
Abstract
Even though genetic perturbations and mutations are important for the development of myeloid malignancies, the effects of an inflammatory microenvironment are a critical modulator of carcinogenesis. Activation of the innate immune system through various ligands and signaling pathways is an important driver of myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). The DAMPs, or alarmins, which activate the inflammasome pathway via the TLR4/NLR signaling cascade causes the lytic cell death of hematopoietic stem and progenitor cells (HSPCs), ineffective hematopoiesis, and β-catenin-induced proliferation of cancer cells, leading to the development of MDS/AML phenotype. It is also associated with other myeloid malignancies and involved in the pathogenesis of associated cytopenias. Ongoing research suggests the interplay of inflammasome mediators with immune modulators and transcription factors to have a significant role in the development of myeloid diseases, and possibly therapy resistance. This review discusses the role and importance of inflammasomes and immune pathways in myeloid malignancies, particularly MDS/AML, to better understand the disease pathophysiology and decipher the scope of therapeutic interventions.
Similar content being viewed by others

Introduction
Hematopoiesis is the process of blood cell formation that occurs during embryonic development and across adulthood. It is a complex multifactorial process that operates in response to stimuli from cytokines and transcription factors under stringent regulatory checks [1, 2]. However, perturbations owing to genetic or acquired events that affect any step of the pathway can trigger the development of hematologic disorders, including cancers [3,4,5]. In particular, uncontrolled proliferation and expansion of abnormal myeloid progenitor cells lead to the development of myeloid malignancies including myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPNs), and acute myeloid leukemia (AML). Depending on factors such as the subtype of a proliferative cell, response to therapeutics, the necessity for transplantation, etc., patients are categorized into different groups of severity, with AML being the most aggressive with poor long-term survival outcomes. A clinical subtype of AML, secondary AML (sAML), defined as AML occurring after an antecedent myeloid disease, has previously been associated with inferior outcomes compared with de novo AML [6, 7].
Chromosomal abnormalities and gene mutations are known to have strong associations with myeloid malignancies. One-third of AML patients harbor mutations in the FLT3 gene. Despite the nature of mutation-internal tandem duplication (FLT3-ITD) or point mutation (FLT3-TKD), both causes a constitutive activation of tyrosine kinase, leading to proliferation and survival of AML. Patients with FLT3 mutations show poor prognosis, increased risk of relapse, and lower OS [8, 9]. Despite the recent FDA approval of several active agents in myeloid malignancies, including in high-risk MDS and AML, patients with these aggressive diseases eventually relapse unless bridged to an allogeneic stem cell transplant. There is a large unmet need for the development of newer therapeutic agents and rational combinations of these drugs.
Enhanced expression of proinflammatory cytokines such as TNF-α, IL-6, TGF-β, IL-8, and IL-1 in bone marrow (BM) of patients are known to be responsible for ineffective hematopoiesis in MDS [10,11,12,13,14]. The role of the innate immune system in driving these inflammatory signals has now been recognized. The innate immune system is thought to act as a bridge in mediating the effect of the mutations in myeloid progenitor cells to the development of the MDS phenotype with the help of accessory effector proteins [15,16,17]. Recent studies have emphasized the importance of alarmins to trigger BM expansion of hematopoietic inhibitory myeloid-derived suppressor cells (MDSCs) to activate the inflammasome pathway in MDS [18]. This, in turn, activates a caspase-1 dependent, novel, pro-inflammatory, pyroptosis-mediated lytic cell death in the BM, resulting in the death of healthy hematopoietic stem and progenitor cells (HSPCs) [19]. This pathway is now recognized as an important driver for the development of the MDS phenotype.
Multiple reports have extensively discussed the different classes of myeloid malignancies and mechanisms associated with the pathophysiology of these diseases [20]. This review focuses on highlighting the role of immune dysregulation, and in particular, inflammasomes in the pathogenesis of myeloid malignancies, especially MDS/AML, while touching upon the emergence of promising therapeutic interventions from these pathways.
The 3I paradigm in myeloid malignancies- immune system, inflammation, and inflammasomes
The role of immune players and inflammation in MDS/AML has been widely studied. While increased apoptosis has been implicated to cause ineffective hematopoiesis in MDS, the cytokine profile and cellular milieu suggests an aberrant innate immune activation in MDS. Elevated levels of pro-inflammatory cytokines such as TNF-α and IL-1 in MDS patients are known to cause the cell death of BM progenitor cells [3, 21,22,23,24]. Overall, most studies point to the crucial role of the immune system, inflammation, and inflammasomes in the pathogenesis of myeloid malignancies.
Immune system and inflammation in MDS
The innate immune system is activated through the interactions of pathogen-associated molecular patterns (PAMPS) against exogenous signals and by damage-associated molecular patterns (DAMPS/Alarmins) against endogenous signals, both of which operate via the activation of pattern recognition receptors (PRRs). Of the known PRRs, Toll-like receptors (TLRs) are the most widely studied, having roles in eliciting innate and adaptive immune reaction and shows an increased expression in inflammatory and autoimmune diseases. TLRs and pro-inflammatory cytokines can activate the hematopoietic stem cells (HSCs) directly [16, 17, 25, 26]. In MDS patients, TLRs are over expressed in HSPCs with respect to age-matched control subjects [27, 28]. In particular, TLR4 is seen to be upregulated in HSPCs of MDS patients and correlates with increased apoptosis of BM-mononuclear cells (BM-MNCs) and CD34+ cells. Also, TLR1, TLR2, and TLR6 are significantly overexpressed in the BM CD34+ cells of MDS subjects with a higher expression of TLR2 in the BM CD34+ cells of low-risk MDS subjects that induces an increase in the level of β-arrestin-1 and cell death. Studies have shown restoration of effective erythropoiesis upon transcriptional silencing of TLR2 [19, 28].
