
Abstract
Introduction
Oculo-dento-digital dysplasia (ODDD, OMIM# 164200) is a rare genetic disorder caused by mutation in Gap junction alpha gene that encodes connexin 43 (Cx43) protein. In this paper, the case of a 16-year-old boy is reported who presented with the complaint of toothache. Examination revealed unusual facial features, i.e., long narrow nose, hypertelorism, prominent epicanthal folds along with syndactyly and camptodactyly. We have also compiled available dental literature on ODDD that will help clinicians in early diagnosis and management of this condition.
Materials and methods
A literature search was performed in PubMed NLM, EBSCO Dentistry & Oral Sciences Source, and EBSCO CINAHL Plus.
Results
A total of 309 articles were identified in the literature search. Only 17 articles were included based on the predetermined inclusion and exclusion criteria in the review synthesis. The included articles were case reports (n = 15), a case report and review (n = 1), and an original article (n = 1). Enamel hypoplasia, hypomineralization, microdontia, pulp stones, curved roots, and taurodontism were common dental findings in ODDD.
Conclusions
After establishing definitive diagnosis, a multidisciplinary team should work in cohesion to improve the quality of life of patients. Immediate treatment should be focused on the correction of current oral condition and symptomatic treatment. In the long term, attention should be diverted to prevent tooth wear and maintaining the occlusal vertical dimension to establish adequate function.
Similar content being viewed by others
Introduction
Oculo-dento-digital dysplasia (ODDD, OMIM# 164200) is a rare congenital genetic disorder characterized by craniofacial, ocular, dental, and digital abnormalities [1]. It was initially recognized by Lohman in 1920 [2]. It is primarily an autosomal dominant disorder but, in a few cases, recessive forms of the disease have been identified [3]. ODDD is caused by a missense mutation in gap junction alpha 1 (GJA1) gene on chromosome 6q22.31 [3]. This gene encodes for connexin 43 (Cx43), a transmembrane protein [3]. In 2003, ODDD became known as the first human disease to be linked to germline Cx43 gene (GJA1) mutations [4]. Cx43 is one of the 20 members of the human connexin protein family [3]. It has been diagnosed in fewer than 300 people worldwide with an incidence of around 1 in 10 million [2]. ODDD has high penetrance and its phenotypic expression is variable [5, 6], ODDD has typical features (Table 1) of syndactyly [3], digital camptodactyly [7], ophthalmic [8], nasal [3], and dental abnormalities [9].
Other than these mentioned features, ODDD also shows neurological [10,11,12], and cardiological involvement [13]. Some features of ODDD are evident at birth, while others may appear with increasing age. Despite undergoing medical treatment for eyesight, and multiple surgeries for syndactyly and camptodactyly in hands in childhood, the case presented here was first diagnosed in our dental practice. This shows that rare syndromes like ODDD can remain either undiagnosed or misdiagnosed. Therefore, this review aims to summarize the available dental literature on ODDD as it will enable better management of associated diseases to improve the quality of life of the patient.
Case report
A 16-year-old male patient visited the dental clinic in February 2020 with complaints of pain in the lower right posterior tooth and sensitivity to cold in all posterior teeth. On physical examination, the patient had syndactyly of fourth-fifth fingers of the right hand and third-fourth fingers of the left hand with camptodactyly and webbing (Fig. 1). Multiple surgeries were carried out from birth to 12 years of age, but digital abnormalities were still present. Another corrective surgery was done in 2021 which resulted in a successful resolution of syndactyly of left-hand digits (Fig. 2). On extra-oral examination patient’s nose was thin, elongated with hypoplastic alae nasi, anteverted nares and ocular findings included prominent epicanthic folds and hypertelorism (Fig. 3).

a–c Syndactyly of fourth-fifth fingers of right hand and third-fourth fingers of left hand with camptodactyly.

Post-operative x-ray showing separation of third and fourth fingers of left hand.

Arrows showing prominent epicanthal folds (blue), Hypertelorism (yellow), Hypoplastic and anteverted nares (red).
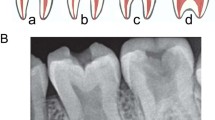
Upon intra-oral examination, findings were generalized staining, hypoplastic, hypomineralized enamel with pitting (most obvious on posterior teeth), multiple carious teeth: # 16, 17, 26, 27, 36, 37, 45, 46, 47, deep fissures in tooth # 25, 34, 35 (Fig. 4). Tooth # 46 was non-vital on electric pulp testing (EPT) and cold test and tender to percussion.

