNAIL could repress E-cadherin and activate the transcription of mesenchymal genes, such as vimentin and αSMA. In addition, SNAIL could promote collagen I synthesis/deposition and upregulate pro-inflammatory interleukins such as IL-1, -6, and -8,513,514 and thus enhances the invasiveness of tumor.
On the basis of the aforementioned oncogenic activity of TGF-β, a feed-forward loop has been proposed to describe skeletal metastasis. Tumor cells in bone secrete osteolytic factors, such as PTHrP and IL-11, leading to osteolytic bone resorption. Active TGF-β is released from bone matrix by osteoclastic resorption and further induces tumor production of osteolytic and pro-metastatic factors including PTHrP and IL-11. Of note, PTHrP is a central mediator of TGF-β-induced osteolytic metastases. Increased expression of PTHrP has been observed in human breast cancer with bone metastases compared with primary breast cancers.515 PTHrP can stimulate RANKL and inhibit OPG expression in osteoblasts to favor osteoclastogenesis.516 TGF-β induces PTHrP secretion from MDA-MB-231 cells via Smad and p38 MAP kinase pathways.517 Stable inactivation of TβRII in the breast cancer cell line MDA-MB-231 inhibits TGF-β-induced PTHrP secretion and suppresses bone metastases in a mouse model.518 Direct neutralization of PTHrP with PTHrP-neutralizing antibodies inhibits development and progression of breast cancer bone metastases in mouse models.519 TGF-β released during bone resorption can also directly act on bone cells. Within a given range of concentrations, TGF-β stimulates osteoclastic bone resorption while inhibits osteoblastic differentiation. Transcriptional profiling of human breast cancer cells with an aggressive bone metastatic phenotype has identified the upregulation of several genes, such as IL-11, CTGF, CXCR4, and MMP1, which are associated with bone metastases. These genes act cooperatively to cause skeletal metastasis by promoting homing to bone, angiogenesis and invasion of tumor. Notably, these genes are all regulated by TGF-β via the canonical TGF-β/Smad pathway in metastatic cells.503,520,521 Either inhibition of TGF-β signaling with small-molecule inhibitors or inhibition of bone resorption with bisphosphonate is effective in decreasing TGF-β signaling activity in the bone metastases.522 This indicates that TGF-β released by osteoclastic bone resorption is the major source of TGF-β acting on tumor cells in bone. Inhibition of either the TGF-β pathway or osteoclastic bone resorption may represent a novel therapeutic for the treatment of skeletal metastasis.
Several preclinical studies have shown that the TGF-β signaling pathway is a potential target for the inhibition of bone metastases. Knockdown of Smad4 expression in breast cancer cells reduces growth of bone metastases,523,524 whereas overexpression of Smad7 reduces bone metastases of melanoma.525 Small-molecule inhibitors of the TβRI kinase have been used to reduce bone metastasis through blockage of TGF-β signaling. Systemic administration of small molecule [3-(pyridine-2yl)-4-(4-quinonyl)]-1H pyrazole] that is able to inhibit TβRI kinase activity has been shown to effectively reduce the number and size of lung metastases and the incidence of skeletal metastases in experimental mice model of bone and lung metastasis.526 Systemic administration of Ki26894 (a TβRI kinase inhibitor) decreased skeletal metastasis and prolonged survival of a nude mouse model with bone metastasis.527 Consistently, preventive treatment with TβRI kinase inhibitor LY2109761 led to reduction of the number of bone lesions and skeletal tumor burden in bone metastatic mice model. Although LY2109761 was less effective in the treatment of established bone metastases,522 SD-208, a more potent TβRI kinase inhibitor, was effective in the treatment of mice with established bone metastases.520 The use of TGF-β neutralizing antibodies is another possible modality for the treatment of bone metastases.528 Treatment with a neutralizing pan-TGF-β antibody (1D11, Genzyme) has been shown to decrease metastases to the lungs in a transplantable 4T1 mice model of metastatic breast cancer,529 and reduce skeletal tumor burden in mice while also increasing the bone volume.530
Combination of anti-TGF-β therapies with other therapeutics is promising for the treatment of patients with bone metastases. Bone is a hypoxic microenvironment, and hypoxia-inducible factor 1α (HIF-1α) has been implicated in enhancing tumor growth and metastasis.520,531 Hypoxia also stimulates the expression of CXCR4 and DUSP1,532 whose upregulation is associated with bone metastases.503 TGF-β stabilizes HIF-1α by inhibiting its degradation.533 TGF-β and hypoxia signaling pathways in breast cancer cells are additive to induce VEGF and CXCR4.520,533 Inhibition of both TGF-β and hypoxia signaling pathways decreases bone metastases more than inhibition of either alone, resulting in enhanced osteoblastic activity and suppressed osteoclastic bone resorption as well as reduced tumor growth.520 Other possible therapies include halofuginone, a natural product derivative that inhibits TGF-β signaling possibly via induction of Smad7. Halofuginone treatment can significantly suppress osteolysis and skeletal tumor burden in mice with established bone metastases.534
In conclusion, bone is the most common site of cancer metastases. Active TGF-β released from bone matrix is the major component of the bone microenvironment, functioning to drive a feed-forward cycle of tumor growth and osteolysis in bone. Modulation of TGF-β signaling in cancer cells has been proven effective to decrease bone metastases in either in vitro or animal models. Development of anti-TGF-β therapies administered alone or supplementing other therapies for bone metastasis of cancer is promising. As TGF-β can be either tumor suppressive or pro-metastatic, a long-term global blockade of this signaling pathway may result in off-target effects. Hence, a delicate modulation of TGF-β signaling during tumor onset and progression is necessary for the development of the most effective antineoplastic therapy with minimal toxicity but potent efficacy.
Modulation of TGF-β signaling is promising for the treatment of disorders associated with TGF-β abnormalities
As the abnormalities in TGF-β signaling will cause a plethora of local or systemic disorders, the development of anti-TGF-β therapies is intriguing. Although many TGF-β targeting approaches have been developed and quite of few of these treatments have undergone clinical trials, concerns remain that long-term blockade of this pathway may have other off-target effects due to the dual functions of TGF-β in maintaining tissue homeostasis. It should always be kept in mind that a transient temporal and spatial activation of TGF-β is necessary for the maintenance of tissue homeostasis, whereas excessive production and sustained activation of TGF-β signaling will inevitably lead to TGF-β-mediated pathologies. Thereby, how to intricately tune the signaling to the optimal magnitude in the right place at the right time is becoming increasingly essential for TGF-β-targeted therapy. Here we discuss the recent updates in potential treatment for disorders with aberrant TGF-β signaling.
TGF-β-modulation has shown significant clinical potential
Theoretically, every component of the TGF-β pathway can be a potential target for drug intervention. In reality, however, most of the treatments have not been validated in clinical trials yet. Currently, antibodies,529,535,536 antisense oligonucleotides (ASOs),537,538,539,540,541 ligand competitive peptides,542,543,544,545 and small-molecule inhibitors against particular component of the TGF-β pathway are being tested in clinic.
Monoclonal antibodies
Monoclonal antibodies against TGF-β can specifically neutralize excessive extracellular ligand. Monoclonal TGF-β1 antibody metelimumab (CAT-192),546 TGF-β2 antibody Lerdelimumab (CAT-152),547,548 as well as TGF-β1–3 pan-specific antibodies such as fresolimumab (GC-1008)549,550 have been well developed by Cambridge Antibody Technologies and Genzyme. Clinical trials on fresolimumab have been conducted for both neoplastic551 (ClinicalTrials.gov Identifier: NCT00923169) and non-neoplastic applications.550 (ClinicalTrials.gov Identifier: NCT01284322) Fresolimumab has been found to be well tolerated and safe at a single-dose infusion up to 4 mg/kg for the treatment of the fibrotic disorder focal segmental glomerulosclerosis,550 and at 15 mg/kg for the treatment of advanced malignant melanoma and renal cell carcinoma.551
Other antibodies that have been developed and tested in clinical trials include TβRII-blocking antibody and anti-integrin β6 antibody. Eli Lilly and Company developed a TβRII-blocking antibody, IMC-TR1, which is being evaluated in clinical trials for the treatment of breast and colon cancer (ClinicalTrials.gov identifier: NCT01646203). Another antibody against integrin β6 has also shown effectiveness in the treatment of fibrosis and cancer in animal models,552 and is currently in a Phase II clinical trial for the treatment of idiopathic pulmonary fibrosis (ClinicalTrials.gov identifier: NCT01371305).
Antisense oligonucleotides and antisense RNA
Antisense oligonucleotides have been developed to target mRNA translation thus downregulating ligand synthesis.537,553 Trabedersen, a synthetic 18-mer phosphorothioate-modified ASO, binds specifically to the human TGF-β encoding gene (TGFB2), and this drug has proven to be effective in the treatment of glioma. Preclinical and clinical studies.537,538,540 indicate that neutralization of TGF-β2-mediated immunosuppression can activate tumor-infiltrating natural killer cells, which suppress tumor proliferation. Trabedersen can bypass the blood–brain–barrier and achieve a homogeneous distribution throughout the tumor, resulting in shrinkage of the targeted tumor as well as tumors elsewhere in the brain. Phase I/II studies of trabedersen for the treatment of anaplastic astrocytoma (grade III glioma) and glioblastoma (grade IV glioma) showed survival benefit compared with conventional chemotherapy.554 More importantly, patients with glioblastoma on trabedersen treatment experienced less adverse events and showed significantly improved cognitive function 2–3 years after therapy compared to standard chemotherapy.541
One of the challenges of ASO is target-delivery to avoid off-target toxicity. In the case of glioblastoma, ASO was delivered directly into the tumor using an intrathecal catheter.541 Intravenous delivery of ASO has also been developed for pancreatic cancer in mouse models,540 as well as in humans.555 An anti-TGF-β2 antisense strategy has been used to augment tumor vaccines. Belagenpumatucel-L is such a tumor vaccine, in which a ~900-nucleotide TGF-β2 antisense construct is transfected into allogeneic non-small cell lung cancer (NSCLC) cells to enhance its tumor-suppressive efficacy. This tumor vaccine shows enhanced activity compared to conventional vaccines.556,557 A recent phase III study showed that Belagenpumatucel-L failed to improve the overall survival of the enrolled NSCLC patients compared to placebo, but an improved survival in patients who were randomized within 12 weeks of completion of chemotherapy and in those who had received prior radiation has been suggested. Further studies are still needed to validate the effectiveness of belagenpumatucel-L in the treatment of NSCLC.558
Ligand traps and peptides
Genzyme has developed a ligand trap by fusing Fcγ to the extracellular domain of TβRII. Although this construct can inhibit mammary tumor cell viability, migration, and metastases in the animal model,559 it has not been tested in clinical trials. A different ligand trap approach, using peptide mimetics of TβRIII (also known as betaglycan),542,543,544,545 has been investigated in a Phase IIa clinical trial for the treatment of scleroderma and skin fibrosis. Data from that trial show safety and efficacy of this ligand trap when topically applied to skin.
Small-molecule inhibitors
Small-molecule inhibitors (SMIs) are designed to specifically target the type I receptor of TGF-β to block the canonical Smad2/3 pathway while keeping other non-canonical responses such as TAK1 activation relatively intact. SMIs in general are ATP mimetics, which completely bind the hydrophobic ATP binding pocket of the receptor kinase.560,561 SMIs have proven to be effective in the control of cancer metastasis in preclinical studies.562,563 The advantages of SMIs include economical production, relative stability, and easy oral administration. A possible disadvantage of SMIs is the cross-inhibition of other kinases, resulting in off-target effects. The short half-life of SMIs favors a predictable inhibition of TGF-β signaling in a desired time frame, and provides the possibility of rapid drug withdrawal should adversary effects arise.
Pre-existing drugs that inhibit TGF-β signaling
Although the precise molecular mechanisms are still unclear, Losartan and Candesartan that are originally developed as angiotensin type II receptor inhibitors for the treatment of hypertension, are able to reduce aneurysm growth in both MFS and LDS likely by downregulating TGF-β signaling.405,564 In addition, angiotensin type II receptor inhibitors have also shown protective effect on patients afflicted with TGF-β over-activation-induced brain, lung, and muscle injuries.565,566,567 Pirfenidone, an approved drug for the treatment of idiopathic pulmonary fibrosis in Europe and Mexico, has shown inhibitory effect on TGF-β activity via unknown targets.568 Pirfenidone is currently in a Phase III trial in the United States.569,570
Other potential approaches
Other potential approaches to suppress TGF-β signaling include gene transfer of inhibitory Smad7, which has been tested in animal models for vascular remodeling, diabetic kidney disease, and colonic and hepatic fibrosis.571,572 The major concern that limits the clinical translation of this approach is the barriers that face all gene therapies.573 TGF-β activation may also be blocked by small peptides. An LSKL (Leu-Ser-Lys-Leu) peptide, which specifically binds to a conserved sequence in the LAP region of the TGF-β latent complex has shown effectiveness in suppressing TGF-β signaling in vitro.71 However, its TGF-β blockade efficacy has not been demonstrated in vivo yet.
TGF-β-modulation represents a promising treatment for osteoarthritis
Excessive activation of TGF-β in subchondral bone has been reported at the onset of OA in animal models.6 Active TGF-β1 concentrations are also high in the subchondral bone of people affected by osteoarthritis. All these data suggest that modulation of TGF-β signaling may provide a promising disease-modifying approach for OA. In the ACLT rodent models, systemic inhibition of TGF-β signaling with intraperiotoneal administration of TβRI inhibitor or specific blockade of active TGF-β ligand in the tibial subchondral bone with TGF-β neutralizing antibody at the time of injury was sufficient to attenuate the degeneration of articular cartilage.6 Furthermore, TGF-β signaling activated in the subchondral bone through osteoclast bone resorption in these animal OA models suggests that inhibition of osteoclast bone resorption may also delay progression of OA. Several clinical trials and preclinical studies support this hypothesis. In addition to many animal studies that reveal the positive effect of bisphosphonates for delaying OA progression,574,575,576,577,578 in the most recent prospective 2-year trial, alendronate treatment successfully improved the Western Ontario and McMaster Universities osteoarthritis (WOMAC) pain scores and decreased biochemical markers in hip OA patients.579 In a cross-sectional study, elderly women who were being treated with alendronate were found to have significantly decreased prevalence of knee OA, as assessed by WOMAC pain scales and subchondral bone lesions by MR imaging compared to elderly women not taking alendronate.580 Randomized controlled trials are needed to assess the efficacy of bisphosphonates as a potential OA treatment. The detailed mechanisms also still need to be further elucidated, anticipating that at least part of the effect is through inhibition of osteoclast activity and subsequent reduction in TGF-β levels. As many musculoskeletal disorders also involve excessive TGF-β signaling, attenuating the TGF-β signaling pathway could also benefit the management of these diseases.
