
Abstract
Carfilzomib (CFZ) is a non-reversible proteasome inhibitor approved for the treatment of patients with relapsed and refractory myeloma (RRMM). Its use has been associated with cardiovascular toxicity but although recently a signal of clinically significant renal complications has also been identified, it is less extensively investigated. We analyzed data of 114 consecutive patients with RRMM who received CFZ-based regimens. Renal complications not related to MM progression were observed in 19 (17%) patients; thrombotic microangiopathy (TMA) was seen in 6 (5%) patients, albuminuria >1 gr/day in 7 patients (6%) and at least grade 3 acute kidney injury (AKI) which could not be otherwise explained in 6 patients (5%). A total of 15 patients discontinued CFZ and dosing was reinitiated at a lower level in one patient with AKI. Albuminuria was associated with focal segmental glomerulosclerosis in the renal biopsy (performed in a total of 6 patients). Renal complications during CFZ therapy are common, occur mostly early and are unpredictable. A potential effect of CFZ on the renal endothelium could be implicated in the pathogenesis of these complications and may also share common pathophysiology with cardiovascular effects of CFZ.
Similar content being viewed by others
Carfilzomib (CFZ) is a next-generation selective and nonreversible proteasome inhibitor (PI), approved at different dosing schedules for patients with relapsed/refractory multiple myeloma (RRMM)1,2. Compared to bortezomib, carfilzomib has a different safety profile, with a low risk of neurotoxicity but higher for cardiovascular toxicity2. Renal clearance is not a significant pathway for carfilzomib elimination and no dose or treatment schedule adjustments are required in patients with renal impairment3 but renal toxicity has been described4. In addition, an association with thrombotic micorangiopathy (TMA) has been reported and has been included in the drug’s Summary of product characteristics. There is, however, limited characterization of the spectrum of renal toxicities associated with carfilzomib, and, given its potent antimyeloma activity and increasing use, a thorough characterization of these toxicities is clinically relevant. We report data for 114 consecutive, unselected, RRMM patients who were treated with carfilzomib-based regimens in our center (Department of Clinical Therapeutics, Athens), closely monitored and evaluated for renal complications, including renal biopsies. Renal events related to myeloma progression were not rated as carfilzomib-related. They were rated according to National Cancer Institute Common Terminology Criteria for Adverse Events, v4.03. Approval was obtained from the institutional review board/Ethics Committee of our hospital. Renal events of interest and discontinuation of carfilzomib as a result of progression of disease or for other reasons was treated as a competing event and the respective cumulative incidence function curves were plotted.
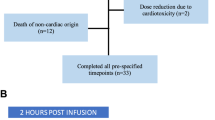
Patients’ characteristics are shown in Table 1. The median follow-up from the start of carfilzomib is 27 months, median duration of carfilzomib therapy was 5.5 months (IQR 3.2−11.5) and 28 (24.5%) patients continued carfilzomib therapy at the time of analysis. During treatment and up to 30 days after the last carfilzomib dose, 19 (17%) patients developed renal complications (not related to myeloma progression) (Fig. 1). These included TMA in 6 (5%) patients, albuminuria > 1 g/day with very low serum free light chains (sFLCs) or negative urine immunofixation in 7 (6%) patients and acute kidney injury (AKI) ≥ grade 3 in 6 (5%) patients which could not be otherwise explained. Median time from start of carfilzomib to development of any renal complication was 62 days (~2 months) (IQR 35−272) and in 15/19 patients carfilzomib was discontinued due to renal complications.

Upper panel: Percentage of patients with progressive disease and any carfilzomib-related complications plotted over time of months on Carfilzomib (CFZ) treatment. Lower panel: Renal complications observed in MM patients. TMA thrombotic microangiopathy, AKI acute kidney injury.
Median time from carfilzomib start to TMA was 3 months (0.5–19.5). At diagnosis of TMA, median platelet counts were 20 × 109/L (range 11–30), median hemoglobin 8 g/dL, median Lactate dehydrogenase (LDH) 449 IU/L (ULN < 225, range 371–619) and median blood schistocytes were 2.5% (range 2–6.5%). All six patients received plasmapheresis and steroids, rituximab was administered in one patient; no patient received eculizumab. Renal function and platelets recovered fully in five patients while one died of sepsis. No patient had progressive myeloma at the time of the event. ADAMTS-13 was evaluated in two patients and was within normal levels. No patient was re-exposed to carfilzomib after TMA.
Median time to development of proteinuria >1 g/d was 6 months (range 2–59 months). Median proteinuria was 3.7 g/d (range 1–4.5) and in all cases >90% of urine protein was albumin. All seven patients were in myeloma remission. Median eGFR was 53 mL/min/1.73 m2 (range 41–92 mL/min/1.73 m2) and one had proteinuria prior to carfilzomib, which was selective Bence−Jones proteinuria. Following carfilzomib interruption there was a gradual decrease of proteinuria to normal levels in two of the seven patients. Carfilzomib was resumed in one of the patients at a reduced dose with no recurrence of the proteinuria. Median time to CFZ-induced AKI was 1.1 months and median creatinine level was 2.5 mg/d. The injury was reversed in three patients who reinitiated treatment at lower doses.
