
Abstract
Epstein–Barr virus (EBV)-associated diffuse large B-cell lymphoma not otherwise specified (DLBCL NOS) constitute a distinct clinicopathological entity in the current World Health Organization (WHO) classification. However, its genomic features remain sparsely characterized. Here, we combine whole-genome sequencing (WGS), targeted amplicon sequencing (tNGS), and fluorescence in situ hybridization (FISH) from 47 EBV + DLBCL (NOS) cases to delineate the genomic landscape of this rare disease. Integrated WGS and tNGS analysis clearly distinguished this tumor type from EBV-negative DLBCL due to frequent mutations in ARID1A (45%), KMT2A/KMT2D (32/30%), ANKRD11 (32%), or NOTCH2 (32%). WGS uncovered structural aberrations including 6q deletions (5/8 patients), which were subsequently validated by FISH (14/32 cases). Expanding on previous reports, we identified recurrent alterations in CCR6 (15%), DAPK1 (15%), TNFRSF21 (13%), CCR7 (11%), and YY1 (6%). Lastly, functional annotation of the mutational landscape by sequential gene set enrichment and network propagation predicted an effect on the nuclear factor κB (NFκB) pathway (CSNK2A2, CARD10), IL6/JAK/STAT (SOCS1/3, STAT3), and WNT signaling (FRAT1, SFRP5) alongside aberrations in immunological processes, such as interferon response. This first comprehensive description of EBV + DLBCL (NOS) tumors substantiates the evidence of its pathobiological independence and helps stratify the molecular taxonomy of aggressive lymphomas in the effort for future therapeutic strategies.
Similar content being viewed by others

Introduction
Epstein–Barr virus-positive (EBV + ) diffuse large B-cell lymphoma not otherwise specified (DLBCL NOS) was first described in a pivotal study by Oyama et al. in 2003, which reported on 22 senile patients having a predominantly dismal clinical outcome and poor response to established therapeutic regimens. Further retrospective clinicopathological case series led to the introduction of the provisional entity of EBV + DLBCL (NOS) of the elderly in the 2008 version of the World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissues. Subsequent studies further refined the perception of EBV + DLBCL (NOS) due to its occasional occurrence in young, immunocompetent individuals with evidence for a tolerogenic immune environment1,2,3. These observations led to an adapted definition of the entity in the 2016 revision of the WHO classification4,5.
While EBV-positive patients account for 8–10% of DLBCL cases in the East Asian population, data on the incidence in the western hemisphere is scarce, yet suggests similar to slightly lower numbers. The multifaceted western spectrum of EBV-related lesions, ranging from reactive lymphoid hyperplasia to DLBCL, was, however, found to be phenotypically similar to the spectrum encountered in Asian countries6,7,8,9.
Although recent observations relativized EBV as an independent risk factor, several studies found EBV + DLBCL (NOS) patients to be significantly enriched for adverse confounding clinical factors, such as stage, impaired performance status, advanced age, and significant canonical and alternative nuclear factor κB (NFκB)-pathway activation10. Altogether this resulted in a high number of cases with non-germinal center B-cell-like immunophenotype2,7,11,12,13,14. These circumstances render the entity a preferable subject for the study of molecularly targeted therapies.
The significant phenotypical overlap between EBV + DLBCL (NOS) and immunodeficiency or transplant-associated lymphoproliferative disorders (LPDs/PTLDs), including similar EBV-latency patterns, was found to be an additional characteristic of the entity12.
Intriguingly, NFκB-activation in EBV + DLBCL (NOS) was shown to result from EBV transcriptional reprogramming together with advanced B-cell differentiation13. Genomic profiling of MYD88-mediated TLR-signaling, as well as the B-cell receptor signaling pathway, revealed infrequent mutations, unlike the ABC-type DLBCL. Beyond these preliminary observations, the genomic features of EBV + DLBCL (NOS) are hitherto sparsely characterized. An initial study using exome sequencing of EBV + DLBCL (NOS) in a Chinese cohort of eleven patients without matched germline DNA proposed a heterogeneous mutational landscape dominated by a mutational signature associated with failure of DNA double-strand break–repair by homologous recombination15.
More recently, a targeted sequencing study of nine patients proposed an elevated frequency of MYC and RHOA mutations together with other genetic aberrations including mutations in MEF2B and MYD8816. Kataoka et al. addressed the overall impact of an underlying EBV infection on the mutational signature with a predefined 140-gene panel in an extended cohort of different lymphoma subtypes including 27 cases of DLBCL, contrasting their observations with 48 EBV-negative cases17. From their study, the authors derived a significant enrichment of mutations in TET2 and DNMT3A in EBV + DLBCL (NOS). Additionally, Sarkozy et al. recently reported whole-exome sequencing data of seven exclusively polymorphic EBV + DLBCL (NOS) with 13 additional cases investigated by panel-based sequencing. Hereby they identified SOCS1, GNA13, and CSF2RB mutations as potential drivers in EBV + DLBCL (NOS) pathogenesis, specifically in its polymorphic subtype, which is morphologically closely related to classical Hodgkin lymphoma18.