TLR signaling involves two main factors—interleukin receptor-associated kinases (IRAK1 and IRAK4), and TNF receptor-associated factor 6 (TRAF6) that leads to NF-κB and MAPK activation [29]. IRAK4 is a serine-threonine kinase that leads to IRAK1 and TRAF6 activation. We and others have shown that IRAK4 is overactivated in MDS and leads to activation of downstream proliferative pathways. IRAK4 can exist in various isoforms. The longer IRAK4 isoform contains the death domain that interacts with Myd88 allowing signaling downstream of TLR activation and is expressed in higher amounts in MDS patients with U2AF1 splicing factor mutations. Interestingly, IRAK4 activation has also been shown to be involved in resistance to FLT3 inhibitors in MDS/AML [30]. There is an activation of the innate immune pathway via IRAK1/4 complexes that contribute to adaptive immune resistance in FLT3 mutant AML cells [30, 31]. Increased TLR9 expression post-FLT3i treatment activates IRAK1/4 signaling. This increased expression of TLR’s, especially TLR9, has been implicated in the activation of innate immune pathways in adaptively resistant FLT3-ITD AML cells [30].
TRAF6 is an adapter protein that possesses nonconventional E3 ubiquitin ligase activity that mediates signaling from several innate immune receptors and is also reported to be overexpressed in MDS patients with (del)5q mutations [32]. Studies have shown that in (del)5q MDS-HSPCs, deletion of miRNA 146a activates TRAF6 [33, 34]. Such an overexpression of TRAF6 caused hematopoietic defects in a mouse model of MDS, suggesting a connection between immune pathway genes with the pathogenesis of MDS.
A recent report published by Muto et al. [35] studied a cohort of MDS patients in which 40% of MDS patients show overexpression of TRAF6 mRNA with deletion or repression of its negative regulators in MDS-HSPCs. The MDS-HSPCs showed an elevated expression of A20, a dual-ubiquitin (DUB) editing protein that activates the noncanonical NF-κB pathway by terminating the activation of canonical NF-κB via the TLR signaling pathway. This shift is believed to sustain the myeloid expansion and provide a selective advantage for disease cell proliferation as compared to WT-HSPCs, which operates on the canonical NF-κB pathway [35].
Another component of the innate immune system that is overactivated in MDS is the IL-8/CXCR2 pathway. IL-8 has been shown to be overexpressed in MDS stem and progenitor cells and acts via an autocrine manner using CXCR2 receptors. Overexpression of the pro-inflammatory cytokine IL-8 and its receptor CXCR2 is well known for promoting tumor growth and survival, and also as a predictor of adverse prognosis in MDS/AML [36, 37]. Targeting the IL-8/CXCR2 axis in MDS/AML patient cohorts have shown promising results, with decreased viability of primary patients’ HSCs without affecting healthy controls [9]. Also, inhibition of IL-8/CXCR2 signaling has been shown to inhibit MDS stem and progenitors [14]. Furthermore, the interleukin 1 receptor accessory protein (IL1RAP) is overactivated in MDS/AML HSPCs and is enriched in high-risk disease with worse prognosis [3]. Recent data shows that IL1RAP can act as a coactivator for FLT3 signaling thus playing a stimulatory role in malignant myeloid expansion [38]. Overall, these studies show that overexpression of genes involved in innate immune pathways is reported in over 50% of MDS patients [32].
Alarmins and NLRP3 activation in MDS
S100A8/S100A9 are cytosolic DAMPS, also known as alarmins, that activates the immune signaling system via the TLR-4/NLR pathway and play an important role in inflammatory diseases, including hematologic malignancies. In MDS, the MDSCs are recognized as the key effectors in the development of cytopenias and are associated with the cation binding DAMP heterodimer S100A8/S100A9 complex. The binding of this complex to the CD33 receptor on MDSCs causes activation and expansion of MDSCs, leading to secretion of immunosuppressive cytokines such as IL-10 and TGF-β, along with the further secretion of S100A8/S100A9, thereby initiating a vicious cycle of inflammatory cytokine generation and suppression of hematopoiesis [39]. Transgenic mice expressing S100A9 (S100A9Tg) mimic the features of human MDS, which could be reversed by depletion of MDSCs or by using short hairpin RNA-based (shRNA) silencing of the CD33 receptor, thereby inhibiting the TLR signaling cascade [40].
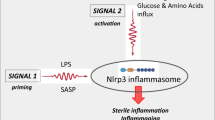
The role of alarmins in MDS is twofold: Activating MDSC expansion along with the NOD-like receptor protein 3 (NLRP3) receptor, via the TLR-4 receptor signaling pathway. NLRP3, a cytosolic redox-sensitive sensor, once activated, recruits an apoptosis-associated speck-like protein (ASC) triggering its polymerization and nucleation of large cytoplasmic aggregates to form ASC specks. This complex, termed the inflammasome, facilitates the recruitment and catalytic conversion of pro-Caspase 1 to active Caspase 1, which in turn activates IL-18 and IL-1β. This further enhances the proinflammatory milieu of the cell, activating proliferation via the β-catenin pathway while triggering lytic cell death via a process termed pyroptosis [39]. Caspase 1 activates a pore-forming protein gasdermin D (GSDMD) that oligomerizes and binds to the plasma membrane of myeloid cells to form pores. These pores compromise the membrane integrity, serving as an entry point for cations that release ROS and pro-inflammatory cytokines into the cytosol causing cell swelling and death (Fig. 1). NLRP3 inflammasomes, therefore, leads to multiple faces of MDS pathogenesis—ineffective hematopoiesis, cytopenias, and β-catenin induced proliferation of cancer cells.

MDSCs and NLRP3 are activated when DAMPS such as S100A8/S100A9 bind to CD33 and TLR4 receptors, respectively. Activated redox-sensitive NOD-like receptor protein 3 (NLRP3) recruits and causes polymerization of adapter apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC) proteins to form a complex termed as the inflammasome. This complex serves as a platform for recruitment and autocatalysis of pro-caspase 1 to give rise to active Caspase 1 and thereafter IL-18 and IL-1β from their precursors. Together with heightened ROS level, these cytokines add to the proinflammatory milieu of the cell. This process is followed by the release of proinflammatory cytokines in the cytosol via a lytic cell death called pyroptosis.
The level of S100A9 in plasma is higher in low-risk MDS as compared to high-risk patients [18]. MDS HSPCs and BM plasma reports higher S100A9 protein level as compared to age-matched controls which further increases with hematopoietic lineage progression in MDS BM-MNCs. Furthermore, treatment of normal BM-MNCs with recombinant S100A9 (rhS100A9) is sufficient to induce inflammasome assembly, activation of Caspase 1, and increase in ROS levels. S100A9Tg mouse model shows higher expression of WNT/β-catenin target genes, which is partially reversed by ICTA, an in vivo inhibitor of the NLRP3 inflammasome. Inhibition of inflammasome signaling by either S100A9 neutralization or pharmacological inhibition restores effective hematopoiesis in the S100A9Tg transgenic mouse model [18]. In addition, S100A9 is also known to suppress erythropoietin production, thereby inhibiting erythropoiesis. Lenalidomide treatment is seen to reduce the steady-state generation of S100A9, thereby increasing the levels of Epo and promoting erythropoiesis [41]. This further corroborates the importance of S100A9 in the pathogenesis of MDS.
