
Abstract
Allogeneic hematopoietic stem cell transplantation (HSCT) is curative for a variety of nonmalignant disorders including osteopetrosis, bone marrow failures, and immune deficiencies. Haploidentical HSCT is a readily available option in the absence of a matched donor, but engraftment failure and other post-transplant complications are a concern. Post-transplant cyclophosphamide (PT-Cy) regimens are gaining popularity and recent reports show promising results. We report our experience with nine pediatric patients with nonmalignant diseases who were transplanted from a haploidentical donor with PT-Cy. From 2015 to 2019, nine children with nonmalignant diseases underwent haploidentical HSCT with PT-Cy, two as a second transplant and seven as primary grafts after upfront serotherapy and busulfan-based myeloablative conditioning. Patient’s diseases included osteopetrosis (n = 5), congenital amegakaryocytic thrombocytopenia (n = 2), hemophagocytic lymphohistiocytosis (n = 1), and Wiskott Aldrich syndrome (n = 1). Two patients failed to engraft following upfront PT-Cy transplants, one was salvaged with a second PT-Cy transplant, and the other with a CD34+ selected graft. None of the patients suffered from graft-versus-host disease. Three patients died from early posttransplant infectious complications and six patients are alive and well. In conclusion, haploidentical HSCT with PT-Cy is a feasible option for pediatric patients with nonmalignant diseases lacking a matched donor.
Similar content being viewed by others

Introduction
Allogeneic hematopoietic stem cell transplantation (HSCT) is a curative treatment option for children with a variety of genetic nonmalignant disorders including osteopetrosis, bone marrow failure, and immune deficiencies [1,2,3]. However, only 30–35% of the patients have an available matched sibling donor, and the odds of finding a matched unrelated donor in international registries varies from 75% for white patients of European descent to as low as 16% in some ethnic groups [4].
Haploidentical HSCT is a readily available alternative option for patients who do not have a matched donor, but engraftment failure, high rates of graft-versus-host disease (GvHD), delayed immune reconstitution and other post-transplant complications are of significant concern. The use of post-transplant cyclophosphamide (PT-Cy) for in vivo T-cell depletion has emerged as a promising strategy in the setting of haploidentical HSCT. The availability, safety profile, effectiveness in reducing GvHD, and low cost of the procedure have contributed to the increasing popularity of this modality all over the world [5]. For pediatric nonmalignant diseases, regimens with PT-Cy for haploidentical HSCT are increasingly adapted as well, and recent reports of haploidentical HSCT with PT-Cy for nonmalignant diseases show feasibility of this approach and promising results [6,7,8,9,10].
Our center introduced PT-Cy regimens for pediatric haploidentical HSCT in 2015. In this case series, we report our experience with unmanipulated haploidentical HSCT with PT-Cy for nine pediatric patients with nonmalignant diseases, including five patients with osteopetrosis, two patients with congenital amegakaryocytic thrombocytopenia (CAMT), one patient with hemophagocytic lymphohistiocytosis (HLH), and one patient with Wiskott Aldrich syndrome (WAS).
Subjects and methods
This retrospective study was performed on pediatric patients who underwent a haploidentical HSCT with PT-Cy for nonmalignant diseases from February 2015 to August 2019 at Hadassah Medical Center. The study was approved by the institutional review board and informed consent was obtained from the families according to the declaration of Helsinki.
The collected data included patient age at HSCT, gender, conditioning regimen, GvHD prophylaxis, time to neutrophil and platelet engraftment, signs and symptoms of GvHD, infectious complications, last chimerism status, and survival. Neutrophil engraftment was defined as the first day of an absolute neutrophil count > 500/μL of 3 consecutive days. Platelet engraftment was defined as a platelet count > 50,000/μL for at least 7 days without transfusion support.
The diagnosis of the patients was confirmed by gene mutation analysis with whole exome sequencing and confirmed in all cases by Sanger sequencing. The diagnosis of osteopetrosis was supported by radiological studies.
