
Abstract
Between October 2007 and July 2013, 183 Mayo Clinic patients (median age 65 years; 58% males) with high/intermediate risk myelofibrosis (MF) were enrolled in consecutive phase 1/2 JAK2 inhibitor (JAKi) clinical trials with momelotinib (n = 79), ruxolitinib (n = 50), fedratinib (n = 23) and BMS-911543 (n = 31). Using conventional criteria, the respective response rates for spleen and “transfusion-dependent anemia” were 47%, 32%, 83%, 62% and 51%, 30%, 10%, 44%, respectively, favoring momelotinib for anemia response (p = 0.02) and fedratinib for spleen response (p < 0.01). All study patients were followed to death or 2022, during which time 177 (97%) drug discontinuations, 27 (15%) leukemic transformations, and 22 (12%) allogeneic stem cell transplants (ASCT) were recorded. 5/10-year survival rate for all 183 patients was 41%/16% and not significantly different across the four drug cohorts (p = 0.33). Multivariable analysis of pre-treatment variables identified age >65 years (HR 3.5), absence of type 1/like CALR mutation (HR 2.8), baseline transfusion need (HR 2.1), and presence of ASXL1/SRSF2 mutation (HR 1.6) as risk factors for overall survival; subsequent HR-based modeling segregated three risk categories with 5/10-year survival rates of 84%/60%, 44%/14%, and 21%/5% (p < 0.01). In addition, spleen (p < 0.01) and anemia (p = 0.01) responses were independently associated with improved short-term survival while long-term survival was secured only by ASCT (5/10-year survival rate 91%/45% vs 47%/19% in non-transplanted patients; p < 0.01). The current retrospective study suggests the value of specific pre-treatment variables in identifying long-lived MF patients receiving JAKi and also confirms recent observations on the favorable impact of treatment response on short-term and of ASCT on long-term survival.
Similar content being viewed by others

Introduction
Currently, ruxolitinib, fedratinib and pacritinib are three FDA-approved JAK2 inhibitors (JAKi) for treatment of myelofibrosis (MF) [1]. In addition, FDA-approval for momelotinib is awaited. All JAKi provide palliation through similar reduction of splenomegaly and relief of constitutional symptoms but differ in their ability to ameliorate anemia. In a phase-1/2 study of ruxolitinib in MF, ruxolitinib effectively reduced spleen size in 52% of patients [2]. The corresponding spleen response rates in the phase-3 COMFORT-1 (ruxolitinib vs placebo) [3], and COMFORT-2 (ruxolitinib vs best available therapy) [4], studies were 41.9 and 28%, while symptom response was achieved in 45.9% of ruxolitinib treated patients in the COMFORT-1 study [3]. However, in both studies, patients on ruxolitinib frequently developed anemia (45.2%/42%) [3, 4]. On the other hand, in a phase-1/2 study of momelotinib in MF, momelotinib therapy produced anemia response (54%) and reduction in splenomegaly (40%) [5, 6]. These observations were confirmed in the phase-3 SIMPLIFY-1 [7] (momelotinib vs ruxolitinib) and SIMPLIFY-2 studies [8] (momelotinib vs best available therapy) with week 24 transfusion independent rates of 66.5 and 43% in the momelotinib arm, respectively. Moreover, spleen response was comparable with momelotinib and ruxolitinib therapy (26.5% vs 29%) [7]. Similar spleen and symptom responses were also observed in phase-1/2 studies of fedratinib (39%/>50%) [9], and BMS-911543 (73%>50%) [10], in MF. These findings were confirmed in the phase-3 JAKARTA-1 study of fedratinib, with spleen and symptom response rates of 36 and 34%, respectively, however, 43% of fedratinib treated patients experienced grade 3 or 4 anemia [11].