Although preliminary studies hint at a heterogeneous spectrum of potential genetic drivers in EBV + DLBCL (NOS), a comprehensive characterization of the genomic landscape in a representative cohort of patients is lacking. To this end, we combined whole-genome (n = 8) and targeted sequencing approaches (additional 39 patients) with FISH for MYC, BCL2, and BCL6 as well as 6q aberrations. Through this integrated analysis, we outline oncogenic drivers, copy number alterations, and pathway perturbations, hereby refining the molecular taxonomy of aggressive B-cell malignancies.
Materials and methods
Case selection and clinicopathological characteristics
In a retrospective approach, we reviewed our institutional database to identify EBV + DLBCL (NOS) patients whose primary diagnostic biopsy specimen had been referred to the Reference center for Hematopathology at University Hospital Schleswig Holstein Campus Lübeck and Hämatopathologie Lübeck for centralized histopathological panel evaluation between 2008 and April 2018. From 80 cases meeting diagnostic and clinical criteria for EBV + DLBCL, nine were selected for subsequent whole-genome analysis, based on tumor DNA quality and availability of paired germline DNA. One case was dismissed from analysis due to quality control issues following library preparation. An additional 39 cases with available tumor DNA of sufficient quality were selected for targeted sequencing. Tissue sections for confirmatory FISH diagnostics were available in 32 patients. For further details on clinicopathological workup, see Supplementary Materials and methods as well as Supplementary Table 1. A summary in the format of a flow diagram depicting the selection of cases for the respective analyses alongside a description of drop-outs is provided in Supplementary Fig. 1.
This retrospective study was approved by the ethics committee of the University of Lübeck (reference no. 18-356) and conducted in accordance with the declaration of Helsinki.
Extraction of nucleic acids
Genomic DNA was extracted from two to four FFPE tissue sections of 5-µm thickness using Maxwell® RSC DNA FFPE kit (Promega, Wiesloch, Germany) or the QiaAmp mini kit 250 (Qiagen, Hilden, Germany), according to the manufacturers’ instructions. DNA samples were subsequently quantified by spectrophotometry utilizing a Nanodrop ND-1000 (Nanodrop®).
Whole-genome sequencing
Whole-genome sequencing (WGS) was performed employing a HiSeq2500 platform (Illumina, San Diego, CA, USA). All samples with successful library preparation (8/9) were taken forward to sequencing. Raw fastq files have been deposited in the European genome-phenome archive (EGA) under the accession number EGAS00001004941.
EBV + DLBCL (NOS) samples were sequenced to a median depth of 30×.
Variant calling
Raw FASTQ reads were trimmed (adapter and quality values) using fastp19 (v0.20.0; minimum length 50 bp, max. unqualified bases 30%, trim tail set to 1), and trimmed reads were mapped to GRCh37/hg19 using bwa mem (v0.7.15)20. The resulting SAM files were cleaned and sorted and converted into BAM format using Picard Tools (v2.18.4). Next, mate-pair information was fixed, duplicates were removed and base quality recalibration was performed using Picard Tools21 and dbSNP v138. Single-nucleotide variants (SNVs) and short insertions and deletions (indels) were identified following the best practices for somatic mutations calling provided by GATK22. Briefly, GATKs Mutect223 (v4.1.5.0) algorithm was applied to all BAM files in tumor-normal matched mode with gnomAD variants as germline resource and the b37 whole-genome panel data as the panel of normal. Afterward, FFPE read orientation artefacts were identified and removed according to GATKs guidelines. Filtered variants were annotated using ANNOVAR24 (v2019Oct24). Coverage for reference and alternative alleles for each variant was extracted using vcf-query (VCFtools v0.1.1325). The top 20 frequently mutated genes (FLAGS26) were removed from further analysis. For further analysis, exonic somatic variants were selected and filtered as follows: minimum coverage of 10, the minimum variant allele frequency of 5%, population allele frequency <0.001 in gnomAD or PopFreqMax database. To identify genes that are more often mutated than expected, MutSigCV (v1.41)27 was applied and potential driver genes were identified using P < 0.05.
Mutational somatic signatures were estimated genome-wide using variants with at least 10× coverage (total; min cov. For variant 3×). Variants with a variant allele frequency <5% were removed as well. The remaining variants were used to estimate somatic signatures against COSMIC signatures using YAPSA (v1.16.0)28. Ploidy and microsatellite stability were assessed applying AMBER, COBALT, and PURPLE on base-recalibrated bam files.
Comparative analysis within the framework of clusters c1–c5 according to Chapuy et al. was performed extracting information about mutated genes and clustering of detected variants.
Targeted next-generation sequencing
In order to validate the initial delineation of the mutational landscape in EBV + DLBCL (NOS), we employed our in-house custom AmpliSeq panel (Thermo Fisher Scientific, Waltham, MA, USA) for targeted amplicon sequencing (tNGS), encompassing all coding exons of 43 genes (see Supplementary Table 2) in both the discovery cohort as well as an additional expansion cohort of 39 cases. Library preparation was carried out according to the manufacturers’ instructions, and sequencing was performed on the Illumina MiSeq platform (Illumina, San Diego, CA, USA) to a median depth of 2416× (s.d. +/− 1057). Raw fastq files have been deposited in the European genome-phenome archive (EGA) under the accession number EGAS00001004941.