Conditioning regimen and GvHD prophylaxis
All patients in the cohort received myeloablative regimens (Table 1). Busulfan was given every 6 h over 4 days and the dose was adjusted according to blood levels to target an area under the curve (AUC) of 1150 µM min/L. Fludarabine was given at a dose of 30 mg/m2/day for 5 days. For three of the patients thiotepa was added to the conditioning regimen in two doses of 5 mg/kg on day −4 pre-HSCT, and for one patient a low dose of cyclophosphamide (14.5 mg/kg) was added on days −3 and −2. Serotherapy was given to all patients. For the first two patients, thymoglobulin was given at a dose of 2.5 mg/kg/day for 4 days (day −4 to −1) and for the rest of the patients alemtuzumab was given for 3 days (day −10 to −8) at a dose of 0.2 mg/kg/day. For five of the patients Rituximab was given at day −10 pre-HSCT at a dose of 375 mg/m2.
In two cases, we used the PT-Cy modality for a salvage transplant and in two cases engraftment failure followed an upfront PT-Cy transplant. For the salvage transplants, a treosulfan-based regimen was used in three of the cases, and in one case, due to the clinical status of the patient, a reduced intensity conditioning (RIC) with fludarabine and cyclophosphamide was used (Table 1).
For the PT-Cy transplants, all patients received an unmanipulated bone marrow graft and PT-Cy was employed at a dose of 50 mg/kg/day on days +3 and +4.
Cyclosporine A and mycophenolate mofetil were started on day +5 for additional GvHD prophylaxis.
Supportive care
All patients were treated with prophylactic trimethoprim/sulfamethoxazole, acyclovir, and fluconazole according to our institutional guidelines. Ursodeoxycholic acid was given for prevention of veno-occlusive disease (VOD). Viral loads of cytomegalovirus (CMV), Epstein–Barr virus, and adenovirus were followed weekly by blood PCR tests. CMV viremia was treated preemptively with ganciclovir or foscarnet. Antiemetic treatment and pain control were given as necessary.
Chimerism
Donor chimerism was assessed weekly after engraftment by molecular testing of peripheral blood short tandem repeats (STR) at the tissue laboratory.
Results
Patients
A total of nine pediatric patients underwent HSCT from haploidentical donors with PT-Cy for nonmalignant diseases during the years 2015–2019 at Hadassah Medical Center. The clinical characteristics of the patients are depicted in Table 1. The underlying diseases of the patients included osteopetrosis (n = 5), CAMT (n = 2), HLH (n = 1), and WAS (n = 1). The median age of the patients at the time of HSCT was 10.7 months (range 4.5 months to 4 years).
Graft characteristics
All grafts used for the PT-Cy haplo-transplants were from bone marrow source and were collected from the donor at the operating room under general anesthesia. The median total nucleated cell count of the collected grafts was 1 × 109 cells/kg (range 4.35–13.98 × 108 cells/kg) and the median CD34+ cell count was 8.23 × 106 cells/kg (range 4.27–19.8 × 106 cells/kg).
Two of the HSCT in the cohort were transplanted with upfront CD34+ selected haploidentical grafts that after engraftment failure were salvaged with a PT-Cy transplant. One additional CD34+ selected graft was used as a salvage transplant after engraftment failure of a PT-Cy transplant. The grafts used for all three CD34+ selected haploidentical transplants were PBSC after mobilization with GCSF. The cell counts of the CD34+ depleted grafts are depicted in Table 1.
Engraftment and chimerism
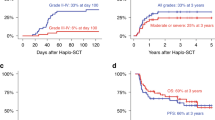
The first two PT-Cy transplants done at our center, both for osteopetrosis patients, were second transplants following primary engraftment failure after CD34+ selected haploidentical transplants. The PT-Cy modality was chosen for these two salvage transplants with the hope to achieve engraftment using a different approach. One of the patients survived and has complete donor chimerism and the other died 12 days post second transplant from sepsis. Another two patients failed to engraft following upfront haploidentical HSCT with PT-Cy. One, a patient with CAMT, successfully engrafted after a second transplant from the other parent, which was performed on day +50 after the first procedure. In this case, considering the primary and complete failure to engraft following the first transplant we opted to use the other parent for the second transplant while repeating the PT-Cy modality. At the time of publication, almost 2 years post his second HSCT, he is well, with complete donor chimerism and cured from his disease. The other, a patient with osteopetrosis, initially engrafted after his first transplant but soon after developed a secondary graft failure with chimerism rapidly dropping down to 4% 3 months post HSCT despite rapid weaning from immunosuppressive treatment. He underwent a second transplant 108 days after the first HSCT with a peripheral stem cell collected graft after CD34+ selection from the same donor and successfully engrafted. In this case, we opted to use the same donor for the second transplant since there were no signs of rejection of the graft, rather a process of secondary graft failure, and in order to enhance the engraftment process we decided to change the transplant modality and give a robust amount of stem cells using a CD34+ selected PBSC graft. At the time of publication, 13 months post his second transplant he is alive and well and has mixed chimerism with 90% donor cells on his latest STR test.