Six patients with persistent renal dysfunction, with no obvious etiology and no contraindications had renal biopsies (Supplementary Table S1 and Supplementary Fig. S1); four had albuminuria and two had AKI. None had findings consistent with immunoglobulin-mediated pathology (cast nephropathy, monoclonal immunoglobulin deposition disease (MIDD) or amyloidosis) or pathology related to activation of the alternative complement pathway. The most consistent finding was a pattern of focal segmental glomerulosclerosis (FSGS) of various subtypes. Additionally, coexistent with the lesions of FSGS, in four renal biopsies, a pattern of acute and/or organized TMA was observed, with intraglomerular and/or arteriolar fibrin microthrombi and/or mucoid degeneration of the arteriolar/arterial wall and/or reduplication of the glomerular basement membranes with swelling of the endothelial cells.
We found no association of baseline proteinuria (immunoglobulin or albumin), sFLC, myeloma subtype, patient age, number of prior lines of therapy, the CFZ dose, carfilzomib regimen, baseline eGFR and prior history of cardiovascular disease, diabetes mellitus or hypertension with the occurrence of renal complications. At start of carfilzomib, 33 patients had an eGFR <60 mL/min/1.73 m2 and 18 (54.5%) improved their eGFR to >60 mL/min/1.73 m2.
Carfilzomib-associated renal complications were observed in 17% of patients in our cohort. They mostly occurred early, within the first 2–3 months in more than half of the patients, although albuminuria developed later (median time of 6 months). These complications seem unpredictable, as we were not able to identify any predictive factors. As a consequence, carfilzomib was discontinued in 15 (13%) patients, establishing carfilzomib-associated nephrotoxicity as a major cause of treatment delay and discontinuation. However, as previously demonstrated, renal impairment is often transient and carfilzomib was effective even for patients with renal dysfunction.
In contrast to previous reports, our evaluation of renal function was not limited to evaluation of creatinine levels alone3,4. Thus, we observed a relatively high rate of renal complications, by reporting renal adverse events that cover the whole spectrum of renal toxicities observed with carfilzomib. According to clinical trial data, grade 3 or higher events of AKI associated with CFZ are seen in approximately 3–5% of patients and in a recent meta-analysis of four phase II studies, the cumulative incidence of grade 3–5 renal toxicities was 8.3%1,2,4. In most datasets no distinction was made between renal impairment due to carfilzomib and secondary to disease progression but we were able to decipher these clinical conditions through detailed and meticulous evaluation in all cases.
The pathophysiology underlying carfilzomib-induced renal toxicity is not clear yet3. The mechanisms proposed range from prerenal insults, to tumor lysis-like phenomena, to biopsy-proven TM. The transient rise in creatinine, which is quite common, implies that the process might be prerenal or vascular and therefore reversible5. Endothelial dysfunction via an increased resting vasoconstricting tone and amplification of the spasmogenic effect of different agents could partly explain the renal effects of carfilzomib according to some animal and clinical data6. It has been demonstrated that proteasomal inhibition causes a shift in favor of endothelium-dependent vasodilation7 but inhibition of chymotrypsin-like (CT-L) subunit of the 20S proteasome by carfilzomib may be key to the vasoconstriction observed.
TMA was observed in several of our patients. The term TMA is used to describe a histopathological lesion characterized by microvascular occlusion secondary to endothelial injury and coagulation cascade activation. The clinical syndrome associated with this entity includes microangiopathic hemolytic anemia (MAHA), thrombocytopenia and end-organ damage8. In our cohort, clinically overt TMA was observed in six (5%) patients but was also a frequent pathological lesion in renal biopsies of patients that presented only with albuminuria, lacking the associated clinical features. Renal failure with biopsy-proven TMA in the absence of hematologic criteria has previously been reported. A recent case report describes renal-limited TMA in a patient who developed bevacizumab-induced proteinuria9. Drug-induced TMA (DITMA), as a clinical syndrome, is a relatively rare complication of several drugs, including PIs10. Two mechanisms have been postulated: immune-mediated antibody damage accounts for early events and direct toxicity which is time and dose-dependent and accounts for events at later timepoints10,11. Case reports have also highlighted the association between carfilzomib and AKI with evidence of underlying TMA (also biopsy-proven in some cases)5,12. In a case series on PI-induced TMA, patients had normal ADAMTS-13 levels and two underwent a biopsy confirming TMA10. Two mechanisms have been proposed for the direct nephrotoxic effects of carfilzomib. The first involves microvascular toxicity to the endothelium mediated by carfilzomib-induced reduction in VEGF levels (via NFκB inhibition)13. VEGF is essential for the functional and structural integrity of the glomerural endothelium and disruption of VEGF production can lead to TMA and podocytopathy, proteinuria and hypertension. The second mechanism involves a potential overactivation of the complement membrane attack complex. One group recently demonstrated increased deposition of C5b-9 on cultured endothelial cells when exposed to plasma of four patients during the acute phase of carfilzomib-induced TMA14.
