
Abstract
Ebola virus (EBOV) causes hemorrhagic fever in humans with high morbidity and fatality. Although over 45 years have passed since the first EBOV outbreak, small molecule drugs are not yet available. Ebola viral protein VP30 is a unique RNA synthesis cofactor, and the VP30/NP interaction plays a critical role in initiating the transcription and propagation of EBOV. Here, we designed a high-throughput screening technique based on a competitive binding assay to bind VP30 between an NP-derived peptide and a chemical compound. By screening a library of 8004 compounds, we obtained two lead compounds, Embelin and Kobe2602. The binding of these compounds to the VP30-NP interface was validated by dose-dependent competitive binding assay, surface plasmon resonance, and thermal shift assay. Moreover, the compounds were confirmed to inhibit the transcription and replication of the Ebola genome by a minigenome assay. Similar results were obtained for their two respective analogs (8-gingerol and Kobe0065). Interestingly, these two structurally different molecules exhibit synergistic binding to the VP30/NP interface. The antiviral efficacy (EC50) increased from 1 μM by Kobe0065 alone to 351 nM when Kobe0065 and Embelin were combined in a 4:1 ratio. The synergistic anti-EBOV effect provides a strong incentive for further developing these lead compounds in future studies.
Similar content being viewed by others

Introduction
Ebola virus (EBOV) is a pathogen in the Filoviridae family [1, 2]. EBOV causes a rapidly progressing hemorrhagic fever named Ebola virus disease (EVD). This highly infectious disease causes high fatality in humans and nonhuman primates [3]. Since the first outbreak in 1976, the Ebola virus has continued to disrupt people’s normal lives and caused numerous outbreaks. The most serious and complex EBOV epidemic in West Africa during 2013–2016 led to more than 28,000 cases and more than 11,000 deaths [4], and some cases were imported to non-West African countries. These multiple outbreaks emphasized that Ebola not only threatens global public health but also threatens society and the economy. According to statistics from the World Bank, the EVD epidemic caused a direct loss of US$2.2 billion in GDP in the three West African countries in 2015. In addition, the epidemic led to a large reduction in regional investment, agricultural production, international trade and tourism [5]. Although antibodies Inmazeb and Ebanga have been approved by the FDA, small molecular therapeutics remain unavailable [6, 7]. In clinical practice, oral rehydration or intravenous infusion and other supportive therapies are the main tools employed to improve the survival rate.
Ebola virus is a single, negative-stranded RNA virus, and its genome encodes one nonstructural and seven structural proteins. The seven structural proteins include the nucleoprotein (NP), the virion protein (VP24), VP35, VP30, VP40, the transmembrane glycoprotein (GP) and the RNA-dependent RNA polymerase (L) [8]. In addition to its structural role, VP30 is uniquely essential for RNA synthesis, unlike Marburg virus [9]. As a result, the Ebola VP30 protein is a potential drug target [10, 11].
Previous studies found that VP30 directly interacts with nucleoprotein (NP), VP35 and RNA-dependent RNA polymerse (L) [12, 13]. Additionally, EBOV VP30 contains an RNA binding domain associated with viral RNA transcription and RNA synthesis machinery [14]. EBOV VP30 is critical for viral transcription (mRNA synthesis) because it is necessary for initiation at EBOV NP, the first gene of the seven gene genomes [15]. VP30 is also involved in regulating the cotranscriptional editing of viral glycoprotein mRNAs and in the modulation of viral transcription reinitiation [16, 17]. Previously, we determined the complex structure between an NP-derived peptide and VP30, indicating that NP binds to a shallow hydrophobic cleft on VP30 [12]. Here, we established a drug screening method that targets this specific “molecular pocket”. After screening a small-molecule compound library, we discovered two compounds, Embelin and Kobe2602, that can disrupt the VP30 and NP interaction. Those compounds and their analogs exhibit anti-Ebola virus activities when employed alone or in combination.
Materials and methods
Cell culture
Huh7-4P cells (obtained from Institut Pasteur of Shanghai, CAS) were cultured in complete growth medium containing Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 g/mL streptomycin (P/S) and 0.1 mg/ml hygromycin at 37 °C in 5% CO2.
Plasmid constructs
The fusion gene MBP-VP30110–272 was synthesized by Genscript and then inserted into the pET28 vector by overlap PCR. The resulting ligation product was verified by sequencing before use.
Protein expression and purification
Ebola VP30110–272 protein was expressed as a maltose-binding protein (MBP) fusion protein in BL21(DE3) Escherichia coli cells in lysogeny broth medium. After the cells were cultured with shaking at 37 °C for 4 h, the optical density of OD600 reached approximately 0.6–0.7. At this point, 0.2 mM isopropylthiogalactoside (IPTG) was added to induce protein expression, and bacterial cultures were grown for 16 h at 16 °C. The bacterial cells were harvested by centrifugation and then resuspended in lysis buffer containing 20 mM Tris-HCl at pH 7.5, 150 mM NaCl, and 5 mM 2-hydroxy-1-ethanethiol (β-ME). The resuspended cells were crushed with an ultrasonic crusher and clarified by centrifugation at 20,000g at 4 °C for 30 min.
The VP30110–272 protein was purified by using multiple affinity and ion exchange chromatographic columns. The supernatant was loaded onto Ni-NTA resin, and the elution from Ni-NTA resin was purified by ion exchange chromatography. The MBP tag was cleaved using TEV protease. SDS‒PAGE was used to verify the removal of the MBP tag. The product was purified by Ni-NTA resin column again to remove the MBP protein and collect the target protein. Finally, the protein was loaded on a size-exclusion column (Superdex 200, Cytivas) with buffer containing 20 mM Tris-HCl (pH 7.5) and 150 mM NaCl. The protein purity was determined by SDS‒PAGE and concentrated by a 3 kDa MWCO concentrator (Millipore).
Compound library
The compound library used for high-throughput screening was obtained from TOPSCIENCE (Shanghai, China). This library consists of 8004 small-molecule compounds with diverse structures and pharmacological activities, including clinically approved drugs, natural products, and enzyme inhibitors. All compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM.
High-throughput screening (HTS)
We employed a fluorescence polarization assay (FPA) for high-throughput screening. FPA was performed on a Tecan Spark instrument. NP polypeptide labeled with FITC fluorescein, namely, NP-FITC, was synthesized by Genscript. In this assay, 50 μl of NP-FITC peptide at a final concentration of 0.5 μM was loaded into 50 μl of VP30110–272 protein samples at different concentrations at a 2.5-fold dilution (0.031–120 μM) with pH 7.0 buffer containing 10 mM HEPES, 150 mM NaCl, and 2 mM DTT in a 96-well plate. After 10 min of incubation, FP signals were measured at an excitation wavelength of 485 nm and an emission wavelength of 528 nm, with a bandpass of 20 nm. The read height and G factor were set to 8.5 mm and 1.26, respectively, using the autogain function.
In the competitive FP assay, the unlabeled NP polypeptide was employed as a competitive inhibitor of NP-FITC. Fifty microliters of 0.78 μM VP30110–272 protein was incubated with 25 μl of unlabeled NP peptide at various concentrations from 0.048 to 100 μM for 30 min. Then, 50 μl of NP-FITC was loaded into each well at a final concentration of 0.3 μM for a 10 min incubation. The FP signals were measured by a Tecan Spark instrument using the same parameters as above.