Panel resequencing data analysis
Resequencing data was processed as described above for whole-genome data, but the remove duplicates step was omitted. Variant calling was done using freebayes (v1.3.2-46-g2c1e395), variants were annotated using ANNOVAR, and coverage for each variant was extracted using vcf-query29. Afterward, variants were filtered and only variants with a minimum coverage of 100, minimum variant allele frequency of 5%, population allele frequency <0.001 in gnomAD or PopFreqMax database were kept for further analysis.
Fluorescence in situ hybridization (FISH) for MYC, BCL2, BCL6, and 6q aberrations
Chromosomal breakpoints were analyzed by means of FISH using commercially available dual-color break-apart probes for 8q24 (MYC), 18q21 (BCL2), and 3q27 (BCL6) (Abbott Vysis, Des Plaines, IL, USA) according to the manufacturer’s instructions as well as a custom-designed (enumeration-)probe targeting 6q deletions found in our exploratory WGS cohort encompassing the genes PRDM1 in the region 6q21 and A20 in the region 6q23 prepared at the Institut für Tumorgenetik Nord.
Structural variants and copy number variations
Genomic rearrangements were detected using a matched tumor-normal approach following best practices of GRIDSS. The resulting structural variations were filtered using GRIPSS and final variation calls were checked for artefacts30. Moreover, in order to detect somatic copy number alterations a fragment-based GC- and mappability correction algorithm was applied, followed by a subsequent analysis of these normalized fragment counts in ~10-kbp windows (window sizes chosen based on the regional coverage)31. We hereby screened for deviations from the expected copy number 2 and classified events as gains or losses based on the read depth. For CN2LOH (copy number neutral loss-of-heterozygosity) assessment, we utilized 1000 genomes SNPs that were genotyped across all samples. Results were phased using Eagle2 resulting in haplotype blocks which were then used to average individual SNP B-allele frequencies across the haplotype blocks, thereby essentially “de-noising” of the raw SNP B-allele frequencies in our FFPE samples32.
Network propagation and gene set enrichment analysis
The effect of potential driver genes identified by MutSigCV on neighboring genes was assessed using a network propagation (network diffusion33) approach with a regularized Laplacian kernel based on STRINGdb v1134 protein–protein interaction network as implemented in the diffuStats R package (v1.10.0)35. Mutated genes were set to 1, whereas non-mutated genes were set to 0 to model the behavior of the mutation. Network diffusion was performed using a parametric method with statistical normalization (z-scores). Afterward, gene set variation analysis was performed on diffusion scores using the GSVA R package (v1.38.0)36 with a Gaussian kernel for continuous values against HALLMARK gene sets (R package msigdf37 with minimum/maximum size of the resulting gene sets set to 10/500; MSigDB (v7.1)38 and the NFκB-signaling pathway (genes were retrieved from KEGG; entry ID hsa04064). The resulting z-scores were used as pre-ranked input for a rank-MANOVA based statistical approach to detect enriched gene sets (mitch R packages)39,40.
Statistical analyses
If not stated differently, all statistical analyses were performed using R (v4.0.3) and tidyverse (v1.3.0)41 for data handling. Filtering of genomic regions was performed using the GenomicRanges R (v1.42.0)42 package and data was visualized using maftools (v2.6.0)43. Progression-free survival and overall survival (PFS, OS) were calculated from the date of diagnosis and censored at the last clinical contact. Survival (PFS and OS) according to potential prognostic factors was estimated by means of the Kaplan–Meier method and univariate log-rank test. Survival analysis was carried out employing the R packages survival (3.2-7) and survminer (v0.4.8).
Results
Clinical characteristics of the study group
We collected 47 cases of treatment naive de novo EBV + DLBCL (NOS) with sufficient FFPE material for molecular studies. The median age of the study cohort was 74 years (range 19–90 years). An underlying HIV infection had to be clinically excluded prior to recruitment in all cases and EBV + DLBCL (NOS) tumor cells were shown to express EBER in >50% of large cell infiltrates in accordance with the current WHO definition of the entity. Clinical data retrospectively collected from our institutional database was available in all patients. The majority of patients in our study were male (29/47; 62%) and many presented with advanced-stage disease (23/47 stage III/IV; 49%) and exhibited an adverse prognostic constellation (22/47; 47% R-IPI > 2). Despite their advanced age, most patients received an intensive CHOP-like therapeutic frontline approach (33/47; 70%), resulting in an overall response rate (ORR) of 41/47; 87%.
Baseline characteristics of EBV + DLBCL (NOS) cases included in this study are briefly summarized in Table 1. Most samples of the current study were previously investigated for the impact of treatment variability and clinicopathological baseline characteristics on clinical outcome in EBV + DLBCL (NOS)10.