The favorable impact of JAKi on splenomegaly and constitutional symptoms, and of momelotinib on anemia in patients with MF is well-established, but whether achievement of response following JAKi therapy positively influences long-term survival is unclear. Accordingly, in the current study, which includes MF patients treated on JAKi clinical trials at the Mayo Clinic, our primary objective was to i) retrospectively compare long-term treatment outcomes in momelotinib, ruxolitinib, fedratinib, and BMS-911543 treated patients with MF, ii) identify clinical and molecular predictors of response, overall and leukemia-free survival, and iii) examine the impact of treatment response on survival.
Methods
The current study includes JAKi naïve patients with primary myelofibrosis (PMF), post-polycythemia vera and post-essential thrombocythemia MF enrolled in consecutive phase-1/2 momelotinib (NCT00935987), ruxolitinib (NCT00509899), fedratinib (NCT00631462, NCT01420770), and BMS-911543 (NCT01236352) clinical trials between October 2007 and July 2013. The latter clinical trial was terminated by the sponsor in November 2015. Study patients were retrospectively recruited after institutional review board approval with follow-up updated in July 2022. Patient eligibility, study design, drug doses and schedule have previously been published [2, 5, 9, 10]. Diagnosis of PMF, post-ET and post-PV MF were according to conventional criteria [12]. Baseline transfusion status, spleen and anemia response were assessed according to the modified revised international working group for myelofibrosis research and treatment (IWG-MRT) criteria [13]. Screening for JAK2, MPL, CALR, ASXL1, SRSF2, IDH1/2, and U2AF1 mutations and dynamic international scoring system (DIPSS)-plus risk stratification and unfavorable karyotype categorization were as previously described [14, 15]. All statistical analyses considered clinical and laboratory variables obtained at the time of study initiation. Conventional statistical methods were applied for response and survival analysis. Survival was calculated from the time of study drug initiation to last follow-up or death and survival curves prepared by the Kaplan-Meier method and compared by the log-rank test. Cox proportional hazard model was used for multivariable analysis. JMP Pro 16.0.0 software package, SAS Institute, Cary, NC was used for statistical analysis.
Results
Patient characteristics
A total of 183 JAKi naïve patients with MF (median age 65 years, range 34–89; 58% males, 60% PMF) received momelotinib (n = 79, 43%, median dose, 300 mg daily), ruxolitinib (n = 50, 27%, median dose, 22.5 mg twice daily), fedratinib (n = 23, 13%, median dose, 680 mg daily), and BMS-911543 (n = 31, 17%, median dose, 320 mg daily) at a median of 27 months following diagnosis. Driver mutation profile included JAK2 in 80%, CALR in 11% (type 1/like CALR in 9%), MPL in 5%, triple negative in 4%; other mutations included ASXL1 in 58/124 (47%), SRSF2 in 16/84 (19%), U2AF1 in 3/47 (6%) and IDH1/2 in 2/74 (3%) of evaluable patients. Karyotype was abnormal in 97 (53%); among the latter, 53% were unfavorable. DIPSS-plus risk distribution was intermediate-1 (15%), intermediate-2 (46%), and high (39%). At the time of study drug initiation, 166 (91%), 66 (36%), and 136 (74%) of patients demonstrated palpable splenomegaly, transfusion-dependent anemia, and constitutional symptoms, respectively. Table 1 provides a comparison of clinical and laboratory characteristics at the time of treatment initiation and response outcomes for MF patients receiving JAKi in the context of a clinical trial. Patients on the momelotinib arm were more likely to be older (p < 0.01), transfusion-dependent (p = 0.01), harbor type 1/like CALR mutations (p = 0.09) and belong to DIPSS-plus high-risk category (p < 0.01), and less likely to be affected by constitutional symptoms (p < 0.01), compared to patients not receiving momelotinib. On the other hand, patients receiving ruxolitinib were more likely to be younger (p = 0.02), males (p = 0.01), JAK2 mutated (p = 0.04), belong to DIPSS-plus intermediate risk group (p < 0.01), and less likely to present with transfusion-dependent anemia (p < 0.01). Fedratinib treated patients were more likely to present with leukocytosis (p = 0.02) and thrombocytosis (p = 0.02) while patients receiving BMS-911543 were more likely to be females (p = 0.04) and affected by constitutional symptoms (p < 0.01). However, ASXL1/SRSF2 mutation distribution and median time from diagnosis to initiation of therapy was similar for each treatment group (p > 0.1).