Our data suggest that the spectrum of carfilzomib-related renal adverse effects extends beyond increase of serum creatinine, to TMA as a clinical syndrome, and albuminuria associated with FSGS but also with histopathological-only findings of TMA. Importantly, carfilzomib-induced renal toxicity is unpredictable. Thus, in our practice, we follow serum creatinine almost weekly and in case of significant creatinine increase we assess platelet counts and serum LDH. We also assess proteinuria and urine protein electrophoresis regularly, even in patients without Bence−Jones proteinuria at the start of carfilzomib therapy. Further investigation of the underlying mechanisms of carfilzomib-induced nephrotoxicity is needed, in order to predict and manage these complications and provide this active drug with safety.
References
Stewart, A. K. et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 372, 142–152 (2015).
Dimopoulos, M. A. et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 17, 27–38 (2016).
Badros, A. Z. et al. Carfilzomib in multiple myeloma patients with renal impairment: pharmacokinetics and safety. Leukemia 27, 1707–1714 (2013).
Siegel, D. et al. Integrated safety profile of single-agent carfilzomib: experience from 526 patients enrolled in 4 phase II clinical studies. Haematologica 98, 1753–1761 (2013).
Jhaveri, K. D., Chidella, S., Varghese, J., Mailloux, L. & Devoe, C. Carfilzomib-related acute kidney injury. Clin. Adv. Hematol. Oncol. 11, 604–605 (2013).
Stangl, V. et al. Long-term up-regulation of eNOS and improvement of endothelial function by inhibition of the ubiquitin-proteasome pathway. FASEB J. 18, 272–279 (2004).
Lorenz, M. et al. Proteasome inhibition prevents experimentally-induced endothelial dysfunction. Life Sci. 84, 929–934 (2009).
George, J. N. & Nester, C. M. Syndromes of thrombotic microangiopathy. N. Engl. J. Med. 371, 1847–1848 (2014).
Toriu, N. et al. Renal-limited thrombotic microangiopathy due to bevacizumab therapy for metastatic colorectal cancer: a case report. Case Rep. Oncol. 12, 391–400 (2019).
Yui, J. C. et al. Proteasome inhibitor associated thrombotic microangiopathy. Am. J. Hematol. 91, E348–E352 (2016).
Al-Nouri, Z. L., Reese, J. A., Terrell, D. R., Vesely, S. K. & George, J. N. Drug-induced thrombotic microangiopathy: a systematic review of published reports. Blood 125, 616–618 (2015).
Hobeika, L., Self, S. E. & Velez, J. C. Renal thrombotic microangiopathy and podocytopathy associated with the use of carfilzomib in a patient with multiple myeloma. BMC Nephrol. 15, 156 (2014).
Usui, J. et al. Clinicopathological spectrum of kidney diseases in cancer patients treated with vascular endothelial growth factor inhibitors: a report of 5 cases and review of literature. Hum. Pathol. 45, 1918–1927 (2014).
Portuguese, A. J. & Lipe, B. Carfilzomib-induced aHUS responds to early eculizumab and may be associated with heterozygous CFHR3-CFHR1 deletion. Blood Adv. 2, 3443–3446 (2018).
Acknowledgements
The research would not have been possible without the input and expertise of Dr. Charikleia Gakiopoulou and Dr. Erasmia Psimenou.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fotiou, D., Roussou, M., Gakiopoulou, C. et al. Carfilzomib-associated renal toxicity is common and unpredictable: a comprehensive analysis of 114 multiple myeloma patients. Blood Cancer J. 10, 109 (2020). https://doi.org/10.1038/s41408-020-00381-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-020-00381-4
This article is cited by
-
Multiple myeloma with acute light chain cast nephropathy
Blood Cancer Journal (2023)
-
Mutations in the alternative complement pathway in multiple myeloma patients with carfilzomib-induced thrombotic microangiopathy
Blood Cancer Journal (2023)
-
Carfilzomib-induced thrombotic microangiopathy (TMA): an under-recognised spectrum of disease from microangiopathic haemolysis to subclinical TMA
Blood Cancer Journal (2023)
-
Reporting of adverse events of treatment interventions in multiple myeloma: an overview of systematic reviews
Annals of Hematology (2023)
-
Influenza-induced thrombotic microangiopathy in a patient with cancer on proteasome inhibitor: a diagnostic dilemma
CEN Case Reports (2022)