For the HTS experiments based on FPA, 1 μl of testing compounds at a final concentration of 100 μM was mixed with 50 μl of 15.36 μM VP30110–272 protein samples in a 96-well plate. Incubation was performed at least 10 min before 50 μl of 600 nM NP-FITC peptide was loaded into each well of a 96-well plate. After 30 min of incubation, FP signals were read. Data were normalized to signals from the negative control (DMSO only), and a composite evaluation of inhibition rate and Z score was applied for locating the hits. The inhibition rate can be defined as the percentage of effectiveness of a compound. Meanwhile, the Z score can be described as a value to evaluate the fluorescence polarization normal distribution of the compound. In the results of this experiment, a |Z| value greater than 4 indicates that the fluorescence polarization of a sample was at both ends of the normal distribution. This means that the drug either exhibited no effect or was relatively effective. Z scores were calculated to judge the quality of the tested compounds [18]. The active compounds selected by a combination analysis of inhibition rate and Z score were reformatted into a new 96-well plate and tested again with concentration gradient experiments to confirm the results of high-throughput screening. The hit drugs were serially diluted twofold for dose response titration, and the data were analyzed with GraphPad Prism 8.0.2 to determine the IC50 values [19].
Thermal shift assay (TSA)
A thermal shift assay (TSA) using SYPRO Orange fluorescent dye was carried out first to assess the thermal stability of the VP30110–272 protein when binding with potential compounds under various pH conditions. In this assay, 10 μl of 15.6 μM testing compounds was mixed with 10 μl of 13 μM VP30110–272 protein in various pH buffers (molar ratio of compound to protein was 1.2:1). After 10 min of incubation, 1 μl of SYPRO Orange fluorescent dye was loaded into each sample. Two groups of controls were set up in the experiment. VP30110–272 protein and the same amount of DMSO were added to the buffer corresponding to the pH conditions to replace the compound as a negative control; the compounds and the same amount of buffer were added as a blank control. All treatments were performed in triplicate. After 10 min of coincubation, 1 μl of SYPRO Orange fluorescent dye was added to each sample. The change curves for the melting temperature (Tm) were measured on an ABI real-time fluorescence quantitative instrument. The instrument parameters were set as follows: the temperature was increased from 25 °C to 95 °C at a rate of 1 °C/min and held at 95 °C for 3 min.
Surface plasmon resonance (SPR)
The binding capacity and kinetics between potential small molecules and VP30110–272 protein were determined with Plexera PlexArray™ HT based on surface plasmon resonance (SPR) technology. Prior to the binding experiments, small molecule compounds with different concentrations were labeled on the 3D optical crosslinking chip according to the chip manual. PBS was used as the binding buffer, and 0.01 M Gly-HCl (pH 2.0) was the regeneration buffer. VP30 protein was diluted to various concentrations with PBS as the mobile phase at 5.00, 2.50, 1.25, 0.62, and 0.31 μΜ. All experimental operations were performed according to the protocol provided for the instrument. The assay method was set as follows: 300 s baseline, 300 s association, 300 s dissociation. For each baseline step throughout the experiment, fresh PBS buffer was used. The whole data collection process was automated and consisted of recording successive cycles of measurement. Each independent analysis cycle included baseline, association, dissociation, regeneration and return to baseline steps [20]. Data were processed with the Plexera analysis program. The data collected by the instrument were plotted with GraphPad Prism 8.0.2 according to the corresponding relationship between time and intensity, which is the association and dissociation response curve. In addition, the affinity constant (KD) was also fitted by BIAeval software.
Minigenome (MG) assay
EBOV minigenome were carried out to validate the antiviral activity. The minigenome system for EBOV replicon reconstitutes EBOV polymerase activity in Huh7 cells and can stably replicate and transcribe the viral genome [21]. The system is used as a genome model of Ebola virus for antiviral activity assays. Prior to the MG assay, Huh7-4P cells were seeded in 96-well plates at a density of 1 × 104 cells/well overnight in 100 μl of DMEM containing 10% (v/v) FBS and 0.1 mg/ml hygromycin. The cells were treated with effective drugs at various concentrations (0.097-200 μM), which were diluted with 100 μl of serum-free medium. Plates were incubated at 37 °C and 5% CO2 for 48 h prior to assessing luciferase activity using the Renilla-LumiTM, Luciferase Reporter Gene Assay Kit (Beyotime, China). Huh7-4P cells treated with PBS were used as a negative control. All treatments were performed in triplicate. Data analysis and IC50 values of the samples were performed in GraphPad Prism 8.0.2.
Cytotoxicity assay
The cytotoxicity of drugs to cells was also examined on the EBOV minigenome system and detected by Cell Counting Kit-8 (CCK-8) (Topscience, Shanghai). The drug treatment procedure was similar to that of the MG assay. Briefly, 1 × 104 Huh7-4P cells per well were grown overnight in 96-well plates at 37 °C. A total of 100 μl of serum-free medium containing the indicated concentrations of drugs was added to the cells. After 48 h of incubation, 10 μl of CCK-8 solution was mixed into each well 4 h prior measuring the absorbance at 450 nm on a microplate reader (Biotek, USA). Data analysis and CC50 values of the samples were performed in GraphPad Prism 8.0.2.
Pull-down assays
A pull-down assay was performed in a buffer containing 20 mM Tris-HCl (pH 7.5), 200 mM NaCl, 1 mM EDTA and 1 mM DTT at 25 °C. MBP-His-tagged VP30110–272 protein was incubated with purified NP600 protein for 1 h with MBP beads as a control group. For small-molecule competition assays, MBP-His-tagged VP30110–272 protein was incubated with a final concentration of 500 μM small molecule compound prior to incubation with NP600 protein. The same procedure was performed for inhibitors different from those in the experimental groups. To wash away unbound material, beads were subsequently eluted with buffer containing 20 mM Tris-HCl (pH 7.5), 200 mM NaCl, 1 mM EDTA, 1 mM DTT and 20 mM maltose. Each component sampled was verified by SDS‒PAGE.
Coimmunoprecipitation assays
The full-length viral plasmids of HA-VP30 and FLAG-NP were reconstructed. Forty-eight hours posttransfection with plasmids, HEK293T cells were lysed in NP-40 lysis buffer containing 50 mM Tris-HCl (pH 7.5), 250 mM NaCl, 0.5% Nonidet P-40, 1 mM EDTA, 10% glycerol, and protease inhibitor (Beyotime) for 30 min at 4 °C. Anti-HA beads (Beyotime) were incubated with the above lysates for 1 h at 4 °C. For small-molecule competition assays, an additional 500 μM of different small molecule inhibitors was added and incubated in the same way. Following washing eight times in wash buffer (50 mM Tris-HCl (pH 7.5), 250 mM NaCl, 1 mM EDTA, 10% glycerol), the final beads were then boiled in PBS and Western blotted with mouse anti-FLAG antibody and anti-HA antibody, followed by horseradish peroxidase-conjugated goat anti-mouse antibody (Bioss).