Response
At a median follow up of 3.7 years (0.1–14.4 years), 178 (97%) patients have discontinued therapy. 3 and 5-year discontinuation rates were 77 and 92%, respectively with median treatment duration of 17 months for all patients. Notably, patients receiving momelotinib remained on therapy longer than patients on ruxolitinib (21 vs 10 months; p < 0.01). Reasons for drug discontinuation included suboptimal response or progressive disease (61%), toxicity (19%), study discontinuation by sponsor (9%), leukemic transformation (4%), death (3%) or secondary malignancy (2%). 108 (59%) of patients received subsequent therapy following discontinuation of study drug, which included JAKi therapy in 61(33%), hydrea in 16 (9%), imetelstat clinical trial in 11 (6%), thalidomide/lenalidomide/pomalidomide in 8 (4%), cladribine in 4 (2%), hypomethylating agent in 4 (2%), alisertib clinical trial in 3 (2%) and SL-401 clinical trial in 1 (1%). Overall, treatment-related grade 3 or 4 hematologic and non-hematologic adverse events were reported in 88 (48%) and 34 (19%) of patients, respectively.
Spleen response was achieved in 83 of 166 (50%) evaluable patients with median response duration of 22 months (2–132 months) and was more likely with fedratinib (83% vs 47%, in those receiving vs not receiving fedratinib, p < 0.01), and in the absence of ASXL1/SRSF2 mutations (58% vs 40%, in absence vs presence of ASXL1/SRSF2, p = 0.04). Multivariable analysis confirmed superior spleen response with fedratinib and absence of ASXL1/SRSF2 mutations (Table 2). Transfusion independence was achieved in 27 of 66 (47%) transfusion-dependent patients for a median of 14 months (6–82 months) and was more likely with momelotinib (51% vs 28% in those receiving vs not receiving momelotinib, p = 0.02) and in the absence of thrombocytopenia < 100 × 109/l (50% vs 0%, in absence vs presence of thrombocytopenia, p < 0.01) (Table 2). Neither spleen nor anemia response was influenced by driver mutation status, unfavorable karyotype, leukocyte count >25 × 109/l, peripheral blasts ≥1% and hemoglobin <10 g/dl (p > 0.1). In addition, symptom response was documented in 83 of 136 (60%) of patients with constitutional symptoms at baseline, response rates were 48, 57, 65, and 85% in patients receiving momelotinib, ruxolitinib, fedratinib, and BMS-911543, respectively.