a Right lateral view. b Frontal view. c Left lateral view. d Maxillary occlusal view. e Mandibular occlusal view showing generalized staining, hypoplastic enamel (most obvious on posterior teeth) and deep fissure and multiple carious teeth. Tooth #46 root canal treated and 47 was filled.
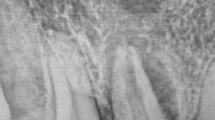
On panoramic (A) and right (B) and left (C) bitewing radiographs, all upper and lower second and third molars had taurodontism and pulp stones (red arrows) along with curved roots (Fig. 5).

Orthopantomogram (a) showing taurodontism in all upper, lower second and third molars with curved roots in lower first molars. Right (b) and left (c) bitewings showing pulp stones (red arrows) in all first molars and lower right, left second molars.
Management and follow-up
Orthograde endodontic treatment was initiated in tooth # 46, after copious sodium hypochlorite irrigation and intracanal medicament, temporary restoration was placed. After 1 week, endodontic treatment of tooth # 46 was completed followed by permanent restoration. Restoration of all carious teeth # 16, 17, 26, 27, 36, 37, 45, 46, 47 was done with amalgam filling material (Fig. 6). Deep fissures in tooth #25, 34, 35 were sealed with light cure resin composite (Fig. 6). Dietary modification and oral hygiene instruction were reinforced. Impressions were obtained for diagnostic casts to monitor tooth wear on follow-up visits (Fig. 7). A 3-monthly follow-up was scheduled to evaluate the further enamel loss of the unrestored dentition and check the restored teeth status. The patient was asymptomatic and tooth sensitivity had diminished. No further deterioration of enamel was apparent.

a, b Showing maxillary and mandibular teeth after completion of dental treatment.

a–e Diagnostic cast to monitor tooth wear on follow-up visits.
Review questions
We decide to systematically look into these cases as no such review has been published till now. For this review the authors (KDVH & FUMR) compiled the available dental literature on ODDD patients based on the following questions:
-
1.
What are the diagnostic measures, i.e., clinical features or gene analysis?
-
2.
What are the dental manifestations commonly seen in patients with ODDD?
-
3.
What are the management options for dental abnormalities?
Materials and methods
Search strategy
The authors (KDVH & FUMR) conducted a pilot search based on various combinations of key search terms. The final search strategy was formulated based on this pilot search. A comprehensive online literature search was performed (in June 2021) in three major health sciences databases, PubMed NLM, EBSCO Dentistry & Oral Sciences Source, EBSCO CINAHL Plus, and a hand search was also done in collaboration with a medical information specialist (Librarian, Aga Khan University Hospital, Pakistan).
Search terms
(“Oculodentodigital Dysplasia” [Supplementary Concept] OR Oculodentodigital dysplasia OR Oculo-dento-digital dysplasia OR Oculodentodigital syndrome OR Oculo-dento-digital syndrome OR Oculo-dento-osseous syndrome OR Oculo-dento-osseous syndrome OR Oculo-dento-osseous dysplasia OR Oculo-dento-osseous dysplasia OR ODDD OR ODOD).
Screening process
Endnote 20 reference manager was used for article citations. After removing duplicate references, all the remaining articles were screened by two authors (KDVH & FUMR) according to the predetermined inclusion criteria. Later, data was extracted by KDVH on a calibrated predetermined proforma independently, which was rechecked by FUMR.
Inclusion criteria
-
Dental case reports
-
Dentistry-related original articles
Exclusion criteria
-
Animal studies/In vitro studies
-
Molecular studies (gene analysis)
-
Medical case reports/Original articles
-
Abstracts only
-
Letter to Editors
-
Conference proceedings
Data extraction
For data extraction customized proforma was designed by the authors to extract the required data from included studies:
-
1.
Study details (title, type of study, authors, journal of publication, year of publication).
-
2.
Study characteristics (specialty field, no. of cases included).
-
3.
Age, gender, Oral manifestations, treatment provided, follow-up time.
-
4.
Diagnostic measure (genetic analysis).
Results
A total of 309 studies were identified after a detailed literature search. After removing duplicates, the number of studies was reduced to 223. After screening these studies by the authors (KDVH & FUMR) following the predetermined inclusion and exclusion criteria, 17 articles were included in this study for final analysis (full text of 6 articles could not be retrieved due to their non-availability in the database). The full screening process is shown in the PRISMA flowchart shown in Fig. 8.