Synergy calculation for compound combination
The antiviral activities of the compound combination were equal to those obtained by the minigenome method previously described. One day prior to compound treatment, 1 × 104 Huh7-4P cells were seeded in each well of a 96-well plate. After 24 h, the drugs diluted with 100 μl of serum-free medium were added into the cells in a 7 × 7 matrix for 7 doses (49 combinations in total). An equal volume of PBS was utilized as a negative control. Plates were then incubated at 37 °C and 5% CO2 for 48 h prior to assessing luciferase activity using the Renilla-LumiTM, Luciferase Reporter Gene Assay Kit (Beyotime, China).
Before calculating the synergy scores with SynergyFinderPlus, the dose‒response data are represented in a table format where each row represents one observation in the dose‒response matrix. Then, the resulting dose‒response matrix data were analyzed with SynergyFinder for the detection of synergistic drug combinations. The results provided the dose‒response curve, dose‒response map, and synergy scores of four major models, including HSA, Bliss, Loewe and ZIP. The synergistic antiviral effect of each combination was found to be significantly better than the monotherapy effect of each drug. The proportions of drugs with high synergy scores under different models were selected, and the antiviral activities were revalidated in the minigenome system. To quantify the degree of compound interactions, the combination index (CI) was calculated by using the Compusyn program to assess the synergistic effect of each drug combination. In this study, the 50% inhibitory concentration (IC50) was used as the combination index of the synergistic effect. According to the values of CI, they were divided into strong synergies, moderate synergies, and weak synergies.
Synergy SPR for compound combination
The synergy SPR here is basically equivalent to that of the SPR approach described above. Two different chips must be prepared to solidify the small molecules and the protein. The basic idea of the experiment is similar to the sandwich binding method. In the first method, small molecule A is used as the stationary phase, VP30 is used as the first mobile phase to associate with A, and then small molecule B is used as the second mobile phase to associate with VP30, and the change trend of the binding curve is observed. The second method is to inversely monitor the binding curves by using VP30 protein as the stationary phase and then using different small molecules as the mobile phase in turn. The whole process includes baseline, association, reassociation and dissociation, regeneration and return to baseline. Compared with ordinary SPR, competitive SPR is equivalent to the data combination of two independent cycles, but there is no dissociation and regeneration step between the two cycles.
Preparation of protein and ligands for docking and molecular dynamics simulations
The 3D structure of VP30 (PDB ID: 5vao, chain ID: B) was downloaded from the PDB database. Since there are some missing atoms and alternative atoms in the protein 3D structure, we used DeepViewer [22] and MGLTools [23] to process it for docking preparation. We employed Open Babel [24] and MGLTools [23] to process the ligand structures of Embelin (CID: 3218), 8-gingerol (CID: 168114), Kobe0065 (CID: 3827663) and Kobe2602 (CID: 3827738) downloaded from PubChem. These procedures are similar to those employed previously [25].
Docking and molecular dynamics simulations
After preparing the protein and ligands, we docked each ligand to the protein with AutoDock Vina [26]. For each ligand, AutoDock Vina reported 9 possible binding conformations. We treated each conformation as a candidate conformation for further molecular dynamics analysis. The topologies of the protein and ligands were prepared by GROMACS [27] and ACPYPE [28] with the Amber recommended force field (ff19SB for proteins and gaff2 for ligands [29, 30], respectively). The system with a dodecahedron box extending 1 nm from the solute and other parameters were similar to those employed previously by us [25]. All simulations set up with the running parameters were performed for at least 200 ns on the high-performance cluster of Shenzhen Bay Laboratory.
Antibodies
Mouse anti-HA antibody and anti-FLAG antibody were purchased from Abclonal Technology. Horseradish peroxidase-conjugated goat anti-mouse antibody was purchased from Bioss.
Results
High-throughput screening of small compounds targeting EBOV VP30
Expression and Purification of VP30
The sequences of VP30110–272 were codon-optimized and harbored an N-terminal MBP tag, and a 6 × His tag and TEV digestive sequence were inserted between MBP and VP30. The recombinant plasmid pET28a-MBP-VP30110–272 was transformed into E. coli to express the VP30110‒272 protein. After the cells were crushed and centrifuged, the supernatant was successively purified by nickel column affinity, anion exchange, nickel column affinity and size exclusion chromatography (SEC) to high purity. The analysis of SEC and SDS‒PAGE confirmed the purity of expressed VP30 with a molecular weight of 18.3 kDa (Fig. 1a).

a Purification profile from size exclusion chromatography on a Superdex 200 column and SDS-PAGE with EBOV VP30110-272 at a molecular weight of 18.3 kDa. b The schematic diagram to illustrate the principle of fluorescence polarization (FP) assay and competitive fluorescence polarization-based assay for screening inhibitors that compete with NP-derived peptide for binding with VP30. Unbound fluorescent labeled NP peptide (NP-FITC) has a rapidly rotating of small molecule fluorophore and gives a low FP signal. The binding of NP-FITC with a large VP30 protein slows down the rotation of the fluorophore, leading to an increased FP signal. The competitive binding to other small molecules to VP30 will free NP-FITC from binding and result in a decreased FP signal. c Fluorescence polarization curves given by VP30 in the presence of NP-FITC at different concentrations (0.031–120 μM). d Fluorescence polarization curves at 300 nM of NP-FITC mixed with various concentrations of the VP30 protein. e Fluorescence polarization changes in competition by adding unlabeled NP peptide in the presence of 300 nM NP-FITC and 7.68 μM of VP30. f Compound libraries of 8004 compounds at 100 μM were screened against VP30-NP binding by fluorescence polarization (FP) assay. Z scores indicate the prominence of potential inhibitors discovered. The percentage of effect is the inhibition rate of the interface activity of VP30/NP by the compounds.
Competitive binding assay between the NP-derived peptide and small molecules for binding to VP30
To screen for small molecules binding to the same interaction pocket between NP and VP30, we established a high-throughput competitive assay using fluorescence polarization (FP). The binding competition between the fluorescently labeled NP-derived peptide and a small molecule can be monitored by changes in polarization and measured by the generation of a fluctuating FP signal [31] (Fig. 1b). EBOV VP30 binding peptide (NP) was conjugated to fluorescein 5-isothiocyanate (FITC), which could bind to VP30 and lead to a slower tumbling speed, resulting in an increase in fluorescence. When the tested compounds competitively bind to the VP30 protein, the fluorescent small molecule NP-FITC is in a free state, and the rotation speed becomes faster, resulting in a decrease in the fluorescence signal. To establish this assay, we first determined the optimal concentration for fluorescein 5-isothiocyanate (FITC)-labeled NPs (NP-FITC). Using various concentrations of NP-FITC as the substrate and VP30 as the target protein, 300 nM NP-FITC was selected as the optimal concentration in this assay (Fig. 1c). The trend observed for the curve is better under this condition, and the concentration of NP-FITC is moderate. Then, we determined the optimal concentration of VP30 after using various concentrations of VP30 protein to react with 300 nM NP-FITC, and the IC50 was 3.07 μM (Fig. 1d). The optimal concentration of VP30 was determined to be 7.68 μM by concentration titration, which is one gradient value above the IC50 value as the optimum concentration. Afterward, we employed unlabeled NP peptide to confirm the reliability of this competitive binding assay. A constant concentration of VP30 was incubated with increasing concentrations of unlabeled NP peptide as a competitor. After incubation, NP-FITC was added to measure the changes in FP signals, which successfully reproduced the binding between NP-derived peptide and VP30 (Fig. 1e). The IC50 value of the NP polypeptide in the competitive FP assay was 0.33 μM. The above established FP method was performed to conduct high-throughput screening for compound libraries including 8004 compounds containing clinically approved drugs and natural products. Compounds were screened at a final concentration of 100 μM in each well. The results of high-throughput screening based on the FP method are displayed in a volcano plot (Fig. 1f). In this screening experiment, compounds with Z scores less than −4 and inhibitory effect values greater than 80% were defined as potential antiviral inhibitors [18].