High levels of active TGF-β also alter the microenvironment of subchondral bone, leading to aggregation of osteoprogenitors and increased angiogenesis in bone marrow. PTH is FDA-approved as an anabolic therapy for osteoporosis. Daily injection of PTH increases bone formation with normal micro-architecture.185,355,356,357,358,359,360,361,362 PTH can improve the bone marrow microenvironment by orchestrating the signaling of local factors for bone remodeling, reducing reactive oxygen species, and stimulating Wnt signaling in bone of senescent mice.185,313,355,356,357,358,359,360,361,362,581,582 In addition, PTH has demonstrated potent chondroprotective and chondroregenerative effects by inducing cartilage matrix synthesis and suppressing chondrocyte hypertrophy and matrix metalloproteinase 13 expression in different OA animal models.583,584 The dual beneficiary effects of PTH on both cartilage and subchondral bone make PTH another promising candidate for the treatment of OA. A recent study using a rabbit model of osteoarthritis preceded by osteoporosis (OPOA) has demonstrated that PTH treatment on the early stage of OA is able to improve microstructure and quality of subchondral bone, and thus attenuates subsequent cartilage damage.585 These data support the relevance of the role of subchondral bone osteopenia in the pathogenesis of OA, and indicate the potential application of anabolic agents (such as PTH) to the treatment in early stages of OA associated with osteoporosis. The osteogenic microenvironment created by PTH has also been shown to expand HSPC niches,323,352 and could also potentially be translated into therapies for hematologic diseases such as cytopenias, myelodysplastic syndromes, or myeloproliferative disorders.586
In conclusion, articular cartilage and subchondral bone constantly interact as a functional unit. TGF-β has a critical role in maintaining both bone and articular cartilage homeostasis. Joint instability causes increased subchondral bone remodeling, which releases excessive active TGF-β1 through osteoclastic bone resorption. The pathologically high level of TGF-β1 in the subchondral bone leads to aberrant bone remodeling and formation of marrow osteoid islets. The abnormal subchondral bone microstructure in turn likely alters the stress distribution on the articular cartilage, eventually resulting in cartilage degeneration. The concept of the holism is essential for exploring therapeutic strategies for OA. Improving mechanical properties of subchondral bone and its physiological function is at least equally important as directly targeting articular cartilage. Therapies that can normalize TGF-β signaling, either directly by neutralizing TGF-β over-activation or indirectly by PTH-mediated modulation of the bone marrow microenvironment, may serve as a potential approach to the management of joint disorders.
Conclusion
TGF-β as a dual functional growth factor has been attracting major research efforts ever since its discovery three decades ago. Unlike most of the growth factors that are ready to function upon secretion, TGF-β is unique in that it is secreted as part of a latent complex that is stored in the ECM for activation and action at a later time point. Thereby, the magnitude and duration of TGF-β signaling are mainly dependent on, and meticulously controlled by the temporal and spatial activation of its ligand. If activated properly, TGF-β signaling has an essential role in normal physiology ranging from embryonic development to adult tissue homeostasis, whereas sustained activation or functional deletion via genetic mutations or environmental stimuli will exacerbate its adversary effects, and contribute to the pathophysiology of major diseases such as musculoskeletal disorders, cancer progression and organs fibrosis. Therefore, TGF-β has been described as “an excellent servant but a bad master”,587 in reference to its paradoxical characteristics.
Precise temporal and spatial activation of TGF-β signaling is necessary to counter tissue perturbations, either by suppressing excessive adversary cellular responses such as inflammation, or by recruiting adult stem cells to participate in tissues repair/remodeling process. The bone remodeling process clearly underscores the importance of proper activation of TGF-β in the maintenance of tissue homeostasis. The physiological gradient of active TGF-β formed by normal bone remodeling functions as a coupling factor that directs the MSCs/osteoprogenitors to the bone resorbing sites for a coupled bone formation, whereas sustained activation of TGF-β by either genetic mutations or pathologically elevated osteoclastic activity recruits excessive MSCs/osteoprogenitors for an aberrant bone remodeling. The same theory can be applied to other pathologies with TGF-β involvement, where the normal functions of this growth factor are switched to an off-target activity that disrupts tissue homeostasis.
Therefore, a comprehensive understanding of the mechanisms that underlie the physiological or pathological effects of TGF-β, and full interpretation of how cells integrate these signals into coherent responses in a context-dependent way, can lead to promising therapeutics for TGF-β-involved pathologies. Novel strategies that intricately tune TGF-β signaling to properly respond to specific contexts are being developed with anticipated clinical implications.
References
Crane, J. L. & Cao, X. Bone marrow mesenchymal stem cells and TGF-beta signaling in bone remodeling. J. Clin. Invest. 124, 466–472 (2014).
Annes, J. P., Munger, J. S. & Rifkin, D. B. Making sense of latent TGF beta activation. J. Cell Sci. 116, 217–224 (2003).
Pfeilschifter, J., Bonewald, L. & Mundy, G. R. Characterization of the latent transforming growth factor beta complex in bone. J. Bone Miner. Res. 5, 49–58 (1990).
Pedrozo, H. A. et al. Potential mechanisms for the plasmin-mediated release and activation of latent transforming growth factor-beta1 from the extracellular matrix of growth plate chondrocytes. Endocrinology 140, 5806–5816 (1999).
Gordon, K. J. & Blobe, G. C. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim. Biophys. Acta 1782, 197–228 (2008).
Zhen, G. et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 19, 704–712 (2013).
Xu, J., Lamouille, S. & Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172 (2009).
Tang, Y. et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 15, 757–765 (2009).
Kingsley, D. M. The TGF-beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 8, 133–146 (1994).
Javelaud, D. & Mauviel, A. Mammalian transforming growth factor-betas: Smad signaling and physio-pathological roles. Int. J. Biochem. Cell Biol. 36, 1161–1165 (2004).
Chang, H., Brown, C. W. & Matzuk, M. M. Genetic analysis of the mammalian transforming growth factor-beta superfamily. Endocr. Rev. 23, 787–823 (2002).
Proetzel, G. et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 11, 409–414 (1995).
Letterio, J. J. & Roberts, A. B. Transforming growth factor-beta1-deficient mice: identification of isoform-specific activities in vivo. J. Leukoc. Biol. 59, 769–774 (1996).
Sanford, L. P. et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGF beta knockout phenotypes. Development 124, 2659–2670 (1997).
Derynck, R. et al. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature 316, 701–705 (1985).
Dickinson, M. E. et al. Chromosomal localization of seven members of the murine TGF-beta superfamily suggests close linkage to several morphogenetic mutant loci. Genomics 6, 505–520 (1990).
Flanders, K. C. et al. Localization and actions of transforming growth factor-beta s in the embryonic nervous system. Development 113, 183–191 (1991).
de Martin, R. et al. Complementary DNA for human glioblastoma-derived T cell suppressor factor, a novel member of the transforming growth factor-beta gene family. EMBO J. 6, 3673–3677 (1987).
Kaartinen, V. et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial–mesenchymal interaction. Nat. Genet. 11, 415–421 (1995).
Bandyopadhyay, B. et al. A “traffic control” role for TGFbeta3: orchestrating dermal and epidermal cell motility during wound healing. J. Cell Biol. 172, 1093–1105 (2006).
Kondaiah, P. et al. cDNA cloning of porcine transforming growth factor-beta 1 mRNAs. Evidence for alternate splicing and polyadenylation. J. Biol. Chem. 263, 18313–18317 (1988).
Clark, D. A. & Coker, R. Transforming growth factor-beta (TGF-beta). Int. J. Biochem. Cell Biol. 30, 293–298 (1998).
Qian, S. W., Kondaiah, P., Roberts, A. B. & Sporn, M. B. cDNA cloning by PCR of rat transforming growth factor beta-1. Nucleic Acids Res. 18, 3059 (1990).
Derynck, R., Jarrett, J. A., Chen, E. Y. & Goeddel, D. V. The murine transforming growth factor-beta precursor. J. Biol. Chem. 261, 4377–4379 (1986).
Dubois, C. M., Laprise, M. H., Blanchette, F., Gentry, L. E. & Leduc, R. Processing of transforming growth factor beta 1 precursor by human furin convertase. J. Biol. Chem. 270, 10618–10624 (1995).
Manning, A. M., Auchampach, J. A., Drong, R. F. & Slightom, J. L. Cloning of a canine cDNA homologous to the human transforming growth factor-beta 1-encoding gene. Gene 155, 307–308 (1995).
Sengle, G., Ono, R. N., Sasaki, T. & Sakai, L. Y. Prodomains of transforming growth factor beta (TGF beta) superfamily members specify different functions: extracellular matrix interactions and growth factor bioavailability. J. Biol. Chem. 286, 5087–5099 (2011).
Brunner, A. M., Marquardt, H., Malacko, A. R., Lioubin, M. N. & Purchio, A. F. Site-directed mutagenesis of cysteine residues in the pro region of the transforming growth factor beta 1 precursor. Expression and characterization of mutant proteins. J. Biol. Chem. 264, 13660–13664 (1989).
Mittl, P. R. et al. The crystal structure of TGF-beta 3 and comparison to TGF-beta 2: implications for receptor binding. Protein Sci. 5, 1261–1271 (1996).
Gray, A. M. & Mason, A. J. Requirement for activin A and transforming growth factor—beta 1 pro-regions in homodimer assembly. Science 247, 1328–1330 (1990).
Oklu, R. & Hesketh, R. The latent transforming growth factor beta binding protein (LTBP) family. Biochem. J. 352, 601–610 (2000).
Saharinen, J., Taipale, J. & Keski-Oja, J. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J. 15, 245–253 (1996).
Mangasser-Stephan, K. & Gressner, A. M. Molecular and functional aspects of latent transforming growth factor-beta binding protein: just a masking protein? Cell Tissue Res. 297, 363–370 (1999).
Koli, K., Saharinen, J., Karkkainen, M. & Keski-Oja, J. Novel non-TGF-beta-binding splice variant of LTBP-4 in human cells and tissues provides means to decrease TGF-beta deposition. J. Cell Sci. 114, 2869–2878 (2001).
Bismar, H. et al. Transforming growth factor beta (TGF-beta) levels in the conditioned media of human bone cells: relationship to donor age, bone volume, and concentration of TGF-beta in human bone matrix in vivo. Bone 24, 565–569 (1999).
Roberts, A. B., Frolik, C. A., Anzano, M. A. & Sporn, M. B. Transforming growth factors from neoplastic and nonneoplastic tissues. Fed. Proc. 42, 2621–2626 (1983).
Seyedin, S. M., Thomas, T. C., Thompson, A. Y., Rosen, D. M. & Piez, K. A. Purification and characterization of two cartilage-inducing factors from bovine demineralized bone. Proc. Natl. Acad. Sci. USA 82, 2267–2271 (1985).
Jenkins, G. The role of proteases in transforming growth factor-beta activation. Int. J. Biochem. Cell Biol. 40, 1068–1078 (2008).
Munger, J. S. et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328 (1999).
Shi, M. et al. Latent TGF-beta structure and activation. Nature 474, 343–349 (2011).
Derynck R., Miyazono K. The TGF-[beta] Family (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 2008.
Sato, Y. & Rifkin, D. B. Inhibition of endothelial cell movement by pericytes and smooth muscle cells: activation of a latent transforming growth factor-beta 1-like molecule by plasmin during co-culture. J. Cell Biol. 109, 309–315 (1989).
Yu, Q. & Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 14, 163–176 (2000).
Werb, Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell 91, 439–442 (1997).
Taipale, J., Miyazono, K., Heldin, C. H. & Keski-Oja, J. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. J. Cell Biol. 124, 171–181 (1994).
Lyons, R. M., Keski-Oja, J. & Moses, H. L. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J. Cell Biol. 106, 1659–1665 (1988).
Ignotz, R. A. & Massague, J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 261, 4337–4345 (1986).
Verrecchia, F., Chu, M. L. & Mauviel, A. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 276, 17058–17062 (2001).
Bugge, T. H. et al. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell 87, 709–719 (1996).
Murphy-Ullrich, J. E. & Mosher, D. F. Localization of thrombospondin in clots formed in situ. Blood 66, 1098–1104 (1985).
Raugi, G. J., Olerud, J. E. & Gown, A. M. Thrombospondin in early human wound tissue. J. Invest. Dermatol. 89, 551–554 (1987).
Reed, M. J. et al. Differential expression of SPARC and thrombospondin 1 in wound repair: immunolocalization and in situ hybridization. J. Histochem. Cytochem. 41, 1467–1477 (1993).
DiPietro, L. A. et al. Thrombospondin 1 synthesis and function in wound repair. Am. J. Pathol. 148, 1851–1860 (1996).
Agah, A., Kyriakides, T. R., Lawler, J. & Bornstein, P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am. J. Pathol. 161, 831–839 (2002).
Adams, J. C. & Lawler, J. The thrombospondins. Int. J. Biochem. Cell Biol. 36, 961–968 (2004).
Poczatek, M. H., Hugo, C., Darley-Usmar, V. & Murphy-Ullrich, J. E. Glucose stimulation of transforming growth factor-beta bioactivity in mesangial cells is mediated by thrombospondin-1. Am. J. Pathol. 157, 1353–1363 (2000).