Inflammasome activity in AML and other myeloid malignancies
The importance of inflammasomes is also reported in other myeloid malignancies besides MDS. In primary pediatric ALL cells, NLRP3 inflammasome was found to be activated in response to doxorubicin-induced chemotherapy. The p20 subunit of caspase 1 was found to be transcriptionally active in the ALL cells while the NLRP3 expression was found to be modulated by endogenous expression of a cellular DAMP- HMGB1 [42].
A report published by Höckendorf et al., in 2016 suggested a tumor suppressor role of the inflammasome in AML [43]. Failure of inflammasome activation due to loss of RIPK3, a protein kinase, led to the progression from myeloproliferation in FLT3-ITD mutated mice to the development of AML. Contrary to this, subsequent studies reported elevated plasma levels of IL-1β and IL-18 in AML patients as compared to controls [44, 45]. In a panel of 94 cytokines, IL-1β showed the highest effect on the growth of primary AML cells, clustering with GM-CSF and IL-3. In fact, the expression of IL1RAP is now considered a prognostic marker of AML [3] as it is consistently expressed across multiple genetic subtypes of AML and even at the stem cell level. IL1RAP has emerged as an important therapeutic target. It interacts and mediates pro-proliferative effects in AML stem cells through FLT3 kinases. This interaction can be further exploited therapeutically [38]. Interestingly, mRNA expression of NLRP3 and ASC in the BM-MNCs and plasma IL-18 levels show a significant decline in AML patients under complete remission as compared to newly diagnosed ones [44].
A recent article by Hamarsheh et al. [46] shows the activation of an inflammasome pathway in AML patients with KRAS mutation, KrasG12D. A mouse model expressing active KrasG12D mutation in the hematopoietic system showed myeloproliferation and cytopenia, which can be reversed in KrasG12D mice with NLRP3 deficiency. The gene expression profile of bone marrow-derived dendritic cells (BMDCs) from either WT or KrasG12D mice following treatment by tamoxifen have identified NLRP3/caspase1/IL-1β to be a major contributing axis. Oncogenic KRAS is found to produce ROS and thereby activate the inflammasome pathway via RAC1 protein. The findings were confirmed in AML, CMML, and JMML patient samples harboring the KRAS mutation. It is, therefore, possible for inflammasomes to have a diverse role in disease progression, depending on genetic and epigenetic factors.
The cross-talk between myeloid transcription factors and the inflammasome
Definitive hematopoiesis, which occurs during postembryonic development, engages multipotent HSCs that migrate to the BM and give rise to all blood lineages. HSC maturation involves the differentiation from the blast stage to the diversification of the lineages, giving rise to the lymphoid (T, B, and NK cells), myeloid (granulocytes and macrophages), and erythroid cell lineages (megakaryocytes and erythrocytes) [47, 48].
Two transcription factors, GATA1 and Spi-1 (also known as PU.1), show a cross-inhibitory relationship and are thought to be responsible for the decision of erythroid and myeloid fates [49,50,51]. Nonetheless, the additional factors and pathways are debated to be responsible for terminal erythroid and myeloid differentiation and their regulation [52,53,54,55]. As hematopoietic lineage bias is associated with increased incidence of diseases with prominent inflammatory components, the pathways and players hitherto untapped are believed to hold clinical significance.
In the lineage progression of HSCs, the presence of Spi-1 and GATA-1 transcription factors are necessary for the formation of common myeloid progenitor cells [52]. Interestingly, in the next step, their levels separately dictate the decision of differentiation into either megakaryocyte-erythrocyte progenitor (MEP) or granulocyte-monocyte progenitor (GMP), as shown in Fig. 2. Tyrkalska et al. [56] suggest that an underlying inflammatory background is responsible for the imbalance in the ratio of GATA-1: Spi-1 and subsequently, the decision of the lineage and its bias. Inflammasomes are thought to be prerequisite for myeloid differentiation that operates in an evolutionarily conserved mechanism by regulating the GATA1: Spi-1 ratio in the cells. In a zebrafish model, inflammasome deficient larvae express higher GATA1 at transcript and protein levels, inhibiting myeloid differentiation and enforcing erythropoiesis. As expected, pharmacological inhibition of the inflammasome rescues anemia and neutrophilic inflammation in the disease model. In mouse HSCs, Caspase 1 inhibitor upregulates GATA1 levels, with a direct increase in megakaryocyte-erythrocyte (MegE) colonies and a decrease in granulocyte-monocyte (GM) colonies. Pharmacological inhibition of the inflammasome in human erythroleukemic K562 cells leads to the suppression of erythroid differentiation with a decline in hemoglobin accumulation and decreased GATA1 levels as compared to DMSO treated control cells. In human HEK293 cells, Caspase 1 directly cleaves GATA1 at residue D300 thereby causing its degradation, indicating the likely reason of GATA1 accumulation on Caspase 1 inhibition. This evolutionarily conserved role of the inflammasome in the regulation of erythroid versus myeloid fate suggests a potential new area of drug development [56].

Hematopoietic stem cells (HSC) evolve from long-term (LT), short term (ST) to multipotent progenitor cells (MPP) from which myeloid and lymphoid cells are formed. At this stage, multiple cytokines along with transcription factors— Spi-1 and GATA1 provide necessary cues for the development of common myeloid progenitor (CMP). However, the next line of myeloid differentiation to megakaryocyte-erythrocyte progenitor (MEP) and granulocyte-monocyte progenitor (GMP) is carried by distinct signaling of GATA1 and Spi-1, respectively. This is followed by the formation of all types of blood cells namely, megakaryocytes (platelets), erythrocytes (RBCs), neutrophils, basophils, eosinophils, and monocytes.