Genomic landscape of EBV + DLBCL (NOS) patients identified by WGS
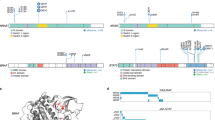
To delineate the entire spectrum of genetic alterations in this rare subtype of aggressive B-cell lymphoma, we performed WGS of paired tumor and normal samples from eight EBV + DLBCL (NOS) patients. Clinicopathological baseline characteristics of these specific patients are summarized in Table 1. We identified single-nucleotide variants and indels in individual samples after applying a filtering algorithm to correct for FFPE-derived artefacts and spurious mutations (“Methods”). All EBV + DLBCL (NOS) cases harbored mutations in genes implicated in oncogenesis according to our bioinformatic annotations44. We hereby detected on average 89 exonic somatic mutations (median: 69; resulting in a low tumor mutational burden of 2.318 mutations/Mb/sample on average), including 675 SNVs and 39 insertions and deletions (indels) (Fig. 1), alongside 132 SVs (Figs. 2A and 3A). Assessment of tumor mutational signatures identified a variety of constellations, which, however, lacked an apparent EBV-driven predominance (Supplementary Fig. 2). Polymorphic cases harbored significantly more subclones (ANOVA: P = 0.045), while showing no enrichment in the number of mutations (albeit at similar relative tumor cell content following microdissection/macrodissection) (Supplementary Fig. 3).

Panel (A) shows the mutational burden of EBV + DLBCL (NOS) against TCGA cohorts, the number of coding variants stratified by variant classification is depicted in panel (B), and the number of non-silent mutations per sample is shown in (C).

Panel (A) depicts whole genomic CNAs, whereas panel (B) illustrates the recurrent 6q losses subsequently validated by FISH (A20 in spectrum orange, PRDM1 in spectrum green) with panel displaying a wild-type constellation (C) and panel showing a case with a deletion of the A20 and PRDM1 gene locus in 6q (D).

Panel (A) depicts an oncoplot of all genes harboring mutations in at least two samples of our discovery WGS cohort. Panel (B) illustrates the mutational landscape in our discovery and extension cohort of EBV + DLBCL (NOS) by tNGS. Mutations in ANKRD11 were identified as indicators of superior relapse-free but not overall survival (C). Moreover, distinct molecular subtypes exhibiting combined alterations of ARID1A and DAPK1 are shown to exhibit inferior overall survival (D). The corresponding endpoint analysis for (C) and (D) (overall survival for ANKRD11 mutation status and progression-free survival for our combined mutation set of ARID1A and DAPK1) is provided in Supplementary Fig. 7. For information on outcome according to these two mutational markers in the subset of R-CHOP treated patients, please see Supplementary Fig. 8. Further, we found mutations in both KMT2D as well as NOTCH2 to be significantly enriched in 6q wild-type patients (E). Lollipop plots illustrating the localized distribution of mutational patterns affecting ARID1A, KMT2A, ANKRD11, and NOTCH2 in our combined cohorts; green dots refer to missense mutations; gray to truncating mutations (F–I).
Significantly mutated candidate driver genes in NFκB, WNT, and IL6/JAK/STAT dominate the mutational landscape of EBV + DLBCL (NOS)
We next screened for significantly mutated genes and potential oncogenic drivers via MutSig2CV (Fig. 4A). A gene set enrichment analysis allocated recurrent mutations to their respective biological pathways (Supplementary Fig. 4). Hereby, we unveiled a potentially perturbing role of several recurrent alterations in NFκB (75%; CSNK2A2, CARD10, LYN, SYK), IL6/JAK/STAT (38%; SOCS1, CXCL10, PIM1, SOCS3, and STAT3) as well as WNT signaling (62%; FRAT1, FRAT2, LRP5). A network propagation approach (Fig. 4B) using potential driver mutations as seeds uncovered a significant enrichment in the mutational signature of EBV + DLBCL (NOS) for immunological processes, including allograft rejection, interferon-alpha and gamma response, as well as IL6/JAK/STAT3 signaling. Yet, cellular metabolism processes like oxidative phosphorylation as well as fatty acid metabolism were significantly spared from oncogenic mutations. In order to evaluate our cohort in the context of the molecularly restructured landscape of DLBCL, we cross-referenced our WGS mutational data with the recently described molecular clusters proposed by Chapuy et al. and thereby found no significant affiliation to a singular subgroup but rather a wide distribution onto the different clusters (Supplementary Fig. 5).

A Significance levels for all EBV + DLBCL (NOS) MutSigCV genes (P < 0.05) and UpSet plot (B) showing the overlap of MutSigCV genes using EBV + DLBCL (NOS), all-type DLBCL and ABC-type DLBCL data; C shows the overlap between the three tumor entities. D HALLMARK gene sets enrichment for network diffusion analysis of significant MutSigCV genes (MutSigCV P < 0.05).