Hugo, C. The thrombospondin 1-TGF-beta axis in fibrotic renal disease. Nephrol. Dial. Transplant. 18, 1241–1245 (2003).
Daniel, C., Schaub, K., Amann, K., Lawler, J. & Hugo, C. Thrombospondin-1 is an endogenous activator of TGF-beta in experimental diabetic nephropathy in vivo. Diabetes 56, 2982–2989 (2007).
Schultz-Cherry, S. & Murphy-Ullrich, J. E. Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J. Cell Biol. 122, 923–932 (1993).
Murphy-Ullrich, J. E. & Poczatek, M. Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor. Rev. 11, 59–69 (2000).
Young, G. D. & Murphy-Ullrich, J. E. Molecular interactions that confer latency to transforming growth factor-beta. J. Biol. Chem. 279, 38032–38039 (2004).
Walton, K. L. et al. Two distinct regions of latency-associated peptide coordinate stability of the latent transforming growth factor-beta1 complex. J. Biol. Chem. 285, 17029–17037 (2010).
Sweetwyne, M. T. & Murphy-Ullrich, J. E. Thrombospondin1 in tissue repair and fibrosis: TGF-beta-dependent and independent mechanisms. Matrix Biol. 31, 178–186 (2012).
Crawford, S. E. et al. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 93, 1159–1170 (1998).
Ludlow, A. et al. Characterization of integrin beta6 and thrombospondin-1 double-null mice. J. Cell Mol. Med. 9, 421–437 (2005).
Yevdokimova, N., Wahab, N. A. & Mason, R. M. Thrombospondin-1 is the key activator of TGF-beta1 in human mesangial cells exposed to high glucose. J. Am. Soc. Nephrol. 12, 703–712 (2001).
Wang, S., Shiva, S., Poczatek, M. H., Darley-Usmar, V. & Murphy-Ullrich, J. E. Nitric oxide and cGMP-dependent protein kinase regulation of glucose-mediated thrombospondin 1-dependent transforming growth factor-beta activation in mesangial cells. J. Biol. Chem. 277, 9880–9888 (2002).
Wang, S., Skorczewski, J., Feng, X., Mei, L. & Murphy-Ullrich, J. E. Glucose upregulates thrombospondin 1 gene transcription and transforming growth factor-beta activity through antagonism of cGMP-dependent protein kinase repression via upstream stimulatory factor 2. J. Biol. Chem. 279, 34311–34322 (2004).
Zhou, Y., Poczatek, M. H., Berecek, K. H. & Murphy-Ullrich, J. E. Thrombospondin 1 mediates angiotensin II induction of TGF-beta activation by cardiac and renal cells under both high and low glucose conditions. Biochem. Biophys. Res. Commun. 339, 633–641 (2006).
Belmadani, S. et al. A thrombospondin-1 antagonist of transforming growth factor-beta activation blocks cardiomyopathy in rats with diabetes and elevated angiotensin II. Am. J. Pathol. 171, 777–789 (2007).
Lu, A., Miao, M., Schoeb, T. R., Agarwal, A. & Murphy-Ullrich, J. E. Blockade of TSP1-dependent TGF-beta activity reduces renal injury and proteinuria in a murine model of diabetic nephropathy. Am. J. Pathol. 178, 2573–2586 (2011).
Yehualaeshet, T. et al. A CD36 synthetic peptide inhibits bleomycin-induced pulmonary inflammation and connective tissue synthesis in the rat. Am. J. Respir. Cell Mol. Biol. 23, 204–212 (2000).
Chen, Y. et al. A TSP-1 synthetic peptide inhibits bleomycin-induced lung fibrosis in mice. Exp. Toxicol. Pathol. 61, 59–65 (2009).
Nor, J. E. et al. Activation of latent TGF-beta1 by thrombospondin-1 is a major component of wound repair. Oral. Biosci. Med. 2, 153–161 (2005).
Sakai, K. et al. Thrombospondin-1 promotes fibroblast-mediated collagen gel contraction caused by activation of latent transforming growth factor beta-1. J. Dermatol. Sci. 31, 99–109 (2003).
Chipev, C. C. et al. Myofibroblast phenotype and apoptosis in keloid and palmar fibroblasts in vitro. Cell Death Differ. 7, 166–176 (2000).
Mimura, Y. et al. Constitutive thrombospondin-1 overexpression contributes to autocrine transforming growth factor-beta signaling in cultured scleroderma fibroblasts. Am. J. Pathol. 166, 1451–1463 (2005).
Chen, Y. et al. Thrombospondin 1 is a key mediator of transforming growth factor beta-mediated cell contractility in systemic sclerosis via a mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK)-dependent mechanism. Fibrogenes. Tissue Repair 4, 9 (2011).
van der Flier, A. & Sonnenberg, A. Function and interactions of integrins. Cell Tissue Res. 305, 285–298 (2001).
Nishimura, S. L. Integrin-mediated transforming growth factor-beta activation, a potential therapeutic target in fibrogenic disorders. Am. J. Pathol. 175, 1362–1370 (2009).
Wipff, P. J. & Hinz, B. Integrins and the activation of latent transforming growth factor beta1 - an intimate relationship. Eur. J. Cell Biol. 87, 601–615 (2008).
Yang, Z. et al. Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J. Cell Biol. 176, 787–793 (2007).
Breuss, J. M., Gillett, N., Lu, L., Sheppard, D. & Pytela, R. Restricted distribution of integrin beta 6 mRNA in primate epithelial tissues. J. Histochem. Cytochem. 41, 1521–1527 (1993).
Breuss, J. M. et al. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J. Cell Sci. 108, 2241–2251 (1995).
Miller, L. A., Barnett, N. L., Sheppard, D. & Hyde, D. M. Expression of the beta6 integrin subunit is associated with sites of neutrophil influx in lung epithelium. J. Histochem. Cytochem. 49, 41–48 (2001).
Border, W. A. & Ruoslahti, E. Transforming growth factor-beta in disease: the dark side of tissue repair. J. Clin. Invest. 90, 1–7 (1992).
Wang, A., Yokosaki, Y., Ferrando, R., Balmes, J. & Sheppard, D. Differential regulation of airway epithelial integrins by growth factors. Am. J. Respir. Cell Mol. Biol. 15, 664–672 (1996).
Annes, J. P., Chen, Y., Munger, J. S. & Rifkin, D. B. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J. Cell Biol. 165, 723–734 (2004).
Yoshinaga, K. et al. Perturbation of transforming growth factor (TGF)-beta1 association with latent TGF-beta binding protein yields inflammation and tumors. Proc. Natl. Acad. Sci. USA 105, 18758–18763 (2008).
Ahamed, J. et al. In vitro and in vivo evidence for shear-induced activation of latent transforming growth factor-beta1. Blood 112, 3650–3660 (2008).
Pretorius, M. et al. Plasminogen activator inhibitor-1 as a predictor of postoperative atrial fibrillation after cardiopulmonary bypass. Circulation 116, I1–7 (2007).
Aoki, K. et al. Elevation of plasma free PAI-1 levels as an integrated endothelial response to severe burns. . Burns 27, 569–575 (2001).
Christ, G. et al. Predictive value of plasma plasminogen activator inhibitor-1 for coronary restenosis: dependence on stent implantation and antithrombotic medication. J. Thromb. Haemost. 3, 233–239 (2005).
Gando, S. Disseminated intravascular coagulation in trauma patients. Semin. Thromb. Hemost. 27, 585–592 (2001).
Kluft, C. et al. The postoperative fibrinolytic shutdown: a rapidly reverting acute phase pattern for the fast-acting inhibitor of tissue-type plasminogen activator after trauma. Scand. J. Clin. Lab. Invest. 45, 605–610 (1985).
Rahr, H. B., Sorensen, J. V., Larsen, J. F., Jensen, F. S. & Bredahl, C. Plasminogen activators and plasminogen activator inhibitor before and after surgery in patients with and without gastric malignancy. Haemostasis 25, 248–256 (1995).
Seeber, C., Hiller, E., Holler, E. & Kolb, H. J. Increased levels of tissue plasminogen activator (t-PA) and tissue plasminogen activator inhibitor (PAI) correlate with tumor necrosis factor alpha (TNF alpha)-release in patients suffering from microangiopathy following allogeneic bone marrow transplantation (BMT). Thromb. Res. 66, 373–383 (1992).
Malek, A. M., Alper, S. L. & Izumo, S. Hemodynamic shear stress and its role in atherosclerosis. JAMA 282, 2035–2042 (1999).
Mu, D. et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J. Cell Biol. 157, 493–507 (2002).
Rolli, M., Fransvea, E., Pilch, J., Saven, A. & Felding-Habermann, B. Activated integrin alphavbeta3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc. Natl. Acad. Sci. USA 100, 9482–9487 (2003).
Brooks, P. C. et al. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 85, 683–693 (1996).
Scaffidi, A. K. et al. alpha(v)beta(3) Integrin interacts with the transforming growth factor beta (TGF beta) type II receptor to potentiate the proliferative effects of TGFbeta1 in living human lung fibroblasts. J. Biol. Chem. 279, 37726–37733 (2004).
Yehualaeshet, T. et al. Activation of rat alveolar macrophage-derived latent transforming growth factor beta-1 by plasmin requires interaction with thrombospondin-1 and its cell surface receptor, CD36. Am. J. Pathol. 155, 841–851 (1999).
Wang, B. et al. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology 46, 1404–1412 (2007).
Henderson, N. C. et al. Targeting of alpha V integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 19, 1617–1624 (2013).
Teitelbaum, S. L. Bone resorption by osteoclasts. Science 289, 1504–1508 (2000).
Hering, S. et al. TGFbeta1 and TGFbeta2 mRNA and protein expression in human bone samples. Exp. Clin. Endocrinol. Diabetes 109, 217–226 (2001).
Oreffo, R. O., Mundy, G. R., Seyedin, S. M. & Bonewald, L. F. Activation of the bone-derived latent TGF beta complex by isolated osteoclasts. Biochem. Biophys. Res. Commun. 158, 817–823 (1989).
Oursler, M. J. Osteoclast synthesis and secretion and activation of latent transforming growth factor beta. J. Bone Miner. Res. 9, 443–452 (1994).
Pfeilschifter, J. & Mundy, G. R. Modulation of type beta transforming growth factor activity in bone cultures by osteotropic hormones. Proc. Natl. Acad. Sci. USA 84, 2024–2028 (1987).
Wu, X. et al. Inhibition of Sca-1-positive skeletal stem cell recruitment by alendronate blunts the anabolic effects of parathyroid hormone on bone remodeling. Cell Stem Cell 7, 571–580 (2010).
Barcellos-Hoff, M. H., Derynck, R., Tsang, M. L. & Weatherbee, J. A. Transforming growth factor-beta activation in irradiated murine mammary gland. J. Clin. Invest. 93, 892–899 (1994).
Barcellos-Hoff, M. H. & Dix, T. A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 10, 1077–1083 (1996).
Proell, V. et al. TGF-beta dependent regulation of oxygen radicals during transdifferentiation of activated hepatic stellate cells to myofibroblastoid cells. Comp. Hepatol. 6, 1 (2007).
Boudreau, H. E., Emerson, S. U., Korzeniowska, A., Jendrysik, M. A. & Leto, T. L. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: a new contributor to HCV-induced oxidative stress. J. Virol. 83, 12934–12946 (2009).
Statius van Eps, R. G. & LaMuraglia, G. M. Photodynamic therapy inhibits transforming growth factor beta activity associated with vascular smooth muscle cell injury. J. Vasc. Surg. 25, 1044–1052 (1997).
Wang, L., Clutter, S., Benincosa, J., Fortney, J. & Gibson, L. F. Activation of transforming growth factor-beta1/p38/Smad3 signaling in stromal cells requires reactive oxygen species-mediated MMP-2 activity during bone marrow damage. Stem Cells 23, 1122–1134 (2005).
Jobling, M. F. et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat. Res. 166, 839–848 (2006).
Derynck, R. TGF-beta-receptor-mediated signaling. Trends Biochem. Sci. 19, 548–553 (1994).
Schmierer, B. & Hill, C. S. TGF beta-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8, 970–982 (2007).
Derynck, R. & Feng, X. H. TGF-beta receptor signaling. Biochim. Biophys. Acta 1333, F105–150 (1997).
ten Dijke, P., Miyazono, K. & Heldin, C. H. Signaling via hetero-oligomeric complexes of type I and type II serine/threonine kinase receptors. Curr. Opin. Cell Biol. 8, 139–145 (1996).
Chai, Y., Ito, Y. & Han, J. TGF-beta signaling and its functional significance in regulating the fate of cranial neural crest cells. Crit. Rev. Oral. Biol. Med. 14, 78–88 (2003).
de Caestecker, M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 15, 1–11 (2004).
Esparza-Lopez, J. et al. Ligand binding and functional properties of betaglycan, a co-receptor of the transforming growth factor-beta superfamily. Specialized binding regions for transforming growth factor-beta and inhibin A. J. Biol. Chem. 276, 14588–14596 (2001).
Lopez-Casillas, F., Wrana, J. L. & Massague, J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell 73, 1435–1444 (1993).
Yamashita, H. et al. Endoglin forms a heteromeric complex with the signaling receptors for transforming growth factor-beta. J. Biol. Chem. 269, 1995–2001 (1994).
Cheifetz, S. et al. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J. Biol. Chem. 267, 19027–19030 (1992).
Massague, J., Seoane, J. & Wotton, D. Smad transcription factors. Genes Dev. 19, 2783–2810 (2005).
Moustakas, A. & Heldin, C. H. The regulation of TGF beta signal transduction. Development 136, 3699–3714 (2009).
Derynck, R. & Zhang, Y. E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584 (2003).
Feng, X. H. & Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693 (2005).
Dennler, S. et al. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17, 3091–3100 (1998).
Zawel, L. et al. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1, 611–617 (1998).
Katagiri, T. et al. Identification of a BMP-responsive element in Id1, the gene for inhibition of myogenesis. Genes Cells 7, 949–960 (2002).