Confirmation of Embelin and Kobe2602 inhibitors identified from high-throughput screening
Dose-dependent competitive assay
The above screening led to a total of 70 compounds with Z scores < −4 and inhibitory effect values > 80% (Fig. 1f). Among them, we first excluded self-fluorescence interference according to the properties of the compounds and then removed those small molecules with poor medicinal properties according to the pharmacological effects. Finally, we chose 15 potential inhibitors for low-throughput dose-dependent experiments to determine their IC50 values. The Z values and inhibition rates of these compounds are moderate but do not deviate too much from the median to exclude the fluorescence interference of the drug themselves. Among them, Embelin and Kobe2602 were prominent due to their relatively strong inhibitory activity in the FP assay. Embelin is an active benzoquinone compound that is extracted from an herb of the Myrsinaceae family [32] (Fig. 2a). Kobe2602 generally serves as an effective inhibitor of the Ras signaling pathway and is a novel antitumoral agent [33, 34] (Fig. 2b). The IC50 values of Embelin and Kobe2602 from the competitive assay were 33.19 and 26.95 μM, respectively (Fig. 2c, d).

a Chemical structure of Embelin. b Chemical structure of Kobe2602. c, d The dose‒response curve of Embelin and Kobe2602 in fluorescence polarization (FP) assay. The IC50 values of Embelin and Kobe2602 are 33.19 and 26.95 μM, respectively. e, f Sensorgrams of Embelin and Kobe2602 in the SPR assay at various compound concentrations. The affinity constant (KD) of Embelin and Kobe2602 are 4.62 and 0.88 μM, respectively. g, h Tm changes of VP30 in the absence or presence of small molecule inhibitors in the thermal shift assay (TSA) at various pH. Under various pH conditions, Embelin and Kobe2602 both can specifically bind to VP30 and change its melting temperature Tm.
Binding assay
The binding between VP30 and small molecules can be confirmed by surface plasmon resonance (SPR). The binding affinity (KD) of Embelin and Kobe2602 were found to be 4.62 and 0.88 μM, respectively (Fig. 2e, f).
Thermal shift assay
TSA experiments were performed to characterize the melting temperature (Tm) of a protein after adding a potential inhibitor to further confirm the binding interaction [35]. As shown in Fig. 2g, Embelin increased the stability of VP30 at pH 6.5, 7.0, 7.5 and 8.0. Kobe2602, by comparison, can increase the stability of VP30 at all pH ranges tested (from 5.5 to 8.5) (Fig. 2h). Notably, under different pH conditions, the number of ions in the solution alters the surface charge of the protein, which affects the binding affinity between the protein and the small molecule compound and produces different changes in Tm values. Moreover, some compounds exert different effects on protein stability and decrease or increase the melting temperature (Tm) under different pH conditions. Zhao et al. reported similar results in Nature, 2016 [36].
Interaction mechanism of Embelin and Kobe2602 with EBOV VP30
To further clarify the interaction mechanism between VP30 and the small molecule discovered (Embelin and Kobe2602), we performed docking and molecular dynamics (MD). Previously, we reported that there are two key residues in the interaction interface of VP30 and NP [12]. Mutating one of these two key residues (Glu197, Trp230) to alanine will result in complete loss of binding between VP30 and NP. It is interesting to know if the binding locations of Embelin and Kobe2602 are near Glu197 and Trp230. We employed AutoDock Vina to generate initial conformations and subsequent long 200 ns MD simulations to examine the stable binding conformations. As shown in Fig. 3, the average distance between the center of mass (COM) of Embelin and the COM of the sidechain of Trp230 is approximately 6 Å, suggesting that both Embelin and Kobe2602 bind in or near the NP-VP30 binding pocket. The 2D diagrams describing the distance between small molecules and the key amino acid residues of the VP30 protein are now presented in the supplementary materials (Fig. S1).

a, b Embelin/Kobe2602 RMSD represents the heavy-atom RMSD of Embelin/Kobe2602 with the docking conformation as the reference. Embelin/Kobe2602 and Glu197/Trp230 represent the distance between the center of mass (COM) of Embelin/Kobe2602 and the COM of Glu197/Trp230 side chain. c, d The stable conformation of Embelin and Kobe2602 binding with VP30 found by MD simulations, along with the NP-derived peptides. VP30 is shown in gray, Embelin and Kobe2602 in magenta, the VP30 residues within 5 Å of NP peptide in cyan.
Inhibiting the transcription and replication of Ebola virus in the minigenome system
Due to the high lethality of EBOV, the minigenome system was used as a surrogate of the authentic virus for use in BSL-2 laboratories. The MG system can replicate for months and stably express critical key proteins of the virus life cycle. We observed that Embelin and Kobe2602 have a similar capability to inhibit virus reproduction, with EC50 values of 16.85 μM and 22.93 μM, respectively (Fig. 4a, b). Simultantously, the cytotoxicity of these drugs was negligible in Huh7-4P cells, with a 50% cytotoxic concentration (CC50 value) of 494.1 μM for Embelin and 382.5 μM for Kobe2602. Thus, these two small molecule drugs exhibit anti-Ebola virus activities, making them attractive candidates for the development of antiviral inhibitors.

a, b The inhibitory curve and cytotoxic measurement in the minigenome assay. The inhibition concentration on virus transcription and replication of Embelin and Kobe2602 (EC50) are 16.85 μM and 22.25 μM, respectively. Cell viabilities (green) were determined by CCK-8 assay. The toxicities of the two compounds are all greater than 200 μM.
Embelin and Kobe2602 analogs
Embelin analog
Based on the relationship between drug core structural frameworks and pharmacological activities, we tested a structural analog of Embelin, 8-gingerol (Fig. 5a). It has been reported that Embelin is an inhibitor of XIAP (inhibitor of apoptosis) and simultaneously modulates multiple signaling pathways, such as NF-κB, PI3K/AKT, P53 and JAK/STAT3, to induce apoptosis [32, 37, 38]. As a structural analog of Embelin, 8-gingerol can inhibit the generation of reactive oxygen species (ROS) and shows good antioxidant and anti-inflammatory effects [39]. More interestingly, in terms of the mechanism of pharmacological activity, 8-gingerol is consistent with multiple signaling pathways regulated by Embelin, which increases the credibility of its study as an analog [40]. Competitive binding assays based on fluorescence polarization indicated that 8-gingerol has an IC50 value of 61.95 μM (Fig. 5b). 8-Gingerol has a much weaker IC50 than Embelin (33.19 μM). Thus, it is not surprising that 8-gingerol showed weaker antiviral activity in the MG assay with an EC50 of 32.00 μM (Fig. 5c). The cytotoxicity of this small molecule displayed negligible toxicity to Huh7-4P cells with a CC50 of 274.50 μM for 8-gingerol. Unlike Embelin, 8-gingerol negatively affects the stability of VP30 for all pH ranges tested (Fig. 5d) with a poorer binding affinity at 17.90 μM, compared to 4.62 μM for Embelin (Fig. 5e). This reduction in binding affinity may be associated with the reduction of the carbonyl groups on the benzene ring in 8-gingerol.