Survival
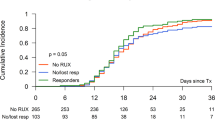
All study patients were followed to death or 2022, during which time 149 (81%) deaths, 27 (15%) leukemic transformations, and 22 (12%) allogeneic stem cell transplants (ASCT) were recorded. Median overall survival was 3.7 years with 3/5/10-year survival rates of 60, 41 and 16%, respectively. Survival was similar with momelotinib, ruxolitinib, fedratinib and BMS-911543 (3.5, 4, 4.4, and 5.9 years, respectively, p = 0.33) (Fig. 1a.) but was prolonged in patients that underwent ASCT vs those not transplanted (not reached vs 3.3 years; p < 0.001, 5/10-year survival, 91%45% vs 47%19%) (Fig. 1b). In univariate analysis of pre-treatment variables, age > 65 years (p < 0.01), absence of type 1/like CALR mutation (p < 0.01), baseline transfusion need (p < 0.01), unfavorable karyotype (p = 0.02), and presence of ASXL/SRSF2 mutations (p = 0.01) predicted inferior survival (Table 2). Multivariable analysis confirmed age >65 years (p < 0.01, HR 3.5), absence of type 1/like CALR mutation (p < 0.01, HR 2.8), baseline transfusion need (p < 0.01, HR 2.1), and presence of ASXL1/SRSF2 mutations (p = 0.04, HR 1.6) as risk factors for inferior survival (Table 2). A subsequent HR-based survival prediction model was generated by allocation of two adverse points for age > 65 years and absence of type 1/like CALR mutation and one adverse point each for transfusion dependence and presence of ASXL1/SRSF2 mutations which segregated three risk categories; low risk (0–2 points), intermediate risk (3 points) and high risk (>3 points) with 5/10-year survival rates of 84%/60%, 44%/14%, and 21%/5%, respectively (p < 0.01) (Fig. 2a). In addition, both spleen (6 vs 2.8 years in responders vs non-responders; p < 0.01, 5/10-year survival, 54%24% vs 29%12%) and anemia response (3.6 vs 2.3 years in responders vs non-responders; p = 0.01, 5/10-year survival, 33%19% vs 21%3%) were independently associated with improved short-term survival (Fig. 2b, c). On the other hand, in univariate analysis, leukemia-free survival was not influenced by driver mutation status, ASXL1/SRSF2 mutations, unfavorable karyotype, leukocyte count >25 × 109/l, peripheral blasts ≥1%, hemoglobin <10 g/dl, or thrombocytopenia <100 × 109/l (p > 0.1) (Table 2). Moreover, leukemic transformation rates were similar in patients receiving momelotinib (16%), ruxolitinib (18%), fedratinib (4%), and BMS-911543 (13%) (p = 0.33).

a Post-treatment survival of 183 JAK2 inhibitor-naive patients with myelofibrosis receiving JAK2 inhibitors in the context of clinical trials, stratified by specific drug, b Post-treatment survival of 183 inhibitor-naive patients with myelofibrosis receiving JAK2 inhibitors in the context of clinical trials, stratified by allogeneic transplantation.

a Post-treatment survival of 124 JAK2 inhibitor-naive patients with myelofibrosis receiving JAK2 inhibitors in the context of clinical trials and informative for high-risk mutations, stratified by HR-weighted scoring system. b Post-treatment survival of 166 JAK2 inhibitor-naive patients with myelofibrosis receiving JAK2 inhibitors in the context of clinical trials and evaluable for spleen response, stratified by spleen response. c Post-treatment survival of 66 JAK2 inhibitor-naive patients with myelofibrosis receiving JAK2 inhibitors in the context of clinical trials and transfusion-dependent anemia, stratified by anemia response.
Discussion
We pioneered early clinical development of momelotinib, ruxolitinib, fedratinib and BMS-911543 JAKi for the treatment of MF, therefore the current study is uniquely poised to provide comparative data on long-term outcomes for patients treated with each JAKi [2, 5, 6, 9, 10]. The vast majority of patients were followed until death, and follow-up for living patients exceeded 14 years. The findings from the current study confirm the palliative role of JAKi therapy through reduction of splenomegaly, achievement of transfusion-independence and relief of constitutional symptoms [16]. In that regard, a novel retrospective comparison of anemia and spleen response across all four JAKi is unveiled; fedratinib was associated with higher spleen response, while anemia response was superior with momelotinib. In addition, spleen response was found to be superior in the absence of ASXL1/SRSF2 mutations. Previous studies on the impact of mutations on spleen response in JAKi treated patients with MF have yielded conflicting results. In an analysis of MF patients treated with ruxolitinib in the COMFORT-2 study, spleen response was not influenced by mutational profile [17], while another study showed superior spleen response in the absence of ASXL1/EZH2/IDH1/2 mutations in ruxolitinib treated patients [18]. On the other hand, we have previously reported superior spleen and anemia response in the absence of ASXL1 mutations in momelotinib treated patients with MF [19, 20].