Korchynskyi, O. & ten Dijke, P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 277, 4883–4891 (2002).
Morikawa, M. et al. ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res. 39, 8712–8727 (2011).
Descargues, P. et al. IKKalpha is a critical coregulator of a Smad4-independent TGF beta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc. Natl. Acad. Sci. USA 105, 2487–2492 (2008).
Davis, B. N., Hilyard, A. C., Nguyen, P. H., Lagna, G. & Hata, A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol. Cell 39, 373–384 (2010).
Davis, B. N., Hilyard, A. C., Lagna, G. & Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 454, 56–61 (2008).
Hinck, A. P. Structural studies of the TGF-betas and their receptors—insights into evolution of the TGF-beta superfamily. FEBS Lett. 586, 1860–1870 (2012).
Attisano, L., Wrana, J. L., Montalvo, E. & Massague, J. Activation of signalling by the activin receptor complex. Mol. Cell Biol. 16, 1066–1073 (1996).
Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 67, 753–791 (1998).
Liu, F., Ventura, F., Doody, J. & Massague, J. Human type II receptor for bone morphogenic proteins (BMPs): extension of the two-kinase receptor model to the BMPs. Mol. Cell Biol. 15, 3479–3486 (1995).
Nishitoh, H. et al. Identification of type I and type II serine/threonine kinase receptors for growth/differentiation factor-5. J. Biol. Chem. 271, 21345–21352 (1996).
Nohno, T. et al. Identification of a human type II receptor for bone morphogenetic protein-4 that forms differential heteromeric complexes with bone morphogenetic protein type I receptors. J. Biol. Chem. 270, 22522–22526 (1995).
Koenig, B. B. et al. Characterization and cloning of a receptor for BMP-2 and BMP-4 from NIH 3T3 cells. Mol. Cell Biol. 14, 5961–5974 (1994).
Penton, A. et al. Identification of two bone morphogenetic protein type I receptors in Drosophila and evidence that Brk25D is a decapentaplegic receptor. Cell 78, 239–250 (1994).
Yamashita, H. et al. Osteogenic protein-1 binds to activin type II receptors and induces certain activin-like effects. J. Cell Biol. 130, 217–226 (1995).
Bharathy, S., Xie, W., Yingling, J. M. & Reiss, M. Cancer-associated transforming growth factor beta type II receptor gene mutant causes activation of bone morphogenic protein-Smads and invasive phenotype. Cancer Res. 68, 1656–1666 (2008).
Daly, A. C., Randall, R. A. & Hill, C. S. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol. Cell Biol. 28, 6889–6902 (2008).
Goumans, M. J. et al. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 21, 1743–1753 (2002).
Liu, I. M. et al. TGF beta-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGF beta switch. EMBO J. 28, 88–98 (2009).
Wrighton, K. H., Lin, X., Yu, P. B. & Feng, X. H. Transforming growth factor {beta} can stimulate Smad1 phosphorylation independently of bone morphogenic protein receptors. J. Biol. Chem. 284, 9755–9763 (2009).
Gronroos, E. et al. Transforming growth factor beta inhibits bone morphogenetic protein-induced transcription through novel phosphorylated Smad1/5-Smad3 complexes. Mol. Cell Biol. 32, 2904–2916 (2012).
Massague, J. & Wotton, D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 19, 1745–1754 (2000).
Wrana, J. L. & Attisano, L. The Smad pathway. Cytokine Growth Factor Rev. 11, 5–13 (2000).
Silvestri, C. et al. Genome-wide identification of Smad/Foxh1 targets reveals a role for Foxh1 in retinoic acid regulation and forebrain development. Dev. Cell 14, 411–423 (2008).
Massague, J. & Gomis, R. R. The logic of TGF beta signaling. FEBS Lett. 580, 2811–2820 (2006).
Labbe, E., Silvestri, C., Hoodless, P. A., Wrana, J. L. & Attisano, L. Smad2 and Smad3 positively and negatively regulate TGF beta-dependent transcription through the forkhead DNA-binding protein FAST2. Mol. Cell 2, 109–120 (1998).
Teo, A. K. et al. Pluripotency factors regulate definitive endoderm specification through eomesodermin. Genes Dev. 25, 238–250 (2011).
Mullen, A. C. et al. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell 147, 565–576 (2011).
Suzuki, A. et al. Nanog binds to Smad1 and blocks bone morphogenetic protein-induced differentiation of embryonic stem cells. Proc. Natl. Acad. Sci. USA 103, 10294–10299 (2006).
Vallier, L. et al. Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development 136, 1339–1349 (2009).
Ross, S. & Hill, C. S. How the Smads regulate transcription. Int. J. Biochem. Cell Biol. 40, 383–408 (2008).
van Grunsven, L. A., Verstappen, G., Huylebroeck, D. & Verschueren, K. Smads and chromatin modulation. Cytokine Growth Factor Rev. 16, 495–512 (2005).
Feng, X. H., Zhang, Y., Wu, R. Y. & Derynck, R. The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for smad3 in TGF-beta-induced transcriptional activation. Genes Dev. 12, 2153–2163 (1998).
Janknecht, R., Wells, N. J. & Hunter, T. TGF-beta-stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes Dev. 12, 2114–2119 (1998).
Beyer, T. A., Narimatsu, M., Weiss, A., David, L. & Wrana, J. L. The TGF beta superfamily in stem cell biology and early mammalian embryonic development. Biochim. Biophys. Acta 1830, 2268–2279 (2013).
Watanabe, M., Masuyama, N., Fukuda, M. & Nishida, E. Regulation of intracellular dynamics of Smad4 by its leucine-rich nuclear export signal. EMBO Rep. 1, 176–182 (2000).
Xu, L., Kang, Y., Col, S. & Massague, J. Smad2 nucleocytoplasmic shuttling by nucleoporins CAN/Nup214 and Nup153 feeds TGF beta signaling complexes in the cytoplasm and nucleus. Mol. Cell 10, 271–282 (2002).
Hill, C. S. Nucleocytoplasmic shuttling of Smad proteins. Cell Res. 19, 36–46 (2009).
Varelas, X. et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 10, 837–848 (2008).
Varelas, X. et al. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev. Cell 19, 831–844 (2010).
Lin, X. et al. PPM1A functions as a Smad phosphatase to terminate TGF beta signaling. Cell 125, 915–928 (2006).
Wrighton, K. H., Lin, X. & Feng, X. H. Phospho-control of TGF-beta superfamily signaling. Cell Res. 19, 8–20 (2009).
Di Guglielmo, G. M., Le Roy, C., Goodfellow, A. F. & Wrana, J. L. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat. Cell Biol. 5, 410–421 (2003).
Hayashi, H. et al. The MAD-related protein Smad7 associates with the TGF beta receptor and functions as an antagonist of TGF beta signaling. Cell 89, 1165–1173 (1997).
Nakao, A. et al. Identification of Smad7, a TGF beta-inducible antagonist of TGF-beta signalling. Nature 389, 631–635 (1997).
Zhu, H., Kavsak, P., Abdollah, S., Wrana, J. L. & Thomsen, G. H. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 400, 687–693 (1999).
Gao, S. et al. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol. Cell 36, 457–468 (2009).
Soond, S. M. & Chantry, A. Selective targeting of activating and inhibitory Smads by distinct WWP2 ubiquitin ligase isoforms differentially modulates TGF beta signalling and EMT. Oncogene 30, 2451–2462 (2011).
Fuentealba, L. C. et al. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 131, 980–993 (2007).
Alarcon, C. et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 139, 757–769 (2009).
Qiu, T. et al. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat. Cell Biol. 12, 224–234 (2010).
Moustakas, A. & Heldin, C. H. Non-Smad TGF-beta signals. J. Cell Sci. 118, 3573–3584 (2005).
Zhang, Y. E. Non-Smad pathways in TGF-beta signaling. Cell Res. 19, 128–139 (2009).
Lee, M. K. et al. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 26, 3957–3967 (2007).
Sorrentino, A. et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 10, 1199–1207 (2008).
Yamashita, M. et al. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 31, 918–924 (2008).
Bhowmick, N. A. et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 12, 27–36 (2001).
Edlund, S., Landstrom, M., Heldin, C. H. & Aspenstrom, P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 13, 902–914 (2002).
Ozdamar, B. et al. Regulation of the polarity protein Par6 by TGF beta receptors controls epithelial cell plasticity. Science 307, 1603–1609 (2005).
Vardouli, L., Moustakas, A. & Stournaras, C. LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-beta. J. Biol. Chem. 280, 11448–11457 (2005).
Bakin, A. V., Tomlinson, A. K., Bhowmick, N. A., Moses, H. L. & Arteaga, C. L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 275, 36803–36810 (2000).
Shin, I., Bakin, A. V., Rodeck, U., Brunet, A. & Arteaga, C. L. Transforming growth factor beta enhances epithelial cell survival via Akt-dependent regulation of FKHRL1. Mol. Biol. Cell 12, 3328–3339 (2001).
Lamouille, S. & Derynck, R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 178, 437–451 (2007).
Lamouille, S., Connolly, E., Smyth, J. W., Akhurst, R. J. & Derynck, R. TGF-beta-induced activation of mTOR complex 2 drives epithelial–mesenchymal transition and cell invasion. J. Cell Sci. 125, 1259–1273 (2012).
Ikushima, H. & Miyazono, K. TGF-beta signal transduction spreading to a wider field: a broad variety of mechanisms for context-dependent effects of TGF-beta. Cell Tissue Res. 347, 37–49 (2012).
Vincent, T. et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial–mesenchymal transition. Nat. Cell Biol. 11, 943–950 (2009).
Kim, K. K. et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Invest. 119, 213–224 (2009).
Zhou, B. et al. Interactions between beta-catenin and transforming growth factor-beta signaling pathways mediate epithelial–mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J. Biol. Chem. 287, 7026–7038 (2012).
Edlund, S. et al. Interaction between Smad7 and beta-catenin: importance for transforming growth factor beta-induced apoptosis. Mol. Cell Biol. 25, 1475–1488 (2005).
Singh, A. M. et al. Signaling network cross talk in human pluripotent cells: a Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem. Cell 10, 312–326 (2012).
Niimi, H., Pardali, K., Vanlandewijck, M., Heldin, C. H. & Moustakas, A. Notch signaling is necessary for epithelial growth arrest by TGF-beta. J. Cell Biol. 176, 695–707 (2007).
Zavadil, J., Cermak, L., Soto-Nieves, N. & Bottinger, E. P. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 23, 1155–1165 (2004).
Aoyagi-Ikeda, K. et al. Notch induces myofibroblast differentiation of alveolar epithelial cells via transforming growth factor-{beta}-Smad3 pathway. Am. J. Respir. Cell Mol. Biol. 45, 136–144 (2011).
Ohashi, S. et al. A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer Res. 71, 6836–6847 (2011).
Gotzmann, J. et al. A crucial function of PDGF in TGF-beta-mediated cancer progression of hepatocytes. Oncogene 25, 3170–3185 (2006).
Fischer, A. N. et al. PDGF essentially links TGF-beta signaling to nuclear beta-catenin accumulation in hepatocellular carcinoma progression. Oncogene 26, 3395–3405 (2007).
Xue, G. et al. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-beta signaling axes. Cancer Discov. 2, 248–259 (2012).
Ferrari, G. et al. Muscle regeneration by bone marrow-derived myogenic progenitors. Science 279, 1528–1530 (1998).
Takahashi, T. et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat. Med. 5, 434–438 (1999).
Lagasse, E. et al. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat. Med. 6, 1229–1234 (2000).
Orlic, D. et al. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc. Natl. Acad. Sci. USA 98, 10344–10349 (2001).
Kale, S. et al. Bone marrow stem cells contribute to repair of the ischemically injured renal tubule. J. Clin. Invest. 112, 42–49 (2003).
Wojakowski, W., Landmesser, U., Bachowski, R., Jadczyk, T. & Tendera, M. Mobilization of stem and progenitor cells in cardiovascular diseases. Leukemia 26, 23–33 (2012).
Krankel, N., Spinetti, G., Amadesi, S. & Madeddu, P. Targeting stem cell niches and trafficking for cardiovascular therapy. Pharmacol. Ther. 129, 62–81 (2011).
Caplan, A. I. & Correa, D. The MSC: an injury drugstore. Cell Stem Cell 9, 11–15 (2011).
Zaidi, M. Skeletal remodeling in health and disease. Nat. Med. 13, 791–801 (2007).
Bianco, P. et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat. Med. 19, 35–42 (2013).
Xian, L. et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 18, 1095–1101 (2012).
Janssens, K., ten Dijke, P., Janssens, S. & Van Hul, W. Transforming growth factor-beta1 to the bone. Endocr. Rev. 26, 743–774 (2005).
Wan, M. et al. Injury-activated transforming growth factor beta controls mobilization of mesenchymal stem cells for tissue remodeling. Stem Cells 30, 2498–2511 (2012).
Gao, P. et al. Functional effects of TGF-beta1 on mesenchymal stem cell mobilization in cockroach allergen-induced asthma. J. Immunol. 192, 4560–4570 (2014).
Lee, C. H. et al. Regeneration of the articular surface of the rabbit synovial joint by cell homing: a proof of concept study. Lancet 376, 440–448 (2010).
Shinojima, N. et al. TGF-beta mediates homing of bone marrow-derived human mesenchymal stem cells to glioma stem cells. Cancer Res. 73, 2333–2344 (2013).
Thomson, J. A. et al. Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147 (1998).
Ying, Q. L., Nichols, J., Chambers, I. & Smith, A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 115, 281–292 (2003).
Chen, X. et al. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature 389, 85–89 (1997).
Alliston, T., Choy, L., Ducy, P., Karsenty, G. & Derynck, R. TGF-beta-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J. 20, 2254–2272 (2001).
Liu, D., Black, B. L. & Derynck, R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 15, 2950–2966 (2001).
Choy, L. & Derynck, R. Transforming growth factor-beta inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J. Biol. Chem. 278, 9609–9619 (2003).