PRISMA Flow Chart for literature search.
Characteristics of included studies
The selected articles included dental case reports (n = 15), one case report with a review, and one original article. A total of 22 patients (8 = females, 14 = males) are reported with ODDD with dental malformations. Ten out of 17 studies confirmed gene analysis of reported patients. A summary of selected studies is given in Table 2. Below are brief results of the selected studies based on the diagnostic measure, dental manifestations, and their management options.
Diagnostic measures
As previously mentioned, ODDD is a genetic disorder caused by a missense mutation in the GJA1 gene that encodes for a transmembrane protein (Cx43). Eight studies reported diagnosis based on clinical features [7, 14,15,16,17,18,19,20], while nine studies confirmed missense mutation in GJA1 at chromosome 6q22-q23 by genetic analysis [8, 21,22,23,24,25,26,27,28].
Dental manifestations
Enamel hypoplasia, hypomineralization, microdontia, pulp stones, curved roots, taurodontism, discolored teeth, tooth loss with or without caries, and peri-apical abscess are common findings in ODDD patients [7, 8, 15,16,17,18,19, 21,22,23, 25,26,27,28]. Other rare findings, i.e., cleft lip and palate [20, 24] short mandibular ramus and body, absent frontal sinus [7], distally inclined condyles [22], and hypoplastic maxilla [14, 17, 22] may also be present.
Management and follow-up
Management described below was reported by selected articles and was primarily based on the minimum invasive options:
Primary teeth
Extraction of grossly carious teeth, deep fissure sealants, early restoration of dental caries, prevention of tooth wear with stainless steel crowns (SSCs) in posterior teeth with or without pulpectomy was carried out in studies [8, 21, 23].
Permanent teeth
Extraction of unrestorable teeth, restoration of caries, pulpotomy in immature asymptomatic teeth, root canal treatment (apexification in immature teeth) in symptomatic teeth followed by full coverage restorations were done in studies [22]. A regular, usually 3-monthly dental evaluation of restored teeth and unrestored dentition is recommended to monitor and early management of any abnormality [8, 22].
Discussion
Oculo-dento-digital dysplasia is an uncommon condition that is rarely recognized by dentists,
For this reason, we decided to write this report along with a literature search so that the readers can become more familiar with this condition to better serve their patients.
In our review, we found that ODDD is an autosomal dominant genetic disorder, which is characterized by abnormal ocular, dental, and digital findings. It is caused by a mutation in GJA1 gene encoding Cx43 [9, 29]. In our case, the patient presented with multiple dental caries, enamel hypoplasia, pulp stones, and taurodontism and syndactyly, which are typically digital and dental manifestations of this syndrome. Genetic analysis of 178 genes (List given in genetic analysis report) responsible for limb and digital malformations was done for this patient. An uncertain significance of heterozygous variant for DLX6 (Distal-less homeobox) [99_119del (p.Gln38_Gln44del)] which is responsible for autosomal dominant split-hand/foot malformation type 1 [30] and GJA1 [c.196 T > C (p.Tyr66His)] gene involved in autosomal dominant and recessive oculodentodigital dysplasia was identified (9). DLX6 gene abnormality rarely shows dental involvement usually crowding [31]. However, the facial (extra-oral) features were mild but consistent with ODDD (autosomal dominant), showing a thin nose with hypoplastic alae nasi, short palpebral fissures. The present case suggests, ODDD should be considered even when ocular symptoms are un-remarkable and this correlates with the previous literature that there is approximately 70% chance of ocular manifestations in ODDD patients [9].
The other conditions that have similar features to ODDD are amelogenesis imperfecta (AI), oral-facial-digital syndrome, Hallerman-Streiff syndrome (HSS) [32], and Saethre-Chotzen syndrome. ODDD can be differentiated from AI, as the later condition shows little systematic involvement [33]. Oral-facial-digital dysplasia involves the renal system and has features like lobulated tongue without ocular manifestations which differentiates it from ODDD [34]. HSS may share similar clinical ocular and dental features with ODDD but the presence of the skin conditions, dwarfism differentiate it from ODDD [35]. Saethre-chotzen syndrome has features of the characteristic craniosynostosis, ptosis, and absence of any dental manifestations that differentiate it from ODDD [36]. Further details are given in Table 3.