Additional noteworthy observations include high treatment discontinuation rates, with less than 5% of study patients remaining on long-term therapy (>10 years). Importantly, two-thirds of patients discontinued therapy due to suboptimal response or disease progression, while death or leukemic transformation on-study was documented in a minority (<5%) of patients. In our series, the high discontinuation rates secondary to toxicity was likely a result of exposure to higher drug dosage in the phase-1 studies. The high treatment discontinuation rates attest to the transient benefit from JAKi therapy and although short-term survival was superior in spleen and anemia responders, long-term survival was not impacted by neither achievement of spleen nor anemia response with 10-year survival rates of <25%. Moreover, we have recently shown that the short-term survival benefit associated with anemia response in momelotinib treated MF patients might be limited to those with unfavorable genetic profile [21]. It is to be noted that patients remained on momelotinib longer and had higher anemia response rates compared to ruxolitinib, despite which survival was similar, which is likely due to momelotinib treated patients being older and belonging to higher DIPSS-plus risk.
The subject of whether MF patients derive long-term survival benefit from JAKi therapy remains contentious. A pooled analysis of the COMFORT-1 and 2 studies and a recently published prospective real-world series suggested prolonged survival in ruxolitinib-treated patients [22, 23]. In contrast, patients treated on momelotinib and ruxolitinib clinical trials at the Mayo Clinic did not show a significant survival advantage when compared to risk-adjusted patient cohorts not receiving JAKi therapy [19, 24]. The current study reinforces the prognostic impact of previously established clinical and molecular risk factors for survival in MF patients treated with JAKi, namely age >65 years, absence of type 1/like CALR mutation, presence of ASXL1/SRSF2 mutations, and transfusion dependence [15, 25]. Based on the aforementioned variables, we propose a practical three-tiered survival prediction model for JAKi-treated MF patients, which enables identification of long-lived patients with 10 year survival of 60%. On the other hand, MF patients categorized as “high-risk” had considerably shortened long-term survival with 10-year survival of 5%, despite receiving JAKi therapy. Although achievement of anemia and spleen response improved short-term survival, a durable survival benefit was secured only by ASCT, which could have possibly resulted from ASCT candidacy itself by favoring younger patients with fewer medical comorbidities. Limitations of the current study include the retrospective design, which resulted in our inability to account for the impact of subsequent therapies on survival. Taken together, our findings underscore the transient benefit derived from JAKi therapy in MF in terms of palliation of splenomegaly and constitutional symptoms, however, impact on long-term survival was limited and primarily determined by pre-treatment clinical and molecular risk profile. Accordingly, early ASCT referral for patients with higher risk MF is advised.
Data availability
Please email the corresponding author.
References
Tefferi A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification, and management. Am J Hematol. 2021;96:145–62.
Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in Myelofibrosis. N Engl J Med. 2010;363:1117–27.
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807.
Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–98.
Pardanani A, Laborde RR, Lasho TL, Finke C, Begna K, Al-Kali A, et al. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia. 2013;27:1322–7.
Pardanani A, Gotlib J, Roberts AW, Wadleigh M, Sirhan S, Kawashima J, et al. Long-term efficacy and safety of momelotinib, a JAK1 and JAK2 inhibitor, for the treatment of myelofibrosis. Leukemia. 2018;32:1035–8.
Mesa RA, Kiladjian JJ, Catalano JV, Devos T, Egyed M, Hellmann A, et al. SIMPLIFY-1: a phase III randomized trial of momelotinib versus ruxolitinib in janus kinase inhibitor-naive patients with myelofibrosis. J Clin Oncol. 2017;35:3844–50.
Harrison CN, Vannucchi AM, Platzbecker U, Cervantes F, Gupta V, Lavie D, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol. 2018;5:e73–e81.
Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29:789–96.