Innate immune players such as TNF-α and IL-1β are known to upregulate Spi-1 protein in HSC in vitro and in vivo, via activation of inflammatory signaling [56,57,58]. As these proinflammatory cytokines are the released byproducts of inflammasome formation, it is hypothesized that the mechanism of upregulation of Spi-1 is superseded by GATA1 downregulation. While few studies have addressed the missing links in this area of research, the underlying mechanism of immune regulation, inflammation, and lineage bias in myeloid malignancies are not well understood and is still in its infancy.
Is inflammasome pathway the converging point for the development of MDS phenotype?
A recent review discussed the impact of genetic abnormalities and their relation to immune and inflammasome signaling in MDS [19]. For multiple genetic events, be it somatic mutations or chromosomal abnormalities, a different player of the immune system comes into action. For example, epigenetic modifications in different genes activate distinct immune signaling cascade. TET2 mutants increase IL-6 production via a decrease in HDAC2 recruitment with a concomitant increase in IL-1β while ASXL1 mutation activates TLR4 signaling and increases NADPH oxidase, and in turn ROS levels [18, 59, 60]. A similar trend can be observed for mutation in spliceosome genes. For example, NF-κB activation in the SF3B1 mutant occurs by downregulation of MAP3K7, but for the SRSF2 mutant, the activation is due to caspase 8 isoform, along with an increase in alarmin levels [18, 61]. Chromosomal abnormalities such as (del)5q mutation causes haploinsufficiency, increasing alarmins and miR-145/146 levels and subsequently activating TRAF6/IRAK1 signaling [62]. All these mutations and diverse effectors that trigger the activation of different immune signaling pathways point towards an interesting observation, a unanimous activation of pyroptosis and the β-catenin signaling pathway. This suggests the possibility of inflammasomes playing a central role in the pathogenesis of MDS/AML and the importance of exploiting this pathway to identify novel therapeutic targets in MDS/AML.
Therapeutic avenues for inhibiting the myeloid inflammasome
Previous sections of this review have established that innate immune signaling and subsequent inflammasome activation via NLRP3 plays a major role in the proliferation and sustenance of MDS HSPCs. Inflammasome activation has also been linked to several inflammatory diseases, including MDS. Thus, players involved in the inflammasome pathway can work as excellent pharmacological targets against MDS/AML. In this section, we discuss multiple mechanisms by which inflammasome formation and activity can be inhibited, and the early-phase clinical trials showing promise for future clinical practice.
As described before, the alarmins- S100A8/S100A9 bind the CD33 receptor on MDSCs leading to their activation, expansion, and ultimate suppression of hematopoiesis. Several monoclonal antibodies, antibody-drug conjugates (ADC), a bispecific T-cell engager (BiTE), and a trispecific killer engaged molecule (TriKE) against CD33, have been developed in hopes of suppressing MDSC proliferation and renewing hematopoiesis in MDS/AML.
Lintuzumab, a CD33 humanized monoclonal antibody was studied in older patients with untreated AML. However, the randomized Phase IIb trial failed to show improvement in overall survival (OS) in a cohort of 211 patients treated with Lintuzumab in combination with cytarabine as compared to cytarabine alone (4.7 months versus 5.1 months), thereby suspending its further clinical development [63]. A novel radioimmunoconjugate using Lintuzumab, 225Ac-lintuzumab, however, has shown early promise in preliminary phase II data in older AML patients unfit for induction chemotherapy who express CD33 on >25% of blasts [64]. 225Ac-lintuzumab links Lintuzumab to a short-range, high-energy, cytotoxic alpha-particle emitter (225Ac) which uses radiotherapy to elicit single and double-strand DNA breaks in selectively targeted CD33 cells. Preliminary data has been reported on nine patients treated with 225Ac-lintuzumab monotherapy showing an overall response rate (ORR) of 56% with two complete remissions with incomplete platelet recovery (CRp) and three complete remissions with incomplete hematologic recovery (CRi) [64]. 225Ac-lintuzumab in combination with Venetoclax is also being studied in an early phase I/II trial in relapsed/refractory AML (NCT03867682).
BI 836858, another CD33 monoclonal antibody glycoengineered against CD33 has also not shown much promise in untreated patients with MDS/AML, with only 18% patients achieving a CR/CRi among 28 patients with untreated AML in the phase I/II dose-escalation study of BI 836858 in combination with azacitidine (AZA) as part of the Beat AML dataset [65]. A similar phase I/II study (NCT02240706) using BI 836858 in patients with low or intermediate-1 risk MDS has been terminated as the company decided to stop the clinical development of BI 836858 prematurely for strategic reasons. As per the study results listed on clinicaltrials.gov, it appears this drug was limited by both efficacy and toxicity.
Two CD33 ADC have been developed, vadastuximab talirine and IMGN779, however, all clinical trials of vadastuximab talirine were suspended after the phase III CASCADE trial (NCT02785900) was terminated in 2017 after reporting a higher rate of deaths including fatal infections compared to the control arm despite promising initial phase I data showing an ORR of 70% [66]. IMGN779, however, is an ADC using an indolinobenzodiazepine pseudodimer payload, with encouraging initial phase I results showing manageable tolerability and anti-leukemia activity in patients with relapsed/refractory AML [67].
Although CD33 targeting has not shown much promise in humans thus far, the newest BiTe and TriKE agents are using innovative approaches to target MDSC production. AMV564 is a novel bivalent, bispecific, CD33/CD3 T-cell engager that binds CD33 on target cells and CD3 on T-cells leading to T-cell-directed lysis of CD33+ leukemic blasts and MDSCs, as well as T-cell expansion, differentiation, and proliferation [68]. GTB-3550 is a novel CD16/IL-15/CD33 TriKE that induces natural killer cell function by targeting malignant cells as well as the CD33+ MDSC, which contribute to tumor-induced immunosuppression. Because CD16 is the most potent activating receptor on NK cells, this single agent may induce a targeted cytolytic anti-CD33 tumor response [69]. Both agents are currently in early phase clinical trials in MDS/AML patients (Table 1).