Five patients in the cohort underwent a single haploidentical transplant with PT-Cy, three of them survived and have complete donor chimerism, and two died from early post-transplant infectious complications.
Post-transplant complications and outcomes
A total of five patients had CMV reactivations, but only one progressed to a clinically significant disease. Three patients had bacterial infections during the post-transplant neutropenia, two of which progressed to severe sepsis and death. None of the patients suffered from any form of GvHD. The six survivors are all cured of their disease, alive and well, with a median follow-up time of 34 months (range 13–63 months).
Of the three surviving patients with osteopetrosis, one was already blind prior to HSCT (patient #1), and on latest follow-up he is 6 years old, developing well and attending a school for children with special needs. The second patient (patient #3) has severe optic damage with blindness in one eye and decreased vision in the other eye on his latest ophthalmologic assessment. Otherwise, his neurological examination is normal. The third patient (patient #9) has preserved vision and no nystagmus despite significant bilateral optic nerve atrophy. He was 2 years old on his latest follow-up and achieved all developmental milestones. All three patients have no known hearing deficits.
Three patients died, all from early post-transplant infectious complications (Table 2). Two of the deceased patients had osteopetrosis, both died from overwhelming Klebsiella pneumonia sepsis shortly after transplant; one on day +11 post his second transplant following engraftment failure and the other on day +12 post HSCT. The third deceased patient, a patient with HLH, suffered from VOD and severe CMV pneumonitis after transplant and eventually died on day +20 post HSCT from pulmonary bleeding and respiratory failure.
Discussion
In this paper, we report our experience with haploidentical HSCT with upfront serotherapy, busulfan-based myeloablative conditioning and PT-Cy in nine pediatric patients with nonmalignant diseases. The modality of PT-Cy for in vivo T-cell depletion without need for an additional graft manipulation increased the availability and popularity of the haploidentical option over the world [5]. Furthermore, the advantages of this cost-effective modality have set alight the debate of preference of an haploidentical family donor over an acceptably matched unrelated donor for various HSCT indications [11].
Recent reports focusing on haploidentical HSCT with PT-Cy in pediatric patients with nonmalignant diseases offer different promising approaches using a variety of conditioning regimens [6,7,8,9,10]. Klein et al. reported HSCT of 11 children with nonmalignant disorders showing very good outcomes with limited GvHD, excellent engraftment outcomes and no treatment related mortality using a RIC regimen with fludarabine, melphalan and upfront serotherapy with alemtuzumab [9]. Another recent report by Mallhi et al. of 23 pediatric nonmalignant patients also used a RIC regimen including low dose cyclophosphamide (50 mg/kg), fludarabine and a low dose of total body irradiation (2–4 Gy) and showed favorable outcomes but relatively high rates of GvHD [10]. Kurzay et al. used a treosulfan-based myeloablative conditioning regimen with upfront serotherapy and an addition of thiotepa and low dose cyclophosphamide in a recent report of 13 pediatric patients with nonmalignant diseases showing good overall survival, low rate of GvHD and few infectious complications [7]. Finally, Neven et al. recently reported 27 haploidentical HSCT with PT-Cy for pediatric nonmalignant diseases using a busulfan-based myeloablative conditioning regimen with upfront alemtuzumab showing a high engraftment rate (24 of 27 patients engrafted) and a 2 years overall survival rate of 77.7% [6].
Since successful engraftment is a major concern in haploidentical transplants, especially in osteopetrosis patients with a damaged bone marrow, we elected to adopt a similar approach to Neven et al. using a busulfan-based myeloablative regimen rather than a RIC or a treosulfan-based regimen. In view of the relatively high treatment related mortality in our cohort (33%), a less aggressive conditioning regimen may be considered for this patient population. As our patient population is very young, we opted not to expose them to irradiation. Rather, we gave serotherapy (thymoglobulin or alemtuzumab) for additional immune suppression (Table 1). During 2016, we switched from thymoglobulin to alemtuzumab following personal communications with other European centers with an aim to establish a standard practice for haploidentical transplants for pediatric inborn errors. The highly immunosuppressive properties of alemtuzumab are particularly appealing in the setting of nonmalignant HSCT, where GvHD provides no clinical benefit and relapse of malignancy is not applicable. Indeed, none of our patients developed any signs of acute or chronic GvHD as compared to the relatively high rates of acute and chronic GvHD reported by Mallhi et al. using a RIC conditioning without serotherapy [10]. Commonly used pediatric transplantation regimens include alemtuzumab doses of 0.5–1 mg/kg given over 3–5 days [12]. We gave a relatively low dose of alemtuzumab (0.6 mg/kg), and distal to the transplant day (days −10 to −8) in order not to compromise T-cell immunity and allow a swift immune reconstitution. However, considering the high rate of engraftment failure in our cohort, a higher dose may be considered for augmenting immune ablation.