Additional insights into EBV + DLBCL (NOS) mutational landscape revealed by deep targeted-amplicon-based sequencing
To expand on our data derived from WGS, we performed targeted resequencing on a tumor-only extension cohort comprising the patient samples from WGS for validation purposes. A custom in-house AmpliSeq panel for tNGS, encompassing genes previously implicated in B-cell lymphoma pathogenesis combined with mutational targets first identified from the WGS cohort, identified putative oncogenic driver mutations in 40/47 patients (Fig. 3B, F–I, lollipop plots) and detected 250 mutations (resulting in a median of four mutations/sample), including 245 SNVs and 5 insertions and deletions (indels). The mutational signature was dominated by variants affecting ARID1A (45%), KMT2A (32%), ANKRD11 (32%), NOTCH2 (32%), KMT2D (30%) CCR6 (15%) in frequencies, differing significantly from previous findings in EBV-negative DLBCL. Upon comparative analysis of WGS and targeted resequencing data, we were able to demonstrate a concordance rate of 84% (21/25) of mutational calls (when considering genes with a mutational frequency >10% in our present cohort).
CCR6, CCR7, DAPK1, TNFRSF21, and YY1 mutations occur recurrently and appear to be specific for EBV + DLBCL (NOS)
To further elucidate the genomic taxonomy of B-cell malignancies and to clarify the mutational similarities and distinctions between EBV + DLBCL (NOS) and the closely related unselected ABC-type DLBCL) as well as (all-type) de novo DLBCL (NOS (n = 115145,46,47,48), we compared candidate mutational drivers.
In accordance with previous reports, ARID1A (45%), KMT2A (32%), ANKRD11 (32%), NOTCH2 (32%), and KMT2D (30%) were recurrently mutated in our EBV + DLBCL (NOS) cohort, albeit at partially elevated frequencies compared with EBV-negative DLBCL. Intriguingly, our integrated WGS and tNGS data analysis found CCR6 (15%), DAPK1 (15%), TNFRSF21 (13%), and CCR7 (11%) mutations as reoccurring and specific features of EBV + DLBCL (NOS), when compared with other subtypes of DLBCL. Further genes that were significantly enriched for mutations compared to EBV- DLBCL (NOS) included YY1, SOCS3, NELL2, CSNK2B. Our data revealed similarities to the recently published cohort of strictly polymorphic EBV + DLBCL NOS (Poly-EBV-L) by Sarkozy et al. in terms of oncogenic mutations in SOCS1, STAT6 and KMT2D, and differences with respect to the mutational frequencies in GNA13, CSF2RB, CSMD3, CD58 and PRKDC (prominent in Poly-EBV-L), the typical DLBCL mutations in ARID1A, KMT2A, ANKRD11, and NOTCH2 and lastly the exclusive mutations for EBV + DLBCL (NOS) in the genes CCR6, CCR7, DAPK1, TNFRSF21, CSNK2B, and YY118 (Fig. 4C).
6q deletions identified by WGS and FISH are a recurrent feature of EBV + DLBCL (NOS)
Structural variants detected by WGS are summarized in Supplementary Table 3 and depicted in Supplementary Fig. 6. Upon additional interrogation for CNVs, we identified several recurrent alterations, most prominent of which was a 6q deletion of variable extent (Fig. 2A, B; 5/8 cases), that encompassed known driver genes of B-cell lymphomagenesis PRDM1 and A20 in 4/5 cases.
FISH analysis of all cases having available tissue sections in both the WGS and the extension cohort, identified 6q deletions in 14/32 (44%) cases (concurrence rate of 100% between FISH and WGS) (Fig. 2C, D). Interestingly, these cases harbored significantly fewer mutations for the oncogenic drivers ANKRD11 and NOTCH2 (Fig. 3E), yet no further differences with respect to clinical or pathological characteristics were found in patients with 6q deletion (Supplementary Table 4). Other recurrent and previously described cytogenetic alterations in EBV + DLBCL (NOS) include 17p del (2/8) resulting in a deletion of TP53.
Survival analysis
Upon combined analysis of molecular and clinical data, we interrogated genomic alterations regarding their clinical impact. Due to the explorative nature of the study, Bonferroni’s correction for multiple testing was omitted. Hereby, we identified a significant impact (P < 0.05) of a distinct molecular subtype exhibiting combined alterations of ARID1A and DAPK1, which correlated with inferior overall survival. Moreover, mutations in ANKRD11 were identified as indicators of superior relapse-free but not overall survival (Fig. 3C, D). These effects were still visible as trends bordering on statistical significance when restricting the analysis on R-CHOP-treated patients. By means of a Chi-squared-test, we determined the statistical independence of mutations in ARID1A (P = 0.3782), DAPK1 (P = 0.2615), and ANKRD11 (P = 0.369) from 6q deletion status.
Discussion
Our whole-genome and targeted resequencing study in the hitherto largest cohort of EBV + DLBCL (NOS) in which all cases underwent rigorous central hematopathological panel review and cytogenetic workup prior to inclusion, we find a significant perturbing role of several recurrent alterations in NFκB, WNT, and IL6/JAK/STAT signaling. In line with previous publications, the overall mutational frequency was lower than in EBV-negative de novo DLBCL (NOS), which is compatible with the hypothesis that EBV-driven pathogenesis relies on a limited number of additional driver mutations and requires fewer oncogenic events than EBV-negative malignancies in order to shape a similar phenotype49. Further, our results support the preliminary notion on EBV + DLBCL (NOS) proposed by Liu et al., derived from a small Asian cohort, which emphasized a heterogeneous mutational profile with only few recurrent mutational events15. From our study, we deduce two essential insights into the molecular pathobiology of EBV + DLBCL (NOS).