Xu, R. H. et al. NANOG is a direct target of TGF beta/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell 3, 196–206 (2008).
Yoon, S. J., Wills, A. E., Chuong, E., Gupta, R. & Baker, J. C. HEB and E2A function as SMAD/FOXH1 cofactors. Genes Dev. 25, 1654–1661 (2011).
Xi, Q. et al. A poised chromatin platform for TGF-beta access to master regulators. Cell 147, 1511–1524 (2011).
Agricola, E., Randall, R. A., Gaarenstroom, T., Dupont, S. & Hill, C. S. Recruitment of TIF1gamma to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol. Cell 43, 85–96 (2011).
Dupont, S. et al. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 121, 87–99 (2005).
Aragon, E. et al. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 25, 1275–1288 (2011).
Lonn, P. et al. PARP-1 attenuates Smad-mediated transcription. Mol. Cell 40, 521–532 (2010).
Orkin, S. H. & Hochedlinger, K. Chromatin connections to pluripotency and cellular reprogramming. Cell 145, 835–850 (2011).
Young, R. A. Control of the embryonic stem cell state. Cell 144, 940–954 (2011).
Chen, X. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117 (2008).
Oshimori, N. & Fuchs, E. Paracrine TGF-beta signaling counterbalances BMP-mediated repression in hair follicle stem cell activation. Cell Stem Cell 10, 63–75 (2012).
Phinney, D. G. & Prockop, D. J. Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair—current views. Stem Cells 25, 2896–2902 (2007).
Shi, S. et al. The efficacy of mesenchymal stem cells to regenerate and repair dental structures. Orthod. Craniofac. Res. 8, 191–199 (2005).
Boland, G. M., Perkins, G., Hall, D. J. & Tuan, R. S. Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J. Cell Biochem. 93, 1210–1230 (2004).
Jian, H. et al. Smad3-dependent nuclear translocation of beta-catenin is required for TGF-beta1-induced proliferation of bone marrow-derived adult human mesenchymal stem cells. Genes Dev. 20, 666–674 (2006).
Baksh, D., Boland, G. M. & Tuan, R. S. Cross-talk between Wnt signaling pathways in human mesenchymal stem cells leads to functional antagonism during osteogenic differentiation. J. Cell Biochem. 101, 1109–1124 (2007).
Liu, Z., Tang, Y., Qiu, T., Cao, X. & Clemens, T. L. A dishevelled-1/Smad1 interaction couples WNT and bone morphogenetic protein signaling pathways in uncommitted bone marrow stromal cells. J. Biol. Chem. 281, 17156–17163 (2006).
Roelen, B. A. & Dijke, P. Controlling mesenchymal stem cell differentiation by TGF Beta family members. J. Orthop. Sci. 8, 740–748 (2003).
Maeda, S., Hayashi, M., Komiya, S., Imamura, T. & Miyazono, K. Endogenous TGF-beta signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J. 23, 552–563 (2004).
Lin, X. et al. Smad6 recruits transcription corepressor CtBP to repress bone morphogenetic protein-induced transcription. Mol. Cell Biol. 23, 9081–9093 (2003).
Tseng, Y. H. et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 454, 1000–1004 (2008).
Kurpinski, K. et al. Transforming growth factor-beta and notch signaling mediate stem cell differentiation into smooth muscle cells. Stem Cells 28, 734–742 (2010).
Yamazaki, S. et al. TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood 113, 1250–1256 (2009).
Larsson, J. et al. TGF-beta signaling-deficient hematopoietic stem cells have normal self-renewal and regenerative ability in vivo despite increased proliferative capacity in vitro. Blood 102, 3129–3135 (2003).
Larsson, J., Blank, U., Klintman, J., Magnusson, M. & Karlsson, S. Quiescence of hematopoietic stem cells and maintenance of the stem cell pool is not dependent on TGF-beta signaling in vivo. Exp. Hematol. 33, 592–596 (2005).
Kale, V. P. & Vaidya, A. A. Molecular mechanisms behind the dose-dependent differential activation of MAPK pathways induced by transforming growth factor-beta1 in hematopoietic cells. Stem Cells Dev. 13, 536–547 (2004).
Yamazaki, S. et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 147, 1146–1158 (2011).
Challen, G. A., Boles, N. C., Chambers, S. M. & Goodell, M. A. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell 6, 265–278 (2010).
Reynolds, B. A. & Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255, 1707–1710 (1992).
Falk, S. et al. Brain area-specific effect of TGF-beta signaling on Wnt-dependent neural stem cell expansion. Cell Stem Cell 2, 472–483 (2008).
Gage, F. H. Mammalian neural stem cells. Science 287, 1433–1438 (2000).
Flanders, K. C., Ren, R. F. & Lippa, C. F. Transforming growth factor-betas in neurodegenerative disease. Prog. Neurobiol. 54, 71–85 (1998).
Bottner, M., Krieglstein, K. & Unsicker, K. The transforming growth factor-betas: structure, signaling, and roles in nervous system development and functions. J. Neurochem. 75, 2227–2240 (2000).
Aigner, L. & Bogdahn, U. TGF-beta in neural stem cells and in tumors of the central nervous system. Cell Tissue Res 331, 225–241 (2008).
Dhandapani, K. M. & Brann, D. W. Transforming growth factor-beta: a neuroprotective factor in cerebral ischemia. Cell Biochem. Biophys. 39, 13–22 (2003).
Krieglstein, K., Strelau, J., Schober, A., Sullivan, A. & Unsicker, K. TGF-beta and the regulation of neuron survival and death. J. Physiol. 96, 25–30 (2002).
Wachs, F. P. et al. Transforming growth factor-beta1 is a negative modulator of adult neurogenesis. J. Neuropathol. Exp. Neurol. 65, 358–370 (2006).
Battista, D., Ferrari, C. C., Gage, F. H. & Pitossi, F. J. Neurogenic niche modulation by activated microglia: transforming growth factor beta increases neurogenesis in the adult dentate gyrus. Eur. J. Neurosci. 23, 83–93 (2006).
Docagne, F. et al. Smad3-dependent induction of plasminogen activator inhibitor-1 in astrocytes mediates neuroprotective activity of transforming growth factor-beta 1 against NMDA-induced necrosis. Mol. Cell Neurosci. 21, 634–644 (2002).
Fuchs, E. Scratching the surface of skin development. Nature 445, 834–842 (2007).
Hsu, Y. C., Pasolli, H. A. & Fuchs, E. Dynamics between stem cells, niche, and progeny in the hair follicle. Cell 144, 92–105 (2011).
Plikus, M. V. et al. Cyclic dermal BMP signalling regulates stem cell activation during hair regeneration. Nature 451, 340–344 (2008).
Foitzik, K. et al. Control of murine hair follicle regression (catagen) by TGF-beta1 in vivo. FASEB J. 14, 752–760 (2000).
Soma, T., Dohrmann, C. E., Hibino, T. & Raftery, L. A. Profile of transforming growth factor-beta responses during the murine hair cycle. J. Invest. Dermatol. 121, 969–975 (2003).
Foitzik, K., Paus, R., Doetschman, T. & Dotto, G. P. The TGF-beta2 isoform is both a required and sufficient inducer of murine hair follicle morphogenesis. Dev. Biol. 212, 278–289 (1999).
Chang, C., Eggen, B. J., Weinstein, D. C. & Brivanlou, A. H. Regulation of nodal and BMP signaling by tomoregulin-1 (X7365) through novel mechanisms. Dev. Biol. 255, 1–11 (2003).
Campion, D. R. The muscle satellite cell: a review. Int. Rev. Cytol. 87, 225–251 (1984).
Charge, S. B. & Rudnicki, M. A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209–238 (2004).
Peault, B. et al. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol. Ther. 15, 867–877 (2007).
Wang, H. et al. Bmp signaling at the tips of skeletal muscles regulates the number of fetal muscle progenitors and satellite cells during development. Dev. Cell 18, 643–654 (2010).
Collins, C. A. et al. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 122, 289–301 (2005).
Conboy, I. M., Conboy, M. J., Smythe, G. M. & Rando, T. A. Notch-mediated restoration of regenerative potential to aged muscle. Science 302, 1575–1577 (2003).
Brack, A. S. et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 317, 807–810 (2007).
Carlson, M. E., Hsu, M. & Conboy, I. M. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 454, 528–532 (2008).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Ichida, J. K. et al. A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell Stem Cell 5, 491–503 (2009).
Maherali, N. & Hochedlinger, K. Tgf beta signal inhibition cooperates in the induction of iPSCs and replaces Sox2 and cMyc. Curr. Biol. 19, 1718–1723 (2009).
Lin, T. et al. A chemical platform for improved induction of human iPSCs. Nat. Methods 6, 805–808 (2009).
Li, R. et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7, 51–63 (2010).
Li, Z., Yang, C. S., Nakashima, K. & Rana, T. M. Small RNA-mediated regulation of iPS cell generation. EMBO J. 30, 823–834 (2011).
Miyoshi, N. et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 8, 633–638 (2011).
Subramanyam, D. et al. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 29, 443–448 (2011).
Samavarchi-Tehrani, P. et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7, 64–77 (2010).
Moss, M. L. Studies of the acellular bone of teleost fish. V. Histology and mineral homeostasis of fresh-water species. Acta Anat. 60, 262–276 (1965).
Weiss, R. E. & Watabe, N. Studies on the biology of fish bone. III. Ultrastructure of osteogenesis and resorption in osteocytic (cellular) and anosteocytic (acellular) bones. Calcif. Tissue Int. 28, 43–56 (1979).
Glowacki, J., Cox, K. A., O'Sullivan, J., Wilkie, D. & Deftos, L. J. Osteoclasts can be induced in fish having an acellular bony skeleton. Proc. Natl. Acad. Sci. USA 83, 4104–4107 (1986).
Witten, P. E. & Huysseune, A. A comparative view on mechanisms and functions of skeletal remodelling in teleost fish, with special emphasis on osteoclasts and their function. Biol. Rev. Camb. Philos. Soc. 84, 315–346 (2009).
Bonewald, L. F. Osteocytes as dynamic multifunctional cells. Ann. N. Y. Acad. Sci. 1116, 281–290 (2007).
Kamioka, H., Honjo, T. & Takano-Yamamoto, T. A three-dimensional distribution of osteocyte processes revealed by the combination of confocal laser scanning microscopy and differential interference contrast microscopy. Bone 28, 145–149 (2001).
Verborgt, O., Tatton, N. A., Majeska, R. J. & Schaffler, M. B. Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: complementary roles in bone remodeling regulation? J. Bone Miner. Res. 17, 907–914 (2002).
Xiong, J. et al. Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241 (2011).
Nakashima, T. et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 (2011).
van Bezooijen, R. L. et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J. Exp. Med. 199, 805–814 (2004).
Bellido, T. et al. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146, 4577–4583 (2005).
Robling, A. G. et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J. Biol. Chem. 283, 5866–5875 (2008).
Keller, H. & Kneissel, M. SOST is a target gene for PTH in bone. Bone 37, 148–158 (2005).
Krause, C. et al. Distinct modes of inhibition by sclerostin on bone morphogenetic protein and Wnt signaling pathways. J. Biol. Chem. 285, 41614–41626 (2010).
Li, X. et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 280, 19883–19887 (2005).
Semenov, M., Tamai, K. & He, X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 280, 26770–26775 (2005).
Raggatt, L. J. & Partridge, N. C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 285, 25103–25108 (2010).
Li, Y. et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 109, 3839–3848 (2007).
Kacena, M. A. et al. Megakaryocyte-osteoblast interaction revealed in mice deficient in transcription factors GATA-1 and NF-E2. J. Bone Miner. Res. 19, 652–660 (2004).
Hume, D. A. Differentiation and heterogeneity in the mononuclear phagocyte system. Mucosal Immunol. 1, 432–441 (2008).
Andersen, T. L. et al. A physical mechanism for coupling bone resorption and formation in adult human bone. Am. J. Pathol. 174, 239–247 (2009).
Chang, M. K. et al. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J. Immunol. 181, 1232–1244 (2008).
Morikawa, S. et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J. Exp. Med. 206, 2483–2496 (2009).
Park, D. et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 10, 259–272 (2012).
Omatsu, Y. et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 33, 387–399 (2010).
Kunisaki, Y. et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643 (2013).
Mendez-Ferrer, S. et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829–834 (2010).
Ono, N. et al. Vasculature-associated cells expressing nestin in developing bones encompass early cells in the osteoblast and endothelial lineage. Dev. Cell 29, 330–339 (2014).
Zheng, Y. & Geiger, H. HSPCs get their motors running for asymmetric fate choice. Cell Stem Cell 14, 1–2 (2014).
Ding, L., Saunders, T. L., Enikolopov, G. & Morrison, S. J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462 (2012).
Lapraz, F. et al. RTK and TGF-beta signaling pathways genes in the sea urchin genome. Dev. Biol. 300, 132–152 (2006).
Neidlinger-Wilke, C. et al. Human osteoblasts from younger normal and osteoporotic donors show differences in proliferation and TGF beta-release in response to cyclic strain. J. Biomech. 28, 1411–1418 (1995).
Dallas, S. L., Rosser, J. L., Mundy, G. R. & Bonewald, L. F. Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem. 277, 21352–21360 (2002).
Engler, A. J., Sen, S., Sweeney, H. L. & Discher, D. E. Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 (2006).
Kim, J. S. et al. Transforming growth factor-beta1 regulates macrophage migration via RhoA. Blood 108, 1821–1829 (2006).
Martin, T. J. & Sims, N. A. Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol. Med. 11, 76–81 (2005).
Pederson, L., Ruan, M., Westendorf, J. J., Khosla, S. & Oursler, M. J. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc. Natl. Acad. Sci. USA 105, 20764–20769 (2008).
Kreja, L. et al. Non-resorbing osteoclasts induce migration and osteogenic differentiation of mesenchymal stem cells. J. Cell Biochem. 109, 347–355 (2010).