Our cohort included a variety of nonmalignant diseases; five patients with infantile osteopetrosis, two patients with CAMT, one patient with HLH, and one patient with WAS.
Patients with Infantile osteopetrosis usually presents in early infancy with a rapidly progressing disease, which in most cases may be curable by HSCT [13, 14]. We have recently published our experience with HSCT for osteopetrosis patients with HLA matched or one-locus mismatched donors, showing favorable outcomes [15]. However, not all patients have an available matched donor or an acceptable mismatched donor. For these patients, haploidentical HSCT may be a readily available option.
Historically, outcomes of haploidentical HSCT for osteopetrosis patients were significantly worse than HSCT from matched donors [16]. Improved outcomes were reported using mega-doses of CD34+ selected haploidentical grafts with an intensive, busulfan-based myeloablative conditioning, but post-transplant complications such as VOD and respiratory failure were common [17]. Recent reports of successful haploidentical transplantations with alpha–beta T-cell depletion for osteopetrosis are promising [18], but this modality is not readily available in many centers due to its high cost, especially in developing countries with limited resources [19].
Reports of haploidentical HSCT with PT-Cy for osteopetrosis are scarce. In one report of three osteopetrosis patients by Bahr et al. only one of the three had a successful transplant course and the other two failed to engraft [20]. A recent publication by Neven et al. of haploidentical HSCT with PT-Cy included five patients with osteopetrosis, all of which engrafted successfully and are alive and well [6].
In our cohort, three out of the five osteopetrosis patients failed to engraft after the first haploidentical transplant. Two failed to engraft after a CD34+ selected graft and the third failed after a PT-Cy transplant. The high engraftment failure rate may be attributed to the damaged and sclerotic bone marrow niches of these patients. It is still too early to draw conclusions from these scarce number of patients, but engraftment failure seems to be a grave concern in these transplants. Despite the positive experience we have with reduced toxicity treosulfan-based conditioning in matched donor transplants for osteopetrosis, a myeloablative busulfan-based regimen was chosen for the haplo-transplants because of the concern of engraftment. However, weighed against the relatively high treatment related mortality in our cohort (33%), a less aggressive conditioning regimen may be considered for this patient population. Furthermore, until optimal timing and dosing of alemtuzumab are determined in prospective trials, a higher dose should be considered to augment engraftment, especially for patients with osteopetrosis. Finally, in view of these results, our current institutional policy is to prefer an unrelated donor if available, even with a one-locus mismatch, over a haploidentical donor for these patients.
Two of the patients in our cohort were transplanted for CAMT, an inherited bone marrow failure syndrome characterized by the absence of megakaryocytes in the bone marrow [21, 22]. Literature reporting HSCT with alternative donors for CAMT is very scarce. A report of five patients with CAMT who underwent umbilical cord blood transplantations showed promising results [23]. To our knowledge there are no reports of haploidentical transplants with PT-Cy for this indication yet. Our two CAMT patients are both cured of their disease, alive and well. The first engrafted well with no complications but the second required a second transplant due to engraftment failure. Of note, a second successful transplant was performed using PT-Cy from another donor. It is difficult to draw conclusions from only two cases but haploidentical HSCT with PT-Cy seems a feasible option for CAMT patients. Furthermore, this case demonstrated the feasibility of re-using the same modality of in vivo T-cell depletion using another haploidentical donor for a salvage, second transplant.
One patient in our cohort was transplanted for familial HLH, a life-threatening systemic hyperinflammatory syndrome [24]. HSCT for HLH patients is challenging and associated with high rates of toxicities and mortality, likely related to the underlying inflammatory state of these patients [25]. Our patient did not survive and died 20 days post HSCT from VOD and CMV pneumonitis. However, there are some recent reports with promising results of successful haploidentical transplants with PT-Cy for patients with HLH [8, 26].