First, we have delineated its mutational landscape, which is partially consistent with the literature as outlined by preliminary studies, recently published by Sarkozy and Zhou in terms of oncogenic mutations in KMT2D, CD58, BCL6, and NOTCH216,18. Previously reported mutations in TET2, DNMT3A, and TNFRSF14 were, however, not detectable in a significant subset of WGS patients. These observations do however require interpretation in the light of diverging compositions of selected NGS panels and the potential geographic heterogeneity between Asian and western DLBCL cohorts17. We identified no significant impact of mono- or polymorphic subtype of EBV + DLBCL on mutational signatures or mutational burden and no overall EBV-associated patterns or signatures were observed. Polymorphic lymphomas were, however, shown to harbor significantly more subclones, reflecting the morphological impression on a mutational level.
Our current investigation expands on previous efforts, not only through the largest number of cases studied so far, or a tumor-normal controlled WGS, rather than a tumor-only exome-focused approach but through the inclusion of both monomorphic as well as polymorphic (recurrently exhibiting Hodgkin and/or Reed/Sternberg cells) EBV + DLBCL (NOS). The genomic taxonomy of B-cell malignancies is, however, significantly refined in the current study through the comparative analysis of our data and the mutational studies in both unselected de novo DLBCL (NOS) as well as ABC-type DLBCL, as we reveal the mutational spectrum of aggressive EBV-driven lymphomas to exclusively encompass oncogenic mutations in CCR6, CCR7, DAPK1, TNFRSF21, CSNK2B, and YY1. Previous studies propose an oncogenetic role for mutations affecting CCR6. This is especially true for MALT-type lymphoma, where mutations are implicated in defective β-arrestin-mediated receptor desensitization and internalization, which is hypothesized to result in impaired regulation of transduction of extracellular stimulation into intracellular signaling50. Further studies implicated a (mutational) deregulation of the CCL20/CCR6 axis not only in inflammatory and infectious diseases but in disease progression across a variety of cancers, including pancreatic, colorectal, and breast cancer alongside hepatocellular carcinoma and others51. Moreover, CCR7 induction in EBV-infected cells was recurrently proposed to enable homing of lymphoid cells to secondary lymphoid tissue, where the virus in turn propagates infection or establishes latency, thereby driving lymphomagenesis52. It is further tempting to speculate on a potential role of oncogenic CCR7 mutations in EBV + DLBCL (NOS), prompting cellular proliferation and migration upon the binding of the cognate chemokine receptors, as was recently shown in breast cancer and other solid tumors53. Of note, we describe oncogenic mutations in DAPK1 as an exclusive feature of EBV + DLBCL (NOS). In a recent study, hypermethylation of DAPK1, speculatively resulting in a similar loss of function of DAPK1 as a regulatory partner of TP53 was shown to predict inferior clinical outcome54. Intriguingly, impairment of TNFRSF21/DR6 was previously implicated in increased cellular division and reduced apoptosis rates in B-cells and T-cell malignancies, including angioimmunoblastic T-cell lymphoma, another, recurrently EBV-driven type of cancer55,56. The known oncogenic driver YY1 was previously implicated in DLBCL pathogenesis irrespective of EBV status. Its overexpression was found to result in B-cell transformation and tumor progression, while mutations affecting CSNK2B, as another exclusive feature of EBV + DLBCL (NOS), remain insufficiently characterized to date57. Further, we sought to allocate our EBV + DLBCL (NOS) WGS cohort within the system of molecular clusters proposed by Chapuy et al. by means of cross-referencing evaluation of mutational patterns. This, however, yielded no predominant association with a singular cluster, most likely marking EBV positivity as an independent oncogenic driver, not restricted to a particular DLBCL subtype. We hereby offer substantial and novel insights into the molecular pathogenesis of this rare entity, potentially providing novel therapeutic targets, which now require functional validation.
Second, analyzing the entity on the scale of whole genomic structural variants and CNVs and thereby expanding on previous array comparative genomic hybridization findings by our group, we were able to identify recurrent 6q deletions. These observations are in line with the post-germinal phenotype of EBV + DLBCL (NOS) given that recurrent 6q deletions, affecting both PRDM1 and A20, have previously been implicated in ABC-type DLBCL pathogenesis as adverse prognosticators58. Intriguingly, we found 6q del cases to harbor significantly fewer mutations in oncogenic drivers such as ANKRD11 and NOTCH2, hinting at an independent oncogenic effect of this aberration, requiring even fewer additional genomic hits than 6q wild-type EBV + DLBCL (NOS) to achieve the malignant phenotype of the disease. This has been described before in EBV-negative ABC-type DLBCL, where the loss of PRDM1/BLIMP-1 was revealed to result in significantly reduced expression of genes implicated in plasma-cell differentiation while favoring the expression of genes involved in B-cell receptor signaling and proliferation58.