Instead of targeting MDSC’s directly through CD33, inhibitors that specifically target TLR signaling and their downstream effectors, IRAK and the NLRP3 inflammasome, may provide another avenue to disrupt pyroptosis-mediated cell death of HSPC’s and β-catenin-induced proliferation of cancer cells. Most promising has been the TLR2 humanized monoclonal antibody Tomaralimab (OPN-305). In a phase II trial using Tomaralimab in heavily pretreated, transfusion-dependent patients with low or intermediate-1 risk MDS after failure with prior hypomethylating agents, 50% (6/12) patients had an overall response in the form of hematologic improvement with 17% (2/12) patients achieving transfusion independence. This therapy was well tolerated without significant toxicities [70]. Another potential way of targeting TLR signaling may be through proteasome inhibition. In an exploratory clinical trial using bortezomib in 15 patients with low-risk MDS, investigators noted that proteosome inhibition with bortezomib modulated TLR signaling. This occurs by decreasing levels of phosphorylated p65, a surrogate for NF-κB activation that leads to a significant p65 downregulation in 54% of patients, which correlated with clinical response, albeit only 20% of patients had evidence of short-lived hematologic improvement [71]. The investigators argued that using a drug with more potent and specific inhibition of the NF- κB pathway could lead to a more significant and long-lasting clinical response. One potentially attractive target is the IRAK4 inhibitor CA-4948, which directly inhibits NF-κB activation through its interaction with MyD88/IRAK1/TRAF6 [72]. A phase I dose-escalation trial using CA-4948 in adult patients with AML or high-risk MDS is currently enrolling and an early report shows the drug to be safe and well tolerated (NCT04278768) [73]. IRAK1/4 is also an attractive immune target in therapy-resistant myeloid malignancies and as previously mentioned, contributes to adaptive immune resistance in FLT3 mutant AML cells that can be therapeutically exploited. A small molecule dual inhibitor of FLT3/IRAK4 can overcome adaptive resistance in FLT3-ITD AML preclinical models by inhibiting compensatory IRAK 1/4 activation and downstream immune activation in FLT3-ITD AML [30]. Furthermore, a combinatory inhibitor targeting the FLT3/IL-8-CXCR2 axis may also serve to overcome the FLT3 resistance in AML.
In addition, a phase I study evaluating the small molecule CWP232291, an inhibitor of Wnt signaling that leads to direct degradation of β-catenin, has shown proof of concept that directly targeting downstream β-catenin cell proliferation maybe another novel mechanism for eradication of early leukemic progenitors. Further exploration as combination therapy is likely required [74].
Sustained levels of IL-1β have also been shown to activate NF-κB and MAPK signaling leading to an increase in IL-6 production. Furthermore, it supports MDSC accumulation as observed by the IL-1 receptor-deficient mouse model that shows inhibition of tumor progression due to delayed MDSC accumulation. Anti-IL-1β targets such as IL-1β neutralizing antibody Canakinumab [75], soluble decoy IL-1 receptor Rilonacept, and recombinant IL-1 receptor antagonist Anakinra [76] have been approved for autoinflammatory diseases. These can also be promising clinical targets against MDS, [77] with two early phase trials using Canakinumab in low-risk MDS currently recruiting (NCT04810611 and NCT04239157). However, targeting IL-1β alone may be insufficient as other cytokines such as IL-18 are also involved in adding to the pro-inflammatory milieu.
As previously mentioned, IL1RAP, a surface molecule that is consistently overexpressed on AML stem cells interacts with FLT3 kinases and is an important therapeutic target. Anti IL1RAP/CD3 bispecific antibodies have demonstrated a high degree of specificity and IL1RAP targeting therapies are now going into first-in-human clinical trials (NCT02842320) in leukemias [78].
Inhibitors that specifically target NLRP3 have not yet made it to the clinic for MDS/AML, but are currently under preclinical development, and have shown promising results with lesser toxicity. The NLRP3 inhibitor MCC950 showed high specificity for both canonical and noncanonical inflammasome pathway activation and significant efficacy at nanomolar concentrations [79, 80]. However, anecdotal reports of hepatotoxicity in patients with rheumatoid arthritis in early phase trials blocked its further development [67]. Other direct inhibitors of NLRP3 such as 3,4-methylenedioxy-β-nitrostyrene (MNS) and CY-09 (an analog of CFTR inhibitor-172 (C172) which inhibits the CFTR channel), specifically inhibits ATP binding of NLRP3 without affecting other NOD-like receptors. Tranilast and OLT1177 have shown promising results in animal models and ex vivo studies and seem to show significant potential as drug targets against NLRP3 related diseases [81,82,83].
Bruton tyrosine kinase (BTK) also regulates NLRP3 inflammasome activity by direct interaction with ASC and NLRP3. Ibrutinib-a BTK inhibitor prevents the formation of ASC specks and Caspase 1 activation. It was shown to suppress IL-1β maturation and caspase 1 activation in PBMCs of Ibrutinib treated cancer patients and is currently under phase 1 trials in combination with lenalidomide and 5′-Azacytidine for MDS (NCT03359460 and NCT02553941). Unfortunately, a phase II trial of ibrutinib alone or in combination with cytarabine or AZA in AML patients unfit for standard therapy or with relapsed/refractory disease, did not show any clinically relevant anti-leukemia activity [84]. Orally active Caspase 1 inhibitor such as VX-765 [85] and other caspase 1 inhibitors such as soluble analogs of Parthenolide (anti-inflammatory sesquiterpene lactone compound) can also be potential drug targets in hematological lineage bias disorders such as MDS and AML, which have only been studied in epilepsy and dermatologic conditions so far. Interestingly, recent reports have identified GSDMD to be a specific effector protein triggering pyroptosis, making it an attractive target which could have an important future role in immune therapeutic approaches designed to target specifically pyroptosis as an inflammasome activation downstream effect [81, 86].
Inflammasomes and immune response pathways have opened avenues for several exciting new drug targets for MDS and AML. As inflammasomes appears to play a role in MDS originating from multiple genetic defects, promising outcomes are expected from its drug targets. Table 1 and Fig. 3 shows the potential therapeutic agents targeting the players of the inflammasome pathway in myeloid malignancies.

The figure depicts the various biomolecules from the inflammasome pathway that are targeted by the novel therapeutic agents in MDS and AML.