a Chemical structure of 8-gingerol, compared to Embelin. b The inhibitory curve and cytotoxic effect of 8-gingerol in minigenome assay. The inhibition activity of 8-gingerol (red) are 32.00 μM. Cell viabilities (green) were determined by CCK-8 assay. The toxicity of 8-gingerol is greater than 200.00 μM based on CC50. c The dose-response curve of 8-gingerol in the competitive fluorescence polarization (FP) assay. The IC50 of 8-gingerol is 61.96 μM. d Changes in melting temperatures (Tm) of VP30 in the absence or presence of small molecule inhibitors in the thermal shift assay (TSA) at various pH. Unlike Embelin, 8-gingerol destabilizes, rather than stabilizes VP30. e Sensorgrams of 8-gingerol in the SPR assay at different compound concentrations. The affinity constant (KD) of 8-gingerol is 17.90 μM.
Kobe2602 analog
We also examined a structural analog of Kobe2602, Kobe0065 (Fig. 6a). Kobe2602 and Kobe0065 are both inhibitors of Ras proteins that were discovered by structure-based molecular drug design in silico [33]. Competitive binding assays based on fluorescence polarization indicated that Kobe0065 has an IC50 value of 29.57 μM (Fig. 6b), a value similar to that of Kobe2602 (26.95 μM). However, Kobe0065 exhibited a weaker binding affinity to VP30 (KD = 65.60 μM) than that of Kobe2602 (0.88 μM) (Fig. 6c). Kobe0065 caused a similar stabilization effect on VP30 in TSA analysis across all pH ranges (Fig. 6d). Despite a similar IC50 for the competitive binding assay, similar stabilization effect, and weaker binding affinity to VP30, Kobe0065 exhibits a much stronger MG activity with an EC50 of 1.33 μM compared to 22.25 μM for Kobe2602 (Fig. 6e). Kobe0065 is also relatively nontoxic, with a CC50 of 278.20 μM.

a Chemical structure of Kobe0065, compared to Kobe2602. b The inhibitory curve and cytotoxic effect of 8-gingerol in minigenome assay. The inhibition activity of Kobe0065 (red) are 1.33 μM. Cell viabilities (green) were determined by CCK-8 assay. The toxicity of Kobe0065 is greater than 200.00 μM based on CC50. c The dose‒response curve of 8-gingerol in fluorescence polarization (FP) assay. The IC50 of Kobe0065 is 29.57 μM. d Changes in melting temperatures (Tm) of VP30 in the absence or presence of small molecule inhibitors in the thermal shift assay (TSA) at various pH. Similar to Kobe2602, Kobe0065 stabilizes VP30. e Sensorgrams of Kobe0065 in the SPR assay at different compound concentrations. The affinity constant (KD) of Kobe0065 is 65.60 μM.
Compounds that disrupt the VP30-NP interaction in vitro
We tested the abilities of selected small molecule inhibitors to block the binding of VP30 and NP using purified MBP-His-VP30110–272 and NP600 proteins by pull-down assays with MBP beads [41]. As shown in Fig. S2, selected compounds can effectively abolish or attenuate the NP-VP30 interaction. In small-molecule competition assays, different small molecule inhibitors competitively bind to the VP30 protein; as a result, NP proteins are in a free state and washed out. Thus, there was no corresponding NP protein band in the final beads. We further reconstructed the full-length viral plasmids of HA-VP30 and FLAG-NP. Coimmunoprecipitation experiments with anti-HA beads were performed on lysates of HEK293T cells cotransfected with plasmids of HA-VP30 and FLAG-NP [42], Western blotted with mouse anti-FLAG antibody and anti-HA antibody, followed by horseradish peroxidase-conjugated goat anti-mouse antibody. Different levels of FLAG-NP noted in the control group relative to experimental groups may reflect inhibition levels (Fig. S3). Selected compounds can abolish or attenuate the NP-VP30 interaction with full-length viral proteins according to a coimmunoprecipitation assay.
Synergy of inhibitor combinations in inhibiting Ebola virus transcription and replication
Drug combination therapeutic strategies aim to reduce toxicity, enhance efficacy and improve selectivity. These strategies play an important role in antiviral therapies [43]. In the drug combination experiment, we combined two structural types of compounds to evaluate the synergistic effect of their antiviral effects. The synergy scores of two-drug combinations were calculated using the SynergyFinder tool [44, 45]. Considering the antiviral EC50 of the four drugs alone in the minigenome system, we established a drug cross design to evaluate the synergies and sensitivity of antiviral activity of the two drug combinations. Huh7-4P cells were treated with increasing doses of combination drugs in a 7 × 7 concentration checkerboard format [46, 47]. To fully understand the synergy of the drug combination, avoid the calculation deviation caused by a certain calculation mode, HSA, Bliss, Loewe and ZIP synergy metrics were compared for combinations and single agents calculated with the SynergyFinder tool [48].
The dose‒response map of Embelin and Kobe2602 is shown in Fig. 7a. The abscissa and ordinate represent the concentration gradients of the two drugs, respectively, and each grid contains two drugs at different doses. The darker the color, the higher the synergy score. In the experimental results, we observed the dose and corresponding antiviral responses of Embelin and Kobe2602 alone or in combination. In the presence of Kobe2602, Embelin can reach or even exceed its highest response at lower concentrations. Similarly, due to Embelin, Kobe2602 could produce a higher response at lower doses. Fig 7b further shows that the toxicity of the combination on Huh7-4P cells continues to show low toxicity. Loewe in 3D showed an obvious synergistic effect in Fig. 7c. Four synergy metrics of HSA, Bliss, Loewe and ZIP are also reported in Fig. S4a–d and provided a consistent trend. Notably, in different calculation models, Embelin and Kobe2602 show the highest synergistic effects in the condition of 1:1 or 1:2. These results provided a reference for our follow-up studies on the antiviral synergistic effects of combining the two drugs.

a The dose–response table of Embelin and Kobe2602 mixture in a 7 × 7 dose matrix with a series of one blank and six half-dilution concentrations. b Cytotoxicity assay of the drug combination of Embelin and Kobe2602. Cell viability matric shows that the two drug combinations have low cytotoxicity. c The synergistic score of Embelin and Kobe2602 mixture in Loewe 3D matrix. d The dose response table of Embelin and Kobe0065 mixture in a 7 × 7 dose matrix with a series of one blank and six half-dilution concentration. e Cytotoxicity assay of the drug combination of Embelin and Kobe0065. Cell viability matric shows that the two drug combinations have low cytotoxicity. f The synergistic score of Embelin and Kobe0065 mixture in Loewe 3D matrix.