Sanchez-Fernandez, M. A., Gallois, A., Riedl, T., Jurdic, P. & Hoflack, B. Osteoclasts control osteoblast chemotaxis via PDGF-BB/PDGF receptor beta signaling. PLoS ONE 3, e3537 (2008).
Xie, H. et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat. Med. 20, 1270–1278 (2014).
Zhao, C. et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 4, 111–121 (2006).
Owen, M. & Friedenstein, A. J. Stromal stem cells: marrow-derived osteogenic precursors. Ciba. Found. Symp. 136, 42–60 (1988).
Sacchetti, B. et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131, 324–336 (2007).
Woodbury, D., Schwarz, E. J., Prockop, D. J. & Black, I. B. Adult rat and human bone marrow stromal cells differentiate into neurons. J. Neurosci. Res. 61, 364–370 (2000).
Negishi-Koga, T. et al. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 17, 1473–1480 (2011).
Chim, S. M. et al. Angiogenic factors in bone local environment. Cytokine Growth Factor Rev. 24, 297–310 (2013).
Seghezzi, G. et al. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J. Cell Biol. 141, 1659–1673 (1998).
Beck, L. Jr. & D'Amore, P. A. Vascular development: cellular and molecular regulation. FASEB J. 11, 365–373 (1997).
Klein, S. et al. Basic fibroblast growth factor modulates integrin expression in microvascular endothelial cells. Mol. Biol. Cell 4, 973–982 (1993).
Klein, S., Bikfalvi, A., Birkenmeier, T. M., Giancotti, F. G. & Rifkin, D. B. Integrin regulation by endogenous expression of 18-kDa fibroblast growth factor-2. J. Biol. Chem. 271, 22583–22590 (1996).
Sabbieti, M. G. et al. Prostaglandins differently regulate FGF-2 and FGF receptor expression and induce nuclear translocation in osteoblasts via MAPK kinase. Cell Tissue Res. 319, 267–278 (2005).
Sobue, T. et al. Regulation of fibroblast growth factor 2 and fibroblast growth factor receptors by transforming growth factor beta in human osteoblastic MG-63 cells. J. Bone Miner. Res. 17, 502–512 (2002).
Sabbieti, M. G. et al. Prostaglandins regulate the expression of fibroblast growth factor-2 in bone. Endocrinology 140, 434–444 (1999).
Shao, E. S., Lin, L., Yao, Y. & Bostrom, K. I. Expression of vascular endothelial growth factor is coordinately regulated by the activin-like kinase receptors 1 and 5 in endothelial cells. Blood 114, 2197–2206 (2009).
Morrison, S. J. & Scadden, D. T. The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334 (2014).
Calvi, L. M. et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846 (2003).
Hsu, Y. C. & Fuchs, E. A family business: stem cell progeny join the niche to regulate homeostasis. Nat. Rev. Mol. Cell Biol. 13, 103–114 (2012).
Okabe, M. & Graham, A. The origin of the parathyroid gland. Proc. Natl. Acad. Sci. USA 101, 17716–17719 (2004).
Bikle, D. D. et al. Insulin-like growth factor I is required for the anabolic actions of parathyroid hormone on mouse bone. J. Bone Miner. Res. 17, 1570–1578 (2002).
Canalis, E., Centrella, M., Burch, W. & McCarthy, T. L. Insulin-like growth factor I mediates selective anabolic effects of parathyroid hormone in bone cultures. J. Clin. Invest. 83, 60–65 (1989).
Lombardi, G. et al. Role of IGF-I on PTH effects on bone. J. Endocrinol. Invest. 33, 22–26 (2010).
Miyakoshi, N., Kasukawa, Y., Linkhart, T. A., Baylink, D. J. & Mohan, S. Evidence that anabolic effects of PTH on bone require IGF-I in growing mice. Endocrinology 142, 4349–4356 (2001).
Pfeilschifter, J. et al. Parathyroid hormone increases the concentration of insulin-like growth factor-I and transforming growth factor beta 1 in rat bone. J. Clin. Invest. 96, 767–774 (1995).
Wan, M. et al. Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 22, 2968–2979 (2008).
Watson, P. et al. Parathyroid hormone restores bone mass and enhances osteoblast insulin-like growth factor I gene expression in ovariectomized rats. Bone 16, 357–365 (1995).
Yu, B. et al. Parathyroid hormone induces differentiation of mesenchymal stromal/stem cells by enhancing bone morphogenetic protein signaling. J. Bone Miner. Res. 27, 2001–2014 (2012).
Polo, S. & Di Fiore, P. P. Endocytosis conducts the cell signaling orchestra. Cell 124, 897–900 (2006).
He, X., Semenov, M., Tamai, K. & Zeng, X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development 131, 1663–1677 (2004).
Itasaki, N. & Hoppler, S. Crosstalk between Wnt and bone morphogenic protein signaling: a turbulent relationship. Dev. Dyn. 239, 16–33 (2010).
Li, C. et al. Disruption of LRP6 in osteoblasts blunts the bone anabolic activity of PTH. J. Bone Miner. Res. 28, 2094–2108 (2013).
Jiang, Y. et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418, 41–49 (2002).
Kim, S. W. et al. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J. Bone Miner. Res. 27, 2075–2084 (2012).
Terauchi, M. et al. T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab. 10, 229–240 (2009).
Prisby, R. et al. Intermittent PTH(1-84) is osteoanabolic but not osteoangiogenic and relocates bone marrow blood vessels closer to bone-forming sites. J. Bone Miner. Res. 26, 2583–2596 (2011).
Goldring, M. B. & Goldring, S. R. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann. N. Y. Acad. Sci. 1192, 230–237 (2010).
Newberry, W. N., Zukosky, D. K. & Haut, R. C. Subfracture insult to a knee joint causes alterations in the bone and in the functional stiffness of overlying cartilage. J. Orthop. Res. 15, 450–455 (1997).
Radin, E. L. & Rose, R. M. Role of subchondral bone in the initiation and progression of cartilage damage. Clin. Orthop. Relat. Res. 213, 34–40 (1986).
Intema, F. et al. Similarities and discrepancies in subchondral bone structure in two differently induced canine models of osteoarthritis. J. Bone Miner. Res. 25, 1650–1657 (2010).
Sniekers, Y. H. et al. A role for subchondral bone changes in the process of osteoarthritis; a micro-CT study of two canine models. BMC Musculoskelet. Disord. 9, 20 (2008).
Burr, D. B. & Gallant, M. A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol. 8, 665–673 (2012).
Karsdal, M. A. et al. Should subchondral bone turnover be targeted when treating osteoarthritis? Osteoarthr. Cartil. 16, 638–646 (2008).
Grynpas, M. D., Alpert, B., Katz, I., Lieberman, I. & Pritzker, K. P. Subchondral bone in osteoarthritis. Calcif. Tissue Int. 49, 20–26 (1991).
Huebner, J. L., Hanes, M. A., Beekman, B., TeKoppele, J. M. & Kraus, V. B. A comparative analysis of bone and cartilage metabolism in two strains of guinea-pig with varying degrees of naturally occurring osteoarthritis. Osteoarthr. Cartil. 10, 758–767 (2002).
Burr, D. B. & Radin, E. L. Microfractures and microcracks in subchondral bone: are they relevant to osteoarthrosis? Rheum. Dis. Clin. North. Am. 29, 675–685 (2003).
Ni, G. X. et al. Matrix metalloproteinase-3 inhibitor retards treadmill running-induced cartilage degradation in rats. Arthritis Res. Ther. 13, R192 (2011).
Farquhar, T. et al. Swelling and fibronectin accumulation in articular cartilage explants after cyclical impact. J. Orthop. Res. 14, 417–423 (1996).
Clements, K. M., Bee, Z. C., Crossingham, G. V., Adams, M. A. & Sharif, M. How severe must repetitive loading be to kill chondrocytes in articular cartilage? Osteoarthr. Cartil. 9, 499–507 (2001).
Chen, C. T., Bhargava, M., Lin, P. M. & Torzilli, P. A. Time, stress, and location dependent chondrocyte death and collagen damage in cyclically loaded articular cartilage. J. Orthop. Res. 21, 888–898 (2003).
Lin, P. M., Chen, C. T. & Torzilli, P. A. Increased stromelysin-1 (MMP-3), proteoglycan degradation (3B3- and 7D4) and collagen damage in cyclically load-injured articular cartilage. Osteoarthr. Cartil. 12, 485–496 (2004).
Serra, R. et al. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J. Cell Biol. 139, 541–552 (1997).
Yang, X. et al. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 153, 35–46 (2001).
van der Kraan, P. M., Goumans, M. J., Blaney Davidson, E. & ten Dijke, P. Age-dependent alteration of TGF-beta signalling in osteoarthritis. Cell Tissue Res. 347, 257–265 (2012).
van Beuningen, H. M., van der Kraan, P. M., Arntz, O. J. & van den Berg, W. B. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab. Invest. 71, 279–290 (1994).
Scharstuhl, A., Vitters, E. L., van der Kraan, P. M. & van den Berg, W. B. Reduction of osteophyte formation and synovial thickening by adenoviral overexpression of transforming growth factor beta/bone morphogenetic protein inhibitors during experimental osteoarthritis. Arthritis Rheum. 48, 3442–3451 (2003).
Warman, M. L. et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A 155A, 943–968 (2011).
Janssens, K. et al. Mutations in the gene encoding the latency-associated peptide of TGF-beta 1 cause Camurati–Engelmann disease. Nat. Genet. 26, 273–275 (2000).
Kinoshita, A. et al. Domain-specific mutations in TGFB1 result in Camurati–Engelmann disease. Nat. Genet. 26, 19–20 (2000).
Janssens, K., ten Dijke, P., Ralston, S. H., Bergmann, C. & Van Hul, W. Transforming growth factor-beta 1 mutations in Camurati–Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J. Biol. Chem. 278, 7718–7724 (2003).
Saito, T. et al. Domain-specific mutations of a transforming growth factor (TGF)-beta 1 latency-associated peptide cause Camurati–Engelmann disease because of the formation of a constitutively active form of TGF-beta 1. J. Biol. Chem. 276, 11469–11472 (2001).
Janssens, K. et al. Camurati–Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J. Med. Genet. 43, 1–11 (2006).
Whyte, M. P. et al. Camurati–Engelmann disease: unique variant featuring a novel mutation in TGFbeta1 encoding transforming growth factor beta 1 and a missense change in TNFSF11 encoding RANK ligand. J. Bone Miner. Res. 26, 920–933 (2011).
Bondestam, J. et al. Bone biopsy and densitometry findings in a child with Camurati–Engelmann disease. Clin. Rheumatol. 26, 1773–1777 (2007).
Kainulainen, K., Karttunen, L., Puhakka, L., Sakai, L. & Peltonen, L. Mutations in the fibrillin gene responsible for dominant ectopia lentis and neonatal Marfan syndrome. Nat. Genet. 6, 64–69 (1994).
Dietz, H. C., Loeys, B., Carta, L. & Ramirez, F. Recent progress towards a molecular understanding of Marfan syndrome. Am. J. Med. Genet. C. Semin. Med. Genet. 139C, 4–9 (2005).
Dietz, H. C. et al. The Marfan syndrome locus: confirmation of assignment to chromosome 15 and identification of tightly linked markers at 15q15-q21.3. Genomics 9, 355–361 (1991).
Dietz, H. C. et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352, 337–339 (1991).
Neptune, E. R. et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat. Genet. 33, 407–411 (2003).
Loeys, B. L. et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 37, 275–281 (2005).
Gallo, E. M. et al. Angiotensin II-dependent TGF-beta signaling contributes to Loeys–Dietz syndrome vascular pathogenesis. J. Clin. Invest. 124, 448–460 (2014).
van de Laar, I. M. et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 43, 121–126 (2011).
Lindsay, M. E. et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 44, 922–927 (2012).
Carmignac, V. et al. In-frame mutations in exon 1 of SKI cause dominant Shprintzen–Goldberg syndrome. Am. J. Hum. Genet. 91, 950–957 (2012).
Doyle, A. J. et al. Mutations in the TGF-beta repressor SKI cause Shprintzen–Goldberg syndrome with aortic aneurysm. Nat. Genet. 44, 1249–1254 (2012).
Prunier, C. et al. The oncoprotein Ski acts as an antagonist of transforming growth factor-beta signaling by suppressing Smad2 phosphorylation. J. Biol. Chem. 278, 26249–26257 (2003).
Reed, J. A. et al. Cytoplasmic localization of the oncogenic protein Ski in human cutaneous melanomas in vivo: functional implications for transforming growth factor beta signaling. Cancer Res. 61, 8074–8078 (2001).
Nomura, T. et al. Ski is a component of the histone deacetylase complex required for transcriptional repression by Mad and thyroid hormone receptor. Genes Dev. 13, 412–423 (1999).
Rhodes, S. D. et al. Hyperactive transforming growth factor-beta1 signaling potentiates skeletal defects in a neurofibromatosis type 1 mouse model. J. Bone Miner. Res. 28, 2476–2489 (2013).
Ray, N. F., Chan, J. K., Thamer, M. & Melton, L. J. III Medical expenditures for the treatment of osteoporotic fractures in the United States in 1995: report from the National Osteoporosis Foundation. J. Bone Miner. Res. 12, 24–35 (1997).
Cummings, S. R., Kelsey, J. L., Nevitt, M. C. & O'Dowd, K. J. Epidemiology of osteoporosis and osteoporotic fractures. Epidemiol. Rev. 7, 178–208 (1985).
Raisz, L. G. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J. Clin. Invest. 115, 3318–3325 (2005).
Xu, X. H. et al. Molecular genetic studies of gene identification for osteoporosis: the 2009 update. Endocr. Rev. 31, 447–505 (2010).
Langdahl, B. L., Knudsen, J. Y., Jensen, H. K., Gregersen, N. & Eriksen, E. F. A sequence variation: 713-8delC in the transforming growth factor-beta 1 gene has higher prevalence in osteoporotic women than in normal women and is associated with very low bone mass in osteoporotic women and increased bone turnover in both osteoporotic and normal women. Bone 20, 289–294 (1997).