Conclusion
Identification of novel pathways that are integral to the myeloid disease pathophysiology can guide the field towards newer targets for therapy. Often, pretransplant candidates with FLT3-ITD mutated AML develop rapid resistance to the FLT3 inhibitors. As pointed out, inflammasome targeting therapies can be explored as combinatorial strategies with FLT3 inhibitors to overcome therapy resistance as ample evidence points towards possible synergistic mechanisms. Underlying genetics and chronic inflammation can activate the innate immune system to trigger the inflammasome pathway, which seems to play a central role in the pathophysiology of myeloid malignancies. While DAMPS such as S100A8/S100A9 are known to activate the inflammasome pathway, the signaling mechanism that increases the DAMP levels in the first place remains unknown. Even though a number of drug targets have been identified from this pathway, it is a relatively new field in the context of myeloid malignancies. We anticipate that understanding the inflammasome pathway, its activators, inhibitors, and effectors will be crucial for further identification of novel and improved therapeutic outcomes against these diseases that currently have a dismal prognosis. With several therapies targeting the inflammasome currently in clinical development, it is our sincere hope that some of them will come to fruition and we will soon have an FDA-approved inflammasome targeting drug in a clinic for our patients with aggressive myeloid malignancies.
References
Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene. 2007;26:6724–37.
Maratheftis CI, Bolaraki PE, Voulgarelis M. GATA-1 transcription factor is up-regulated in bone marrow hematopoietic progenitor CD34(+) and erythroid CD71(+) cells in myelodysplastic syndromes. Am J Hematol. 2007;82:887–92.
Barreyro L, Will B, Bartholdy B, Zhou L, Todorova TI, Stanley RF, et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood. 2012;120:1290–8.
Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87.
Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7.
Hulegårdh E, Nilsson C, Lazarevic V, Garelius H, Antunovic P, Rangert Derolf Å, et al. Characterization and prognostic features of secondary acute myeloid leukemia in a population-based setting: a report from the Swedish Acute Leukemia Registry. Am J Hematol. 2015;90:208–14.
Granfeldt Østgård LS, Medeiros BC, Sengeløv H, Nørgaard M, Andersen MK, Dufva IH, et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: a National Population-Based Cohort Study. J Clin Oncol. 2015;33:3641–9.
Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61:7233–9.
Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–35.
Flores-Figueroa E, Gutiérrez-Espíndola G, Montesinos JJ, Arana-Trejo RM, Mayani H. In vitro characterization of hematopoietic microenvironment cells from patients with myelodysplastic syndrome. Leuk Res. 2002;26:677–86.
Shetty V, Mundle S, Alvi S, Showel M, Broady-Robinson L, Dar S, et al. Measurement of apoptosis, proliferation and three cytokines in 46 patients with myelodysplastic syndromes. Leuk Res. 1996;20:891–900.
Zhou L, Nguyen AN, Sohal D, Ying Ma J, Pahanish P, Gundabolu K, et al. Inhibition of the TGF-beta receptor I kinase promotes hematopoiesis in MDS. Blood. 2008;112:3434–43.
Zhou L, McMahon C, Bhagat T, Alencar C, Yu Y, Fazzari M, et al. Reduced SMAD7 leads to overactivation of TGF-beta signaling in MDS that can be reversed by a specific inhibitor of TGF-beta receptor I kinase. Cancer Res. 2011;71:955–63.
Schinke C, Giricz O, Li W, Shastri A, Gordon S, Barreyro L, et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood. 2015;125:3144–52.
Stifter G, Heiss S, Gastl G, Tzankov A, Stauder R. Over-expression of tumor necrosis factor-alpha in bone marrow biopsies from patients with myelodysplastic syndromes: relationship to anemia and prognosis. Eur J Haematol. 2005;75:485–91.
Maratheftis CI, Andreakos E, Moutsopoulos HM, Voulgarelis M. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clin Cancer Res. 2007;13:1154–60.
Kuninaka N, Kurata M, Yamamoto K, Suzuki S, Umeda S, Kirimura S, et al. Expression of Toll-like receptor 9 in bone marrow cells of myelodysplastic syndromes is down-regulated during transformation to overt leukemia. Exp Mol Pathol. 2010;88:293–8.
Basiorka AA, McGraw KL, Eksioglu EA, Chen X, Johnson J, Zhang L, et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood. 2016;128:2960–75.
Sallman DA, List A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood. 2019;133:1039–48.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Seipelt G, Ganser A, Duranceyk H, Maurer A, Ottmann OG, Hoelzer D. Induction of TNF-alpha in patients with myelodysplastic syndromes undergoing treatment with interleukin-3. Br J Haematol. 1993;84:749–51.
Alexandrakis M, Coulocheri S, Xylouri I, Ganotakis E, Eliakis P, Karkavitsas N, et al. Elevated serum TNF-alpha concentrations are predictive of shortened survival in patients with high-risk myelodysplastic syndromes. Haematol. 1998;29:13–24.
Sawanobori M, Yamaguchi S, Hasegawa M, Inoue M, Suzuki K, Kamiyama R, et al. Expression of TNF receptors and related signaling molecules in the bone marrow from patients with myelodysplastic syndromes. Leuk Res. 2003;27:583–91.
Musto P, Matera R, Minervini MM, Checchia-de Ambrosio C, Bodenizza C, Falcone A, et al. Low serum levels of tumor necrosis factor and interleukin-1 beta in myelodysplastic syndromes responsive to recombinant erythropoietin. Haematologica. 1994;79:265–8.
Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–12.
Sioud M, Floisand Y, Forfang L, Lund-Johansen F. Signaling through toll-like receptor 7/8 induces the differentiation of human bone marrow CD34+ progenitor cells along the myeloid lineage. J Mol Biol. 2006;364:945–54.
Monlish DA, Bhatt ST, Schuettpelz LG. The role of Toll-like receptors in hematopoietic malignancies. Front Immunol. 2016;7:390.
Wei Y, Dimicoli S, Bueso-Ramos C, Chen R, Yang H, Neuberg D, et al. Toll-like receptor alterations in myelodysplastic syndrome. Leukemia. 2013;27:1832–40.
Rhyasen GW, Bolanos L, Fang J, Jerez A, Wunderlich M, Rigolino C, et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell. 2013;24:90–104.
Melgar K, Walker MM, Jones LM, Bolanos LC, Hueneman K, et al. Overcoming adaptive therapy resistance in AML by targeting immune response pathways. Sci Transl Med. 2019;11:eaaw8828.
Smith MA, Choudhary GS, Pellagatti A, Choi K, Bolanos LC, Bhagat TD, et al. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nat Cell Biol. 2019;21:640–50.
Barreyro L, Chlon TM, Starczynowski DT. Chronic immune response dysregulation in MDS pathogenesis. Blood. 2018;132:1553–60.