We also examined the synergistic effect when Kobe0065 was used in combination with Embelin. In the dose‒response table, Kobe0065 at 1.56 μM and Embelin exhibited excellent antiviral responses at concentrations greater than 0.78 μM (Fig. 7d). For example, at a concentration of Kobe0065 of 3.12 μM, the inhibition rate increased by 10 times with the addition of Embelin. It should be mentioned that the combination of these drugs caused no apparent cytotoxicity to Huh7-4P cells; even at the maximal concentration ratio conditions, these two drug combinations exhibited low cytotoxicity (Fig. 7e). Loewe in 3D for Embelin and Kobe0065 showed a prominent synergistic effect in Fig. 7f. This highest score is better than the combination of Embelin and Kobe2602. Notably, the combination of Embelin and Kobe0065 provided a better synergy score at low concentrations, while the synergy at high concentration ratios was not obvious (Fig. S4e–h). These results highlight the advantages of the synergistic effect when the two drugs are used in combination, that is, the maximum therapeutic effect is achieved with the smallest dose.
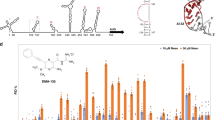
We optimized the drug concentration ratios based on the synergy scores obtained above and calculated the drug combination index (CI) under various concentration ratios using CompuSyn software [49]. When the inhibition rate was 50%, the combination index of Embelin and Kobe2602 and that of Embelin and Kobe0065 at various concentration ratios were less than 1, indicating that they produced synergistic effects with each other. Synergies were divided into different intensities according to CI values, in which CI values between 0.1–0.3 represented strong synergies, 0.3–0.7 represented moderate synergies, and 0.8–0.9 indicated weak synergies [50]. According to the calculated CI values and the synergy scores obtained in SynergyFinderPlus, we chose the optimal concentration ratios of each drug combination. After that, the inhibitory effects and antiviral effects of several drug combinations with the selected optimal concentration ratios were verified on the fluorescence polarization (FP) system and minigenome system, respectively (Fig. 8). In the FP assay, the IC50 of Embelin alone was 33.19 μM. The IC50 value was reduced to 21.89 and 2.68 μM when Embelin was combined with Kobe2602 at a ratio of 1:2 and with Kobe0065 at a ratio of 1:4, respectively (Fig. 8a, b). Similarly, the IC50 values of 8-gingerol decreased from 61.96 μM to 22.54 μM and 3.15 μM when combined with Kobe2602 (1:2) and Kobe0065 (1:2), respectively (Fig. 8c, d). The IC50 of Kobe0065 decreases from more than 20 μM to 2–3 μM when used in combination with Embelin at a ratio of 4:1. A large decrease in the IC50 is summarized in Fig. 8i. The reduction in IC50 when combining Kobe0065 with Embelin or 8-gingerol is larger than when Kobe2602 is combined with the two inhibitors.

a–d Competitive FP assays of the drug combinations of Embelin and Kobe2602 at the ratio of 1:2, Embelin and Kobe0065 at the ration of 1:4, 8- gingerol and Kobe2602 at the ratio of 1:2 and 8-gingerol and Kobe0065 at the ratio of 1:2 on the VP30/NP binding interface. IC50 values are 21.89, 2.68, 22.54, and 3.15 μM, respectively. e–h Different drug combinations inhibitory activities of the transcription and replication of Ebola virus in minigenome assay. EC50 of Embelin and Kobe2602 (1:2) in mixture are 2316 and 463.7 nM respectively. EC50 of Embelin and Kobe0065 in mixture (1:4) are 98.80 and 395.10 nM respectively. EC50 of 8-gingerol and Kobe2602 (1:2) in mixture are 759.80 and 1520.00 nM respectively. EC50 of 8-gingerol and Kobe0065 (1:2) in mixture are 350.60 and 701.20 nM respectively. i Comparison IC50 given by each compound administered alone and in combination with another compound as labeled in FP assay. Embelin:Kobe2602 (1:2), Embelin:Kobe0065 (1:4), 8-gingerol:Kobe2602 (1:2), Embelin:Kobe0065 (1:2). j In MG assay, comparison of the EC50 when each compound administered alone or with another compound. Embelin:Kobe2602 (1:2), Embelin:Kobe0065 (1:4), 8-gingerol:Kobe2602 (1:2), Embelin:Kobe0065 (1:2).
We further examined the synergistic effect of using two compounds by employing the MG assay. When combining Embelin with Kobe2602 (1:2) or Kobe0065 (1:4), the EC50 values decreased from 16.85 μM to 2316 nM and 98.80 nM, respectively (Fig. 8e, f). After mixing 8-gingerol with Kobe2602 or Kobe0065 in a 1:2 proportion, the EC50 was reduced from 32 μM to 759.8 and 350.6 nM, respectively (Fig. 6g, h). The combined use of two compounds in MG analysis is summarized in Fig. 8j, indicating a large reduction in EC50 (as high as 90-fold) occurred. Simultaneously, the antiviral effects of Kobe2602 and Kobe0065 in combination with Embelin or 8-gingerol were better than those obtained with these compounds as single agents, and their EC50 values were all significantly reduced. For example, when combining Kobe2602 with Embelin (2:1) or 8-gingerol (2:1), the EC50 values of Kobe2602 decreased from 22.25 μM to 463.70 nM and 1520 nM, respectively (Fig. 8e, g). The drug combination of Kobe0065 with Embein and 8-gingerol decreased its EC50 from 1.33 μM to 395.1 nM and 701.2 nM, respectively (Fig. 8f, h). In the anti-Ebola virus experiment, the advantages of combined drugs were more prominent. The combined use of each drug combination in MG analysis resulted in several-fold or even dozens-fold improvement in antiviral efficacy (Fig. 8j).
To further confirm that binding to VP30 was synergistic rather than competitive between the two structural types found in this work (Embelin and Kobe2602), we performed the SPR binding assay as before but with a small-molecule chip as the stationary phase along with VP30 and another small molecule as the first and second mobile phases to bind in sequence. The observed changes in binding curves are shown in Fig. S5a. Embelin or 8-gingerol was first bound to VP30. The addition of Kobe2602 or Kobe0065 leads to a further increase in binding (Figure S5a). We also tested using VP30 as the stationary phase of the chip and another compound, GW0742, as a control. GW0742 is also a small molecule inhibitor targeting VP30 that we screened in the FP assay. GW0742 also exhibits good antiviral activity in the MG assay; however, GW0742 does not exhibit obvious synergistic effects in combination with other similar drugs. The results showed that VP30 could still efficiently bind to Kobe2602 and Kobe0065 after binding to Embelin or 8-gingerol (Fig. S5b). However, a reduction in binding was observed when GW0742 was added (Fig. S5b). This clearly confirmed the additive effect of Embelin or 8-gingerol with Kobe2602 or Kobe0065.
Discussion
This study is based on the previous discovery that the interaction interface between VP30 and NP can directly affect the synthesis of Ebola virus RNA [12]. We targeted this specific molecular interface by high-throughput screening. To do that, we established a high-throughput FP assay to examine the competitive binding of VP30 to either the NP-derived peptide or to small molecules. We employed a library of more than 8000 compounds that include natural products, clinically approved drugs, and synthetic small molecules.