One successful transplant in our cohort was of a patient with WAS, a rare X-linked disorder characterized by a triad of immunodeficiency, eczema, and thrombocytopenia. HSCT is a curative treatment for WAS, with excellent results for patients with HLA-matched family or unrelated donors [27]. A recent report by Yue et al. showed promising results in a case series of five patients who underwent haploidentical HSCT with PT-Cy for WAS [28].
In conclusion, in our experience, haploidentical HSCT with PT-Cy is a feasible option for pediatric patients with nonmalignant diseases, but with significant risks of post-transplant infectious complications and engraftment failure.
References
Steward CG. Hematopoietic stem cell transplantation for osteopetrosis. Pediatr Clin NAm. 2010;57:171–80.
Alter BP. Inherited bone marrow failure syndromes: considerations pre- and posttransplant. Hematol Am Soc Hematol Educ Progr. 2017;2017:88–95.
Gennery AR, Albert MH, Slatter MA, Lankester A. Hematopoietic stem cell transplantation for primary immunodeficiencies. Front Pediatr. 2019;7:445.
Gragert L, Eapen M, Williams E, Freeman J, Spellman S, Baitty R, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N Engl J Med. 2014;371:339–48.
Mussetti A, Greco R, Peccatori J, Corradini P. Post-transplant cyclophosphamide, a promising anti-graft versus host disease prophylaxis: where do we stand? Expert Rev Hematol. 2017;10:479–92.
Neven B, Diana JS, Castelle M, Magnani A, Rosain J, Touzot F, et al. Haploidentical hematopoietic stem cell transplantation with post-transplant cyclophosphamide for primary immunodeficiencies and inherited disorders in children. Biol Blood Marrow Transplant. 2019;25:1363–73.
Kurzay M, Hauck F, Schmid I, Wiebking V, Eichinger A, Jung E, et al. T-cell replete haploidentical bone marrow transplantation and post-transplant cyclophosphamide for patients with inborn errors. Haematologica. 2019;104:e478–82.
Uppuluri R, Sivasankaran M, Patel S, Swaminathan VV, Ramanan KM, Ravichandran N, et al. Haploidentical stem cell transplantation with post-transplant cyclophosphamide for primary immune deficiency disorders in children: challenges and outcome from a Tertiary Care Center in South India. J Clin Immunol. 2019;39:182–7.
Klein OR, Chen AR, Gamper C, Loeb D, Zambidis E, Llosa N, et al. Alternative-donor hematopoietic stem cell transplantation with post-transplantation cyclophosphamide for nonmalignant disorders. Biol Blood Marrow Transplant. 2016;22:895–901.
Mallhi KK, Srikanthan MA, Baker KK, Frangoul HA, Torgerson TR, Petrovic A et al. HLA-haploidentical hematopoietic cell transplantation for treatment of non-malignant diseases using nonmyeloablative conditioning and post-transplant cyclophosphamide. Biol Blood Marrow Transplant. 2020. https://doi.org/10.1016/j.bbmt.2020.03.018.
Adhikari J, Gyawali B, Sharma P, Bhatt VR. Outcomes of haploidentical transplant compared with matched donor allogeneic stem cell transplant. Future Oncol. 2017;13:935–44.
Guilcher GMT, Shah R, Shenoy S. Principles of alemtuzumab immunoablation in hematopoietic cell transplantation for non-malignant diseases in children: a review. Pediatr Transplant. 2018; 22. https://doi.org/10.1111/petr.13142.
Tolar J, Teitelbaum SL, Orchard PJ. Osteopetrosis. N Engl J Med. 2004;351:2839–49.
Penna S, Capo V, Palagano E, Sobacchi C, Villa A. One disease, many genes: implications for the treatment of osteopetroses. Front Endocrinol. 2019;10:85.
Shadur B, Zaidman I, NaserEddin A, Lokshin E, Hussein F, Oron HC, et al. Successful hematopoietic stem cell transplantation for osteopetrosis using reduced intensity conditioning. Pediatr Blood Cancer. 2018;65:e27010.
Orchard PJ, Fasth AL, Le Rademacher J, He W, Boelens JJ, Horwitz EM, et al. Hematopoietic stem cell transplantation for infantile osteopetrosis. Blood. 2015;126:270–6.