In the present case, the primary goal was to treat the dental disease (i.e., pulpitis and tooth wear) and seal the other teeth for the preservation of arch integrity for patients well being (nutritional, esthetic, and psychological). Teeth were structurally compromised, thus prone to caries and fractures from trauma. Though the symptomatic management in ODDD patients is the same as non-syndromic patients in a few situations, special considerations are required for long-term prognosis, i.e., preservation of teeth by sealing the deep pits and fissures to prevent caries [22]. Conservative treatment plan was of utmost importance as extraction could have led to compromised development of alveolar bone. In ODDD patients, the remodeling process may not be as efficient as in unaffected population due to lack of coordinating events in the alveolar bone due to alteration in Cx43 (29). This can affect the ossteo-integration in case of implant placement or remodeling process in orthodontic movement, thus preservation of alveolar bone is of great importance [22].
Conclusions
In our patient after genetic analysis, clinical and radiological findings were consistent with ODDD. The primary goals of dental treatment in patients with ODDD should be intended to correct the current oral condition and prevent further tooth loss for maintaining masticatory efficiency, phonetics, and esthetics. In these cases, dentists, pediatric dentists, orthodontists, prosthodontists should work with coordination and multidisciplinary approaches should be provided to improve quality of life.
References
Kumar V, Couser NL, Pandya A, Oculodentodigital. Dysplasia: a case report and major review of the eye and ocular adnexa features of 295 reported cases. Case Rep Ophthalmol Med. 2020;2020:1–16.
Doshi DC, Limdi PK, Parekh NV, Gohil NR. Oculodentodigital dysplasia. Indian J Ophthalmol. 2016;64:227–30.
Judisch GF, Martin-Casals A, Hanson JW, Olin WH. Oculodentodigital dysplasia. Four new reports and a literature review. Arch Ophthalmol. 1979;97:878–84.
Laird DW. Syndromic and non-syndromic disease-linked Cx43 mutations. FEBS Lett. 2014;588:1339–48.
Gorlin RJ, Cohen Jr MM, Hennekam RC. Syndromes of the head and neck. Oxford University Press; 2001.
Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408–18.
Gorlin RJ, Miskin LH. Oculodentodigital dysplasia. J Pediatr. 1963;63:69–75.
Jensen ED. Generalised hypomineralisation of enamel in oculodentodigital dysplasia: comprehensive dental management of a case. BMJ Case Rep. 2021;14:1–4.
Paznekas WA, Karczeski B, Vermeer S, Lowry RB, Delatycki M, Laurence F, et al. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat. 2009;30:724–33.
Barzegar M, Sayadnasiri M, Tabrizi A. Epilepsy as a rare neurologic manifestation of oculodentodigitalis dysplasia. Iran J Child Neurol. 2012;6:39–43.
De Bock M, Kerrebrouck M, Wang N, Leybaert L. Neurological manifestations of oculodentodigital dysplasia: a Cx43 channelopathy of the central nervous system? Front Pharm. 2013;4:120.
Constantinides VC, Paraskevas GP, Kalogera S, Yapijakis C, Kapaki E. Hot cross bun sign and prominent cerebellar peduncle involvement in a patient with oculodentodigital dysplasia. Neurol Sci. 2021;42:343–5.
Wittlieb-Weber CA, Haude KM, Fong CT, Vinocur JM. A novel GJA1 mutation causing familial oculodentodigital dysplasia with dilated cardiomyopathy and arrhythmia. HeartRhythm Case Rep. 2016;2:32–5.
Owlia F, Akhavan Karbassi MH, Hakimian R, Alemrajabi MS. A highlighted case for emphasizing on clinical diagnosis for rare syndrome in third world. Iran J Child Neurol. 2017;11:77–80.
Mills JK, Wheeler L, Oishi SN. A case of familial syndactyly associated with eye and dental abnormalities. JAAPA. 2015;28:40–3.
Scheutzel P. Oculodentodigital syndrome: report of a case. Dentomaxillofac Radiol. 1991;20:175–8.
Schuller MG, Barnett ML, Strassburger K, Friedman DL, Sonnenberg EM. Oculodentodigital dysplasia. Oral Surg Oral Med Oral Pathol. 1986;61:418–21.
Thodén CJ, Ryöppy S, Kuitunen P. Oculodentodigital dysplasia syndrome. Report of four cases. Acta Paediatr Scand. 1977;66:635–8.
Zach GA. Oculodento-osseous dysplasia syndrome. Oral Surg Oral Med Oral Pathol. 1975;40:122–5.
Eidelman E, Chosack A, Wagner ML. Orodigitofacial dysostosis and oculodentodigital dysplasia. Two distinct syndromes with some similarities. Oral Surg Oral Med Oral Pathol. 1967;23:311–9.