Yamada, Y. et al. Association of the C-509-->T polymorphism, alone of in combination with the T869-->C polymorphism, of the transforming growth factor-beta1 gene with bone mineral density and genetic susceptibility to osteoporosis in Japanese women. J. Mol. Med. 79, 149–156 (2001).
Yamada, Y. et al. Association of a polymorphism of the transforming growth factor-beta1 gene with genetic susceptibility to osteoporosis in postmenopausal Japanese women. J. Bone Miner. Res. 13, 1569–1576 (1998).
Kou, I. et al. Common variants in a novel gene, FONG on chromosome 2q33.1 confer risk of osteoporosis in Japanese. PLoS ONE 6, e19641 (2011).
Hubacek, J. A. et al. No associations between genetic polymorphisms of TGF-beta, PAI-1, and COL1A1, and bone mineral density in Caucasian females. Endocr. Regul. 40, 107–112 (2006).
Tural, S. et al. Association between osteoporosis and polymorphisms of the IL-10 and TGF-beta genes in Turkish postmenopausal women. Hum. Immunol. 74, 1179–1183 (2013).
Willis, B. C. & Borok, Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 293, L525–534 (2007).
Gressner, A. M., Weiskirchen, R., Breitkopf, K. & Dooley, S. Roles of TGF-beta in hepatic fibrosis. Front. Biosci. 7, d793–807 (2002).
Zeisberg, E. M. et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13, 952–961 (2007).
Okuda, S., Languino, L. R., Ruoslahti, E. & Border, W. A. Elevated expression of transforming growth factor-beta and proteoglycan production in experimental glomerulonephritis. Possible role in expansion of the mesangial extracellular matrix. J. Clin. Investig. 86, 453–462 (1990).
Tomooka, S., Border, W. A., Marshall, B. C. & Noble, N. A. Glomerular matrix accumulation is linked to inhibition of the plasmin protease system. Kidney Int. 42, 1462–1469 (1992).
Kagami, S., Border, W. A., Ruoslahti, E. & Noble, N. A. Coordinated expression of beta 1 integrins and transforming growth factor-beta-induced matrix proteins in glomerulonephritis. Lab. Invest. 69, 68–76 (1993).
Border, W. A., Okuda, S., Languino, L. R., Sporn, M. B. & Ruoslahti, E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor beta 1. Nature 346, 371–374 (1990).
Border, W. A. et al. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature 360, 361–364 (1992).
Isaka, Y. et al. Glomerulosclerosis induced by in vivo transfection of transforming growth factor-beta or platelet-derived growth factor gene into the rat kidney. J. Clin. Invest. 92, 2597–2601 (1993).
Lee, H. S. Pathogenic role of TGF-beta in the progression of podocyte diseases. Histol. Histopathol. 26, 107–116 (2011).
Chen, H. C. et al. Altering expression of alpha3beta1 integrin on podocytes of human and rats with diabetes. Life Sci. 67, 2345–2353 (2000).
Das, F., Ghosh-Choudhury, N., Kasinath, B. S. & Choudhury, G. G. TGF beta enforces activation of eukaryotic elongation factor-2 (eEF2) via inactivation of eEF2 kinase by p90 ribosomal S6 kinase (p90Rsk) to induce mesangial cell hypertrophy. FEBS Lett. 584, 4268–4272 (2010).
Abdel-Wahab, N., Weston, B. S., Roberts, T. & Mason, R. M. Connective tissue growth factor and regulation of the mesangial cell cycle: role in cellular hypertrophy. J. Am. Soc. Nephrol. 13, 2437–2445 (2002).
Rodriguez-Barbero, A. et al. TGF-beta1 induces COX-2 expression and PGE2 synthesis through MAPK and PI3K pathways in human mesangial cells. Kidney Int. 70, 901–909 (2006).
Zeisberg, E. M., Potenta, S. E., Sugimoto, H., Zeisberg, M. & Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 19, 2282–2287 (2008).
Sato, M., Muragaki, Y., Saika, S., Roberts, A. B. & Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Invest. 112, 1486–1494 (2003).
Li, J. et al. Blockade of endothelial–mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 59, 2612–2624 (2010).
Garcia-Sanchez, O., Lopez-Hernandez, F. J. & Lopez-Novoa, J. M. An integrative view on the role of TGF-beta in the progressive tubular deletion associated with chronic kidney disease. Kidney Int. 77, 950–955 (2010).
Grande, M. T. & Lopez-Novoa, J. M. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat. Rev. Nephrol. 5, 319–328 (2009).
Meran, S. & Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 92, 158–167 (2011).
Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200, 500–503 (2003).
LeBleu, V. S. et al. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19, 1047–1053 (2013).
Brenner, D. A. Molecular pathogenesis of liver fibrosis. Trans. Am. Clin. Climatol. Assoc. 120, 361–368 (2009).
Castilla, A., Prieto, J. & Fausto, N. Transforming growth factors beta 1 and alpha in chronic liver disease. Effects of interferon alfa therapy. N. Engl. J. Med. 324, 933–940 (1991).
Nagy, P., Schaff, Z. & Lapis, K. Immunohistochemical detection of transforming growth factor-beta 1 in fibrotic liver diseases. Hepatology 14, 269–273 (1991).
Czaja, M. J. et al. In vitro and in vivo association of transforming growth factor-beta 1 with hepatic fibrosis. J. Cell Biol. 108, 2477–2482 (1989).
Ueberham, E. et al. Conditional tetracycline-regulated expression of TGF-beta1 in liver of transgenic mice leads to reversible intermediary fibrosis. Hepatology 37, 1067–1078 (2003).
Ueno, H. et al. A soluble transforming growth factor beta receptor expressed in muscle prevents liver fibrogenesis and dysfunction in rats. Hum. Gene Ther. 11, 33–42 (2000).
Dooley, S. & ten Dijke, P. TGF-beta in progression of liver disease. Cell Tissue Res. 347, 245–256 (2012).
Hayashi, H. & Sakai, T. Biological significance of local TGF-beta activation in liver diseases. Front. Physiol. 3, 12 (2012).
Carr, B. I., Hayashi, I., Branum, E. L. & Moses, H. L. Inhibition of DNA synthesis in rat hepatocytes by platelet-derived type beta transforming growth factor. Cancer Res. 46, 2330–2334 (1986).
Oberhammer, F. A. et al. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc. Natl. Acad. Sci. USA 89, 5408–5412 (1992).
Jiang, J. X. et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 139, 1375–1384 (2010).
Dooley, S. et al. Transforming growth factor beta signal transduction in hepatic stellate cells via Smad2/3 phosphorylation, a pathway that is abrogated during in vitro progression to myofibroblasts. TGF beta signal transduction during transdifferentiation of hepatic stellate cells. FEBS Lett. 502, 4–10 (2001).
Hu, T. et al. Reactive oxygen species production via NADPH oxidase mediates TGF-beta-induced cytoskeletal alterations in endothelial cells. Am. J. Physiol. Ren. Physiol. 289, F816–825 (2005).
Westergren-Thorsson, G. et al. Altered expression of small proteoglycans, collagen, and transforming growth factor-beta 1 in developing bleomycin-induced pulmonary fibrosis in rats. J. Clin. Invest. 92, 632–637 (1993).
Broekelmann, T. J., Limper, A. H., Colby, T. V. & McDonald, J. A. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 88, 6642–6646 (1991).
Deguchi, Y. Spontaneous increase of transforming growth factor beta production by bronchoalveolar mononuclear cells of patients with systemic autoimmune diseases affecting the lung. Ann. Rheum. Dis. 51, 362–365 (1992).
Khalil, N., Whitman, C., Zuo, L., Danielpour, D. & Greenberg, A. Regulation of alveolar macrophage transforming growth factor-beta secretion by corticosteroids in bleomycin-induced pulmonary inflammation in the rat. J. Clin. Invest. 92, 1812–1818 (1993).
Hinchcliff, M. & Varga, J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am. Fam. Phys. 78, 961–968 (2008).
Ghahary, A., Shen, Y. J., Scott, P. G., Gong, Y. & Tredget, E. E. Enhanced expression of mRNA for transforming growth factor-beta, type I and type III procollagen in human post-burn hypertrophic scar tissues. J. Lab. Clin. Med. 122, 465–473 (1993).
Border, W. A. & Noble, N. A. Transforming growth factor beta in tissue fibrosis. N. Eng. J. Med. 331, 1286–1292 (1994).
Loeys, B. L. et al. Mutations in fibrillin-1 cause congenital scleroderma: stiff skin syndrome. Sci. Transl. Med. 2, 23ra20 (2010).
Lindsay, M. E. & Dietz, H. C. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 473, 308–316 (2011).
Gerber, E. E. et al. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature 503, 126–130 (2013).
Munger, J. S. & Sheppard, D. Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb. Perspect. Biol. 3, a005017 (2011).
Shah, M., Foreman, D. M. & Ferguson, M. W. Control of scarring in adult wounds by neutralising antibody to transforming growth factor beta. Lancet 339, 213–214 (1992).
Giri, S. N., Hyde, D. M. & Hollinger, M. A. Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice. Thorax 48, 959–966 (1993).
Logan, A. et al. Effects of transforming growth factor beta 1 on scar production in the injured central nervous system of the rat. Eur. J. Neurosci. 6, 355–363 (1994).
Wahl, S. M., Allen, J. B., Costa, G. L., Wong, H. L. & Dasch, J. R. Reversal of acute and chronic synovial inflammation by anti-transforming growth factor beta. J. Exp. Med. 177, 225–230 (1993).
Wolf, Y. G., Rasmussen, L. M. & Ruoslahti, E. Antibodies against transforming growth factor-beta 1 suppress intimal hyperplasia in a rat model. J. Clin. Invest. 93, 1172–1178 (1994).
Gupta, G. P. & Massague, J. Cancer metastasis: building a framework. Cell 127, 679–695 (2006).
van der Pluijm, G. et al. Monitoring metastatic behavior of human tumor cells in mice with species-specific polymerase chain reaction: elevated expression of angiogenesis and bone resorption stimulators by breast cancer in bone metastases. J. Bone Miner. Res. 16, 1077–1091 (2001).
Mohan, S. & Baylink, D. J. Bone growth factors. Clin. Orthop. Relat. Res. 263, 30–48 (1991).
Juarez, P. & Guise, T. A. TGF-beta in cancer and bone: implications for treatment of bone metastases. Bone 48, 23–29 (2011).
Siegel, P. M. & Massague, J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 3, 807–821 (2003).
Derynck, R., Akhurst, R. J. & Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 29, 117–129 (2001).
Massague, J. TGF beta in cancer. Cell 134, 215–230 (2008).
Markowitz, S. D. & Roberts, A. B. Tumor suppressor activity of the TGF-beta pathway in human cancers. Cytokine Growth Factor Rev. 7, 93–102 (1996).
Park, K. et al. Genetic changes in the transforming growth factor beta (TGF-beta) type II receptor gene in human gastric cancer cells: correlation with sensitivity to growth inhibition by TGF-beta. Proc. Natl. Acad. Sci. USA 91, 8772–8776 (1994).
Ohue, M. et al. Mutations of the transforming growth factor beta type II receptor gene and microsatellite instability in gastric cancer. Int. J. Cancer 68, 203–206 (1996).
Izumoto, S. et al. Microsatellite instability and mutated type II transforming growth factor-beta receptor gene in gliomas. Cancer Lett. 112, 251–256 (1997).
Chen, T., Carter, D., Garrigue-Antar, L. & Reiss, M. Transforming growth factor beta type I receptor kinase mutant associated with metastatic breast cancer. Cancer Res. 58, 4805–4810 (1998).
Chen, T. et al. Transforming growth factor-beta receptor type I gene is frequently mutated in ovarian carcinomas. Cancer Res. 61, 4679–4682 (2001).
Goggins, M. et al. Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res. 58, 5329–5332 (1998).
Schiemann, W. P., Pfeifer, W. M., Levi, E., Kadin, M. E. & Lodish, H. F. A deletion in the gene for transforming growth factor beta type I receptor abolishes growth regulation by transforming growth factor beta in a cutaneous T-cell lymphoma. Blood 94, 2854–2861 (1999).
Eppert, K. et al. MADR2 maps to 18q21 and encodes a TGF beta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell 86, 543–552 (1996).
Takagi, Y. et al. Somatic alterations of the SMAD-2 gene in human colorectal cancers. Br. J. Cancer 78, 1152–1155 (1998).
Schutte, M. et al. DPC4 gene in various tumor types. Cancer Res. 56, 2527–2530 (1996).
Howe, J. R. et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 280, 1086–1088 (1998).
Yakicier, M. C., Irmak, M. B., Romano, A., Kew, M. & Ozturk, M. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene 18, 4879–4883 (1999).
Daly, A. C., Vizan, P. & Hill, C. S. Smad3 protein levels are modulated by Ras activity and during the cell cycle to dictate transforming growth factor-beta responses. J. Biol. Chem. 285, 6489–6497 (2010).
Jones, S. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806 (2008).
Leary, R. J. et al. Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers. Proc. Natl. Acad. Sci. USA 105, 16224–16229 (2008).
Iavarone, A. & Lasorella, A. ID proteins as targets in cancer and tools in neurobiology. Trends Mol. Med. 12, 588–594 (2006).
Gold, L. I. The role for transforming growth factor-beta (TGF-beta) in human cancer. Crit. Rev. Oncog. 10, 303–360 (1999).
Padua, D. & Massague, J. Roles of TGF beta in metastasis. Cell Res. 19, 89–102 (2009).
Hagedorn, H. G., Bachmeier, B. E. & Nerlich, A. G. Synthesis and degradation of basement membranes and extracellular matrix and their regulation by TGF-beta in invasive carcinomas (review). Int. J. Oncol. 18, 669–681 (2001).
Pertovaara, L. et al. Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. J. Biol. Chem. 269, 6271–6274 (1994).