Zhao JL, Starczynowski DT. Role of microRNA-146a in normal and malignant hematopoietic stem cell function. Front Genet. 2014;5:219.
Fang J, Barker B, Bolanos L, Liu X, Jerez A, Makishima H, et al. Myeloid malignancies with chromosome 5q deletions acquire a dependency on an intrachromosomal NF-κB gene network. Cell Rep. 2014;8:1328–38.
Muto T, Walker CS, Choi K, Hueneman K, Smith MA, Gul Z, et al. Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome HSCs. Nat Immunol. 2020;21:535–45.
Tang W, Li Z, Li X, Huo Z. High CXCR2 expression predicts poor prognosis in adult patients with acute myeloid leukemia. Ther Adv Hematol. 2020;11:2040620720958586.
Lu C, Zhu J, Chen X, Hu Y, Xie W, Yao J, et al. Risk stratification in acute myeloid leukemia using CXCR gene signatures: a bioinformatics analysis. Front Oncol. 2020;10:584766.
Mitchell K, Barreyro L, Todorova TI, Taylor SJ, Antony-Debré I, Narayanagari SR, et al. IL1RAP potentiates multiple oncogenic signaling pathways in AML. J Exp Med. 2018;215:1709–27.
Sallman DA, Cluzeau T, Basiorka AA, List A. Unraveling the pathogenesis of MDS: the NLRP3 inflammasome and pyroptosis drive the MDS phenotype. Front Oncol. 2016;6:151.
Chen X, Eksioglu EA, Zhou J, Zhang L, Djeu J, Fortenbery N, et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J Clin Invest. 2013;123:4595–611.
Cluzeau T, McGraw KL, Irvine B, Masala E, Ades L, Basiorka AA, et al. Pro-inflammatory proteins S100A9 and tumor necrosis factor-α suppress erythropoietin elaboration in myelodysplastic syndromes. Haematologica. 2017;102:2015–20.
Tola LM, Mariano JL, Winter MW, Shand JC. Intrinsic activation of the NLRP3 inflammasome in primary human ALL cells is regulated by endogenous HMGB1 and bone marrow macrophages. Blood. 2017;130:2559.
Höckendorf U, Yabal M, Herold T, Munkhbaatar E, Rott S, Jilg S, et al. RIPK3 restricts myeloid leukemogenesis by promoting cell death and differentiation of leukemia initiating cells. Cancer Cell. 2016;30:75–91.
Jia Y, Zhang C, Hua M, Wang M, Chen P, Ma D. Aberrant NLRP3 inflammasome associated with aryl hydrocarbon receptor potentially contributes to the imbalance of T-helper cells in patients with acute myeloid leukemia. Oncol Lett. 2017;14:7031–44.
Wang H, Hua M, Wang S, Yu J, Chen C, Zhao X, et al. Genetic polymorphisms of IL-18 rs1946518 and IL-1β rs16944 are associated with prognosis and survival of acute myeloid leukemia. Inflamm Res. 2017;66:249–58.
Hamarsheh S, Osswald L, Saller BS, Unger S, De Feo D, Vinnakota JM, et al. Oncogenic Kras(G12D) causes myeloproliferation via NLRP3 inflammasome activation. Nat Commun. 2020;11:1659.
Bertrand JY, Kim AD, Violette EP, Stachura DL, Cisson JL, Traver D. Definitive hematopoiesis initiates through a committed erythromyeloid progenitor in the zebrafish embryo. Development. 2007;134:4147–56.
McGrath KE, Frame JM, Fromm GJ, Koniski AD, Kingsley PD, Little J, et al. A transient definitive erythroid lineage with unique regulation of the beta-globin locus in the mammalian embryo. Blood. 2011;117:4600–8.
Alagha A, Zaikin A. Asymmetry in erythroid-myeloid differentiation switch and the role of timing in a binary cell-fate decision. Front Immunol. 2013;4:426.
Rekhtman N, Radparvar F, Evans T, Skoultchi AI. Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: functional antagonism in erythroid cells. Genes Dev. 1999;13:1398–411.
Nerlov C, Querfurth E, Kulessa H, Graf T. GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood. 2000;95:2543–51.
Strasser MK, Hoppe PS, Loeffler D, Kokkaliaris KD, Schroeder T, Theis FJ, et al. Early myeloid lineage choice is not initiated by random PU.1 to GATA1 protein ratios. Nature. 2016;535:299–302.
Crispino JD, Lodish MB, MacKay JP, Orkin SH. Use of altered specificity mutants to probe a specific protein-protein interaction in differentiation: the GATA-1:FOG complex. Mol Cell. 1999;3:219–28.
Porcher C, Swat W, Rockwell K, Fujiwara Y, Alt FW, Orkin SH. The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell. 1996;86:47–57.
Robb L, Lyons I, Li R, Hartley L, Köntgen F, Harvey RP, et al. Absence of yolk sac hematopoiesis from mice with a targeted disruption of the scl gene. Proc Natl Acad Sci USA. 1995;92:7075–9.
Tyrkalska SD, Pérez-Oliva AB, Rodríguez-Ruiz L, Martínez-Morcillo FJ, Alcaraz-Pérez F, Martínez-Navarro FJ, et al. Inflammasome regulates hematopoiesis through cleavage of the master erythroid transcription factor GATA1. Immunity. 2019;51:50–63.e5.
Yamashita M, Passegué E. TNF-α coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell. 2019;25:357–72.e7.
Espín-Palazón R, Stachura DL, Campbell CA, García-Moreno D, Del Cid N, Kim AD, et al. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell. 2014;159:1070–85.
Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–93.
Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med. 2013;210:2641–59.
Lee SC, North K, Kim E, Jang E, Obeng E, Lu SX, et al. Synthetic lethal and convergent biological effects of cancer-associated spliceosomal gene mutations. Cancer Cell. 2018;34:225–41.e8.
Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med. 2010;16:49–58.
Sekeres MA, Lancet JE, Wood BL, Grove LE, Sandalic L, Sievers EL, et al. Randomized phase IIb study of low-dose cytarabine and lintuzumab versus low-dose cytarabine and placebo in older adults with untreated acute myeloid leukemia. Haematologica. 2013;98:119–28.