Choi J, Yang A, Song A, Lim M, Kim J, Jang JH, et al. Oculodentodigital dysplasia with a novel mutation in GJA1 diagnosed by targeted gene panel sequencing: a case report and literature review. Ann Clin Lab Sci. 2018;48:776–81.
Hadjichristou C, Christophidou-Anastasiadou V, Bakopoulou A, Tanteles GA, Loizidou MA, Kyriacou K, et al. Oculo-dento-digital dysplasia (ODDD) due to a GJA1 mutation: report of a case with emphasis on dental manifestations. Int J Prosthodont. 2017;30:280–5.
Porntaveetus T, Srichomthong C, Ohazama A, Suphapeetiporn K, Shotelersuk V. A novel GJA1 mutation in oculodentodigital dysplasia with extensive loss of enamel. Oral Dis. 2017;23:795–800.
Amano K, Ishiguchi M, Aikawa T, Kimata M, Kishi N, Fujimaki T, et al. Cleft lip in oculodentodigital dysplasia suggests novel roles for connexin43. J Dent Res. 2012;91:S38–44.
Aminabadi NA, Pourkazemi M, Oskouei SG, Jamali Z. Dental management of oculodentodigital dysplasia: a case report. J Oral Sci. 2010;52:337–42.
Aminabadi NA, Ganji AT, Vafaei A, Pourkazemi M, Oskouei SG. Oculodentodigital dysplasia: disease spectrum in an eight-year-old boy, his parents and a sibling. J Clin Pediatr Dent. 2009;33:337–41.
Feller L, Wood NH, Sluiter MD, Noffke C, Raubenheimer EJ, Lemmer J, et al. Report of a black South African child with oculodentodigital dysplasia and a novel GJA1 gene mutation. Am J Med Genet A. 2008;146A:1350–3.
van Es RJ, Wittebol-Post D, Beemer FA. Oculodentodigital dysplasia with mandibular retrognathism and absence of syndactyly: a case report with a novel mutation in the connexin 43 gene. Int J Oral Maxillofac Surg. 2007;36:858–60.
Civitelli R. Cell-cell communication in the osteoblast/osteocyte lineage. Arch Biochem Biophys. 2008;473:188–92.
Lo Iacono N, Mantero S, Chiarelli A, Garcia E, Mills AA, Morasso MI, et al. Regulation of Dlx5 and Dlx6 gene expression by p63 is involved in EEC and SHFM congenital limb defects. Development 2008;135:1377–88.
Ullah A, Hammid A, Umair M, Ahmad W. A novel heterozygous intragenic sequence variant in DLX6 probably underlies first case of autosomal dominant split-hand/foot malformation type 1. Mol Syndromol. 2017;8:79–84.
Acharya S, Mohanty M, Acharya S. Hallermann Streiff syndrome—the oral manifestations in a child. J Genet Syndr Gene Ther. 2015;6:1–4.
Crawford PJ, Aldred M, Bloch-Zupan A. Amelogenesis imperfecta. Orphanet J Rare Dis. 2007;2:1–11.
Franco B, Thauvin-Robinet C. Update on oral-facial-digital syndromes (OFDS). Cilia 2016;5:1–11.
Mirshekari A, Safar F. Hallermann–Streiff syndrome: a case review. Clin Exp Dermatol. 2004;29:477–9.
de Heer IM, de Klein A, van den Ouweland AM, Vermeij-Keers C, Wouters CH, Vaandrager JM, et al. Clinical and genetic analysis of patients with Saethre-Chotzen syndrome. Plast Reconstr Surg. 2005;115:1894–902.
Acknowledgements
Mr. Khawaja Mustafa, medical information specialist (Librarian, Aga Khan University Hospital, Pakistan). This study is not funded by any research grant or trust.
Author information
Authors and Affiliations
Contributions
The present case was done under care of FUMR, data was extracted by KDVH for systematic analysis from databases on a predetermined proforma, which was reviewed by FUMR. The manuscript has been read and approved by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
The authors have no financial interest or affiliations that would constitute a conflict of interest. Informed written consent was obtained from the patient for participation in this study, who allowed us to use the relevant data and pictures for publication.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hindu, K.D., Umer, F. Oculo-dento-digital dysplasia: a systematic analysis of published dental literature. BDJ Open 9, 13 (2023). https://doi.org/10.1038/s41405-023-00139-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41405-023-00139-7