Two molecules stood out after the screening, which were Embelin and Kobe2602. The binding of these compounds to VP30 was further confirmed by low-throughput SPR binding assay and thermal shifting assay. More importantly, both displayed an ability to inhibit virus transcription and expression, as determined by a minigenome assay, with EC50 values of 16.85 and 22.25 μM, respectively. We further examined two analogs, 8-gingerol and Kobe0065, for Embelin and Kobe2602, respectively. These analogs are also inhibitory, with Kobe0065 having the best EC50 of 1.33 μM.
We performed MD simulations and confirmed that Embelin and Kobe2602 bind with VP30 in a pocket overlapping with the binding pocket of the NP-derived peptide. More importantly, the binding pockets of Embelin and Kobe2602 do not overlap as much. Thus, we performed a synergistic binding assay and found that a combination of Embelin with Kobe0065 at a mixing ratio of 1:4 achieved the best EC50 of 98.8 nM, an approximately 1 order of magnitude reduction from prior to mixing. Synergic, rather than competitive, binding between Embelin and Kobe2602 (or Kobe0065) was confirmed by SPR experiments.
Embelin is a derivative of the natural antioxidant coenzyme Q10, and the presence of two phenolic hydroxyl groups on the benzoquinone ring may increase its antioxidant activity. As a multiple-targeted therapeutic agent, Embelin has been shown to possess a wide spectrum of biological activities, such as antitumoral, anti-inflammatory, antioxidant, anti-infectious, and antithrombotic activities [32, 37, 38, 51]. Kobe0065 is an inhibitor of Ras proteins with excellent antitumor activity. Here, we found that these two compounds exhibit new antiviral activities. When used in combination, the compounds have a potent 100 nM for 50% reduction of Ebola virus transcription and replication. Together, these results provide a comprehensive approach to effectively identify inhibitors acting on the interface of VP30/NP (Fig. 9). Our results provide a new scaffold for further optimizing specificity to fight against the lethal Ebola virus.

When inhibitors or synergistic drug combinations are applied to EBOV, the inhibitors can competitively insert into the molecular pockets of VP30, thereby blocking the binding of VP30 and NP, effectively reducing the transcription and replication of the EBOV genomes.
In this manuscript, Embelin and Kobe2602 were identified to target the VP30/NP interaction interface. Several biochemistry methods were adopted to validate the VP30/NP interface as the target for these compounds, and their antiviral activities were confirmed in the Ebola minigenome system. However, because testing work with Ebola live viruses is restricted to maximum containment laboratories (BSL4), we cannot access laboratories capable of conducting tests with authentic Ebola viruses at present. As a result, it is not possible to test the inhibitory activity in the context of authentic virus infection. The use of the minigenome system to replace the live Ebola virus test is an international evaluation method that has been proven to have high consistency with a live virus test. It is a powerful tool for studying the biology of Ebola viruses as well as screening and developing antiviral drugs in secondary laboratories [52, 53].
One potential issue of this work is the lack of a positive control. However, due to the lack of known small molecule inhibitors targeting the VP30 protein of Ebola virus, we used remdesivir, an RNA-dependent RNA polymerase inhibitor for Ebola virus, as a positive control in this manuscript. The antiviral activity of remdesivir on the Ebola minigenome system is now provided in the supplementary material of Fig. S6, validating the performance of our established minigenome system. For negative controls, 2,4-dihydroxypyrimidine-5-carboxylic acid, which fell into the effective zone in the HT FP drug screening assay but failed in the FP concentration gradient experiment, was selected. The FP assay, binding test (TSA) and MG system-based antiviral assays and cytotoxicity test were performed. These negative control experiments are now presented in Figure S8.
In this paper, we chose the EBOV minigenome over traditional transient transfection. This is because the latter requires six plasmids to be transferred into a cell at the same time. Such a complicated operation demands high transfection efficiency and a precise ratio. The replication level is often low, which mainly reflects the transcription of the virus [54]. In contrast, the stable EBOV minigenome can maintain stable replication and transcription in Huh7-4P cells for several months, which is a cell model that can better mimic the transcription and replication of Ebola virus [21]. The technology can also provide a more effective alternative tool for studying EBOV replication and drug screening in BSL2 laboratories.
References
Messaoudi I, Amarasinghe GK, Basler CF. Filovirus pathogenesis and immune evasion: insights from Ebola virus and Marburg virus. Nat Rev Microbiol. 2015;13:663–76.
Misasi J, Sullivan N. Camouflage and misdirection: the full-on assault of ebola virus disease. Cell. 2014;159:477–86.
Feldmann H, Geisbert TW. Ebola haemorrhagic fever. Lancet. 2011;377:849–62.
Malvy D, Mcelroy AK, Clerck HD, Gunther S, Griensven JV. Ebola virus disease [seminar]. Lancet. 2019;393:936–48.
Zoë M. The cost of Ebola. Lancet Glob Health. 2015;3:e423.
Chakraborty C. Therapeutics development for Ebola virus disease: a recent scenario. Curr Opin Pharmacol. 2021;60:208–15.
Tshiani Mbaya O, Mukumbayi P, Mulangu S. Review: insights on current FDA-approved monoclonal antibodies against Ebola virus infection. Front Immunol. 2021;12:721328.
Baseler L, Chertow DS, Johnson KM, Feldmann H, Morens DM. The pathogenesis of Ebola virus disease. Annu Rev Pathol. 2017;12:387–418.
Mühlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J Virol. 1999;73:2333–42.
Hartlieb B, Modrof J, Mühlberger E, Klenk HD, Becker S. Oligomerization of Ebola virus VP30 is essential for viral transcription and can be inhibited by a synthetic peptide. J Biol Chem. 2003;278:41830–6.
Sun W, Luan F, Wang J, Ma L, Li X, Yang G, et al. Structural insights into the interactions between lloviu virus VP30 and nucleoprotein. Biochem Biophys Res Commun. 2022;616:82–8.
Xu W, Luthra P, Wu C, Batra J, Leung DW, Basler CF, et al. Ebola virus VP30 and nucleoprotein interactions modulate viral RNA synthesis. Nat Commun. 2017;8:15576.
Biedenkopf N, Hartlieb B, Hoenen T, Becker S. Phosphorylation of ebola virus VP30 influences the composition of the viral nucleocapsid complex: Impact on viral transcription and replication. J Biol Chem. 2013;288:11165–74.
Groseth A, Charton JE, Sauerborn M, Feldmann F, Jones SM, Hoenen T, et al. The Ebola virus ribonucleoprotein complex: a novel VP30–L interaction identified. Virus Res. 2009;140:8–14.
Weik M, Modrof J, Klenk HD, Becker S, Mühlberger E. Ebola virus VP30-mediated transcription is regulated by RNA secondary structure formation. J Virol. 2002;76:8532–9.
Martinez MJ, Volchkova VA, Hervé R, Nathalie AD, Olivier R, Volchkov VE. Role of VP30 phosphorylation in the Ebola virus replication cycle. J Infect Dis. 2011;204:S934.
Martínez MJ, Biedenkopf N, Volchkova V, Hartlieb B, Alazard-Dany N, Reynard O, Becker S, Volchkov V. Role of Ebola virus VP30 in transcription reinitiation. J Virol. 2008;82:12569–73.