Finn LE, Levy M, Orozco JJ, Park JH, Atallah E, Craig M, et al. A phase 2 study of actinium-225 (225Ac)-lintuzumab in older patients with previously untreated acute myeloid leukemia (AML) unfit for intensive chemotherapy. Blood. 2017;130 Suppl 1:2638.
Blum W, Ruppert AS, Mims AS, Stein EM, Duong VH, Odenike O, et al. Phase 1b dose escalation study of BI 836858 and azacitidine in previously untreated AML: results from beat AML S2. Blood. 2018;132 Suppl 1:4053.
Stein EM, Walter RB, Erba HP, Fathi AT, Advani AS, Lancet JE, et al. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood. 2018;131:387–96.
Cortes JE, DeAngelo DJ, Erba HP, Traer E, Papadantonakis N, Arana-Yi C, et al. Maturing clinical profile of IMGN779, a next-generation CD33-targeting antibody-drug conjugate, in patients with relapsed or refractory acute myeloid leukemia. Blood. 2018;132 Suppl 1:26.
Smith V, Eckard S, Rettig MP, Gehrs LN, Guenot J, Wei S, et al. Abstract 5699: AMV564, a bivalent, bispecific T-cell engager, depletes myeloid-derived suppressor cells and activates T cells in cancer patients. Cancer Res. 2020;80 Suppl 16:5699.
Warlick ED, Weisdorf DJ, Vallera DA, Wangen R, Lewis D, Knox J, et al. GTB-3550 TriKE™ for the treatment of high-risk myelodysplastic syndromes (MDS) and refractory/relapsed acute myeloid leukemia (AML) safely drives natural killer (NK) cell proliferation at initial dose cohorts. Blood. 2020;136:7–8.
Garcia-Manero G, Montalban-Bravo G, Yang H, Wei Y, Alvarado Y, DiNardo CD, et al. A clinical study of OPN-305, a Toll-like receptor 2 (TLR-2) antibody, in patients with lower risk myelodysplastic syndromes (MDS) that have received prior hypomethylating agent (HMA) therapy. Blood. 2016;128:227.
Daher M, Hidalgo Lopez JE, Randhawa JK, Jabbar KJ, Wei Y, Pemmaraju N, et al. An exploratory clinical trial of bortezomib in patients with lower risk myelodysplastic syndromes. Am J Hematol. 2017;92:674–82.
Gummadi VR, Boruah A, Ainan BR, Vare BR, Manda S, Gondle HP, et al. Discovery of CA-4948, an orally bioavailable IRAK4 inhibitor for treatment of hematologic malignancies. ACS Med Chem Lett. 2020;11:2374–81.
Garcia-Manero G, Tarantolo S, Verma A, Dugan J, Winer ES, Giagounidis, A, et al. Abstract S165: a phase 1, dose escalation trial with novel oral IRAK4 inhibitor CA-4948 in patients with acute myelogenous leukemia or myelodysplastic syndrome – Interim Report. EHA Library; 2021.
Lee JH, Faderl S, Pagel JM, Jung CW, Yoon SS, Pardanani AD, et al. Phase 1 study of CWP232291 in patients with relapsed or refractory acute myeloid leukemia and myelodysplastic syndrome. Blood Adv. 2020;4:2032–43.
Schlesinger N, De Meulemeester M, Pikhlak A, Yücel AE, Richard D, Murphy V, et al. Canakinumab relieves symptoms of acute flares and improves health-related quality of life in patients with difficult-to-treat Gouty Arthritis by suppressing inflammation: results of a randomized, dose-ranging study. Arthritis Res Ther. 2011;13:R53.
Bresnihan B, Newmark R, Robbins S, Genant HK. Effects of anakinra monotherapy on joint damage in patients with rheumatoid arthritis. Extension of a 24-week randomized, placebo-controlled trial. J Rheumatol. 2004;31:1103–11.
Arranz L, Arriero MDM, Villatoro A. Interleukin-1β as emerging therapeutic target in hematological malignancies and potentially in their complications. Blood Rev. 2017;31:306–17.
Meng W, Del Real M, Wei G, Hernandez R, Marcucci E, Lin A, et al. Anti-IL1RAP/CD3 bispecific antibody (BsAb) is a promising novel and effective therapy for acute myeloid leukemia (AML). Blood. 2017;130 Suppl 1:1361.
Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55.
Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS, et al. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep. 2018;8:8618.
Zahid A, Li B, Kombe A, Jin T, Tao J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front Immunol. 2019;10:2538.
Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med. 2018;10:e8689.
Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci USA. 2018;115:E1530–E9.
Cortes JE, Jonas BA, Graef T, Luan Y, Stein AS. Clinical experience with ibrutinib alone or in combination with either cytarabine or azacitidine in patients with acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2019;19:509–15 e1.
Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, et al. S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoy l)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J Pharmacol Exp Ther. 2007;321:509–16.
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8.
Acknowledgements
A.S. is funded by the LLS-TRP grant and the MDS Foundation Young Investigator Award.
Author information
Authors and Affiliations
Contributions
All authors contributed to the review. S.C. and A.S. conceived the idea of the review. S.C. and L.C.S. wrote the review, S.D.O. and B.R.-P. provided critical suggestions, A.S. and A.V. refined the final draft of the review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. A.S. has received research funding from Kymera Therapeutics, Consulting fees from GLG & Guidepoint & honoraria from OncLive. A.V. has received research funding from BMS, Jannsen, MedPacto, Novartis, Curis, Prelude, and Eli Lilly and Company, has received compensation as a scientific advisor to Novartis, Stelexis Therapeutics, Acceleron Pharma, and Celgene, and has equity ownership in Throws Exception and Stelexis Therapeutic.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chakraborty, S., Shapiro, L.C., de Oliveira, S. et al. Therapeutic targeting of the inflammasome in myeloid malignancies. Blood Cancer J. 11, 152 (2021). https://doi.org/10.1038/s41408-021-00547-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-021-00547-8
This article is cited by
-
Bone marrow inflammation in haematological malignancies
Nature Reviews Immunology (2024)
-
Inflammation meets translation in AML
Nature Cancer (2023)
-
Therapeutic Targets in Myelodysplastic Neoplasms: Beyond Hypomethylating Agents
Current Hematologic Malignancy Reports (2023)
-
Role of innate immunological/inflammatory pathways in myelodysplastic syndromes and AML: a narrative review
Experimental Hematology & Oncology (2023)