Integrative analysis of mutational and clinical data revealed combined alterations of ARID1A and DAPK1 as prognosticators of adverse overall survival, while mutations in ANKRD11 appeared to predict superior relapse-free, but not overall survival. These observations were apparently independent of 6q deletion status, a previously described adverse prognosticator in all-type DLBCL undergoing immunochemotherapy59 These preliminary observations do, however, require further validation in a prospective cohort of patients, preferably gathered within the context of a clinical trial.
Limitations of our current study include its retrospective design, implying the potential for fragmentary clinical data. Moreover, paired germline DNA samples for a larger group of EBV + DLBCL (NOS) would have been preferable in order to strengthen the exploratory subgroup of the cohort in our WGS approach. Identification of novel mutational hotspots may have been hindered by the limited number of samples in this group and further studies are needed to validate our findings. Further, the panel, employed in the analysis of our tumor-only extension cohort was limited, resulting in the identification of potentially oncogenic mutations in a mere 40/47 cases of EBV + DLBCL (NOS). Future studies should preferentially rely on a larger panel of genes or whole-exome sequencing.
In summary, we delineate the mutational landscape of EBV + DLBCL (NOS) and comprehensively explore its similarities and distinctions, compared with other types of DLBCL, including several exclusive oncogenic drivers. These provide a valuable starting point for targeted therapy approaches in this entity. In addition, we describe large deletions on chromosome 6 to be a highly recurrent cytogenetic feature of this rare entity, decisively shaping its near-terminally differentiated B-cell phenotype.
References
Beltran, B. E. et al. EBV-positive diffuse large b-cell lymphoma in young immunocompetent individuals. Clin. Lymphoma Myeloma Leuk. 11, 512–516 (2011).
Lu, T. X. et al. Epstein-Barr virus positive diffuse large B-cell lymphoma predict poor outcome, regardless of the age. Sci. Rep. 5, 12168 (2015).
Nicolae, A. et al. EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment. Blood 126, 863–872 (2015).
Oyama, T. et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am. J. Surg. Pathol. 27, 16–26 (2003).
Swerdlow, S. H. et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127, 2375–2390 (2016).
Dojcinov, S. D. et al. Age-related EBV-associated lymphoproliferative disorders in the Western population: a spectrum of reactive lymphoid hyperplasia and lymphoma. Blood 117, 4726–4735 (2011).
Shimoyama, Y. et al. Age-related Epstein-Barr virus-associated B-cell lymphoproliferative disorders: special references to lymphomas surrounding this newly recognized clinicopathologic disease. Cancer Sci. 99, 1085–1091 (2008).
Oyama, T. et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 13, 5124–5132 (2007).
Hofscheier, A. et al. Geographic variation in the prevalence of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: a comparative analysis of a Mexican and a German population. Mod. Pathol. 24, 1046–1054 (2011).
Witte, H. M. et al. Impact of treatment variability and clinicopathological characteristics on survival in patients with Epstein-Barr-virus positive diffuse large B cell lymphoma. Br. J. Haematol. 189, 257–268 (2020).
Klapper, W. et al. Patient age at diagnosis is associated with the molecular characteristics of diffuse large B-cell lymphoma. Blood 119, 1882–1887 (2012).
Ok, C. Y., Papathomas, T. G., Medeiros, L. J. & Young, K. H. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood 122, 328–340 (2013).
Montes-Moreno, S. et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod. Pathol. 25, 968–982 (2012).
Ok, C. Y. et al. Prevalence and clinical implications of Epstein-Barr virus infection in de novo diffuse large B-cell lymphoma in Western countries. Clin. Cancer Res. 20, 2338–2349 (2014).
Liu, F. et al. Genetic heterogeneity and mutational signature in Chinese Epstein-Barr virus-positive diffuse large B-cell lymphoma. PLoS ONE 13, e0201546 (2018).
Zhou, Y. et al. Comprehensive genomic profiling of EBV-positive diffuse large B-cell lymphoma and the expression and clinicopathological correlations of some related genes. Front. Oncol. 9, 683 (2019).
Kataoka, K. et al. Frequent structural variations involving programmed death ligands in Epstein-Barr virus-associated lymphomas. Leukemia 33, 1687–1699 (2019).
Sarkozy, C. et al. Mutational landscape of grey zone lymphoma. Blood 137, 1765–1776 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM 2013. Preprint at https://arxiv.org/abs/1303.3997 (2020).
Institute, B. Picard Toolkit GitHub Repository. http://broadinstitute.github.io/picard/ (2020).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Shyr, C. et al. FLAGS, frequently mutated genes in public exomes. BMC Med. Genomics 7, 64 (2014).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
Hubschmann, D. et al. Analysis of mutational signatures with yet another package for signature analysis. Genes Chromosomes Cancer 60, 314–331 (2021).
O’Fallon, B. D., Wooderchak-Donahue, W. & Crockett, D. K. A support vector machine for identification of single-nucleotide polymorphisms from next-generation sequencing data. Bioinformatics 29, 1361–1366 (2013).