Kang, Y. et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549 (2003).
de Jong, J. S., van Diest, P. J., van der Valk, P. & Baak, J. P. Expression of growth factors, growth-inhibiting factors, and their receptors in invasive breast cancer. II: Correlations with proliferation and angiogenesis. J. Pathol. 184, 53–57 (1998).
Hasegawa, Y. et al. Transforming growth factor-beta1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer 91, 964–971 (2001).
Tuxhorn, J. A., McAlhany, S. J., Yang, F., Dang, T. D. & Rowley, D. R. Inhibition of transforming growth factor-beta activity decreases angiogenesis in a human prostate cancer-reactive stroma xenograft model. Cancer Res. 62, 6021–6025 (2002).
Thomas, D. A. & Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380 (2005).
Gorelik, L. & Flavell, R. A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat. Med. 7, 1118–1122 (2001).
Lopez-Novoa, J. M. & Nieto, M. A. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 1, 303–314 (2009).
Zavadil, J. & Bottinger, E. P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 24, 5764–5774 (2005).
Batlle, E. et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2, 84–89 (2000).
Cano, A. et al. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2, 76–83 (2000).
Boutet, A. et al. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 25, 5603–5613 (2006).
Lyons, J. G. et al. Snail upregulates proinflammatory mediators and inhibits differentiation in oral keratinocytes. Cancer Res. 68, 4525–4530 (2008).
Henderson, M. A. et al. Parathyroid hormone-related protein localization in breast cancers predict improved prognosis. Cancer Res. 66, 2250–2256 (2006).
Thomas, R. J. et al. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology 140, 4451–4458 (1999).
Kakonen, S. M. et al. Transforming growth factor-beta stimulates parathyroid hormone-related protein and osteolytic metastases via Smad and mitogen-activated protein kinase signaling pathways. J. Biol. Chem. 277, 24571–24578 (2002).
Yin, J. J. et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Invest. 103, 197–206 (1999).
Guise, T. A. et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Invest. 98, 1544–1549 (1996).
Dunn, L. K. et al. Hypoxia and TGF-beta drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS ONE 4, e6896 (2009).
Kang, Y. & Massague, J. Epithelial–mesenchymal transitions: twist in development and metastasis. Cell 118, 277–279 (2004).
Korpal, M. et al. Imaging transforming growth factor-beta signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat. Med. 15, 960–966 (2009).
Kang, Y. et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. USA 102, 13909–13914 (2005).
Deckers, M. et al. The tumor suppressor Smad4 is required for transforming growth factor beta-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 66, 2202–2209 (2006).
Javelaud, D. et al. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 67, 2317–2324 (2007).
Bandyopadhyay, A. et al. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 66, 6714–6721 (2006).
Ehata, S. et al. Ki26894, a novel transforming growth factor-beta type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci. 98, 127–133 (2007).
Biswas, S. et al. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J. Clin. Invest. 117, 1305–1313 (2007).
Nam, J. S. et al. An anti-transforming growth factor beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 68, 3835–3843 (2008).
Biswas, S. et al. Anti-transforming growth factor ss antibody treatment rescues bone loss and prevents breast cancer metastasis to bone. PLoS ONE 6, e27090 (2011).
Hiraga, T., Kizaka-Kondoh, S., Hirota, K., Hiraoka, M. & Yoneda, T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 67, 4157–4163 (2007).
Lu, X. et al. In vivo dynamics and distinct functions of hypoxia in primary tumor growth and organotropic metastasis of breast cancer. Cancer Res. 70, 3905–3914 (2010).
McMahon, S., Charbonneau, M., Grandmont, S., Richard, D. E. & Dubois, C. M. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 281, 24171–24181 (2006).
Juarez, P. et al. Halofuginone inhibits the establishment and progression of melanoma bone metastases. Cancer Res. 72, 6247–6256 (2012).
Ganapathy, V. et al. Targeting the transforming growth factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol. Cancer 9, 122 (2010).
Bouquet, F. et al. TGFbeta1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin. Cancer Res. 17, 6754–6765 (2011).
Schlingensiepen, R. et al. Intracerebral and intrathecal infusion of the TGF-beta 2-specific antisense phosphorothioate oligonucleotide AP 12009 in rabbits and primates: toxicology and safety. Oligonucleotides 15, 94–104 (2005).
Schlingensiepen, K. H. et al. Targeted tumor therapy with the TGF-beta 2 antisense compound AP 12009. Cytokine Growth Factor Rev. 17, 129–139 (2006).
Jachimczak, P. et al. The effect of transforming growth factor-beta 2-specific phosphorothioate-anti-sense oligodeoxynucleotides in reversing cellular immunosuppression in malignant glioma. J. Neurosurg. 78, 944–951 (1993).
Schlingensiepen, K. H. et al. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 102, 1193–1200 (2011).
Bogdahn, U. et al. Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. NeuroOncology 13, 132–142 (2011).
Santiago, B. et al. Topical application of a peptide inhibitor of transforming growth factor-beta1 ameliorates bleomycin-induced skin fibrosis. J. Invest. Dermatol. 125, 450–455 (2005).
Llopiz, D. et al. Peptide inhibitors of transforming growth factor-beta enhance the efficacy of antitumor immunotherapy. Int. J. Cancer 125, 2614–2623 (2009).
Hermida, N. et al. A synthetic peptide from transforming growth factor-beta1 type III receptor prevents myocardial fibrosis in spontaneously hypertensive rats. Cardiovasc. Res. 81, 601–609 (2009).
Ezquerro, I. J. et al. A synthetic peptide from transforming growth factor beta type III receptor inhibits liver fibrogenesis in rats with carbon tetrachloride liver injury. Cytokine 22, 12–20 (2003).
Denton, C. P. et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 56, 323–333 (2007).
Mead, A. L., Wong, T. T., Cordeiro, M. F., Anderson, I. K. & Khaw, P. T. Evaluation of anti-TGF-beta2 antibody as a new postoperative anti-scarring agent in glaucoma surgery. Invest. Ophthalmol. Vis. Sci. 44, 3394–3401 (2003).
.. CAT-152 trabeculectomy study. Ophthalmology 114, 1950 (2007).
Lonning, S., Mannick, J. & McPherson, J. M. Antibody targeting of TGF-beta in cancer patients. Curr. Pharm. Biotechnol. 12, 2176–2189 (2011).
Trachtman, H. et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 79, 1236–1243 (2011).
Morris, J. C. et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGF beta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 9, e90353 (2014).
Van Aarsen, L. A. et al. Antibody-mediated blockade of integrin alpha v beta 6 inhibits tumor progression in vivo by a transforming growth factor-beta-regulated mechanism. Cancer Res. 68, 561–570 (2008).
Schlingensiepen, K. H., Fischer-Blass, B., Schmaus, S. & Ludwig, S. Antisense therapeutics for tumor treatment: the TGF-beta2 inhibitor AP 12009 in clinical development against malignant tumors. Recent Results Cancer Res. 177, 137–150 (2008).
Hau, P. et al. Inhibition of TGF-beta2 with AP 12009 in recurrent malignant gliomas: from preclinical to phase I/II studies. Oligonucleotides 17, 201–212 (2007).
Ramfidis, V. S., Strimpakos, A. S., Syrigos, K. N. & Saif, M. W. Clinical studies in the second line setting of advanced pancreatic cancer: are we making any progress? J. Pancreas 13, 358–360 (2012).
Nemunaitis, J. et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J. Clin. Oncol. 24, 4721–4730 (2006).
Nemunaitis, J. et al. Phase II trial of Belagenpumatucel-L, a TGF-beta2 antisense gene modified allogeneic tumor vaccine in advanced non small cell lung cancer (NSCLC) patients. Cancer Gene Ther. 16, 620–624 (2009).
Giaccone, G. et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur. J. Cancer 51, 2321–2329 (2015).
Muraoka, R. S. et al. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J. Clin. Invest. 109, 1551–1559 (2002).
Yingling, J. M., Blanchard, K. L. & Sawyer, J. S. Development of TGF-beta signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 3, 1011–1022 (2004).
Bonafoux, D. & Lee, W. C. Strategies for TGF-beta modulation: a review of recent patents. Expert Opin. Ther. Pat. 19, 1759–1769 (2009).
Easty, D., Gallagher, W. & Bennett, D. C. Protein tyrosine phosphatases, new targets for cancer therapy. Curr. Cancer Drug Targets 6, 519–532 (2006).
Akhurst, R. J. Large- and small-molecule inhibitors of transforming growth factor-beta signaling. Curr. Opin. Investig. Drugs 7, 513–521 (2006).
Habashi, J. P. et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312, 117–121 (2006).
Cohn, R. D. et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 13, 204–210 (2007).
Podowski, M. et al. Angiotensin receptor blockade attenuates cigarette smoke-induced lung injury and rescues lung architecture in mice. J. Clin. Invest. 122, 229–240 (2012).
Lanz, T. V. et al. Angiotensin II sustains brain inflammation in mice via TGF-beta. J. Clin. Invest. 120, 2782–2794 (2010).
Zhou, H., Latham, C. W., Zander, D. S., Margolin, S. B. & Visner, G. A. Pirfenidone inhibits obliterative airway disease in mouse tracheal allografts. J. Heart Lung Transplant. 24, 1577–1585 (2005).
Taniguchi, H. et al. The clinical significance of 5% change in vital capacity in patients with idiopathic pulmonary fibrosis: extended analysis of the pirfenidone trial. Respir. Res. 12, 93 (2011).
Noble, P. W. et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 377, 1760–1769 (2011).
Chen, H. Y. et al. The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes 60, 590–601 (2011).
Kapoor, S. Smad7 gene transfer therapy: therapeutic applications beyond colonic fibrosis. Eur. J. Clin. Invest. 38, 876–877 (2008).
Sheridan, C. Gene therapy finds its niche. Nat. Biotechnol. 29, 121–128 (2011).
Zhu, S. et al. Alendronate protects against articular cartilage erosion by inhibiting subchondral bone loss in ovariectomized rats. Bone 53, 340–349 (2013).
Panahifar, A., Maksymowych, W. P. & Doschak, M. R. Potential mechanism of alendronate inhibition of osteophyte formation in the rat model of post-traumatic osteoarthritis: evaluation of elemental strontium as a molecular tracer of bone formation. Osteoarthr. Cartil. 20, 694–702 (2012).
Shirai, T. et al. Chondroprotective effect of alendronate in a rabbit model of osteoarthritis. J. Orthop. Res. 29, 1572–1577 (2011).
Zhang, L., Hu, H., Tian, F., Song, H. & Zhang, Y. Enhancement of subchondral bone quality by alendronate administration for the reduction of cartilage degeneration in the early phase of experimental osteoarthritis. Clin. Exp. Med. 11, 235–243 (2011).
Hayami, T. et al. The role of subchondral bone remodeling in osteoarthritis: reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheum. 50, 1193–1206 (2004).
Nishii, T., Tamura, S., Shiomi, T., Yoshikawa, H. & Sugano, N. Alendronate treatment for hip osteoarthritis: prospective randomized 2-year trial. Clin. Rheumatol. 32, 1759–1766 (2013).
Carbone, L. D. et al. The relationship of antiresorptive drug use to structural findings and symptoms of knee osteoarthritis. Arthritis Rheum. 50, 3516–3525 (2004).
Jilka, R. L. et al. Decreased oxidative stress and greater bone anabolism in the aged, when compared to the young, murine skeleton with parathyroid hormone administration. Aging Cell 9, 851–867 (2010).
Khosla, S., Westendorf, J. J. & Oursler, M. J. Building bone to reverse osteoporosis and repair fractures. J. Clin. Invest. 118, 421–428 (2008).
Orth, P. et al. Parathyroid hormone [1-34] improves articular cartilage surface architecture and integration and subchondral bone reconstitution in osteochondral defects in vivo. Osteoarthr. Cartil. 21, 614–624 (2013).
Sampson, E. R. et al. Teriparatide as a chondroregenerative therapy for injury-induced osteoarthritis. Sci. Transl. Med. 3, 101ra193 (2011).
Bellido, M. et al. Improving subchondral bone integrity reduces progression of cartilage damage in experimental osteoarthritis preceded by osteoporosis. Osteoarthr. Cartil. 19, 1228–1236 (2011).
Smith, J. N. & Calvi, L. M. Concise review: current concepts in bone marrow microenvironmental regulation of hematopoietic stem and progenitor cells. Stem Cells 31, 1044–1050 (2013).
Kubiczkova, L., Sedlarikova, L., Hajek, R. & Sevcikova, S. TGF-beta - an excellent servant but a bad master. J. Transl. Med. 10, 183 (2012).
Acknowledgements
This work was supported by U.S. National Institutes of Health grants (AR063943 and DK057501 to X.C.; AR064833 to J.L.C.), the National Natural Science Foundation of China (81771099 to X.X.), the Key Project for Frontier Research of Science and Technology Department of Sichuan Province (2016JY0006 to X.Z.), and Sichuan Province Science and Technology Innovation Team Program (2017TD0016 to Q.Y.). X.X. and L.Z. were supported by the visiting scholar fellowship from West China Hospital of Stomatology, Sichuan University. Special thanks to professor Nicola C. Partridge (New York University) for her untiring support and critical editing of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, X., Zheng, L., Yuan, Q. et al. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res 6, 2 (2018). https://doi.org/10.1038/s41413-017-0005-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-017-0005-4
This article is cited by
-
LncRNA GAS5 restrains ISO-induced cardiac fibrosis by modulating mir-217 regulation of SIRT1
Scientific Reports (2024)
-
Kisspeptin-10 binding to Gpr54 in osteoclasts prevents bone loss by activating Dusp18-mediated dephosphorylation of Src
Nature Communications (2024)
-
Fibrosis and bone marrow: understanding causation and pathobiology
Journal of Translational Medicine (2023)
-
Emergent collective organization of bone cells in complex curvature fields
Nature Communications (2023)
-
ASH2L regulates postnatal neurogenesis through Onecut2-mediated inhibition of TGF-β signaling pathway
Cell Death & Differentiation (2023)