Pardanani A, Roberts AW, Seymour JF, Burbury K, Verstovsek S, Kantarjian HM, et al. BMS-911543, a selective JAK2 inhibitor: a multicenter phase 1/2a study in myelofibrosis. Blood. 2013;122:664.
Pardanani A, Harrison C, Cortes JE, Cervantes F, Mesa RA, Milligan D, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1:643–51.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Tefferi A, Cervantes F, Mesa R, Passamonti F, Verstovsek S, Vannucchi AM, et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood. 2013;122:1395–8.
Tefferi A, Nicolosi M, Mudireddy M, Szuber N, Finke CM, Lasho TL, et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1,095 patients. Am J Hematol. 2018;93:348–55.
Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29:392–7.
Tefferi A, Pardanani A, Begna KH, Al-Kali A, Hogan WJ, Litzow MR, et al. Momelotinib for myelofibrosis: 12-year survival data and retrospective comparison to ruxolitinib. Am J Hematol. 2022;97:E433–e5.
Guglielmelli P, Biamonte F, Rotunno G, Artusi V, Artuso L, Bernardis I, et al. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood. 2014;123:2157–60.
Patel KP, Newberry KJ, Luthra R, Jabbour E, Pierce S, Cortes J, et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood. 2015;126:790–7.
Tefferi A, Barraco D, Lasho TL, Shah S, Begna KH, Al-Kali A, et al. Momelotinib therapy for myelofibrosis: a 7-year follow-up. Blood Cancer J. 2018;8:29.
Pardanani A, Abdelrahman RA, Finke C, Lasho TT, Begna KH, Al-Kali A, et al. Genetic determinants of response and survival in momelotinib-treated patients with myelofibrosis. Leukemia. 2015;29:741–4.
Gangat N, Begna KH, Al-Kali A, Hogan W, Litzow M, Pardanani A, et al. Predictors of anemia response to momelotinib therapy in myelofibrosis and impact on survival. Am J Hematol. 2022. https://doi.org/10.1002/ajh.26778.
Vannucchi AM, Kantarjian HM, Kiladjian JJ, Gotlib J, Cervantes F, Mesa RA, et al. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica. 2015;100:1139–45.
Guglielmelli P, Ghirardi A, Carobbio A, Masciulli A, Maccari C, Mora B, et al. Impact of ruxolitinib on survival of patients with myelofibrosis in the real world: update of the ERNEST Study. Blood Adv. 2022;6:373–5.
Tefferi A, Litzow MR, Pardanani A. Long-term outcome of treatment with ruxolitinib in myelofibrosis. N Engl J Med. 2011;365:1455–7.
Tefferi A, Guglielmelli P, Lasho TL, Gangat N, Ketterling RP, Pardanani A, et al. MIPSS70+ version 2.0: mutation and karyotype-enhanced international prognostic scoring system for primary myelofibrosis. J Clin Oncol. 2018;36:1769–70.
Author information
Authors and Affiliations
Contributions
AT, NG, and AP designed the study, compiled data, performed analyses and co-wrote the paper. KHB, AA, WH, and ML contributed patients. All authors reviewed the final draft of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gangat, N., Begna, K.H., Al-Kali, A. et al. Determinants of survival and retrospective comparisons of 183 clinical trial patients with myelofibrosis treated with momelotinib, ruxolitinib, fedratinib or BMS- 911543 JAK2 inhibitor. Blood Cancer J. 13, 3 (2023). https://doi.org/10.1038/s41408-022-00780-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-022-00780-9
This article is cited by
-
Momelotinib for myelofibrosis: our 14 years of experience with 100 clinical trial patients and recent FDA approval
Blood Cancer Journal (2024)
-
Calr type 1/like mutation in myelofibrosis is the most prominent predictor of momelotinib drug survival and longevity without transplant
Blood Cancer Journal (2024)
-
The application of JAK inhibitors in the peri-transplantation period of hematopoietic stem cell transplantation for myelofibrosis
Annals of Hematology (2024)