Cameron, D. L. et al. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 27, 2050–2060 (2017).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Loh, P. R. et al. Reference-based phasing using the haplotype reference consortium panel. Nat Genet. 48, 1443–1448 (2016).
Cowen, L., Ideker, T., Raphael, B. J. & Sharan, R. Network propagation: a universal amplifier of genetic associations. Nat. Rev. Genet. 18, 551–562 (2017).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Picart-Armada, S., Thompson, W. K., Buil, A. & Perera-Lluna, A. diffuStats: an R package to compute diffusion-based scores on biological networks. Bioinformatics 34, 533–534 (2018).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Turner, S. Molecular signatures database (MSigDB) in a data frame 2020. https://github.com/ToledoEM/msigdf (2020).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Ziemann, M. & Kaspi, A. Multi-contrast gene set enrichment analysis. https://github.com/markziemann/mitch (2019).
Cox, J. & Mann, M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinform. 13, S12 (2012).
Wickham, H. et al. Welcome to the tidyverse. J. Open Source Softw. 4, 1686 (2019).
Lawrence, M. et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118 (2013).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756 (2018).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Chapuy, B. et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 24, 679–690 (2018).
Lohr, J. G. et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl Acad. Sci. USA 109, 3879–3884 (2012).
Morin, R. D. et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood 122, 1256–1265 (2013).
Reddy, A. et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell 171, 481–94 e15 (2017).
Grande, B. M. et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 133, 1313–1324 (2019).
Moody, S. et al. Novel GPR34 and CCR6 mutation and distinct genetic profiles in MALT lymphomas of different sites. Haematologica 103, 1329–1336 (2018).
Kadomoto, S., Izumi, K. & Mizokami, A. The CCL20-CCR6 axis in cancer progression. Int. J. Mol. Sci. 21, 5186 (2020).
Kocks, J. R. et al. Chemokine receptor CCR7 contributes to a rapid and efficient clearance of lytic murine gamma-herpes virus 68 from the lung, whereas bronchus-associated lymphoid tissue harbors virus during latency. J. Immunol. 182, 6861–6869 (2009).
Rizeq, B. & Malki, M. I. The role of CCL21/CCR7 chemokine axis in breast cancer progression. Cancers 12, 1036 (2020).
Kristensen, L. S. et al. Hypermethylation of DAPK1 is an independent prognostic factor predicting survival in diffuse large B-cell lymphoma. Oncotarget 5, 9798–9810 (2014).
Schmidt, C. S. et al. Enhanced B cell expansion, survival, and humoral responses by targeting death receptor 6. J. Exp. Med. 197, 51–62 (2003).
Wang, M. et al. Angioimmunoblastic T cell lymphoma: novel molecular insights by mutation profiling. Oncotarget 8, 17763–17770 (2017).
Castellano, G. et al. Yin Yang 1 overexpression in diffuse large B-cell lymphoma is associated with B-cell transformation and tumor progression. Cell Cycle 9, 557–563 (2010).
Xia, Y. et al. Loss of PRDM1/BLIMP-1 function contributes to poor prognosis of activated B-cell-like diffuse large B-cell lymphoma. Leukemia 31, 625–636 (2017).
Novak, A. J. et al. Whole-exome analysis reveals novel somatic genomic alterations associated with outcome in immunochemotherapy-treated diffuse large B-cell lymphoma. Blood Cancer J. 5, e346 (2015).
Acknowledgements
The authors would like to thank Tanja Oeltermann and Biggi Steinfeldt for their skilled technical assistance with immunohistochemical stains and interphase FISH tests. A.K. and H.B. acknowledge computational support from the OMICS compute cluster at the University of Lübeck. The research was supported by a grant to N.G. by the Stefan-Morsch-Foundation alongside infrastructural support. H.B. acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy—EXC 22167-390884018.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Study concept: N.G., A.C.F., and H.B. Data collection: N.G., A.K., H.W., V.B., J.Z., J.G., H.M., V.B., L.H., K.R., S.G., W.P., J.B., and P.T. Data analysis and creation of figures and tables: A.K., J.K., N.G., T.R., V.B., L.H., H.B., N.v.B., and H.B. Initial draft of the manuscript: N.G. Critical revision and approval of final version: all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gebauer, N., Künstner, A., Ketzer, J. et al. Genomic insights into the pathogenesis of Epstein–Barr virus-associated diffuse large B-cell lymphoma by whole-genome and targeted amplicon sequencing. Blood Cancer J. 11, 102 (2021). https://doi.org/10.1038/s41408-021-00493-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-021-00493-5
This article is cited by
-
Lysine demethylase LSD1 is associated with stemness in EBV-positive B cell lymphoma
Scientific Reports (2024)
-
Integrated genetic analyses of immunodeficiency-associated Epstein-Barr virus- (EBV) positive primary CNS lymphomas
Acta Neuropathologica (2023)
-
A genetic profiling guideline to support diagnosis and clinical management of lymphomas
Clinical and Translational Oncology (2023)
-
New concepts in EBV-associated B, T, and NK cell lymphoproliferative disorders
Virchows Archiv (2023)