Schulz AS, Classen CF, Mihatsch WA, Sigl-Kraetzig M, Wiesneth M, Debatin K-M, et al. HLA-haploidentical blood progenitor cell transplantation in osteopetrosis. Blood. 2002;99:3458–60.
Pronk CJ, Turkiewicz D, Vult von Steyern K, Ehinger M, Dykes J, Toporski J. Transplantation of haploidentical TcRab-depleted hematopoietic cells allows for optimal timing and sustained correction of the metabolic defect in children with infantile osteopetrosis. J Bone Min Res. 2017;32:82–5.
Fernandes JF, Nichele S, Daudt LE, Tavares RB, Seber A, Kerbauy FR, et al. Transplantation of hematopoietic stem cells for primary immunodeficiencies in brazil: challenges in treating rare diseases in developing countries. J Clin Immunol. 2018;38:917–26.
Bahr TL, Lund T, Sando NM, Orchard PJ, Miller WP. Haploidentical transplantation with post-transplant cyclophosphamide following reduced-intensity conditioning for osteopetrosis: outcomes in three children. Bone Marrow Transplant. 2016;51:1546–8.
Ballmaier M, Germeshausen M. Congenital amegakaryocytic thrombocytopenia: clinical presentation, diagnosis, and treatment. Semin Thromb Hemost. 2011;37:673–81.
Dalle J-H, Peffault de Latour R. Allogeneic hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Int J Hematol. 2016;103:373–9.
Mahadeo KM, Tewari P, Parikh SH, Driscoll TA, Page K, Martin PL, et al. Durable engraftment and correction of hematological abnormalities in children with congenital amegakaryocytic thrombocytopenia following myeloablative umbilical cord blood transplantation. Pediatr Transplant. 2015;19:753–7.
Canna S, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis (HLH). Blood. 2020. https://doi.org/10.1182/blood.2019000936.
Allen CE, Marsh R, Dawson P, Bollard CM, Shenoy S, Roehrs P, et al. Reduced-intensity conditioning for hematopoietic cell transplant for HLH and primary immune deficiencies. Blood. 2018;132:1438–51.
Kohli S, Rastogi N, Nivargi S, Thakkar D, Katewa S, Yadav SP. Successful haploidentical stem cell transplant with posttransplant cyclophosphamide for hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2019;41:e158–60.
Ngwube A, Hanson IC, Orange J, Rider NL, Seeborg F, Shearer W, et al. Outcomes after allogeneic transplant in patients with Wiskott-Aldrich syndrome. Biol Blood Marrow Transplant. 2018;24:537–41.
Yue Y, Shi X, Song Z, Qin J, Li J, Feng S, et al. Posttransplant cyclophosphamide for haploidentical stem cell transplantation in children with Wiskott-Aldrich syndrome. Pediatr Blood Cancer. 2018;65:e27092.
Acknowledgements
We thank our patients and their families for putting their trust in our care and allowing us to publish this paper. We thank our departmental nursing and administrative staff for their devoted work. We also thank Professor Zeev Rotstein, director of Hadassah Medical Center, for his support of our department. Shadur B’s position is supported by the Australian Government Research Training Program Scholarship and Hadassah, Australia. This work was supported by the Deutsche Forschungsgemeinschaft (Discovery and Evaluation of new Combined Immunodeficiency Disease Entities (DECIDE); grant DFG WA 1597/4-2) and the ERA-Net ERARE Consortium EURO-CID.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Even-Or, E., NaserEddin, A., Dinur Schejter, Y. et al. Haploidentical stem cell transplantation with post-transplant cyclophosphamide for osteopetrosis and other nonmalignant diseases. Bone Marrow Transplant 56, 434–441 (2021). https://doi.org/10.1038/s41409-020-01040-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41409-020-01040-9
This article is cited by
-
Metabolic bone disorders and the promise of marine osteoactive compounds
Cellular and Molecular Life Sciences (2024)
-
HLA-haploidentical donor transplants with post-transplant cyclophosphamide in children with primary immune deficiency disorders
Bone Marrow Transplantation (2022)
-
Allogeneic hematopoietic stem cell transplantation for inherited metabolic disorders
International Journal of Hematology (2022)
-
Haploidentical Hematopoietic Cell Transplantation Using Post-transplant Cyclophosphamide for Children with Non-malignant Diseases
Journal of Clinical Immunology (2021)