Brideau C, Gunter B, Pikounis B, Liaw A. Improved statistical methods for hit selection in high-throughput screening. J Biomol Screen. 2003;8:634.
Cui Q, Du R, Anantpadma M, Schafer A, Hou L, Tian J, et al. Identification of ellagic acid from plant Rhodiola rosea L. as an anti-ebola virus entry inhibitor. Viruses. 2018;10:152.
González-Fernández E, de-los-Santos-Álvarez N, Miranda-Ordieres AJ, Lobo-Castañón MJ. SPR evaluation of binding kinetics and affinity study of modified RNA aptamers towards small molecules. Talanta. 2012;99:767–73.
Tao W, Gan T, Guo M, Xu Y, Zhong J. Novel stable Ebola virus minigenome replicon reveals remarkable stability of the viral genome. J Virol. 2017;91:e01316–17.
Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23.
Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17:57–61.
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33.
Xu Y, Chen K, Pan J, Lei Y, Zhang D, Fang L, et al. Repurposing clinically approved drugs for COVID-19 treatment targeting SARS-CoV-2 papain-like protease. Int J Biol Macromol. 2021;188:137–46.
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61.
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–18.
Sousa da Silva AW, Vranken WF. ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res Notes. 2012;5:367.
Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–8.
He X, Man VH, Yang W, Lee TS, Wang J. A fast and high-quality charge model for the next generation general AMBER force field. J Chem Phys. 2020;153:114502.
Du Y. Fluorescence polarization assay to quantify protein-protein interactions in an HTS format. Methods Mol Biol. 2015;1278:529–44.
Lu H, Wang J, Wang Y, Qiao L, Zhou Y. Embelin and its role in chronic diseases. Adv Exp Med Biol. 2016;928:397–418.
Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc Natl Acad Sci USA. 2013;110:8182–7.
Shima F, Yoshikawa Y, Matsumoto S, Kataoka T. Discovery of small-molecule Ras inhibitors that display antitumor activity by interfering with Ras·GTP-effector interaction. Enzymes. 2013;34:1–23.
Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundbäck T, Nordlund P, et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9:2100–22.
Zhao Y, Ren J, Harlos K, Jones DM, Zeltina A, Bowden TA, et al. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature. 2016;535:169–72.
Ko JH, Lee SG, Yang WM, Um JY, Sethi G, Mishra S, et al. The application of embelin for cancer prevention and therapy. Molecules. 2018;23:621.
Prabhu KS, Achkar IW, Kuttikrishnan S, Akhtar S, Khan AQ, Siveen KS, et al. Embelin: a benzoquinone possesses therapeutic potential for the treatment of human cancer. Future Med Chem. 2018;10:961–76.
Xue Y, Zhang M, Liu M, Liu Y, Li L, Han X, et al. 8-Gingerol ameliorates myocardial fibrosis by attenuating reactive oxygen species, apoptosis, and autophagy via the PI3K/Akt/mTOR signaling pathway. Front Pharmacol. 2021;12:711701.
Hu SM, Yao XH, Hao YH, Pan AH, Zhou XW. 8‑Gingerol regulates colorectal cancer cell proliferation and migration through the EGFR/STAT/ERK pathway. Int J Oncol. 2020;56:390–7.
Brown CS, Lee MS, Leung DW, Wang T, Xu W, Luthra P, et al. In silico derived small molecules bind the filovirus VP35 protein and inhibit its polymerase cofactor activity. J Mol Biol. 2014;426:2045–58.
Xu W, Edwards MR, Borek DM, Feagins AR, Mittal A, Alinger JB, et al. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe. 2014;16:187–200.
Jin W, Stokes JM, Eastman RT, Itkin Z, Zakharov AV, Collins JJ, et al. Deep learning identifies synergistic drug combinations for treating COVID-19. Proc Natl Acad Sci USA. 2021;118:e2105070118.
Ianevski A, He L, Aittokallio T, Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics. 2017;33:2413–5.
Zheng S, Wang W, Aldahdooh J, Malyutina A, Shadbahr T, Tanoli Z, et al. SynergyFinder plus: toward better interpretation and annotation of drug combination screening datasets. Genomics Proteom Bioinform. 2022;20:587–96.
He L, Kulesskiy E, Saarela J, Turunen L, Wennerberg K, Aittokallio T, et al. Methods for high-throughput drug combination screening and synergy scoring. Methods Mol Biol. 2018;1711:351–98.
Mäkelä P, Zhang SM, Rudd SG. Drug synergy scoring using minimal dose response matrices. BMC Res Notes. 2021;14:27.
KalantarMotamedi Y, Choi RJ, Koh SB, Bramhall JL, Fan TP, Bender A. Prediction and identification of synergistic compound combinations against pancreatic cancer cells. iScience. 2021;24:103080.
Bijnsdorp IV, Giovannetti E, Peters GJ. Analysis of drug interactions. Methods Mol Biol. 2011;731:421–34.
Xu W, Wang Q, Yu F, Lu L, Jiang S. Synergistic effect resulting from combinations of a bifunctional HIV-1 antagonist with antiretroviral drugs. J Acquir Immune Defic Syndr. 2014;67:1–6.
Wang D, Yang Y, Wang Y, Proulle V, Andreasen PA, Hong W, et al. Embelin ameliorated sepsis-induced disseminated intravascular coagulation intensities by simultaneously suppressing inflammation and thrombosis. Biomed Pharmacother. 2020;130:110528.
Hoenen T. Minigenome systems for filoviruses. Methods Mol Biol. 2018;1604:237–45.
Edwards MR, Pietzsch C, Vausselin T, Shaw ML, Bukreyev A, Basler CF. High-throughput minigenome system for identifying small-molecule inhibitors of Ebola virus replication. ACS Infect Dis. 2015;1:380–7.
Watanabe S, Watanabe T, Noda T, Takada A, Feldmann H, Jasenosky LD, et al. Production of novel ebola virus-like particles from cDNAs: an alternative to ebola virus generation by reverse genetics. J Virol. 2004;78:999–1005.
Acknowledgements
We appreciate the grant support from the National Natural Science Foundation of China (82173865, 82130101), Foundation of Advanced Customer Cultivation Project of Wuhan National Biosafety Laboratory, Chinese Academy of Sciences, Shenzhen Bay Laboratory Open Fund (SZBL2021080601010) and Major Program of Shenzhen Bay Laboratory (S201101001). The support of the Shenzhen Bay supercomputing facility is also acknowledged.
Author information
Authors and Affiliations
Contributions
YHM cloned, expressed, and purified the proteins, screened the inhibitors, studied the mechanism, and wrote the draft. XH performed the docking and MD simulations. FW, XFX and RL studied the mechanism and edited the draft, J Zhong provided the material, YQZ, SWL, J Zhan and WX. supervised this project. All authors contributed to the writing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ma, Yh., Hong, X., Wu, F. et al. Inhibiting the transcription and replication of Ebola viruses by disrupting the nucleoprotein and VP30 protein interaction with small molecules. Acta Pharmacol Sin 44, 1487–1499 (2023). https://doi.org/10.1038/s41401-023-01055-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-023-01055-0
Keywords
This article is cited by
-
Navigating the Complex Landscape of Ebola Infection Treatment: A Review of Emerging Pharmacological Approaches
Infectious Diseases and Therapy (2024)
